
DNA repair diseases: What do they tell us about cancer and aging?
Carlos FM Menck and Veridiana Munford
Departamento de Microbiologia, Instituto de Ciências Biomédicas, Universidade de São Paulo, São Paulo,
SP, Brazil.
Abstract
The discovery of DNA repair defects in human syndromes, initially in xeroderma pigmentosum (XP) but later in many
others, led to striking observations on the association of molecular defects and patients’ clinical phenotypes. For ex
-
ample, patients with syndromes resulting from defective nucleotide excision repair (NER) or translesion synthesis
(TLS) present high levels of skin cancer in areas exposed to sunlight. However, some defects in NER also lead to
more severe symptoms, such as developmental and neurological impairment and signs of premature aging. Skin
cancer in XP patients is clearly associated with increased mutagenesis and genomic instability, reflecting the defec
-
tive repair of DNA lesions. By analogy, more severe symptoms observed in NER-defective patients have also been
associated with defective repair, likely involving cell death after transcription blockage of damaged templates. En
-
dogenously induced DNA lesions, particularly through oxidative stress, have been identified as responsible for these
severe pathologies. However, this association is not that clear and alternative explanations have been proposed. De
-
spite high levels of exposure to intense sunlight, patients from tropical countries receive little attention or care, which
likely also reflects the lack of understanding of how DNA damage causes cancer and premature aging.
Keywords: xeroderma pigmentosum, ultraviolet, DNA damage, oxidative stress, DNA repair.
Introduction
During evolution, the stability of genetic material
plays a fundamental role in the maintenance of life. The
equilibrium between the benefits of changes in the blue-
print of life, which facilitates diversity, and the perpetua
-
tion of genetic information is fundamental for evolution. In
multicellular organisms, this equilibrium faces other prob
-
lems, such as the transmission of genetic instability to new
generations of cells (gametic cells) and components of the
organism structure (somatic cells). For example, in hu
-
mans, genetic instability in somatic cells has been directly
associated with the formation of cancer, and cells may die
to avoid these deleterious effects. However, cell death may
trigger processes associated with organism aging.
The long DNA molecules, that compose genetic ma
-
terial, are not stable. Exogenous factors (such as ultraviolet,
UV, from daylight), chemical products (such as smoke,
pollution, and natural and artificial drugs), endogenous
agents, and normal byproducts of cell metabolism, includ
-
ing radical oxygen species (ROS) generated during respira
-
tion, constantly threaten DNA stability (Andressoo and
Hoeijmakers, 2005; Iyama and Wilson 3rd, 2013). These
agents damage the DNA molecule, and cells must attend to
these DNA lesions to properly survive and evolve. To
maintain the equilibrium necessary for survival and diver-
sity, evolution has equipped all living beings with the tools
to either remove or tolerate DNA damage. In general, these
tools, called DNA repair mechanisms or pathways, take
care of the DNA molecule, avoiding the deleterious conse
-
quences of the different types of DNA lesions. DNA repair
pathways are highly conserved in nature, indicating these
mechanisms are as old as life on Earth.
The importance of DNA repair mechanisms is dra
-
matically demonstrated in certain human syndromes that
are characterized as being defective in DNA repair pro
-
cesses, exacerbating the clinical effects of the unrepaired
DNA damage, such as xeroderma pigmentosum (XP).
Most XP patients are defective in nucleotide excision re
-
pair (NER), which removes DNA lesions induced by the
UV component of sunlight, although some XP patients
have also shown defects in the replication of UV-damaged
DNA. The main clinical symptoms of XP patients include
the high frequency of tumors in the exposed areas of skin,
indicating a clear causative relationship between
unrepaired DNA lesions, genetic instability (XP cells are
highly mutable) and cancer. Some XP patients, however,
present more severe clinical phenotypes, including devel
-
opmental problems, neurodegeneration and premature ag
-
ing (Lehmann, 2003; Lehmann et al., 2011; DiGiovanna
Genetics and Molecular Biology, 37, 1 (suppl), 220-233 (2014)
Copyright © 2014, Sociedade Brasileira de Genética. Printed in Brazil
www.sbg.org.br
Send correspondence to Carlos F.M. Menck. Departamento de
Microbiologia, Instituto de Ciências Biomédicas, Universidade de
São Paulo, Av. Prof. Lineu Prestes 1374, 05508-900 São Paulo,
SP, Brazil. E-mail: [email protected].
Review Article
and Kraemer, 2012). These severe symptoms have been
associated with other syndromes, such as Cockayne syn
-
drome (CS), that have also been shown to be defective in
NER (see below) (Cleaver et al., 2013). The reasons for
these more severe clinical aspects are still not clearly un
-
derstood, although, in general, these symptoms implicate
unrepaired DNA damage in the process of neurological
degeneration and aging. In this work, we discuss the his
-
torical data associated with the lack of DNA repair and its
deleterious consequences on the human organism, focus
-
ing on the scientific contribution of the findings in XP and
NER-related human diseases associated with cancer and
aging. Other human syndromes, such as ataxia telangiec
-
tasia, Fanconi Anemia, Werner syndrome, among others,
associated with defects in other DNA repair pathways or
DNA damage responses, also show symptoms associated
with cancer, aging and developmental and neurological
problems. These syndromes also support the relevance of
DNA damage in these processes and also indicate that dif
-
ferent DNA repair pathways correspond to networks that
protect the genome. However, these syndromes will not be
reviewed here, and the reader is invited to search for more
information in other recent reviews (Kim and D’Andrea,
2012; Knoch et al., 2012; Marinoglou, 2012; Moraes et
al., 2012; Shiloh and Ziv, 2013). Moreover, mouse mod-
els with defective DNA repair mimic some human clinical
phenotypes, including many aging symptoms. Indeed, an-
imal models have provided several clues and numerous
data to support this debate, but this information will only
be commented in this work when they are applied directly
to some aspects of DNA repair. For more information con-
cerning animal models, the reader is referred to recent re-
views (Diderich et al., 2011; Goss et al., 2011;
Niedernhofer et al., 2011; Gredilla et al., 2012; Jaarsma et
al., 2013).
Most studies concerning XP patients have been per
-
formed in developed countries (North America and Eu
-
rope) at high latitudes, with the exception of African-born
patients studied in Europe. The doses of UV in the sun
-
shine at high latitudes are low compared with tropical
countries, such as Brazil and other Latin American coun
-
tries. Thus, although exposed to a more aggressive envi
-
ronment, the patients in these countries have primarily re
-
mained unattended, except for a few recent studies. More
than just identifying gene mutations that affect XP pa
-
tients in tropical countries, these studies have contributed
to the understanding of how DNA repair defects correlate
with genotype-phenotype associations. A prospective ap
-
proach using next-generation sequencing to diagnose the
mutations of XP and NER-related patients may help these
individuals and their families to address the disease and
may eventually contribute with new data for understand
-
ing how DNA lesions lead to cancer, neurodegeneration
and/or aging.
XP and the Discovery of Molecular Defects in
DNA Repair
XP is a rare autosomal recessive disease; most pa
-
tients exhibit high sensitivity to sunlight. The patients pres
-
ent strong skin freckle-like pigmentation and lesions in
areas exposed to sunlight. These skin-aging symptoms may
also appear as precancerous skin lesions, such as actinic
keratosis, and most XP patients, when unprotected, develop
skin cancer (non melanoma and melanoma) before the age
of 10. Compared with the entire population, XP patients
have a 10,000-fold increase in the incidence of non-mela
-
noma skin tumors and a 2,000-fold increase in the inci
-
dence of melanoma before the age of 20 (Kraemer et al.,
1994). Certain exposed areas of the face, such as the eyes
and mouth, may also suffer from the sensitivity to sunlight
and develop lesions, including tumors. Many XP patients
present photophobia. The frequency of internal tumors also
shows a 10-fold increase in XP patients. Interestingly, 20 to
30% of XP patients develop progressive neurological prob
-
lems (DiGiovanna and Kraemer, 2012; Sepe et al., 2013).
The estimated incidence of XP in the population varies: 1
per 250,000 in USA (Robbins et al., 1974) and 1 per
450,000 in Western Europe (Kleijer et al., 2008). In some
places, such as Japan and North Africa, the incidence of XP
may be higher (1 per 20,000), likely reflecting the high
prevalence of certain mutations due to founder effects
(Hirai et al., 2006; Soufir et al., 2010).
XP was the first syndrome associated with a defect in
the DNA processing pathway. The dermatologists Moritz
Kaposi (Hungarian) and Ferdinand Ritter von Hebra (Aus-
trian) provided the first clinical description of XP patients
(Hebra and Kaposi, 1874). By the nineteenth century, the
syndrome was correctly considered as an inherited disease
and was associated with sunlight exposure. This condition
was referred to as xeroderma pigmentosum (for dry and
pigmented skin, from the Greek words xero and derma; and
Latin word pigmentosum). Subsequently, DeSanctis and
Cacchione (1932) described a more severe form of XP,
DeSanctis-Cacchione syndrome (DSC), which was associ
-
ated with neurological degeneration.
In 1968, James Cleaver identified the molecular de
-
fects associated with XP, showing that XP cells were hy
-
persensitive to UV light and unable to perform unscheduled
DNA synthesis (UDS) after UV irradiation (Cleaver,
1968). As described below, UDS is part of the NER path
-
way after the removal of DNA damage. This outstanding
discovery provided the first evidence that a cell defect in
DNA metabolism is associated with a clinical phenotype,
explaining, in part, the high photosensitivity of XP skin.
