Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
Guidance on the Manufacture of Sterile Pharmaceutical
Products by Aseptic Processing
Task Force
on
Sterile Pharmaceutical Products by Aseptic Processing
With the support of a Grant for Research on Regulatory Science of
Pharmaceuticals and Medical Devices from Ministry of Health, Labour and
Welfare of Japan

Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
Chairred by:
Masashi Muroi Musashino University
Tsuguo Sasaki Pharmaceuticals and Medical Devices Agency
Collaborating researchers:
Yoshimi Urayama Chiyoda Corporation
Hirohito Katayama Bayer Yakuhin, Ltd.
Mamoru Kokubo Shibuya Kogyo Co., Ltd.
Yoshiaki Kogure Towa Pharmaceutical Co., Ltd.
Kazuyuki Kobayashi Eli Lilly Japan K.K.
Yuko Sasaki National Institute of Infectious Diseases
Osamu Shirokizawa Pharma Solutions Co., Ltd.
Mitsuhiro Takahashi Astellas Pharma Tech Co., Ltd.
Nobuo Tateishi Chugai Pharmaceutical Co., Ltd.
Toshikazu Tani C&S Corporation
Takahiro Naitou Shionogi & Co., Ltd
Toshiaki Nishihata Santen Pharmaceutical Co., Ltd.
Yoshiaki Hara Sartorius Stedim Japan K.K.
Toshikazu Harada Santen Pharmaceutical Co., Ltd.
Tsutomu Hinomoto Santen Pharmaceutical Co., Ltd.
Naoki Hirashima Takeda Pharmaceutical Co., Ltd.
Junji Magata Millipore Corporation
Daikichiro Murakami Taikisha Co., Ltd.
Pharmaceuticals and Medical Devices Agency: Hiroshi Kato, Yukio Saito, Singo Sakurai,
Shogo Suzuki, Yutaka Sumi, and Ryoko Naruse
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
[Members of first edition (July 2006)]
Kenichi Tanamoto (chair), Tsuguo Sasaki (co-chair), Kunio Kawamura (co-chair), Yoshimi Urayama,
Mamoru Kokubo, Yoshiaki Kogure, Satoru Sasaki, Yuko Sasaki, Osamu Shirokizawa, Shinji Sugaya,
Toshikazu Tani, Toshiaki Nishihata, Yoshiaki Hara, Tsutomu Hinomoto, Hiroyuki Fujita, Junji
Magata, Taiichi Mizuta, and Daikichiro Murakami
Contributors to the original version:
Research Supported by 2005 Health and Labor Science Grant from the Ministry of Health, Labour
and Welfare of Japan (Research on Regulatory Science of Pharmaceutical and Medical Devices)
Research on the Introduction of International Standards on Aseptic Drug Manufacturing into Japan
Notice: This English version of the Guidance on Sterile Pharmaceutical Products Produced by Aseptic
Processing is prepared for the convenience of users unfamiliar with the Japanese language. When and if
any discrepancy arises between the Japanese original and its English translation, the former is authentic.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- i -
Table of Contents
1. INTRODUCTION ............................................................................................................. 1
2. GLOSSARY ...................................................................................................................... 1
3. QUALITY SYSTEM ......................................................................................................... 5
4. PERSONNEL ................................................................................................................... 9
5. PREVENTION OF CONTAMINATION BY PERSONNEL........................................... 12
6. BUILDINGS AND FACILITIES .................................................................................... 14
7. PROCESSING AREAS FOR STERILE PHARMACEUTICAL PRODUCTS ................. 17
8. CLEANING AND DISINFECTION OF PROCESSING AREAS FOR STERILE
PHARMACEUTICAL PRODUCT MANUFACTURING ....................................................... 22
9. CONTROL OF RAW MATERIALS, CONTAINERS, AND CLOSURES ........................ 25
10. STORAGE AND TRANSPORT OF STERILE INTERMEDIATE PRODUCTS ......... 27
11. ENVIRONMENTAL MONITORING .......................................................................... 30
12. QUALIFICATION OF EQUIPMENT AND UTILITIES ............................................ 36
13. STERILIZATION PROCESS ...................................................................................... 40
14. CLEAN-IN-PLACE SYSTEM ..................................................................................... 48
15. STERILIZATION-IN-PLACE SYSTEM ..................................................................... 50
16. ASEPTIC FILLING PROCESSES ............................................................................. 53
17. FILTRATION STERILIZATION PROCESSES .......................................................... 56
18. FREEZE-DRYING PROCESS .................................................................................... 61
19. ISOLATOR SYSTEM, BARRIER SYSTEM, AND BLOW-FILL SEAL ...................... 65
20. PROCESS SIMULATION........................................................................................... 75
ANNEXES ............................................................................................................................. 79
A1. ACTIVE PHARMACEUTICAL INGREDIENTS (APIS) MANUFACTURED VIA
CELL CULTURE/FERMENTATION.................................................................................... 79
A2 PHARMACEUTICAL WATER ................................................................................... 82
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- ii -
A3 PEST CONTROL OF ASEPTIC MANUFACTURING FACILITIES ......................... 94
A4 BIOSAFETY AND BIOSECURITY MEASURES ...................................................... 97
A5 CHEMICAL HAZARD CONTROL ........................................................................... 103
A6 TESTS AND INSPECTIONS ................................................................................... 107
B REVISION RECORDS ................................................................................................. 111
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 1 -
1. Introduction
This guidance document describes the current basic concepts on sterility assurance and
procedures for manufacturing and controlling sterile pharmaceutical products in order to advise
manufacturers of sterile pharmaceutical products and regulatory personnel responsible for
pharmaceutical inspections on sterility assurance.
This guidance is intended to be applied in the aseptic processing of parenteral drugs; however,
its basic concepts may also be useful when manufacturing ophthalmic solutions and other sterile
pharmaceutical products. The concepts and descriptions contained in this guidance may be
superseded by other processes or procedures of manufacture that are justifiably comparable or more
stringent (except for the Ministerial Ordinance, “Regulations for Manufacturing Control and Quality
Control of Medicinal Products and Quasi-Medicinal Products” [“GMP regulations,” Ordinance No.
179, 2004], and other regulatory requirements, notifications, and issues), as long as the quality of
pharmaceutical products can be ensured.
2. Glossary
2.1 Air lock: A small room that is generally composed of interlocked doors, constructed to
maintain air pressure control between adjoining rooms. The intent of an aseptic processing
airlock is to preclude ingress of particulate matter and microorganism contamination from a
lesser controlled area. The air balance for the bio-safety facility should be established and
maintained to ensure that airflow is from areas of least- to greater contamination.
2.2 Action level: Established criteria of microbial or airborne particle level that, when exceeded,
should trigger appropriate investigation and corrective action based on the investigation.
2.3 Air cleanliness level: A quality which indicates the condition of cleanliness of a monitored
item, expressed as number of particles larger than 0.5 µm permitted per m
3
. It is classified in
grades A, B, C, and D according to the required particulate number in the air.
2.4 Alert level: Established criteria of microbial or airborne particle level (and microbial species
if necessary) giving early warning of potential drift from normal conditions.
2.5 Aseptic filling: A Part of aseptic processing where sterilized products are filled and/or
packaged into sterile containers and closed under Grade A area.
2.6 Aseptic processing: A method of producing sterile products in which sterile bulk product or
sterile raw materials are compounded and filled into sterile containers in a controlled
environment, in which the air supply, materials, equipment and personnel are regulated to
control microbial and particulate contamination to acceptable levels.
2.7 Aseptic processing area (APA): Controlled environments, in which the air supply, materials,
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 2 -
equipment and personnel are regulated to control microbial and particulate number to
acceptable levels. APA is consisted of “critical (processing) area” and “direct support area.”
2.8 Barrier: A physical partition to protect direct intervention of operating personnel in a
controlled environment.
2.9 Bioburden: Population of viable microorganisms which may be present in non-sterile drugs
or materials including intermediate products and raw materials.
2.10 Biological indicator (BI): Microbiological test system providing defined resistance to a
specified sterilization process under defined conditions to be used as an indicator for the
sterilization cycle efficacy.
2.11 Change control system: A formal system planned and designed to assess all changes that
might affect the quality of pharmaceutical product to be intended to ensure the maintenance of
process control
2.12 Chemical indicator (CI): Test system that reveals change in one or more process variables
based on a chemical or physical change resulting from exposure to a sterilization process.