Soon, clever experiments, fusing cells from different
patients, facilitated the identification of complementation
groups (De Weerd-Kastelein et al., 1972). Specifically, if
the fusion of two different cells resulted in the restoration of
UDS, then the cells were deficient in different comple
-
Menck and Munford 221
mentation groups or genes. If the fusion did not restore
UDS, the cells were deficient in the same gene and be
-
longed to the same complementation group. These types of
experiments led to the classification of XP in seven differ
-
ent complementation groups (XP-A to XP-G), correspond
-
ing to seven genes with mutations that might result in XP.
Years later, the cells of some XP patients, however, were
demonstrated as NER proficient and capable of UDS. The
inability of these cells to replicate DNA after UV irradia
-
tion (daughter-strand repair) indicated other problems in
terms of dealing with DNA lesions, although these cells
maintained the ability to remove these lesions (Lehmann et
al., 1975). These cells were referred to as XP variants
(XP-V). The subsequent identification of DNA polymerase
activity associated with the XPV protein (DNA polymerase
eta) confirmed that XP-V patients are deficient in trans
-
lesion synthesis (TLS) (Kannouche and Stary, 2003;
Masutani et al., 2008).
The cells from all XP patients show an increased fre
-
quency of mutagenesis after exposure to UV light that is
correlated with molecular defects in DNA damage process
-
ing, leading to mutagenic consequences in the cells and in-
creased tumorigenesis in patients. Thus, these correlations
were the first clear demonstration that DNA damage and
mutations cause cancer (van Steeg and Kraemer, 1999;
Cleaver, 2000).
Other NER-Defective Genetic Disorders
As described above, approximately 20 to 30% of XP
patients also develop progressive neuronal impairment and
premature aging. The symptoms include hearing loss, swal-
lowing difficulties and mental retardation. Neuronal degen-
eration has been identified as the primary cause for these
clinical features; however, in some cases, severe develop
-
mental problems, such as growth retardation and micro
-
cephaly, have been observed. These more severe symptoms
are associated with the DSC as well as XP in combination
with Cockayne syndrome (CS), and this combined disorder
is referred to as XP/CS. Indeed, CS is a rare autosomal re
-
cessive disorder, first described in 1936 (Cockayne, 1936).
CS patients suffer from sun sensitivity and exhibit many
premature aging features, including cachectic dwarfism,
kyphosis, arrested sexual development, thin hair, subcuta
-
neous fat loss, osteoporosis and a characteristic “bird-like”
face (deep sunken eyes and prominent ears). The neurologi
-
cal symptoms include microcephaly, progressive hearing
and visual loss, progressive ataxia, delayed psychomotor
development, and mental retardation. These features reflect
dysmyelination and neurodegeneration, and the brain
shows prominent calcification (Iyama and Wilson 3rd,
2013; Jaarsma et al., 2013). Similar to XP cells, initial stud
-
ies with CS cells revealed hypersensitivity to UV irradia
-
tion, although normal levels of UDS and daughter strand re
-
pair were observed (Schmickel et al., 1977; Andrews et al.,
1978a). A few years later, the nature of the molecular repair
defect in CS cells was identified as a failure to recover RNA
synthesis after DNA damage induced through
UV-irradiation (Mayne and Lehmann, 1982), suggesting a
problem with the transcription of a damaged template. Im
-
portantly, this RNA synthesis impairment was used to iden
-
tify two different complementation groups for CS cells
(CS-A and CS-B) and an independent complementation
group in XP/CS cells (associated with the XP-B group)
with similar molecular defects (Lehmann, 1982). This im
-
portant discovery revealed two NER sub-pathways associ
-
ated with global genome repair (GGR) and RNA transcrip
-
tion (transcription coupled repair-TCR).
Subsequently, other genetic disorders were described
as the direct results of NER defects. In some cases, the pa
-
tients present neuronal and/or developmental problems,
such as trichothiodystrophy (TTD), where the patients
show features similar to CS (short stature, skeletal abnor
-
malities, impaired intelligence, decreased fertility, and
photosensitivity) and fragile and brittle hair due to the sul
-
fur-deficiency and reduced levels of cysteine-rich matrix
proteins (Stefanini et al., 2010). Cerebro-oculo-facio-ske
-
letal syndrome (COFS) is considered to be a severe form of
CS, with symptoms appearing at birth (Suzumura and Ari-
saka, 2010), and XPF-ERCC1 progeroid (XFE) syndrome
is associated with renal and liver abnormalities (Nieder-
nhofer et al., 2006). However, UV-sensitive syndrome
(UVSS) has been described as a mild genetic disorder lead-
ing to skin photosensitivity (Spivak, 2005; Oh and Spivak,
2010). Curiously, all these syndromes show skin photo-
sensitivity and defects impairing NER, but among their
clinical features, there has been no report of increased can-
cer frequency.
With the advent of DNA recombinant techniques in
the 1980s, many research groups have attempted to clone
the mutated genes in these NER-related diseases, although
this proved to be a difficult task because DNA transfection
in human cells was not trivial. In the 1990s, however, the
use of mutated rodent cells deficient for different DNA re
-
pair pathways facilitated the cloning of XP genes, contrib
-
uting to the understanding of the mechanisms of NER and
TLS, described in more detail below. As human genes
showed cross complementation with rodent cells, these
genes have been, in many cases, referred to as ERCC (for
excision cross-complementing) genes (Boulikas, 1996).
The human genes identified in NER-related diseases and
TLS, with their chromosomal locations and summarized
functions, are shown in Figure 1.
The XP Molecular Defects: Removing DNA
Damage via NER or Replicating Damaged DNA
through TLS
Classical XP (XP-A to XP-G) cells are defective in
the removal of DNA damage through NER, and XP-V cells
tolerate the lesions by replicating damaged templates
222 DNA repair avoids cancer and aging

through TLS. These mechanisms are schematically repre
-
sented in Figures 2 and 3.
Although NER was first described for the removal of
UV-induced lesions (such as cyclobutane pyrimidine di
-
mers, CPDs, and 6-4 pyrimidine-pyrimidone, 6-4PPs), this
pathway is recognized as a versatile and flexible mecha
-
nism. Specifically, NER removes structurally unrelated
bulky lesions, which promote DNA distortions in the dou
-
ble helix. These lesions include DNA adducts or intra-
strand crosslinks induced by chemical agents (such as
benzo(a)pyrene, a product of tobacco smoke, and cisplatin,
an anticancer chemotherapeutic drug) that react with DNA
or injuries induced by endogenous agents, such as lesions
generated by oxidation through ROS produced in the cells
as a potential byproduct of respiration. There are more than
30 proteins that act in a sequential and concerted manner to
remove DNA damage (Costa et al., 2003). Notably, defec
-
tive activity in seven of these proteins (from XPA to XPG)
may result in a clinical XP phenotype. Moreover, the defec
-
tive activity of CSA and CSB may result in severe CS
symptoms (Jaarsma et al., 2013). Other NER proteins have
also been recently described, where mutations cause NER
syndromes: TTDA/p8 (part of TFIIH complex, Giglia-Mari
et al., 2004), causing TTD; ERCC1 (partner of XPF endo
-
nuclease), causing XFE, CS and COFS (Suzumura and
Arisaka, 2010, Kashiyama et al., 2013); UVSSA causing
UVSS (Schwertman et al., 2012; Zhang et al., 2012).
NER can be summarized as having four sequential
steps, including i) lesion recognition, ii) unwinding the
damaged double helix, iii) cleavage and excision of the
damaged strand and iv) filling the gapped molecule, fol
-
lowed by DNA synthesis and final ligation (Figure 2). TLS
through XPV also facilitates tolerance of UV-induced
photoproducts, such as CPDs, and other types of DNA
damage induced through other DNA damaging agents (Fi
-
gure 3).
Two different sub-pathways have been implicated in
the recognition step of NER, depending on the location of
the lesion. The global genome repair (GGR) sub-pathway
involves the recognition of DNA lesions throughout the ge
-
nome, and it depends on the XPC/HR23B protein complex
as the primary DNA damage detector. For some types of le
-
Menck and Munford 223
Figure 1 - Human chromosomal location and general description of the genes encoding proteins involved in nucleotide excision repair (NER) and
translesion synthesis (TLS). Mutations in these genes cause human syndromes, listed in the table.