2.13 Clean area: An area maintained and controlled to prevent contamination of pharmaceutical
products with microorganisms or foreign substances, in compliance with defined particle and
microbiological cleanliness standards. For the purposes of this document, this term is
synonymous with manufacturing area for aseptic products.
2.14 Colony forming unit (CFU): Visible growth of microorganisms arising from a single cell or
multiple cells.
2.15 Critical area: A limited processing area where sterilized containers, raw materials,
intermediate products or the surface of equipment that comes into contact with sterilized
product is exposed to environment. This area is also known as the “critical processing area.”
The level of environmental cleanliness of this area is commonly referred to as Grade A.
2.16 Critical processing: A process that can affect one or more critical quality attributes of a
pharmaceutical product.
2.17 Culture conditions: Stated combination of conditions, including the type of medium and the
period and temperature of incubation, used to promote microbiological growth.
2.18 Decontamination: A process that reduces or removes contaminating substances to a defined
acceptance level using a reproducible method.
2.19 Design qualification (DQ): Documented verification that the proposed design of the facilities,
equipment, or systems is suitable for the intended purpose.
2.20 Direct support area: A background area directly supporting the critical area. Sterilized
products are not directly exposed to the environment in this area. This quality of the
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 3 -
environment is commonly referred to as Grade B.
2.21 Disinfection: A process by which environmental or equipment bioburden is reduced to a safe
level or eliminated.
2.22 D value: A value indicating the extinct rate of microorganism. The time or radiation dosage
required to achieve inactivation of 90% of a population (one tenth of the survival rate) of the
test microorganism under stated exposure conditions.
2.23 Endotoxin: Lipopolysaccharide constituting of outer membrane of Gram negative bacteria
and may have pyrogenic reactions and other biological activities to humans.
2.24 Environmental monitoring program: A system to plan, organize and implement all the
activities to achieve and maintain the required levels of air and surface cleanliness in the
manufacturing areas. The intent is to manufacture aseptic pharmaceutical products in high
quality level, by foreseeing deterioration of environments in manufacturing areas, preventing
bad influence to the quality of products, and performing appropriate cleanliness control
through a proper monitoring of the manufacturing environment.
2.25 Heating ventilation and air condition (HVAC) system: An air handling system including
heating, ventilation, and air conditioning.
2.26 High efficiency particulate air (HEPA) filter: Air filters designed to retain particulates of
larger than a certain size with defined efficiency. The filter retaines particles of ≥ 0.3 µm size
with a minimum efficiency of 99.97%.
2.27 Indirect supporting area: An area where containers, raw materials, and unsterilized
intermediate products are exposed to the environment and where materials and equipment
used for aseptic processing are cleaned.
2.28 Installation qualification (IQ): Documented verification that the equipment or systems, as
installed or modified, comply with the approved design, the manufacturer’s recommendations
and/or user requirements.
2.29 Integrity test for containers: Test for confirming container’s closure integrity as a part of
stability testing for sterile products until the use.
2.30 Integrity test for filter: A non-destructive test which is used to predict the functional
performance of a filter.
2.31 Isolator: A sealed and sterilized enclosure capable of preventing ingress of contaminants by
means of total physical separation of enclosure to the surrounding exterior environment, An
isolator’s air supply is filtered using HEPA or ULPA grade filters.
2.32 Gas filter: Hydrophobic filters equipped in compressed air pipe lines for the porpose of
removing microorganisms and particulates from gases.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 4 -
2.33 Leak test: A test performed to verify that air leak from equipment/ devices and the container
closure system that require to maintain sealing performance remains within the specified
limits.
2.34 Material safety data sheet (MSDS): A specific document that shows important physical and
chemical characteristics of a chemical or product to alert a user, transporter or other interested
party to potential safety hazards that may be associated with the material. An MSDS is a legal
requirement under “Pollutant Release and Transfer Register” for all aspects of commerce
involving chemicals designated in the ordinance as Class I Designated Chemical Substances,
Class II Designated Chemical Substances and products containing these substances.
2.35 Microorganism: General term for bacteria, fungi, protozoa and virus. Microorganism
indicates only bacteria and fungi in this text.
2.36 Operational qualification (OQ): Documented verification that the equipment or systems, as
installed or modified, perform as intended throughout the anticipated operating ranges.
2.37 Overkill sterilization: A process which is sufficient to provide at least a 12 log reduction of
microorganisms having a minimum D value of 1.0 minute, regardless of bioburden count in
the product being sterilized or the resistance of the objective microorganisms to the
sterilization.
2.38 Performance qualification (PQ): Documented verification that the equipment and ancillary
systems, as when operating together, can perform effectively and reproducibly based on the
approved process method and specifications.
2.39 Process parameter: Specified value for a process variable.
2.40 Process simulation test or media fills: One of the processing validations employed to
evaluate the propriety of the aseptic processing of pharmaceutical products using sterile media
instead of actual product.
2.41 Pure steam: Saturated steam that is generally produced using purified water or water of better
quality and will then be condensed into such high grades of water that meet the criteria for water
for injection under Pharmacopoeia.
2.42 Quality system: Organizational structure, procedures, processes and resources needed to
implement quality management.
2.43 Restricted Access Barrier System (RABS): An integrated system that possesses aseptic
processing areas (critical areas) and is composed of some critical elements such as rigid wall
enclosure (often equipped with gloves), unidirectional airflow least- to through HEPA filters and
appropriate operation procedures.
2.44 Sanitation/sanitization: Hygienic means of facilities and equipment by disinfection, cleaning,
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 5 -
hot waters, etc.
2.45 Standard operating procedure (SOP): An authorized written procedure giving instructions
for performing operations not necessarily specific to a given product or material but of a more
general nature (e.g. equipment operation, maintenance and cleaning; validation; cleaning of
premises and environmental control; sampling and inspection). Certain Standard Operating
Procedures (SOPs) may be used to supplement product-specific master and batch production
documentation.
2.46 Sterile: Free from viable microorganisms.
2.47 Sterility assurance level (SAL): Probability of a single viable microorganism being present in
a product unit after exposure to the proper sterilization process, expressed as 10
-n
.
2.48 Sterilization: A process that destroys or eliminates all microorganisms which is used to render
a product free from viable microorganisms.
2.49 Sterilizing filter: Either hydrophilic or hydrophobic filter to perform as required should be
demonstrated through bacterial challenge testing. The filters should retain specified numbers
of indicator bacteria under specified conditions. The nominal pore size of the filters ranges
from 0.20 to 0.22 µm.
2.50 Terminal sterilization: A process whereby a product is sterilized in its final container or
packaging, and which permit the measurement and evaluation of quantifiable microbial
lethality. Typically, the sterility assurance level should be less than 10
-6
.
2.51 Unidirectional airflow: Air flow which has a singular direction of flow and may contain
uniform velocities of air flow along parallel flow lines.
2.52 Working shift: Scheduled period of work or production, usually less than 12 hours in length,
during which operations are conducted by a single defined group of workers.
3. Quality System
The quality system for aseptic manufacturing of sterile pharmaceutical products is
structured to satisfy the requirements for the establishment, documentation, implementation, and
maintenance of an efficient and adequate quality control system in compliance with Sections 1
(General Rules) and 3 (Manufacturing Control and Quality Control of Sterile Pharmaceutical
Products) of Chapter 2 of the current GMP regulations.
3.1 General Requirements
1. General
The written quality system should comprise an organizational structure and description of
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 6 -
operational procedures, manufacturing processes, resources used, and activities necessary to
ensure compliance with the requirements for aseptic processing of sterile pharmaceutical
products stipulated in this guidance document.
All quality control-related activities to be undertaken, including sterility assurance-related
activities, should be identified and documented in detail. Manufacturers of pharmaceutical
products under aseptic conditions are also required to establish and implement an adequate
quality system by setting up quality control standards suitable for the prevention of microbial
product contamination during processing. This quality system should include an investigation
system for identifying deficiencies in sterilization procedures and assessing abnormalities or
deviations in control parameters from the standards, as well as a verification system for
ensuring the acceptability of corrective and preventive measures taken and whether or not the
outcome of these measures was achieved.
2. Scope of application
This guidance is applicable to the quality system governing all processes in sterile
pharmaceutical product manufacturing at facilities where pharmaceutical products are
manufactured under aseptic conditions. In practice, the scope of application includes
environmental control, control of laboratory testing of sterile pharmaceutical products, quality
control of aseptic processing, validation, and systematized control of manufacturing processes
and product quality such as documentation and change control.
3. Document control
The following documents should be prepared, used for fulfilling requirements stipulated in
each provision of this document, and archived to ensure the sterility of sterilized
pharmaceutical products: documents on initial, periodic and change validation; standard
operating procedures (SOPs); area maps with cleanliness levels; movement diagrams of raw
materials, personnel, intermediate products, and finished products; equipment and instrument
layout charts; instructions; records of data; deviation control records; change control records;
out-of-specification (OOS) test results; calibration records; environmental monitoring records;
log books; and computer system data (e.g. records stored on electronic media).
4. Risk management
The concept and procedures for risk management should be included in the quality system,
and contamination preventive measures should be implemented to minimize risks of
contaminating pharmaceutical products with microorganisms, endotoxins, and foreign matters.
The risk management system should be based on risk assessment procedures for analyzing and
evaluating factors affecting product sterility and contamination with endotoxins and foreign
matters as well as based on verification of risk control procedures for demonstrating the
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 7 -
reliability and validity of risk avoidance procedures.
5. Qualification of aseptic processing environment
Environmental parameters of the aseptic processing area should be identified and verified for
qualification. Based on qualification assessment results, a program for HVAC system
maintenance and environmental monitoring should be established and implemented.
6. Qualification of aseptic processing equipment and facilities
Equipment and instruments used for the manufacture of sterile pharmaceutical products in the
aseptic processing area as well as other equipment and facilities that may affect aseptic
processing should be evaluated for qualification. Based on qualification assessment results, a
program for maintaining the equipment and facilities should be established.
7. Prospective validation and periodic review of process control
Process validation that simulates all processes and activities related to sterilization of
pharmaceutical products should be conducted. Such processes and activities are required to
achieve commercial sterility of pharmaceutical products based on scientific evidence-based
designs and operations. A process control program should also be established and validated.
8. Periodic revalidation
Periodic revalidation should include a process simulation program and periodic valuation of
sterilization processes that may affect the sterility of pharmaceutical products.
9. Time limitation for aseptic manufacturing operations
Manufacturing processes of sterile pharmaceutical products from the preparation of drug
solution to filtration and sterilization should be conducted as quickly as possible. The
maximum allowable time from filtration, storage, and filling to sealing should be established
by taking into account the product composition, manufacturing processes, and storage
conditions as well as risks inherent to these processes.
10. Cleaning and disinfection of facilities and equipment
A program for cleaning and disinfecting facilities and equipment should be established taking
into account the potential development of drug-resistant microorganisms. The program should
contain procedures for screening and classifying bacterial isolates in each manufacturing
environment.
11. Pest control
An appropriate pest control program should be directed to aseptic manufacturing facilities to
prevent contamination of sterile pharmaceutical products with insects and other vermin.
12. Flow of raw materials
Flow diagrams of raw materials, parts, and other articles necessary for processing products
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 8 -
into the aseptic processing area should be established and, as the situation may require,
appropriate disinfection and sterilization procedures should be implemented. Appropriate
measures should be takento prevent microbial invasion into the working area during the
transfer of raw materials and other materials.
13. Gowning and flow of personnel
Appropriate procedures should be practiced to prevent microbial invasion into the aseptic
processing area during the entry or exit of personnel. Gowning procedures and flow of
personnel should be standardized.
14. Change control
Changes in standard procedures should be confirmed to have no negative impact on the
sterility of pharmaceutical products based on scientific evidence. Changes implemented
should be evaluated by applicable qualification and validation procedures, and, wherever
possible, control parameters should be established to control risks inherent to such changes
based on risk assessment results.
15. Calibration
A calibration program including calibration frequency and accuracy requirements should be
established and implemented to calibrate analytical equipment used in quality testing and
measuring, inspection, and control devices used in the manufacturing process.
3.2 Routine Monitoring and Control
1. An environmental monitoring program should be established based on results of
environmental tests performed to evaluate the qualification of the aseptic processing area.
2. Cleaning and disinfection of the aseptic processing area should be conducted periodically or
as-needed to verify that the area meets predefined environmental control specifications.
3. A maintenance program should be established and implemented based on results of
qualification and validation tests.
4. A process control program verified by validation experiments should be implemented.
5. Revalidation should be carried out at predetermined intervals.
3.3 Validation
The manufacture of sterile pharmaceutical products by aseptic processing can be achieved by
harmonized application of hardware such as well-designed facilities and equipment and software,
such as SOPs and adequate control systems and programs. In the qualification of an aseptic
processing environment and manufacturing equipment and process validation, not only the safety,
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 9 -
efficacy, and uniformity of the manufacturing process but also the maintenance of required
cleanliness levels of the sterilization procedure, filling and other aseptic processes should be ensured.
In addition, sterility of the processing environment as well as scientific estimation of contamination
risks for commercial production facilities and equipment and commercial manufacturing processes
should be ensured to help avoid product contamination. Validation of sterilization procedures should
include validation of sterility of raw and other materials supplied by external sources and
maintenance of sterility during transport.
The fundamental requirement for manufacturing process control is to control the process based
on validated operating procedures and process control parameters. When attempting to streamline
the manufacturing process, the proposed alterations, which may include omission of one or more
process parameters or shortening of process duration, should be assessed for possible risks, and
proposed changes should be justified by scientific rationale and revalidated as appropriate.
4. Personnel
Humans are the largest source of microbial contamination in aseptic processing areas
(“APAs”) for manufacturing operations. It is essential to minimize personnel intervention as a
possible source of contamination of pharmaceutical products to eliminate the source of
contamination within the APA for manufacturing sterile pharmaceutical products. Appropriate
education and training on the concepts and practical procedures that factory personnel are required to
perform should be provided to maintain high skill levels and improve confidence and morale.
If instruments such as isolators and blow-fill-seals are considered necessary in lowering the
potential for human-related microbial contamination to occur, the importance of personnel education
and training including those on characteristics and operating procedures of instruments should be
taken into account for adequate operation, maintenance, and control of the instruments.
4.1 Personnel Training
1. SOPs for aseptic processing should be developed and in place. The SOPs should contain
detailed descriptions of tasks that personnel are required to perform during aseptic processing.
2. An education and training program should be prepared and implemented for personnel
engaged in the manufacture of sterile pharmaceutical products in the APA. The level of
training should be dependent on the knowledge and skills of individual personnel.
3. At least the following matters should be included in the education and training program on
aseptic processing. While these matters need not be addressed simultaneously, they should be
practiced without fail in accordance with a pre-established training schedule. The contents of
the program and frequency of training should be individualized according to the scope of work
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 10 -
or assignment, skills, knowledge, and experience of personnel.
(1) Hygiene
● Personnel engaged in operations in the APA for manufacturing sterile
pharmaceutical products should not wear make-up or accessories (e.g. rings with
raised settings, earrings, wrist watches) that may damage work gowns, jackets,
gloves, caps, or masks.
(2) Aseptic techniques
● Personnel working in the APA should avoid unnecessary movements and direct
contact with critical surfaces.
● Personnel should minimize movement and conversation in the APA that may
generate airborne particles or create unacceptable turbulence in critical areas.
● Personnel should avoid blocking or disrupting the airflow path directed to
unsealed containers, unprotected pharmaceutical products, and packaging
materials (e.g. rubber closures).
● Personnel should not disrupt airflow directed at the surface of sterilized materials
or pharmaceutical products placed in critical areas.
● Personnel should keep their gloves sanitized by frequent disinfection or other
appropriate procedures.
(3) Knowledge of basic microbiology and skills of microbiological testing
● Understanding the type, properties, and detection methods of microbial species
that are likely to be encountered during manufacture
● Understanding conditions leading to the proliferation or death of microorganisms
as well as generation of endotoxins
● Understanding basic knowledge and skills of sterilization procedures to be used
● Understanding environmental monitoring methods to be employed
(4) Gowning procedures
● Personnel should be trained in proper hand washing, gowning, and degowning
procedures required before entering and after leaving the APA. The supervisor
should periodically evaluate their performance to confirm their adherence to
established rules on gowning, etc.
● Personnel should be trained on appropriate gowning procedures to minimize
contamination risks in the APA.
● Training effectiveness on gowning procedures should be evaluated by a particle
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 11 -
monitoring and microbiological tests. Gowns used during microbiological testing
should not be reused in the APA unless disinfected after testing.
● The supervisor should communicate gowning training results to personnel trained.