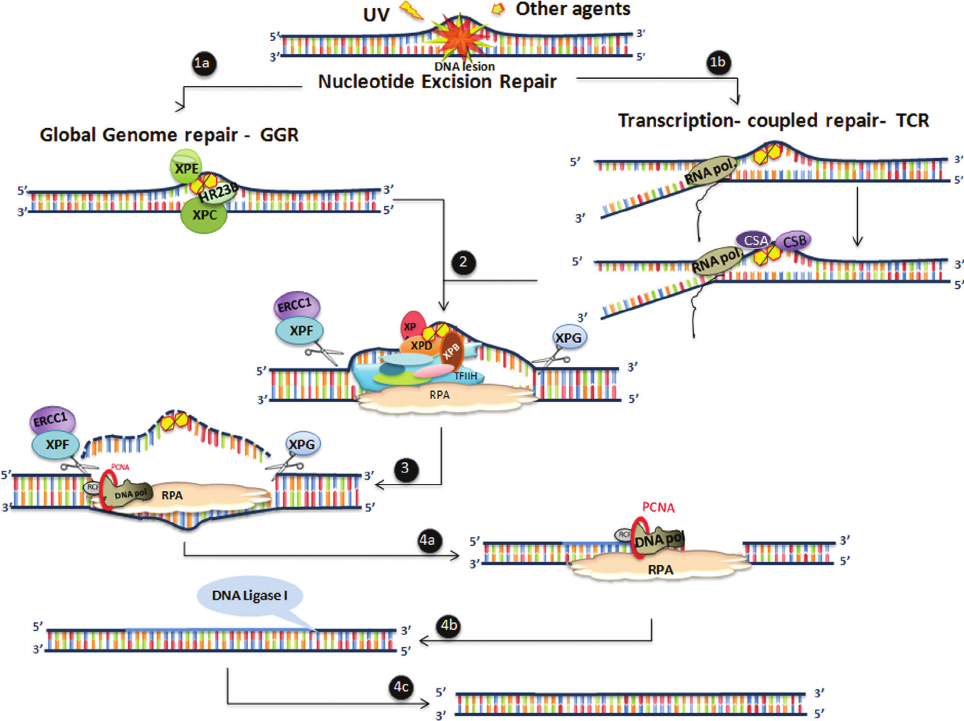
sions, such as CPDs, the damaged DNA binding (DDB)
complex (composed of two proteins, including XPE) is re
-
quired for improving the efficiency of damage recognition
and removal. The transcription-coupled repair (TCR) sub-
pathway acts on the transcribed strand of actively ex
-
pressed genes. The existence of this sub-pathway was first
observed when DNA damage was not homogeneously re
-
moved from the genome of mammalian cells, and the pref
-
erential repair of CPD lesions was detected in active genes
(Bohr et al., 1985). Subsequently, this preferential repair
system was demonstrated as being selective for the tran
-
scribed strand, implicating the transcription machinery in
the recognition of DNA damage (Mellon et al., 1987). Sub
-
sequent studies with CS cells demonstrated that these cells
were deficient in the preferential repair of active genes
(Mullenders et al., 1988; Venema et al., 1990). Indeed, the
CSA and CSB proteins are important players in the recog
-
nition of DNA lesions in the TCR pathway, triggered by the
blockage of RNA polymerase II during transcription.
The next step, common to both GGR and TCR sub-
pathways, is initiated through the recruitment of the tran
-
scription factor TFIIH, a multiprotein complex, which
includes XPB and XPD proteins, 3’-5’ and 5’-3’, respec
-
tively, ATP-dependent DNA helicases. This complex un
-
winds ~30 bp at the damaged DNA site, which is covered
by a single RPA (from Replication Protein A) complex (a
single-stranded DNA binding complex, composed of three
sub-unities). RPA and XPA proteins are likely involved in
the correct three-dimensional assembly of the NER ma
-
chinery prior to the endonucleolytic cleavage step. XPG
proteins and the XPF/ERCC1 complex are the main endo
-
nucleases that nick DNA at both sides, some nucleotides
away from the damage. Although XPG is required for
XPF/ERCC1 activity, this heterodimer makes the initial
5’-incision, followed by the XPG incision at the 3’ side of
the lesion (Fagbemi et al., 2011). The final step of NER is
the gap filling through repair synthesis. Replicative DNA
polymerases (Pol d and Pol e) use the 3’-hydroxyl extrem
-
ity, generated by the XPF/ERCC1 incision, as a primer to
synthesize DNA at the 30-nucleotide-gap. The resulting
nick at the 5’ end of the gap is sealed through DNA ligase I
224 DNA repair avoids cancer and aging
Figure 2 - Schematic representation of nucleotide excision repair (NER): the proteins associated with human diseases are highlighted. The downstream
NER pathway and each of the sub-pathways (GGR and TCR) for lesion recognition are indicated, showing the different steps for DNA lesion removal in
an error-free manner.
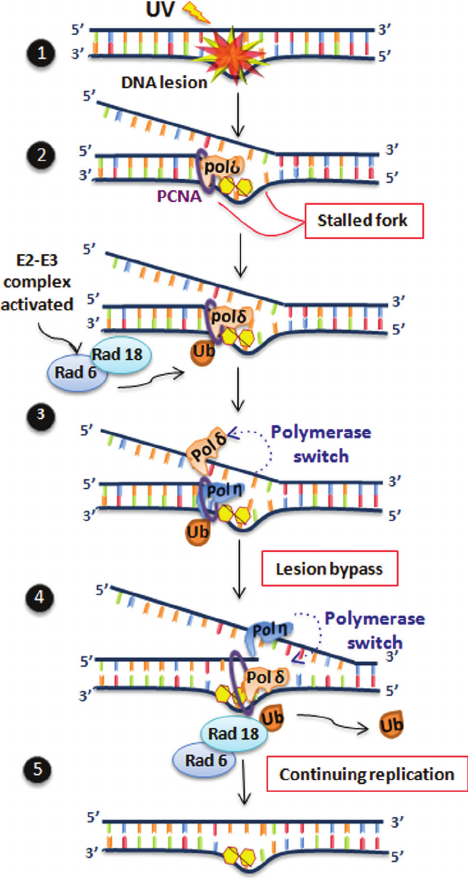
or III (Moser et al., 2007). Thus, normal NER restores the
original DNA molecule in an error-free manner.
The replication machinery may encounter unrepaired
DNA lesions, potentially resulting in a blockage in DNA
synthesis, as replicative DNA polymerases do not have the
capacity to use damaged templates. This important block
-
age may lead to the collapse of the replication fork, produc
-
tion of double-strand breaks, and, ultimately, cell death
(Batista et al., 2009). To tolerate DNA damage in replicat
-
ing templates, nature has evolved several alternative DNA
polymerases that replace the replicating polymerases (also
termed polymerase switching) and replicate the lesions
through TLS (Figure 3). Among the many alternative DNA
polymerases described during the last two decades (Sale et
al., 2012; Sale, 2013), XPV protein, or DNA polymerase
eta (Pol h), deserves special attention. Mutations in this
DNA polymerase have been implicated in the human syn
-
drome XP. The defective activity of this protein in XP-V
cells results in sensitive to UV-light or cisplatin treatment,
particularly when the cells are transformed with SV40 or
HPV, inhibiting the activity of p53 protein. These cells are
also highly mutable after UV-light, which explains the high
incidence of skin tumors in XP-V patients. Pol h bypasses
certain types of lesions, such as the CPDs induced through
UV-light, although the activity of this polymerase through
6-4PPs is controversial. The lack of Pol h leads to a strong
blockage of the replication forks in XP-V cells, but replica
-
tion can still recover through the action of other alternative
polymerases (Kannouche and Stary, 2003; Quinet et al.,
2014). Interestingly, Pol h is an error prone DNA polymer
-
ase, showing reduced replication fidelity. However, this
polymerase seems is capable of bypassing thymidine CPDs
correctly through the insertion of adenosines in the daugh-
ter strand. Thus, the increased mutagenesis observed in
UV-irradiated XP-V cells may reflect the activities of other
polymerases that replicate this lesion in an error prone man-
ner (Kannouche and Stary, 2003; Masutani et al., 2008).
DNA Damage and Repair
vs.
Cancer
Skin photosensitivity is a common clinical feature of
all patients showing defective NER responses. The in-
creased sensitivity to UV-light is also observed in cells
from XP, CS, TTD, or UVSS patients. Thus, the defective
removal of UV photoproducts likely results in this photo
-
sensitivity (Lehmann, 2003; Oh and Spivak, 2010). How
-
ever, although these syndromes reflect defects on the same
DNA processing pathway, apart from photosensitivity,
they are clinically highly heterogeneous, including increa
-
sed carcinogenesis, and more severe clinical features, such
as abnormal development, neurodegeneration and prema
-
ture aging. This clinical heterogeneity raises several unan
-
swered questions concerning how unrepaired DNA damage
leads to a plethora of different symptoms.
The highly increased mutagenesis induced by UV ir
-
radiation in XP cells is well correlated with the high levels
of skin cancer observed in XP patients for all XP comple
-
mentation groups, including NER-defective, and TLS-
defective (Figure 4). Indeed, UV-induced signature muta
-
tions were observed in specific tumor suppressor genes
(such as p53 and patched) in skin tumors from XP patients,
demonstrating the causal role of unrepaired lesions and mu
-
tations in the formation of tumors in these patients (Dumaz
et al., 1993; Bodak et al., 1999; D’Errico et al., 2000;
Giglia-Mari and Sarasin, 2003). Even internal tumors ob
-
served in XP patients carry mutations provoked by unre
-
Menck and Munford 225
Figure 3 - Schematic representation of translesion synthesis (TLS) by Pol
h. The replication fork is blocked by DNA damage, and after a series of
events (including the monoubiquitination of PCNA), a polymerase switch
provides DNA polymerase eta (XP-V) with an opportunity to bypass the
lesion. In this figure, only one strand (leading strand) is represented. At the
end of the process the lesion remains in the DNA, but the replication of this
molecule can proceed.