● Training of gowning procedures should be conducted in an equipment inspection
and maintenance situation or APA entry situation after deregulating aseptic
conditions while production is suspended. The training program should also
include advice on handling instruments to be brought into the APA. When
untrained personnel, including vendor engineers, enter the area, trained personnel
should accompany them to advise on procedures for adequate gowning and
handling of instruments brought in.
(5) Aseptic processing technology necessary for personnel in manufacturing sterile
pharmaceutical products
(6) Cleaning and disinfection of manufacturing environment and equipment
● Properties of cleaning agents and disinfectants as well as materials to be cleaned
or disinfected
● Concentrations, method of preparation, and expiration date of cleaning agents and
disinfectants used
● Points to consider on the use of cleaning agents and disinfectants
(7) Potential hazards that may affect humans if contaminated pharmaceutical products are
administered
4. Personnel (e.g. manufacturing supervisors, QA/QC personnel, maintenance personnel) who
may occasionally enter the APA should be educated and trained on the following matters, as
appropriate:
(1) Hygiene
(2) Microbiology
(3) Gowning procedures
(4) Acceptable behaviors and activities in the APA
5. Education and training topics should be identified in writing, and educational effectiveness of
the training program in increasing knowledge and skills of aseptic processing should be
evaluated.
6. All personnel engaged in aseptic processing operations should participate in a process
simulation test at least once a year, as a rule, and should achieve a predefined level of
performance.
7. Personnel with no experience in aseptic processing operations should participate in a process
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 12 -
simulation test or other similar aseptic processing operations at least once, as a rule, prior to
obtaining permission to engage in operations in the APA. Training of “other similar aseptic
processing operations” may be conducted in a non-APA (e.g. training environment).
8. Inexperienced personnel who have been allowed to enter the APA should be supervised by
experienced personnel for a predefined period and receive on-the-job guidance and subsequent
evaluation of performance.
9. As a rule, entry into the APA should be restricted to personnel who have obtained prior
permission to enter the area. When personnel need to enter the area for any reason, such as
equipment repair, those personnel should obtain entry permission from the supervisor of the
area and be accompanied by authorized personnel throughout their stay in the area.
4.2 Personnel Health Management
1. Personnel should report any clinical signs or symptoms to the supervisory personnel prior to
engagement in operations if affected with fever, skin damage, flu, or diarrhea that may affect
aseptic processing operations in the APA.
2. The supervisor should not permit the entry of the personnel into the APA when informed of
physical abnormalities that may affect aseptic processing operations.
4.3 Personnel Management
1. Personnel who engage in operations in the APA should be subject to personnel management in
accordance with an APA-specific microbiological monitoring program.
2. Microbiological testing should be performed immediately before leaving the APA, if gowns
and other clothing may contact agar during testing.
3. Microbiological monitoring data obtained from individual personnel should be analyzed to
determine a trend of contamination-risk increase for individual personnel at an appropriate
frequency. Personnel who show an undesirable trend in contamination should be educated and
trained repeatedly until acceptable data are obtained.
5. Prevention of Contamination by Personnel
If any personnel show unacceptable microbiological data obtained by monitoring gowns and
other clothing, such personnel should be educated and trained again at the earliest possible occasion.
If re-education and retraining fail to improve microbiological contamination rate, the supervisor
should consider the reassignment of such personnel to non-APAs.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 13 -
5.1 Gowning Requirements
1. Personnel should wear an APA-specific gown and other stuff including shoes before entering
the processing areas for sterile pharmaceutical products. Basic garments include a sterilized or
disinfected gown, shoes, overshoes, gloves, goggles, and mask. The use of clean
undergarments and dual layer gloves should be considered, as the situation may require.
2. A gowning room located before the entrance of the APAs should be separated or partitioned
from the degowning room to avoid cross-contamination. It is recommended that the gowning
procedure be displayed in the gowning room of the APA used for manufacturing sterile
pharmaceutical products by a sequence of pictures to aid in understanding of gowning
procedures and that a mirror be installed to facilitate checking of proper gowning.
3. Gowns and other stuff to be worn in the APA for sterile pharmaceutical products should be
highly functional and suitable for working in the APA and free of generating or discharging
particulate matter into the environment.
4. Personnel entering the APA should not expose any body surfaces to the environment while
working in the APA.
5. Cleanliness of gowns and other stuff should be managed by internal control standards,
including frequency of change and sterilization methods and conditions, established and
implemented to maintain the cleanliness as required.
6. Sterile gowns and other stuff worn in the APA should be changed each time entering the area,
as a rule. If gowns and other stuff are permitted by the internal control standards to be reused
without disinfection or sterilization, the validity of the reuse should be verified with
experimental data. Even if the reuse is supported by data, gowns and other stuff worn for more
than one day or worn during microbiological sampling should not be reused without
disinfection.
7. It is recommended that personnel wear dedicated undergarments (e.g. layered clothing for
complete skin coverage) or over gowns.
5.2 Requirements for Aseptic Processing
1. Personnel should adhere to SOPs for the prevention of microbiological contamination of the
APA.
2. Personnel should check to see if the gowns and other stuff fit properly and are not torn or
defective. If a gown or gloves are found to be defective, necessary counteractions such as
changing or layering of new garments over the defective ones should be immediately taken.
3. Personnel should refrain from speaking after gowning and should avoid direct contact with the
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 14 -
wall, floor, or sanitized surfaces unless necessary.
4. Applicable SOPs should include a provision that restricts unnecessary personnel movement,
such as touching of materials and walls, while staying in the APA.
5. Personnel operating in indirect support areas should not be permitted to enter critical or direct
support areas or rooms if they do not change gown and other stuff or are not adequately
trained on proper gowning procedures.
6. The number of personnel operating in the APA should be set at a minimum for each shift of
manufacturing operations, including the preparatory stage. Personnel handling sterile
pharmaceutical products, containers, or closures and those engaging in operations in an
environment where sterile pharmaceutical products, containers, or closures are exposed should
be identified and recorded.
6. Buildings and Facilities
6.1 Key Features of Facility Design
Clean areas for the manufacture of sterile pharmaceutical products are classified into APAs
(comprising critical and direct support areas) and indirect support areas. These clean areas should be
designed by taking into account the following matters as general requirements:
1. Clean areas should be clearly separated from rest rooms, and eating areas.
2. Clean areas should be well-separated for intended purposes from other processing operations
within a facility, and should have sufficient space to allow proper conduct of all manufacturing
operations that are to be done within them.
3. Clean areas should be designed to achieve efficient flow and control of materials, products, and
personnel within the areas. The location of equipment in the areas should also be carefully
planned to minimize crossing of personnel, products, and materials flows.
4. Material handling procedures or fixed depots should be efficient in preventing a mix-up between
clean and dirty or sterilized and non-sterilized apparatuses and utensils.
5. Facilities should be designed to facilitate ease of cleaning, maintenance, and operations and
periodically inspected to verify that the facilities are maintained as originally designed.
Particular consideration should be given to seals and packing of interior materials such as
doors, walls, ceilings in order to keep processing rooms tightly closed. Insulation materials
to prevent dew drops should be maintained to work well.
6. Ceilings should be effectively sealed.
7. Installation of irregular surfaces and horizontal frames around windows and doors should be
avoided to reduce collection of particulate matter and microorganisms and to avoid
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 15 -
disturbance of airflow. If such designs are unavoidable, their structures should be suitable for
easy cleaning. Sliding doors may be undesirable for this reason.
8. Adequate space should be provided for gowning, storage of gowns, and disposal of used gowns
and other materials.
9. Transparent (e.g. glass) windows or video cameras should be installed in the APA to facilitate
observation and supervision from non-aseptic areas.
10. Layout of equipment in the APA should be designed to minimize environmental exposure of
open containers or finished products and facilitate easy access of personnel to these items during
processing or quipment maintenance.
11. Equipment not essential for processing in the critical area should be installed in non-critical
areas.
12. Corridors should be adequately distributed along working areas in indirect support areas
(Grade C or D) in order to prevent those areas from being used for routine passage of
personnel not directly engaged in processing in the areas.
13. When parenteral and other sterile drug products are manufactured simultaneously in the same
room, manufacturing equipment for preparation, filling, and sealing of drug products should
be dedicated and should be closed system for those operation. If any part of the equipment
structurally is kept open, appropriate measures and activities should be implemented to
prevent contamination.
14. The working areas for preparation, filling, and sealing of sterile drug products and sterile API
should be separated from the areas for processing non-sterile drug and non-sterile API. The
separation is not necessary if there is virtually no risk of contamination of products processed in
the working areas.
15. Facilities should be structurally designed to be efficient in preventing or minimizing risks of
cross contamination if used for processing highly pharmacologically active substances,
pathogenic substances, highly toxic substances, radioactive substances, live viruses, or
bacteria.
16. Walls, floors, and ceilings should be easily cleanable and durable against cleaning agents and
disinfectants.
17. Drains and sinks should be prohibited in the APA. If drains are placed in Grade C areas in
indirect support areas, drains should be fitted with traps or water seals parts which are easy to
clean and disinfect to prevent contamination by back-flow. If floor trenches are located, they
should be shallow to facilitate cleaning.
18. Piping, air ducts, and other utilities in clean areas should be installed so that they do not create
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 16 -
recess and surfaces which are difficult to clean.
19. Clean areas should be supplied with air filtered through an appropriate filter, e.g. a
high-efficiency particulate air (HEPA) filter, to maintain an acceptable level of air quality and
pressure difference between areas. The pressure difference should be monitored to maintain as
specified.
20. Temperature and relative humidity in clean areas should be controlled within ranges compatible
with the properties of materials and products being handled in the areas and also set at levels
suitable for microbiological control.
21. Environmental temperature and relative humidity should be controlled within specified limits
and, wherever feasible, monitored continuously.
22. Air pressure in clean areas should be maintained higher relative to adjacent lower cleanliness
areas through doors, except for containment philosophy facilities for handling potent substances.
23. Airflow patterns in critical areas should be controlled to maintain sterility of critical surfaces and
products.
24. Direct support areas should be separated from adjacent areas by airlocks. Spaces located between
direct support areas and adjacent areas should be equipped with pass-through rooms and/or
pass-through boxes for transfer of sterilized materials. Airlocks should also allow for proper
disinfections or decontamination of wrapped goods, tools and other materials used in the APA
when necessary..
25. Airlock doors should be equipped with a system that prevents simultaneous opening of both sets
of doors (e.g. mechanical and electrical interlocking systems and visual and audible alarm
systems).
26. The gowning room should be equipped with an airlock system and physically portioned into
gowning and degowning areas. Air particulate cleanliness in the gowning room should be
maintained at the same grade as the area (at rest) into which it leads. In order to reduce rapidly
numbers of particles accompanied with gowning activity, volume and/or air change rate of the
room should be adequately considered. Supply air at a relatively high position and exhaust air
at a lower position in the room are desirable. The air cleanliness of the pass box should be
specified according to the intended the purpose of use.
27. The use of separate changing rooms entering and leaving clean areas especially in the direct
supporting areas is desirable. As an alternative measure, it is acceptable to stagger time of
entry and exit.
28. Gowning rooms should be adequately located depending on cleanliness of the working rooms.
Even if the cleanliness level is the same among areas, additional gowning rooms should
preferably be set up depending on potential risks of contamination if there are risks of cross
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 17 -
contamination of raw materials and pharmaceutical products.
29. Rooms for weighing raw materials or washing containers should be carefully designed to secure
seal integrity of doors and maintain appropriate airflow so as to not introduce contaminated air
into adjacent rooms.
7. Processing Areas for Sterile Pharmaceutical Products
7.1 Classification of Manufacturing Areas by Air Cleanliness
Facilities for processing sterile pharmaceutical products comprise clean areas controlled based
on predefined airborne particle and microbiological standards. The areas are classified as critical,
direct support, and indirect support areas depending on the nature of the operation to be conducted.
Generally, the cleanliness of air in processing areas is defined by the number of airborne
particles ≥ 0.5 μm in diameter per unit volume of air. The number of particles ≥ 5 μm in diameter
may serve as a reliable parameter for early detection of environmental deterioration, if regularly
monitored and evaluated by linear trend analysis. Table 1 shows the air cleanliness requirements for
classified areas.
Tab le 1. Categories of Clean Areas
Area
Air cleanliness
Note 1)
Maximum allowable number of airborne particles
(/m
3
)
Count under
non-operating
conditions
Count under operating
conditions
≥ 0.5 μm ≥ 5.0 μm ≥ 0.5 μm ≥ 5.0 μm
Aseptic
processing
area
Critical area Grade A (ISO 5) 3,520 20 3,520 20
Direct support
area
Grade B (ISO 7)
3,520 29 352,000 2,900
Indirect support area
Grade C (ISO 8) 352,000 2,900 3,520,000 29,000
Grade D 3,520,000 29,000
Dependent on process
attributes
Note 2
)
Note 1) The ISO class designation in parenthesis refers to the count during operation.
Note 2) There are cases where maximum allowable number may not be specified.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 18 -
7.1.1 Critical Area (Grade A)
1. The critical area is a processing area where sterilized products and materials as well as their
surfaces are directly exposed to the environment. The environmental conditions should be
specified to be suitable for the virtual elimination of contamination risks and preservation of
the sterility of products. The following processes are conducted in this area: sterilization
activities (e.g. sterile connections, addition of sterile materials) prior to filling, sterile filling,
and sterile closure.
2. The per-cubic-meter content of particles ≥ 0.5 μm in diameter in the critical area should be
controlled to be below 3,520 under both operating and non-operating conditions. This level of
air cleanliness is designated as Grade A, Class 100, or ISO-5 according to domestic and
international standards on air quality.
3. The intervention of personnel into the critical area should always be kept to a minimum.
4. The count of airborne particles and microorganisms should be regularly monitored by
appropriate procedures at sites which are critical for ensuring sterility of pharmaceutical
products.
It is recommended that airborne particles be continuously counted throughout aseptic
processing, including during critical preparatory steps such as assembly of sterile parts that
may contact pharmaceutical products. The location of monitoring should preferably be as
close (≤ 30 cm) as the working place.
The frequency and method of microbiological monitoring should be carefully selected in order
not to break sterility of products by the monitoring.
5. Powder filling operations may generate higher counts of airborne particles than the
specifications. If such a deviation occurs, the count of airborne particles should be obtained by,
for example, sampling air at different locations or monitoring the count in the same room
while no powder filling operation is going, and causes of the deviation should be identified to
maintain air quality in the room at a required level.
7.1.2 Direct Support Area (Grade B)
1. The direct support area is defined as a background area of the critical area when aseptic
processing is conducted using an open clean booth or restricted access barrier system (RABS).
The direct support area is a working area for personnel who operate machines installed in the
critical area and for those who supervise the operation of machines. The direct support area
also serves as a route for the transfer of sterilized products, materials, and equipment to the
critical area or for moving sterilized products from the critical area. In the latter case,
appropriate measures need to be implemented to protect sterilized products or materials from
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 19 -
direct exposure to the environment.
2. The per-cubic-meter count of particles (diameter: ≥ 0.5 μm) in the direct support area should
be controlled below 352,000 and 3,520 under operating and non-operating conditions,
respectively. These levels of air cleanliness are designated as Grade B, Class 10,000, or ISO-7
(under standard operating conditions) according to domestic and international standards on air
quality.
3. The count of airborne particles and microorganisms should be regularly monitored by
appropriate procedures in the direct support area. The frequency and method of monitoring
should be carefully selected based on evaluation results of product contamination risks in the
critical area.
7.1.3 Indirect Support Areas (Grade C or D)
1. The indirect support area is an area used for processing materials and products prior to
sterilization processes and hence materials and products are directly exposed to the
environment. Example indirect support areas include an area for preparing drug solution prior
to sterilization and an area for washing and cleaning sterilization equipment and apparatuses.
2. The cleanliness of the indirect support area needs to be controlled by establishing
specifications for acceptable airborne particle count by taking into account the required level
of contamination control and type of works performed in the area.
3. Air cleanliness of the indirect support area may be either of the following two grades. One of
the grades specifies that the per-cubic-meter particle content (diameter: ≥ 0.5 μm) should not
exceed 3,520,000 and 352,000 under operating and non-operating conditions, respectively.
These levels of cleanliness are designated as Grade C, Class 100,000, or ISO-8 (standard
under operating conditions) according to domestic and international standards on air quality.
The other grade specifies that the per-cubic-meter particle content (diameter: ≥ 0.5 μm) should
not exceed 3,520,000 under non-operating conditions. This level of cleanliness is designated
as Grade D.