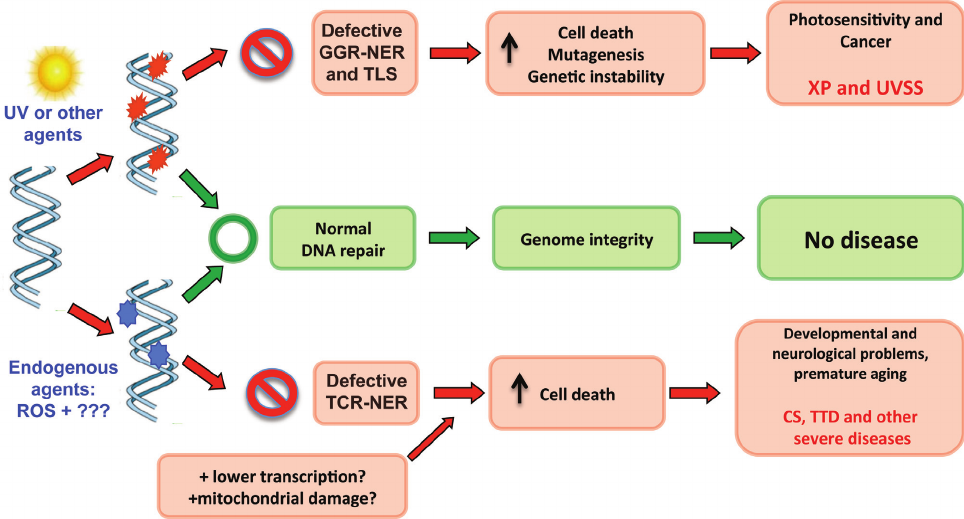
paired lesions, likely originating from oxidative stress
(Giglia et al., 1998).
Thus, there are clear indications that unrepaired DNA
damage induces mutations, and these mutations cause can
-
cer. Nevertheless, the lack of tumors in CS and TTD pa
-
tients is puzzling. Although the cells from CS patients are
defective in a sub-pathway of NER (TCR), the cells from
these patients (Parris and Kraemer, 1993) and rodent cells
with mutations in the CSB homolog (Vreeswijk et al.,
1998) show increased mutations after UV irradiation.
Moreover, mouse models defective for CSA and CSB show
skin cancer predisposition after UVB irradiation (van der
Horst et al., 1997, 2002), unlike their human counterparts.
As GGR is more efficient in human cells compared with ro
-
dent cells (Hanawalt, 2001), this sub-pathway could protect
CS patients from developing skin tumors. Alternatively, it
has been proposed that CS cells have a strong signal for cell
death after DNA damage, likely reflecting higher levels of
transcription blockage (McKay et al., 2001), which may
have a reverse effect on cancer development, as dead cells
cannot induce cancer. However, XP-A cells are also highly
sensitive to DNA lesions, but XP-A patients always de
-
velop skin tumors. Notably, the lifetime of severely af
-
fected CS, TTD, COFS and XFE patients is typically short
and may require living indoors (due to severe clinical fea
-
tures), reducing their exposure to sunlight and the
probability of developing tumors.
Until recently, most mutation studies have investi
-
gated base substitution and small point mutations, deletions
and insertions, although certainly larger changes in the
DNA sequence, including gross rearrangements, copy
number variations and chromosomal anomalies and other
types of genomic instabilities, are affected by unrepaired
DNA damage, representing known alterations in human
cancer (Amiel et al., 2004). Moreover, even with clear evi
-
dence of sunlight being responsible for skin cancer, many
questions remain concerning what is the part of the spec
-
trum acting in tumorigenesis, either in XP patients or the
normal population. Although the results strongly suggest a
role for CPDs, the main lesion induced by UVB (280-320
nm), in skin carcinogenesis, this type of lesion is also gen
-
erated after UVA (320-400 nm) irradiation, and other le
-
sions induced by this spectrum of sunlight should also be
considered (Douki et al., 2003; Cadet et al., 2012; Schuch
et al., 2013). Indeed, a recent study implicated an important
role for both CPDs and 6-4PPs in the biological conse
-
quences of UVA light in XP cells (Cortat et al., 2013). Skin
tumors are the most frequent types of cancer in human pop
-
ulations, particularly in countries near the tropics; thus, a
detailed understanding of how these cancers are induced
226 DNA repair avoids cancer and aging
Figure 4 - The consequences of DNA damage and how defective NER and TLS may explain certain clinical phenotypes. Different types of DNA damage
can be induced by a variety of endogenous and exogenous agents. NER removes several of these lesions and TLS helps to replicate them, aiming mainte-
nance of the genome stability, and guaranteeing normal cell proliferation, and normal human life (represented in green). When these mechanisms fail,er-
rors in DNA replication increase mutagenesis and genetic instability, resulting in higher photosensitivity (observed in UVSS and XP patients) and risk of
cancer development (observed in XP patients). Defective GGR-NER and TLS better explain these phenotypes (in red, superior part of the figure). Alter-
natively, endogenous DNA damage (including those induced by ROS) may disturb the transcription and/or cause replication blockage, leading to cell
death, thus defective TCR-NER better explain the more severe phenotypes (including premature aging) of XP/CS, CS and other syndromes. Other effects,
such as impaired transcription or damaged mitochondria may also be involved in these phenotypes (in red lower part of the figure).
through exposure to sunlight is highly relevant to the gen
-
eral public health. Further investigations with XP patients
and XP cells can provide important contribution to this
field.
Unrepaired DNA Damage and the Aging
Process
Concerning other symptoms, such as developmental
problems, neurological abnormalities and premature aging,
observed in more severely affected XP patients and other
NER-related syndromes (such as CS, COFS, TTD and
XFE), the causal effects remain far from being understood.
This puzzling question was first raised in the 1970s, when
the UV-survival of XP cells from several complementation
groups were compared to understand the neurological ab
-
normalities (Andrews et al., 1976, 1978b; Kraemer et al.,
1976). In general, the cells from patients with more severe
symptoms were more UV sensitive than those with no neu
-
rological abnormalities. Similar results were soon de
-
scribed for CS-derived cells, indicating the sensitivity of
these cells to UV damage (Andrews et al., 1978a). Ulti-
mately, it was concluded that extremely inefficient DNA
repair could not protect these neurons (or supporting glial
cells) from DNA damage in some of the patients, and the
neurons would die prematurely.
Interestingly, in the first reports, XP-C patients were
already noticed as less affected by the severe symptoms. In-
deed, XP patients mutated in the XPC, XPE (both partici-
pate in the initial steps of GGR pathway of NER, see
Figure 2) and XPV (TLS, Figure 3) genes present symp-
toms associated with skin lesions, but not affecting the ner-
vous system. This observation indicates that defects in the
GGR sub-pathway of NER and TLS through Pol h could
explain the increased tumorigenesis observed in the skin of
these patients, which prevents mutagenesis, as described
above. Thus, patients with mutations in these genes and
other genes (XPA, XPB, XPD, XPF and XPG) that partici
-
pate downstream in the GGR sub-pathway may present
only XP phenotypes associated with skin lesions, which
may be avoided by preventing sun exposure.
On the other hand, TCR impairment is clearly associ
-
ated with more severe symptoms in NER-related syndro
-
mes (Figure 4). The high levels of cell death of TCR
impaired cells in response to DNA damage, likely triggered
by a blockage in RNA transcription, have been associated
with the severe symptoms in CS patients (McKay et al.,
2001). A similar explanation may also be associated with
XP/CS, COFS, TTD, and XFE syndromes. However, the
nature of the DNA damage resulting in this increased cell
death remains unknown. Endogenously induced lesions
generated by free radicals, including ROS, are the strongest
candidates for these effects. Indeed, CS-B cells have been
demonstrated as deficient in the repair of DNA damage
generated through oxidative stress under many circum
-
stances. Initially, CSB was shown to be important for the
removal of 8-oxoguanine (8-oxoG), a known lesion pro
-
duced by oxidative stress, in human cells irradiated with
ionizing radiation (Tuo et al., 2001, 2003). Subsequently,
this effect was also described for CS-A human primary
fibroblasts (D’Errico et al., 2007). Mouse models defective
in CSB protein were shown to accumulate oxidized purine
modifications (8-oxoG and (5’S)-8,5’-cyclo 2’-deoxyade
-
nosine) in certain tissues with age (Osterod et al., 2002;
Kirkali et al., 2009). More recently, CS-A and CS-B pri
-
mary human cells and CS-B reprogrammed iPS cells were
shown to have altered metabolic redox balance, with in
-
creased levels of intracellular ROS and basal (and induced)
levels of DNA damage (Andrade et al., 2012; Pascucci et
al., 2012).
Consistent with this model, CS-A and CS-B cells are
more sensitive to hydrogen peroxide treatment compared
with cells derived from UVSS patients, who present only
photosensitivity symptoms. Host cell reactivation after the
transfection of damaged plasmids in human cells yields
similar results. This approach measures the expression of a
reporter gene, and this expression is impaired after DNA
damage, which is recovered through DNA repair in the
transfected cells. Although both CS-B and UVSS cells are
defective in the recovery of UV-irradiated plasmids, only
CS-B cells recover gene expression through plasmids car-
rying oxidized base damage (Spivak and Hanawalt, 2006).
Moreover, a UVSS patient was demonstrated to have a spe-
cific mutation in the CS-A gene, and cells derived from this
patient were highly sensitive to UV irradiation; however, in
contrast to CS-A cells from CS patients, these UVSS cells
were not sensitive to potassium bromate or other agents that
generate oxidative stress (Nardo et al., 2009). Similar re
-
sults were obtained in XP patients carrying missense muta
-
tions in the XPG gene. Specifically, the cells from mildly
affected patients (clinically showing only photosensitivity)
were sensitive to UV-light but not to the treatment of
photoactivated methylene blue (which generates singlet ox
-
ygen), in contrast to XP-G/CS cells that were sensitive to
this oxidative stress. Host cell reactivation also confirmed
that the XPG alleles containing missense mutations fully
reactivated the expression of reporter genes from plasmids
damaged with photoactivated methylene blue in XP-G/CS
deficient cells, while only partial reactivation was obtained
in UV-irradiated plasmids (Soltys et al., 2013).