4. Weighing and preparation processes should preferably be conducted in Grade C or cleaner
areas. If powder handling might elevate the airborne particle count above the specification, air
quality should be maintained below the specification by accurately determining the particle
count that may cause contamination in the area, and for the determination, air should be
sampled, for example, at multiple locations and/or under powder-free conditions.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 20 -
7.2 Heating, Ventilating and Air Conditioning System
Air in clean areas needs to be maintained at appropriate levels by designing, instituting, and
managing a suitable heating, ventilation, and air conditioning (HVAC) system. The integrity of the
system should be ensured with respect to not only temporal variations due to operational activities,
such as door opening and closing and facility equipment operation, but also sustained variations due
to non-operational activities, such as seasonal changes in outdoor conditions or deterioration of
equipment and apparatuses over time.
The HVAC system and its management program are comprised of the following basic
elements: temperature, relative humidity, air flow volume, air exchange rate, unidirection of air flow,
pressure difference relative to adjacent rooms, integrity of HEPA filter, airborne particle count, and
microbacterial count.
7.2.1 Temperature and Relative Humidity
Temperature and relative humidity have a direct impact on the comfort of personnel and
potential for microbial contamination in processing areas; therefore, these environmental parameters
should be appropriately defined, controlled, monitored, and maintained at appropriate levels
throughout processing.
7.2.2 Air
It is critical to secure constant airflow from an area of higher cleanliness level to an area of
lower cleanliness level in order to maintain required environmental conditions of clean areas.
1. Pressure difference between the APA and indirect support areas should be adequately defined,
monitored, and controlled.
2. Air locks should be established between the APA and indirect support areas and pressure
difference between these areas should be maintained at a level sufficient to prevent the
reversal of defined pressure difference or airflow. For example, a desired pressure difference
between areas, when both closed, should be at least 10 to 15 Pa. The air lock design should
meet requirements defined in Item 26 (gowning room) in Article 6.1. Likewise, an appropriate
pressure difference should be established and maintained between indirect support areas of
different cleanliness levels.
3. Wherever pressure difference is an essential part of sterility assurance, it is recommended to
continuously monitor pressure difference between areas and install an alarm system to enable
prompt detection of abnormal pressure differences.
4. Airflow in the critical area (Grade A) should be unidirectional and supplied at velocity and
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 21 -
uniformity sufficient to swiftly remove airborne particles away from the critical area. Airflow
should also be supplied with sufficient care so as not to create reverse currents from adjacent
areas (direct support areas, Grade B) into the critical area to prevent contamination. When
conventional clean benches and RABS are used, the recommended mean flow rate is 0.45
m/sec ± 20%. Lower flow rate may be necessary depending on the type or usage of isolator
system.
5. The airflow requirements stated in the preceding Item 5) should be verified by appropriate
method of validation by smoke test or other qualification tests at the installation of airflow
equipment. Similar validation is also necessary when airflow patterns are changed or
suspected of being changed.
6. Changes in flow velocity can alter flow direction when airflow is specified to be unidirectional.
The velocity should be confirmed to be constant at a predetermined level by monitoring the
velocity of airflow from HEPA filters at time intervals specified in the program.
7. An appropriate air change rate should be established by evaluating the potential of product
contamination for individual processing areas and gowning rooms in the APA to maintain air
cleanliness at specified levels. The generally recommended air change rate is 30 times per
hour in the direct support area and 20 times per hour in Grade C work rooms among indirect
support areas. These change rates should be monitored at regular intervals to verify that the
rates are continuously maintained as specified. Air current should be controlled not to ascend
to prevent deterioration of work environment due to dust and bacteria stirred up from the floor.
The most common method of securing downward current is to install supply vents close to the
ceiling and exhaust vents close to the floor. Similar considerations on ventilation are
applicable to indirect support areas, although the rigidity of specifications depends on
potential risks of contamination with microorganisms and foreign matter.
8. The cleanliness of the work room must be promptly returned to the non-operating level after
completion of processing and workers leave the room. In the direct support area, airborne
particle count should preferably be returned to the non-operating count in 15 to 20 minutes.
9. Intended differential pressure and airflow patterns during processing should be specified and
documented and then validated to be suitable and appropriate for commercial manufacture.
The impact of turbulence created by the movement of personnel on the cleanliness of the
manufacturing environment should be evaluated, and evaluation results should be reflected in
relevant SOPs.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 22 -
7.3 Integrity of HEPA Filters
7.3.1 Certification of Quality
1. HEPA filters should be accompanied by vendor’s certificate of quality verifying that the filter
is capable of eliminating at least 99.97% of particles ≥ 0.3 μm in diameter.
2. Leak test of HEPA filters to be used in critical areas (Grade A) and direct supporting areas
(Grade B) should be performed by using appropriate leak testing aerosols, e.g.
poly-alpha-olefin (PAO). When alternate aerosols are used, such aerosols should be used after
confirming that they do not promote microbial growth.
7.3.2 Testing of HEPA Filters at Installation and at Regular Intervals
1. HEPA filters should be tested for leaks at installation and thereafter at suitable time intervals.
The procedure and frequency of testing should be tailored to the environment, where the filters
are installed, and their intended purpose of use. The integrity of HEPA filters in the critical
area and direct support area should be confirmed at least once a year. The integrity check is
recommended to be twice or more in the case that conditions of use in the critical area are
severe or special considerations are required for the prevention of microbial product
contamination.
2. HEPA filters installed in the critical area (Grade A) should be tested for uniformity of air
velocity across the filter at installation and thereafter at suitable time intervals. The frequency
of integrity check should be determined as stipulated in the preceding Item 1).
3. Pressure difference between the HEPA filter’s initial and final pressure loss should be tested at
installation and thereafter at suitable time intervals. If filter clogging is severe, it is
recommended to include pressure difference monitoring in routine control procedures.
4. When airflow patterns in the APA are altered or suspected of being altered, the patterns should
be evaluated to assess the acceptability of the altered patterns.
5. HEPA filters should be tested by leak test as directed in relevant SOPs when any events or
circumstances that may damage filter integrity occur or when air quality is judged to have
deteriorated.
8. Cleaning and Disinfection of Processing Areas for Sterile Pharmaceutical
Product Manufacturing
Processing areas for manufacturing sterile pharmaceutical products should be cleaned and
disinfected in accordance with relevant SOPs, and results of cleaning and disinfection should be
recorded in writing and retained in an archive.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 23 -
8.1 Cleaning Agents and Disinfectants
1. Cleaning agents and disinfectants should be validated for their appropriateness and reliability
in removing contaminants prior to use. Cleaning and disinfection efficacy should be assessed
and confirmed based on type and count of microorganisms characterized by periodic
environmental monitoring.
2. Cleaning agents and disinfectants used in the APA should be pretreated with filtration or other
appropriate sterilization procedures before use and controlled for the prevention of
microbacterial contamination until use, unless commercial products certified to be sterile are
used by breaking the envelope immediately before use.
3. When prepared in-house, the preparation of cleaning agents and disinfectants should be
pursuant to applicable SOPs, and preparation records should be created in writing and retained
in an archive. When commercial products are used after dilution, details of the dilution
proceduresuch as diluents, dilution ratio, expiration date, storage conditions, and, if
applicable, sterilization methodsshould be recorded in writing and approved by the quality
department.
4. SOPs for the preparation and use of cleaning agents and disinfectants should address the
following matters approved by the quality department: types or brands of cleaning agents and
disinfectants, cleaning and disinfection schedules, directions for the use of disinfectants,
necessity of cleaning following disinfection procedure where necessary, safety precautions for
factory personnel, and procedures for management and storage of cleaning tools.
5. When cleaned or disinfected, the surfaces of facilities and equipment that may come into
direct contact with pharmaceutical products should be verified by appropriate methods to be
free of cleaning agents or disinfectants after the completion of cleaning or disinfection
procedures.
6. Reasonable expiration dates should be established for individual disinfectants, and
disinfectants should be used before that date.
7. The disinfection of the manufacturing environment should not proceed prior to cleaning, as a
rule. If there are any locations in the environment where cleaning agents may reside after
cleaning, the cleaning agents should be verified not to impair the efficiency of disinfectants.
8. Disinfectant containers should not be refilled with disinfectants.
9. The selection and use of disinfectants should take the following matters into account:
(1) The storage and usage of disinfectants should be in accordance with the supplier’s
instructions.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 24 -
(2) The selection of disinfectants should be primarily based on the safety of personnel who
are engaged in disinfection processing.