Thus, defective transcriptional repair of DNA dam
-
age generated through oxidative stress is associated with
more severe clinical features, such as developmental and
neuronal abnormalities and premature aging, while the de
-
fective repair of UV induced DNA damage is associated
with photosensitivity phenotypes (Andressoo and Hoeijm
-
akers, 2005). However, other published data challenge this
model: XPC protein has also been implicated in the repair
of DNA damage induced through oxidative stress, although
XP-C patients present only clinical features associated with
Menck and Munford 227
skin photosensitivity, with no symptoms involving neuro
-
logical or developmental problems. Human XP-C primary
keratinocytes and fibroblasts are hypersensitive to treat
-
ment with oxidizing agents and accumulate oxidized DNA
damage after oxidant exposure (D’Errico et al., 2006). A
similar defect was shown in the host cell reactivation of
photoactivated methylene blue damaged adenovirus vec
-
tors (Kassam and Rainbow, 2007). The participation of
both XPA and XPC proteins in the repair of photoactivated
methylene blue-induced DNA damage was also investi
-
gated in cells treated with this agent or in the host cell reac
-
tivation of damaged plasmids. The results suggest that the
NER pathway directly participates in the repair of DNA le
-
sions generated through oxidative stress (Berra et al.,
2013). However, live imaging data indicates that proteins
that initiate NER (XPC and CSB) are rapidly recruited to
sites of the cell nuclei containing oxidized base damage,
with the absence of the detectable recruitment of other
downstream NER factors. These results indicate that the
binding of XPC and CSB to the sites of DNA damage is in
-
dependent on a further NER reaction (Menoni et al., 2012).
Therefore, as the participation of XPC protein in cell
protection from oxidative stress is confirmed, the lack of
oxidative stress-mediated DNA damage repair may be nec-
essary but not sufficient to explain the severe pathology of
neurological and developmental symptoms and the acceler-
ated aging observed in NER-related syndromes. Further de-
fects are needed to construct these severe symptoms, and
many possibilities are being investigated. Thus, two poten-
tial hypotheses emerge that may help to understand and
eventually answer this question: mitochondrial DNA dam-
age and general deficiency in transcription.
Indeed, the participation of NER factors in mitochon
-
dria is gaining support. CS-B protein was shown to stimu
-
late the repair of 8-oxoG in the mitochondrial DNA
(Stevnsner et al., 2002, Aamann et al., 2010). Moreover,
CS-B cells are deficient in autophagy, leading to increased
oxidative stress and the accumulation of damaged mito
-
chondria. Interestingly, these effects could be reversed
through the addition of autophagy stimulators, such as lith
-
ium chloride and rapamycin, to the cell culture medium
(Scheibye-Knudsen et al., 2012). Moreover, the participa
-
tion of many of the proteins in TCR in the global transcrip
-
tion machinery may also be important. A general reduction
in transcription through RNA polymerase II has been ob
-
served in CSB cells (Balajee et al., 1997). The participation
of CSB in transcription elongation and TFIIH as an initia
-
tion transcription factor should be considered for these
“transcription syndromes” (Compe and Egly, 2012;
Velez-Cruz and Egly, 2013). The need for the repair of
DNA damage to achieve the effects observed in these pa
-
thologies has also been questioned, as the defects on tran
-
scription through RNA polymerases I and II may solely
explain the severe symptoms of CS patients (Brooks,
2013).
Some patient cases still add more confusion to the ge
-
netic basis of carcinogenesis and neurological abnormali
-
ties resulting from NER defects. Although CS patients do
not present skin tumors, at least one case of siblings show
-
ing a severe XP phenotype (diagnosed as the DeSanctis-
Cacchione variant of XP) were identified with a missense
mutation in the CSB gene (Colella et al., 2000). In contrast,
a UVSS patient has been demonstrated as completely null
for this same CSB gene (Horibata et al., 2004). The under
-
standing of the molecular basis underlying these clinical
phenotypes may help to understand the intricate role of
DNA damage and repair in the human pathology of cancer
and aging.
XP Under the Tropical Sun
Although sunlight is much more intense at low lati
-
tudes, with higher intensity of UVB light and more severe
DNA lesions (Schuch et al., 2012), most studies with XP
patients has been performed in Europe, USA and Japan, un
-
der high latitudes. In tropical and poorer countries, these
patients are typically, and unfortunately, unattended. Diffi
-
culties in diagnosis, including the lack of information from
local medical dermatologists and complications in obtain-
ing appropriate rigorous photoprotection, are just some of
the problems faced by these patients and families.
Most of the reports of gene identification and atten-
tion to XP families in these countries depend on studies per-
formed in USA or Europe. These studies showed interest-
ing data of genetic clusters, such as the one identified in
Guatemala for an isolated community, where 12 children
carried a new mutation in the XPC gene, likely reflecting
consanguineous marriage (Cleaver et al., 2007). Another
genetic cluster was also recently described in patients from
a French Comorian Island near the Indian Ocean in West
Africa. Among the black Mayotte population, more than 32
XP patients carried a mutation in the XPC gene (Cartault et
al., 2011), likely resulting from a founder effect from the
African continent. Although black skin protects these pa
-
tients, these individuals still suffer from the heavy sunlight
intensity, with the development of skin tumors and early
ocular injuries. Another important finding was the preva
-
lence of a specific mutation (c.1643_1644delTG,
p.Val548AlafsX25) in the XPC gene in North Africa. This
mutation was observed in 87% of the XP-C patients in this
location, primarily in unrelated families, reflecting a
founder mutation aging 50 generations or 1250 years (Sou
-
fir et al., 2010). It is curious how this deleterious mutation
can still be observed, despite a strong negative selection,
especially in these sunny environments. Certainly, the mu
-
tated allele is kept in heterozygote individuals, as they pres
-
ent normal repair phenotype.
Recently, evidence has been obtained from the direct
assistance of patients in the tropical countries, showing
sporadic cases (Halpern et al., 2008) and more complete
studies in the genetic identification of mutations (mostly
228 DNA repair avoids cancer and aging
novel) in XPC (Leite et al., 2009), XPV (Ortega-Recalde et
al., 2013) and XPG (Soltys et al., 2013) genes. One of the
mutations described for the XPC gene in Brazil (Leite et al.,
2009) is the same as the highly prevalent mutation reported
in North Africa (Soufir et al., 2010), indicating the potential
migration from Africa to Europe and subsequently to Brazil
(likely from Portugal). The two novel XPG missense muta
-
tions observed in the two siblings in Brazil were shown to
produce proteins with low repair of UV-photoproducts, but
normal repair of lesions generated through oxidative stress,
likely explaining the mild phenotype of these patients (Sol
-
tys et al., 2013). Moreover, a pioneering effort for the sys
-
tematic identification of mutations that affect XPC and
XPA genes has recently been underway in Brazilian pa
-
tients, and these results will contribute to both the assis
-
tance to these patients and the scientific knowledge on the
distribution of these pathological mutations (K. Santiago
and MI Achatz, unpublished data). Interestingly, among
those XP patients some have a Native American Indian ori
-
gin, as the XP community described in Guatemala (Cleaver
et al., 2007) and the XP severely affected children belonging
to the Navajo Reservation in Southwest of USA
(http://globalgenes.org/xeroderma-pigmentosum-suddenly-
arises-on-navajo-reservation/). Most likely, these will not be
the only cases for Native Americans, suggesting mutated al
-
leles of XP genes, causing the disease, have possibly existed
before the human migration to the Americas, from Asia.
Moreover, another genetic cluster has been reported
in the Brazilian state of Goias (a sunny central area of this
country), with more than 20 affected XP patients diagnosed
in an isolated community (Chaibub, 2011). Genetic identi
-
fication of the mutation is underway, and preliminary data
indicate that the XPV gene is affected in this population
(unpublished results).
Conclusions and Perspectives
The wide spectrum of symptoms and diseases caused
by NER deficiency reflects the relevance of this repair
Menck and Munford 229
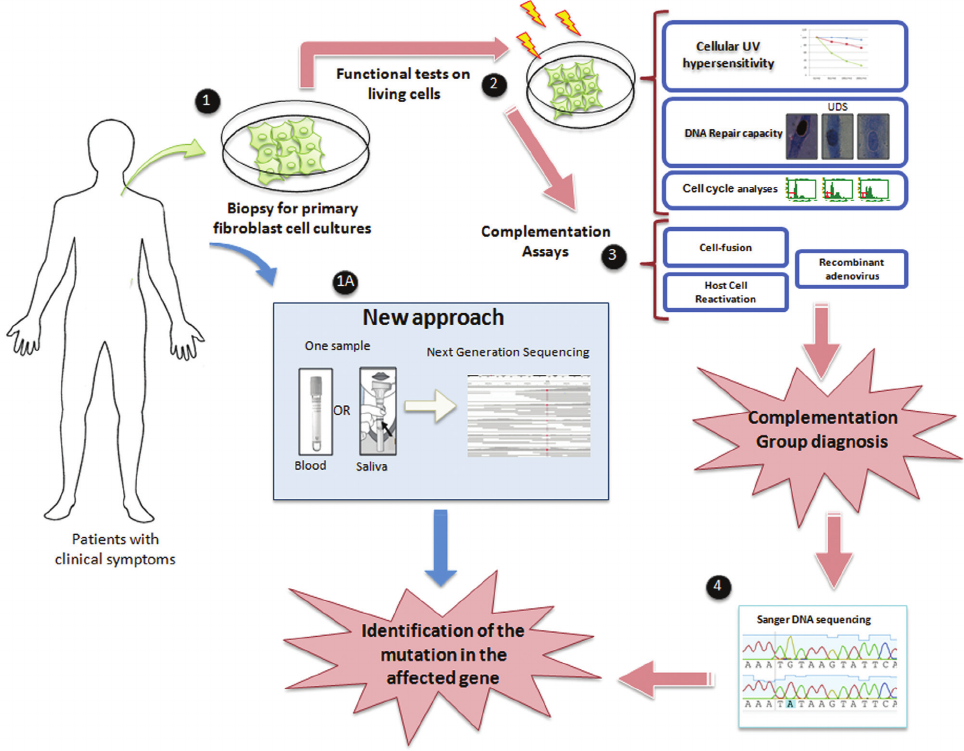
Figure 5 - Strategies for molecular diagnosis of XP patients. Identification of XP gene mutation based on a series of biological and biochemical assays us
-
ing primary cells obtained from patient skin biopsies. Although the information from these cells has always been rich, the direct sequencing of DNA re
-
pair genes using next generation sequencing techniques may facilitate the molecular diagnosis using blood or saliva samples.
pathway in the maintenance of genome stability in humans.