(3) If selected disinfectants might have inferior efficacy against microorganisms isolated
from the environment, the efficacy should be reevaluated and the replacement with or
alternate use of different disinfectants should be considered and implemented, as
appropriate.
(4) If environmental monitoring data indicate or suggest the presence of spore-forming
bacteria or fungi, suitable sporicides or fungicides should be selected for disinfection.
(5) The directions for use of disinfectants should include the method of disinfection,
application site of the agents, and time required for the agents to exert anticipated
effects.
(6) Chemical properties (e.g. corrosivity) which might damage the surface of facilities and
equipment to be treated should be assessed prior to the selection of cleaning agents and
disinfectants.
10. If use of sporicides or fungicides in processing areas for sterile pharmaceutical products is
likely or probable, the type, concentrations, and usage of the agents should be predetermined
and specified in writing.
11. The preceding Item 10 should also be applied to the selection and use of fumigating agents
(including aerosol formulation), although such application is dependent on the properties of
the agents to be used.
12. Cleaning agents, disinfectants, and utensils for applying these agents should not be stored in
critical areas. Materials needed for operations in the critical area such as hand sprays to
disinfect gloves may be stored in critical areas, if well controlled. If cleaning agents and
disinfectants are stored in critical areas, reasons and control procedures for keeping should be
defined in writing.
8.2 Validation of Disinfection Procedures
1. The reliability and frequency of disinfection procedures should be established through an
environmental monitoring program.
2. Disinfectants should be microbiologically assessed prior to use in each facility, and
appropriate control procedures should also be instituted for each facility.
3. The efficacy of disinfectants should be assessed with respect to ensuring that microorganism
counts remain below the count predetermined based on the type and count of isolates collected
from various surfaces through the environmental monitoring program.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 25 -
8.3 Monitoring of Adequacy and Efficacy of Cleaning and Disinfection Procedures
1. The adequacy and efficacy of cleaning and disinfection processes should be established
through the overall environmental monitoring program.
2. The microorganism counts on the surface of equipment and instruments should be periodically
obtained by environmental monitoring and analyzed to detect trends in occurrence and
proliferation. A full investigation is mandatory to determine causes of abnormalities when the
microbial count exceeds the action level, when the species ratio of microorganisms is
obviously different from that routinely reported, or when abnormalities in the count or species
ratio continue for an extended period of time. In addition, corrective and preventative
measures should be implemented, as appropriate whenever considered necessary.
3. If the established disinfection procedure is not found to be effective for certain types or
concentrations of disinfectants, the reliability of such disinfectants should be reevaluated by,
for example, comparing the species and counts of microorganisms obtained before and after
disinfection.
9. Control of Raw Materials, Containers, and Closures
9.1 Control of Raw Materials (API and Additives)
9. 1.1 General Requirements
1. Procedures for receiving, identifying, storing, sampling, and testing raw materials should be
defined as the respective SOPs for control purposes. Acceptance criteria for testing should also
be established.
2. Raw materials should be carefully controlled to avoid contamination from receipt until use
including storage.
3. Raw materials transferred into the critical area should be confirmed to fall in one of the
following categories:
(1) Certified to be
sterile
(2) Adequately sterilized in accordance with their physicochemical properties and
bioburden levels. Their bioburden should be monitored and confirmed to
comply with thier specifications at predetermined intervals.
4. Raw materials should be controlled to meet endotoxin specifications.
(1) If raw materials are not depyrogenated during manufacturing, the endotoxin level of the
materials should be ensured to be below the predetermined level.
(2) If raw materials are depyrogenated during manufacturing, suitable depyrogenation
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 26 -
procedures should be instituted by taking into account physicochemical properties and
endotoxin level. Endotoxin content of the materials prior to depyrogenation is preferred
to be controlled whenever possible.
9.1.2 Validation
1. When raw materials are required to be sterile, validation should be performed to ensure their
sterility.
2. When raw materials need to be sterilized, applicable sterilization procedures should be
validated.
3. When individual raw materials are separately sterilized, not only sterilization processes for
individual materials but also those for final drug solution should be validated to ensure their
sterility.
4. When raw materials are released after sterilization using parametric or dosimetric methods
such as steam sterilization and irradiation, such methods should be validated.
5. When raw materials are depyrogenated, the depyrogenation procedure should be validated.
Generally, the depyrogenation process must achieve at least a 3-log reduction of endotoxins
below challenge.
9.2 Control of Containers and Closures
9.2.1 General Requirements
1. Procedures for receiving, identifying, storing, sampling, and testing containers and closures
should be defined as SOPs for control purposes. Acceptance criteria should also be
established.
2. Containers and closures should be carefully controlled to avoid contamination from receipt
until storage and use.
3. Containers and closures should be washed and cleaned by appropriate and validated
procedures. If water is used for washing, water for injection or water of comparable quality
should be used for final rinsing.
4. Containers and closures transferred into the critical area should be sterilized by appropriate
and validated procedures.
5. Containers and closures should be controlled to meet endotoxin specifications.
(1) If containers and closures are not depyrogenated after transfer into the critical area, their
endotoxin levels should be confirmed prior to transfer to be lower than respective
predetermined levels.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 27 -
(2) If containers and closures are depyrogenated after transfer into the critical area, suitable
depyrogenation procedures should be instituted by taking into account physicochemical
properties of containers and closures.
6. Sterilized containers and closures should be protected from microbial or pyrogenic
contamination by appropriate preventive measures.
9.2.2 Validation
1. Procedures for sterilizing containers and closures should be validated.
2. When containers and closures are depyrogenated, the depyrogenation procedure should be
validated. Generally, the depyrogenation process must achieve at least a 3-log reduction of
endotoxins below challenge.
10. Storage and Transport of Sterile Intermediate Products
Sterile intermediate products referred to in this section are intermediate products in solution or
powder that are stored or transported in a sterile state following aseptic production. This section
describes requirements for containers and procedures necessary for maintaining the sterility of
intermediate products.
10.1 General Requirements
1. Containers used for the storage and transportation of sterile intermediate products
(“containers” in this section refers to cargo transporters, drums, bags, and tanks) should be
capable of isolating the products from the surrounding non-sterile environment and hence
maintaining the sterility of the products. The containers should be durable enough to withstand
handling and environmental conditions encountered during storage and transportation.
2. Containers used for storage and transportation of intermediate products should be cleaned and
sterilized before being filled with and storing or transporting the products. Cleaning and
sterilization are not required for sterile single-use containers. However, the content sterility
must be maintained under all circumstances.
3. SOPs should be established for pouring intermediate products into and discharging them from
the containers in a closed system, as a rule. If adopting a closed system is difficult,
intervention by personnel should be kept to a minimum.
4. The environment to which sterile intermediate products are exposed should be a critical area
(Grade A) free of contamination risks.
5. Sealing performance (tightness) of containers should be checked and confirmed, as required.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 28 -
10.2 Containers for Storage and Transportation
10.2.1 Design of Containers
The choice of containers for maintaining sterility during storage and transportation of
intermediate products should take the following matters into account:
1. The following points should be observed when designing or selecting containers to be used for
isolating contents from the non-sterile environment.
(1) The structure should ensure hermetic sealing.
(2) If the container cannot be hermetically sealed, the inside of the container should be
maintained constantly under positive pressure with sterile gas.
(3) If sealing performance of container cannot be ensured because of changes in external
pressure, air vent filters capable of sterile filtration should be installed and their integrity
tested at appropriate intervals.
(4) Containers should be designed to have a dual structure, as appropriate.
2. If the surface of a container needs to be cleaned and sterilized prior to transferring the
container into the APA, the surface should be able to withstand cleaning and disinfection
agents.
3. Casters and other parts of transport devices should be protected from generating dust and
particulate matter, if such devices are used in the transportation of containers into the APA.
4. If single-use plastic bags are used for storage and transportation, the potential
extractable/leachable of components out of the bags into drug solution and effects of the
components on product quality should be carefully evaluated and discussed.
10.2.2 Confirmation of Hermeticity
1. Whether or not newly designed containers they can be hermetically sealed should be
confirmed.
(1) Eligibility confirmation at designing
Sealing performance of container should be estimated by taking into account projected
use conditions, including worst-case scenarios.
(2) Eligibility confirmation at manufacturing (actual use)
Sealing performance should be tested after storage or transportation under actual use
conditions.
2. Sealing performance can be determined by the following example methods:
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 29 -
(1) Check whether or not leakage occurs in containers on hold status under positive
pressure.