Since 1968, the molecular defects affecting XP patients and
other syndromes have been described. Studies with cells
from these patients have contributed to our current under
-
standing of the causes of cancer and the development of
tools to fight tumors (Moraes et al., 2012). We have also
learned a great deal concerning aging, which might im
-
prove the health and quality of life of the elderly. Thus, the
human society is indebted to NER-defective patients, al
-
though little can really be done to help them. Gene therapy
for these patients is at its infancy (Magnaldo and Sarasin,
2004; Menck et al., 2007), and not even the simple identifi
-
cation of the genetic mutation is provided in many coun
-
tries worldwide, particularly in developing countries. This
identification could, at least, provide some prognosis for
the disease and support for genetic counseling for the af
-
fected families. Until recently, there was a strong need for
biological essays for the identification of the genetic com
-
plementation group in XP patients, requiring skin biopsies,
which are painful for the patient, and cell culture, which is
time consuming and requires specific skills. The direct se
-
quencing of exomes (Ortega-Recalde et al., 2013) or spe-
cific genes using next generation sequencing techniques of
DNA isolated directly from saliva samples provides an ex-
cellent opportunity for the rapid diagnosis of these muta-
tions (Figure 5). Thus, new horizons may bring hope for the
XP patients and their families.
Acknowledgments
This work was financially supported through grants
from the Fundação de Amparo à Pesquisa do Estado de São
Paulo (FAPESP) and Conselho Nacional de Desenvol
-
vimento Científico e Tecnológico (CNPq).
References
Aamann MD, Sorensen MM, Hvitby C, Berquist BR, Muftuoglu
M, Tian J, de Souza-Pinto NC, Scheibye-Knudsen M, Wil
-
son 3rd DM, Stevnsner T, et al. (2010) Cockayne syndrome
group B protein promotes mitochondrial DNA stability by
supporting the DNA repair association with the mitochon
-
drial membrane. FASEB J 24:2334-2346.
Amiel A, Peretz G, Slor H, Weinstein G and Fejgin MD (2004)
Molecular cytogenetic parameters in fibroblasts from pa
-
tients and carriers of xeroderma pigmentosum. Cancer
Genet Cytogenet 149:154-160.
Andrade LN, Nathanson JL, Yeo GW, Menck CF and Muotri AR
(2012) Evidence for premature aging due to oxidative stress
in iPSCs from Cockayne syndrome. Hum Mol Genet
21:3825-3834.
Andressoo JO and Hoeijmakers JH (2005) Transcription-coupled
repair and premature ageing. Mutat Res 577:179-194.
Andrews AD, Barrett SF and Robbins JH (1976) Relation of DNA
repair processes to pathological ageing of the nervous sys
-
tem in xeroderma pigmentosum. Lancet 19:1318-1320.
Andrews AD, Barrett SF, Yoder FW and Robbins JH (1978a)
Cockayne’s syndrome fibroblasts have increased sensitivity
to ultraviolet light but normal rates of unscheduled DNA
synthesis. J Invest Dermatol 70:237-239.
Andrews AD, Barrett SF and Robbins JH (1978b) Xeroderma
pigmentosum neurological abnormalities correlate with col
-
ony-forming ability after ultraviolet radiation. Proc Natl
Acad Sci USA 75:1984-1988.
Balajee AS, May A, Dianov GL, Friedberg EC and Bohr VA
(1997) Reduced RNA polymerase II transcription in intact
and permeabilized Cockayne syndrome group B cells. Proc
Natl Acad Sci USA 94:4306-4311.
Batista LF, Kaina B, Meneghini R and Menck CF (2009) How
DNA lesions are turned into powerful killing structures: In
-
sights from UV-induced apoptosis. Mutat Res 681:197-208.
Berra CM, Oliveira CS, Garcia CCM, Rocha CRR, Lerner LK,
Andrade LCL, Baptista MS and Menck CFM (2013) Nucle
-
otide excision repair activity on DNA damage induced by
photoactivated methylene blue. Free Radic Biol Med
61C:343-356.
Bodak N, Queille S, Avril MF, Bouadjar B, Drougard C, Sarasin
A and Daya-Grosjean L (1999) High levels of patched gene
mutations in basal-cell carcinomas from patients with xero
-
derma pigmentosum. Proc Natl Acad Sci USA 96:5117-
5122.
Bohr VA, Smith CA, Okumoto DS and Hanawalt PC (1985) DNA
repair in an active gene: Removal of pyrimidine dimers from
the DHFR gene of CHO cells is much more efficient than in
the genome overall. Cell 40:359-369.
Boulikas T (1996) Xeroderma pigmentosum and molecular clon-
ing of DNA repair genes. Anticancer Res 16:693-708.
Brooks PJ (2013) Blinded by the UV light: How the focus on tran-
scription-coupled NER has distracted from understanding
the mechanisms of Cockayne syndrome neurologic disease.
DNA Repair 12:656-671.
Cadet J, Mouret S, Ravanat JL and Douki T (2012) Photoinduced
damage to cellular DNA: Direct and photosensitized reac-
tions. Photochem Photobiol 88:1048-1065.
Cartault F, Nava C, Malbrunot AC, Munier P, Hebert JC, N’guyen
P, Djeridi N, Pariaud P, Pariaud J, Dupuy A, et al. (2011) A
new XPC gene splicing mutation has lead to the highest
worldwide prevalence of xeroderma pigmentosum in black
Mahori patients. DNA Repair 10:577-585.
Chaibub SCW (2011) High incidence of xeroderma pigmentosum
in a countryside community in the state of Goiás, Brazil.
Surg Cosmet Dermatol 3:112-115.
Cleaver JE (1968) Defective repair replication of DNA in xero
-
derma pigmentosum. Nature 218:652-656.
Cleaver JE (2000) Common pathways for ultraviolet skin carcino
-
genesis in the repair and replication defective groups of
xeroderma pigmentosum. J Dermatol Sci 23:1-11.
Cleaver JE, Feeney L, Tang JY and Tuttle P (2007) Xeroderma
pigmentosum group C in an isolated region of Guatemala. J
Invest Dermatol 127:493-496.
Cleaver JE, Bezrookove V, Revet I and Huang EJ (2013) Concep
-
tual developments in the causes of Cockayne syndrome.
Mech Ageing 134:284-290.
Cockayne EA (1936) Dwarfism with retinal atrophy and deafness.
Arch Dis Child 11:1-8.
Colella S, Nardo T, Botta E, Lehmann AR and Stefanini M (2000)
Identical mutations in the CSB gene associated with either
Cockayne syndrome or the DeSanctis-cacchione variant of
xeroderma pigmentosum. Hum Mol Genet 9:1171-1175.
230 DNA repair avoids cancer and aging
Compe E and Egly JM (2012) TFIIH: When transcription met
DNA repair. Nat Rev Mol Cell Biol 13:343-354.
Cortat B, Garcia CMC, Quinet A, Schuch AP, de Lima-Bessa KM
and Menck CF (2013) The relative roles of DNA damage in
-
duced by UVA irradiation in human cells. Photochem
Photobiol Sci 12:1483-1495.
Costa RM, Chiganças V, Galhardo R da S, Carvalho H and Menck
CF (2003) The eukaryotic nucleotide excision repair path
-
way. Biochimie 85:1083-1099.
De Weerd-Kastelein EA, Keijzer W and Bootsma D (1972) Ge
-
netic heterogeneity of xeroderma pigmentosum demon
-
strated by somatic cell hybridization. Nat New Biol 238:80-
83.
D’Errico M, Calcagnile A, Canzona F, Didona B, Posteraro P,
Cavalieri R, Corona R, Vorechovsky I, Nardo T, Stefanini
M, et al. (2000) UV mutation signature in tumor suppressor
genes involved in skin carcinogenesis in xeroderma pig
-
mentosum patients. Oncogene 19:463-467.
D’Errico M, Parlanti E, Teson M, de Jesus BM, Degan P, Cal
-
cagnile A, Jaruga P, Bjørås M, Crescenzi M, Pedrini AM, et
al. (2006) New functions of XPC in the protection of human
skin cells from oxidative damage. EMBO J 25:4305-4315.
D’Errico M, Parlanti E, Teson M, Degan P, Lemma T, Calcagnile
A, Iavarone I, Jaruga P, Ropolo M, Pedrini AM, et al. (2007)
The role of CSA in the response to oxidative DNA damage
in human cells. Oncogene 26:4336-4343.
Diderich K, Alanazi M and Hoeijmakers JH (2011) Premature ag-
ing and cancer in nucleotide excision repair-disorders. DNA
Repair 10:772-780.
DiGiovanna JJ and Kraemer KH (2012) Shining a light on xero-
derma pigmentosum. J Invest Dermatol 132:785-796.
Douki T, Reynaud-Angelin A, Cadet J and Sage E (2003) Bipy-
rimidine photoproducts rather than oxidative lesions are the
main type of DNA damage involved in the genotoxic effect
of solar UVA radiation. Biochemistry 42:9221-9226.
DeSanctis C and Cacchione AI (1932) Idiozia xerodermia. Riv
Sper Freniat 56:269-292.
Dumaz N, Drougard C, Sarasin A and Daya-Grosjean L (1993)
Specific UV-induced mutation spectrum in the p53 gene of
skin tumors from DNA-repair-deficient xeroderma pigmen
-
tosum patients. Proc Natl Acad Sci USA 90:10529-10533.
Fagbemi AF, Orelli B and Schärer OD (2011) Regulation of
endonuclease activity in human nucleotide excision repair.
DNA Repair 10:722-729.
Giglia G, Dumaz N, Drougard C, Avril MF, Daya-Grosjean L and
Sarasin A (1998) p53 mutations in skin and internal tumors
of xeroderma pigmentosum patients belonging to the com
-
plementation group C. Cancer Res 58:4402-4409.
Giglia-Mari G, Coin F, Ranish JA, Hoogstraten D, Theil A,
Wijgers N, Jaspers NG, Raams A, Argentini M, van der
Spek PJ, et al. (2004) A new, tenth subunit of TFIIH is re
-
sponsible for the DNA repair syndrome trichothiodystrophy
group A. Nat Genet 36:714-719.
Giglia-Mari G and Sarasin A (2003) TP53 mutations in human
skin cancers. Hum Mutat 21:217-228.
Goss JR, Stolz DB, Robinson AR, Zhang M, Arbujas N, Robbins
PD, Glorioso JC and Niedernhofer LJ (2011) Premature ag
-
ing-related peripheral neuropathy in a mouse model of pro
-
geria. Mech Ageing Dev 132:437-442.
Gredilla R, Garm C and Stevnsner T (2012) Nuclear and mito
-
chondrial DNA repair in selected eukaryotic aging model
systems. Oxid Med Cell Longev 2012:282438.
Halpern J, Hopping B and Brostoff JM (2008) Photosensitivity,
corneal scarring and developmental delay: Xeroderma pig
-
mentosum in a tropical country. Cases J 1:254.
Hanawalt PC (2001) Revisiting the rodent repairadox. Environ
Mol Mutagen 38:89-96.
Hebra F and Kaposi M (1874) On diseases of the skin including
exanthemata, volume III. The New Sydenham Society
61:252-258.
Hirai Y, Kodama Y, Moriwaki S, Noda A, Cullings HM, Macphee
DG, Kodama K, Mabuchi K, Kraemer KH, Land CE, et al.
(2006) Heterozygous individuals bearing a founder muta
-
tion in the XPA DNA repair gene comprise nearly 1% of the
Japanese population. Mutat Res 601:171-178.
Horibata K, Iwamoto Y, Kuraoka I, Jaspers NG, Kurimasa A,
Oshimura M, Ichihashi M and Tanaka K (2004) Complete
absence of Cockayne syndrome group B gene product gives
rise to UV-sensitive syndrome but not Cockayne syndrome.
Proc Natl Acad Sci USA 101:15410-15415.
Iyama T and Wilson 3rd DM (2013) DNA repair mechanisms in
dividing and non-dividing cells. DNA Repair 12:620-636.
Jaarsma D, van der Pluijm I, van der Horst GT and Hoeijmakers
JH (2013) Cockayne syndrome pathogenesis: Lessons from
mouse models. Mech Ageing Dev 134:180-195.
Kannouche P and Stary A (2003) Xeroderma pigmentosum vari-
ant and error-prone DNA polymerases. Biochimie
85:1123-1132.
Kassam SN and Rainbow AJ (2007) Deficient base excision re-
pair of oxidative DNA damage induced by methylene blue
plus visible light in xeroderma pigmentosum group C fibro-
blasts. Biochem Biophys Res Commun 359:1004-1009.
Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki
K, Fawcett H, Wing JF, Lewin SO, Carr L, et al.
(2013) Mal-
function of nuclease ERCC1-XPF results in diverse clinical
manifestations and causes Cockayne syndrome, xeroderma
pigmentosum, and Fanconi anemia. Am J Hum Genet
92:807-819.
Kim H and D’Andrea AD (2012) Regulation of DNA cross-link
repair by the Fanconi anemia /BRCA pathway. Genes Dev
26:1393-1408.
Kleijer WJ, Laugel V, Berneburg M, Nardo T, Fawcett H,
Gratchev A, Jaspers NG, Sarasin A, Stefanini M and
Lehmann AR (2008) Incidence of DNA repair deficiency
disorders in western Europe: Xeroderma pigmentosum,
Cockayne syndrome and trichothiodystrophy. DNA Repair
7:744-750.
Knoch J, Kamenisch Y, Kubisch C and Berneburg M (2012) Rare
hereditary diseases with defects in DNA-repair. Eur J Der
-
matol 22:443-455.
Kirkali G, de Souza-Pinto NC, Jaruga P, Bohr VA and Dizdaroglu
M (2009) Accumulation of (5’S)-8,5’-cyclo-2’-deoxyade
-
nosine in organs of Cockayne syndrome complementation
group B gene knockout mice. DNA Repair 8:274-278.
Kraemer KH, Andrews AD, Barrett SF and Robbins JH (1976)
Colony-forming ability of ultraviolet-irradiated xeroderma
pigmentosum fibroblasts from different DNA repair com
-
plementation groups. Biochim Biophys Acta 442:147-153.
Kraemer KH, Lee MM, Andrews AD and Lambert WC (1994)
The role of sunlight and DNA repair in melanoma and
Menck and Munford 231
nonmelanoma skin cancer. The xeroderma pigmentosum
paradigm. Arch Dermatol 130:1018-1021.
Lehmann AR (1982) Three complementation groups in Cockayne
syndrome. Mutat Res 106:347-356.
Lehmann AR (2003) DNA repair-deficient diseases, xeroderma
pigmentosum, Cockayne syndrome and trichothiodys
-
trophy. Biochimie 85:1101-1111.
Lehmann AR, Kirk-Bell S, Arlett CF, Paterson MC, Lohman PH,
de Weerd-Kastelein EA and Bootsma D (1975) Xeroderma
pigmentosum cells with normal levels of excision repair
have a defect in DNA synthesis after UV-irradiation. Proc
Natl Acad Sci USA 72:219-223.
Lehmann AR, McGibbon D and Stefanini M (2011) Xeroderma
pigmentosum. Orphanet J Rare Dis 6:70.
Leite RA, Marchetto MC, Muotri AR, Vasconcelos D de M, de
Oliveira ZN, Machado MCR and Menck CF (2009) Identifi
-
cation of XP complementation groups by recombinant ade
-
novirus carrying DNA repair genes. J Invest Dermatol
129:502-506.
Magnaldo T and Sarasin A (2004) Xeroderma pigmentosum:
From symptoms and genetics to gene-based skin therapy.
Cells Tissues Organs 177:189-198.
Masutani C, Hanaoka F and Ahmad SI (2008) Xeroderma pig
-
mentosum variant, XP-V: Its product and biological roles.
Adv Exp Med Biol 637:93-102.
Menck CF, Armelini MG and Lima-Bessa KM (2007) On the
search for skin gene therapy strategies of xeroderma pig-
mentosum disease. Curr Gene Ther 7:163-174.
Marinoglou K (2012) The role of the DNA damage response
kinase ataxia telangiectasia mutated in neuroprotection.
Yale J Biol Med 85:469-480.
Mayne LV and Lehmann AR (1982) Failure of RNA synthesis to
recover after UV irradiation: An early defect in cells from
individuals with Cockayne’s syndrome and xeroderma pig-
mentosum. Cancer Res 42:1473-1478.
McKay BC, Becerril C and Ljungman M (2001) P53 plays a pro
-
tective role against UV-and cisplatin-induced apoptosis in
transcription-coupled repair proficient fibroblasts. Onco
-
gene 20:6805-6808.
Mellon I, Spivak G and Hanawalt PC (1987) Selective removal of
transcription-blocking DNA damage from the transcribed
strand of the mammalian DHFR gene. Cell 51:241-249.
Menoni H, Hoeijmakers JH and Vermeulen W (2012) Nucleotide
excision repair-initiating proteins bind to oxidative DNA le
-
sions in vivo. J Cell Biol 199:1037-1046.
Moraes MC, Neto JB and Menck CF (2012) DNA repair mecha
-
nisms protect our genome from carcinogenesis. Front Biosci
17:1362-1388.
Moser J, Kool H, Giakzidis I, Caldecott K, Mullenders LH and
Fousteri MI (2007) Sealing of chromosomal DNA nicks
during nucleotide excision repair requires XRCC1 and DNA
ligase III alpha in a cell-cycle-specific manner. Mol Cell
27:311-323.
Mullenders LH, van Kesteren van Leeuwen AC, van Zeeland AA
and Natarajan AT (1988) Nuclear matrix associated DNA is
preferentially repaired in normal human fibroblasts, ex
-
posed to a low dose of ultraviolet light but not in Cockayne’s
syndrome fibroblasts. Nucleic Acids Res 16:10607-10622.
Nardo T, Oneda R, Spivak G, Vaz B, Mortier L, Thomas P, Orioli
D, Laugel V, Stary A, Hanawalt PC, et al. (2009) A UV-
sensitive syndrome patient with a specific CSA mutation re
-
veals separable roles for CSA in response to UV and oxida
-
tive DNA damage. Proc Natl Acad Sci USA 106:6209-6214.
Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR,
Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van
Leeuwen W, et al. (2006) A new progeroid syndrome re
-
veals that genotoxic stress suppresses the somatotroph axis.
Nature 444:1038-1043.
Niedernhofer LJ, Bohr VA, Sander M and Kraemer KH (2011)
Xeroderma pigmentosum and other diseases of human pre
-
mature aging and DNA repair: Molecules to patients. Mech
Ageing Dev 132:340-347.
Oh DH and Spivak G (2010) Hereditary photodermatoses. Adv
Exp Med Biol 685:95-105.
Ortega-Recalde O, Vergara JI, Fonseca DJ, Ríos X, Mosquera H,
Bermúdez OM, Medina CL, Vargas CI, Pallares AE, Res
-
trepo CM and Laissue P (2013) Whole-exome sequencing
enables rapid determination of xeroderma pigmentosum
molecular etiology. PLoS One 8:e64692.
Osterod M, Larsen E, Le Page F, Hengstler JG, Van Der Horst
GT, Boiteux S, Klungland A and Epe B (2002) A global
DNA repair mechanism involving the Cockayne syndrome
B (CSB) gene product can prevent the in vivo accumulation
of endogenous oxidative DNA base damage. Oncogene
21:8232-8239.
Parris CN and Kraemer KH (1993) Ultraviolet-induced mutations
in Cockayne syndrome cells are primarily caused by cyclo-
butane dimer photoproducts while repair of other photo-
products is normal. Proc Natl Acad Sci USA 90:7260-7264.
Pascucci B, Lemma T, Iorio E, Giovannini S, Vaz B, Iavarone I,
Calcagnile A, Narciso L, Degan P, Podo F, et al. (2012) An
altered redox balance mediates the hypersensitivity of
Cockayne syndrome primary fibroblasts to oxidative stress.
Aging Cell 11:520-529.
Quinet A, Vessoni AT, Rocha CR, Gottifredi V, Biard D, Sarasin
A, Menck CF and Stary A (2014) Gap-filling and bypass at
the replication fork are both active mechanisms for tolerance
of low-dose ultraviolet-induced DNA damage in the human
genome. DNA Repair 14:27-38.
Robbins JH, Kraemer KH, Lutzner MA, Festoff BW and Coon
HG (1974) Xeroderma pigmentosum: An inherited disease
with sun-sensitivity, multiple cutaneous neoplasms, and ab
-
normal DNA repair. Ann Internal Med 80:221-248.
Sale JE, Lehmann AR and Woodgate R (2012) Y-family DNA
polymerases and their role in tolerance of cellular DNA
damage. Nat Rev Mol Cell Biol 13:141-152.
Sale JE (2013) Translesion DNA synthesis and mutagenesis in
eukaryotes. Cold Spring Harb Perspect Biol 5:a012708.
Scheibye-Knudsen M, Ramamoorthy M, Sykora P, Maynard S,
Lin PC, Minor RK, Wilson 3rd DM, Cooper M, Spencer R,
de Cabo R, et al. (2012) Cockayne syndrome group B pro
-
tein prevents the accumulation of damaged mitochondria by
promoting mitochondrial autophagy. J Exp Med 209:855-
869.
Schmickel RD, Chu EH, Trosko JE and Chang CC (1977)
Cockayne syndrome: A cellular sensitivity to ultraviolet
light. Pediatrics 60:135-139.
Schuch AP, Yagura T, Makita K, Yamamoto H, Schuch NJ,
Agnez-Lima LF, Macmahon RM and Menck CF (2012)
DNA damage profiles induced by sunlight at different lati
-
tudes. Environ Mol Mutagen 53:98-206.
232 DNA repair avoids cancer and aging
Schuch AP, Garcia CCM, Makita K and Menck CF (2013) DNA
damage as a biological sensor for environmental sunlight.
Photochem Photobiol Sci 12:1259-1272.
Schwertman P, Lagarou A, Dekkers DH, Raams A, van der Hoek
AC, Laffeber C, Hoeijmakers JH, Demmers JA, Fousteri M,
Vermeulen W, et al. (2012) UV-sensitive syndrome protein
UVSSA recruits USP7 to regulate transcription-coupled re
-
pair. Nat Genet 44:598-602.
Sepe S, Payan-Gomez C, Milanese C, Hoeijmakers JH and Mas
-
troberardino PG (2013) Nucleotide excision repair in
chronic neurodegenerative diseases. DNA Repair 12:568-
577.
Shiloh Y and Ziv Y (2013) The ATM protein kinase: Regulating
the cellular response to genotoxic stress, and more. Nat Rev
Mol Cell Biol 14:197-210.
Soltys DT, Rocha CR, Lerner LK, de Souza TA, Munford V,
Cabral F, Nardo T, Stefanini M, Sarasin A, Cabral-Neto JB,
et al. (2013) Novel XPG (ERCC5) mutations affect DNA re
-
pair and cell survival after ultraviolet but not oxidative
stress. Hum Mutat 34:481-489.
Soufir N, Ged C, Bourillon A, Austerlitz F, Chemin C, Stary A,
Armier J, Pham D, Khadir K, Roume J, et al. (2010) A prev
-
alent mutation with founder effect in xeroderma pigmen
-
tosum group C from north Africa. J Invest Dermatol
130:1537-1542.
Spivak G (2005) UV-sensitive syndrome. Mutat Res 577:162-
169.
Spivak G and Hanawalt PC (2006) Host cell reactivation of
plasmids containing oxidative DNA lesions is defective in
Cockayne syndrome but normal in UV-sensitive syndrome
fibroblasts. DNA Repair 5:13-22.
Stefanini M, Botta E, Lanzafame M and Orioli D (2010) Tricho-
thiodystrophy: From basic mechanisms to clinical implica-
tions. DNA Repair 9:2-10.
Stevnsner T, Nyaga S, de Souza-Pinto NC, van der Horst GT,
Gorgels TG, Hogue BA, Thorslund T and Bohr VA (2002)
Mitochondrial repair of 8-oxoguanine is deficient in
Cockayne syndrome group B. Oncogene 21:8675-8682.
Suzumura H and Arisaka O (2010) Cerebro-oculo-facio-skeletal
syndrome. Adv Exp Med Biol 685:210-214.
Tuo J, Müftüoglu M, Chen C, Jaruga P, Selzer RR, Brosh Jr RM,
Rodriguez H, Dizdaroglu M and Bohr VA (2001) The
Cockayne Syndrome group B gene product is involved in
general genome base excision repair of 8-hydroxyguanine in
DNA. J Biol Chem 276:45772-45779.
Tuo J, Jaruga P, Rodriguez H, Bohr VA and Dizdaroglu M (2003)
Primary fibroblasts of Cockayne syndrome patients are de
-
fective in cellular repair of 8-hydroxyguanine and 8-hydro
-
xyadenine resulting from oxidative stress. FASEB J
17:668-674.
van der Horst GT, van Steeg H, Berg RJ, van Gool AJ, de Wit J,
Weeda G, Morreau H, Beems RB, van Kreijl CF, de Gruijl
FR, et al. (1997) Defective transcription-coupled repair in
Cockayne syndrome B mice is associated with skin cancer
predisposition. Cell 89:425-435.
van der Horst GT, Meira L, Gorgels TG, de Wit J, Velasco-Miguel
S, Richardson JA, Kamp Y, Vreeswijk MP, Smit B,
Bootsma D, et al. (2002) UVB radiation-induced cancer pre
-
disposition in Cockayne syndrome group A (CSA) mutant
mice. DNA Repair 1:143-157.
van Steeg H and Kraemer KH (1999) Xeroderma pigmentosum
and the role of UV-induced DNA damage in skin cancer.
Mol Med Today 52:86-94.
Vélez-Cruz R and Egly JM (2013) Cockayne syndrome group B
(CSB) protein: At the crossroads of transcriptional net
-
works. Mech Ageing Dev 134:234-242.
Venema J, Mullenders LH, Natarajan AT, van Zeeland AA and
Mayne LV (1990) The genetic defect in Cockayne syn-
drome is associated with a defect in repair of UV-induced
DNA damage in transcriptionally active DNA. Proc Natl
Acad Sci USA 87:4707-4711.
Vreeswijk MP, Overkamp MW, Westland BE, van Hees-
Stuivenberg S, Vrieling H, Zdzienicka MZ, van Zeeland AA
and Mullenders LH (1998) Enhanced UV-induced mutagen-
esis in the UV61 cell line, the Chinese hamster homologue
of Cockayne’s syndrome B, is associated with defective
transcription coupled repair of cyclobutane pyrimidine
dimers. Mutat Res 409:49-56.
Zhang X, Horibata K, Saijo M, Ishigami C, Ukai A, Kanno S,
Tahara H, Neilan EG, Honma M, Nohmi T, et al. (2012)
Mutations in UVSSA cause UV-sensitive syndrome and
destabilize ERCC6 in transcription-coupled DNA repair.
Nat Genet 44:593-597.
License information: This is an open-access article distributed under the terms of the
Creative Commons Attribution License, which permits unrestricted use, distribution, and
reproduction in any medium, provided the original work is properly cited.
Menck and Munford 233