(2) Check whether or not leakage occurs in containers on hold status under negative
pressure.
(3) Immerse containers in pigment solution or bacterial suspension and check whether or
not pigment or bacteria are detected in containers.
(4) Inspect containers with a gas leakage detector.
(5) Inspect containers with an electric pin hole testing machine.
10.3 Charging and Discharging Sterile Intermediate Products in and out of Containers
When charging and discharging sterile intermediate products in and out of containers before
and after storage or transportation, the following matters should be taken into account:
1. Automatization
Wherever feasible, automatic charging (including divided charging) and discharging systems
should be instituted to minimize personnel intervention.
2. Minimization of personnel intervention-related risks
If automatic systems cannot be introduced, the following matters should be considered to
manage intervention-related risks:
(1) Working personnel should not physically block or disrupt airflow directed to the
charging and discharging ports.
(2) Charging and discharging operations should be performed in Grade A areas (e.g. clean
booth).
(3) Certain risk reduction measures such as sterile connections using tubing systems that do
not require opening of containers should be examined and evaluated.
3. Process simulation
A process simulation test should be performed to demonstrate the validity of procedures for
charging and discharging sterile intermediate products in and out of containers.
4. Limitation of working hours
Time is always a critical factor for maintaining sterile conditions; the more time required for
charging and discharging operations, the greater the risk of contamination. A maximum time
limit should preferably be set for these operations, and if more than one container is used per
shift, the containers should be marked with identification numbers or other identifiers to
facilitate a first-in, first-out order of operations.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 30 -
10.4 Storage and Transportation Conditions
Potential risks (e.g. temperature, environmental pressure, vibration) that may affect sterility of
intermediate products during storage or transportation should be identified, and acceptable working
conditions or specifications for such risk factors should be specified. Storage and transportation
operations should be conducted with care not to violate established conditions or specifications.
11. Environmental Monitoring
The primary objective of environmental monitoring is to keep manufacturing environments for
sterile pharmaceutical products clean by controlling the levels of microorganisms and airborne
particles within specified limits for individual APAs and indirect support areas, by monitoring
environmental conditions to prevent environmental deterioration and product contamination, and by
continuously assessing the efficiency of cleaning, disinfection, and decontamination procedures.
Environmental monitoring may be classified into two categories: microbiological and particle
control. Microbiological control is not intended to identify and characterize all microorganisms
present in the environment but to scientifically estimate bioburden value of the environment, ensure
that the manufacture of sterile pharmaceutical products is conducted in an appropriately controlled
environment, and implement measures (e.g. disinfection) necessary for maintaining the environment
at the required cleanliness level.
11.1 General Requirements
1. Scope of application
Environmental monitoring should be conducted in critical areas (Grade A) which are APAs,
direct support areas (Grade B), and indirect support areas (Grade C or D) adjacent to APAs.
2. Environmental monitoring programs
An environmental monitoring program and SOPs for implementing the program should be
established, and outcome of the implementation should be adequately recorded. The program
should be developed by assessing and examining properties of substances to be monitored,
frequency of monitoring, sampling locations, and action levels in order to appropriately
estimate environmental contamination risks.
3. Monitoring targets
Monitoring targets are microorganisms and airborne particles.
(1) Target airborne particles are those ≥ 0.5 μm in diameter. Particles of other diameter (e.g.
≥ 5 μm) should be measured as required by a need of environmental monitoring on an
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 31 -
as-needed basis.
(2) Target microorganisms are bacteria and fungi.
(3) Target microorganisms are airborne bacteria and microorganisms on the surface of walls,
floors, fixtures, equipment, gowns, etc.
4. Preparation of environmental monitoring program
An environmental monitoring program should be drawn up prior to performance qualification
(PQ) and finalized after PQ completion. Finalization of the monitoring program may require
reevaluation of the monitoring program initially developed based on PQ and subsequent
inclusion of the monitoring program in the routine control program for routine practice. Since
PQ requires performance testing of the worst-case scenario, the numbers of sites and
measurement of monitoring targets tend to be large. Reduction in number of sampling
locations and frequency is acceptable when the monitoring program is included in the routine
control program, as is reduction in frequency of bacterial monitoring by implementing
adequate inspection, maintenance, and supervision of equipment on regular and occasional
bases, provided the equipment has isolators, RABS, a blow-fill-seal system, or other devices
which prove it robust enough to withstand bacterial contamination. Requirements for routine
monitoring and control such as the number of sampling site may be reduced by referring to
ISO specifications including ISO DIS 14644-1.
5. Monitoring targets and locations
Environmental monitoring targets should also include air in working areas, manufacturing
equipment (and process control equipment, where appropriate), and aseptic environments; air
for keeping the aseptic environment clean; and compressed air or gas that comes in contact
with the environment and equipment. The monitoring frequency of compressed air and gas
necessary for manufacturing equipment or used during manufacturing processes as shown in
Table 2 may be separately set, provided the cleanliness level can be maintained by integrity
test for filters for sterile liquid filtration or other tests.
6. Sampling frequency for environmental monitoring
Sampling frequency should be determined in accordance with air cleanliness level required for
individual working areas under both operating and non-operating conditions. The sampling
procedures should include specifications on the frequency of sample collection from gown and
other stuff. Frequencies of sampling shown in Table 2 may be helpful for establishing the
specifications.
7. Monitoring methods: sampling and testing procedures
Optimal number and locations of monitoring points should be determined for individual
manufacturing areas by taking into account the size of working area, scope of operations, and
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 32 -
process flows of materials or products. The points should be added as they are considered to
be necessary for assessing potential product contamination.
(1) Devices for collecting and counting airborne particles and bacteria should be used after
validated calibration. Results of particle counting should be converted to the count
per-cubic-meter of atmosphere (/m
3
).
(2) Sampling for collecting airborne microorganisms should be conducted via one or more
suitable procedures including settle plate, impact, and filtration methods, and collecting
microorganisms on the surface should be conducted via one or more suitable methods
such as contact plate or swabbing. The size of the area to be sampled should be
determined in accordance with the shape and surface condition of equipment and
apparatuses. In principle, the recommended sampling area of equipment and apparatuses
is 24 to 30 cm
2
. Air volume to be sampled for airborne microorganism monitoring
should be decided by overall considerations and discussion of factors involved, such as
cleanliness of the target area and routine monitoring frequency. If the target area is
Grade A, microbial count monitoring usually uses a circular flat plate of 90 cm in
diameter, and the maximum exposure time is 4 hours.
(3) The culture medium used for the detection and enumeration of airborne and surface
microorganisms should be suitable for the growth of target microorganisms such as
aerobic bacteria, fungi (i.e. yeasts, molds), and anaerobic bacteria. The medium should
be tested for the absence of cell growth inhibitory substances to select a competent
medium suitable for microbacterial monitoring. The objective of testing for cell growth
inhibitory substances is to confirm that the collection and growth of microorganisms
will not be affected by the presence of alcohol, antibiotics, etc. and to ensure that
monitoring results are not altered by the quality of medium used.
(4) The incubation temperature of the medium should be suitable for the growth of target
microorganisms.
8. Alert and action level specifications
Alert and action levels should be established for individual target substances and locations to
be monitored.
(1) Action level specifications may be established by referring to data contained in Table 3.
Caution should be exercised not to underestimate the contamination risk by averaging
particle or microbacterial count. If microorganisms are detected in a Grade A area, the
effect of such microorganisms on product quality should be evaluated even if the count
meets acceptable criteria.
(2) Alert level specifications should be established based on results of PQ tests.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 33 -
(3) The monitoring program should include actions and measures to be taken (i.e.,
investigation of causes of non-compliance, suspension of manufacturing) when alert or
action level specifications are met. In principle, a deviation from the alert level
specifications does not require a suspension of manufacturing but other appropriate
actions or measures should be taken. Deviations from the action level specifications
should be investigated for cause(s) prior to shipment of final products manufactured
through the process where the deviation occurred, and corrective measures should be
taken. The validity of corrective measures taken should be verified to confirm the
recovery of acceptable environmental conditions, as needed. The recovery may be
readily confirmed in some instances by, for example, counting particulate matter, but
not reproducible in other instances, such as with bacteria adherence to gowns. If the
cause(s) cannot be traced, recovery should be established by general approaches
including prohibition of personnel entry for a certain period, retraining of personnel, and
reviewing assigned tasks.
11.2 Routine Monitoring and Control
1. Implementation of the monitoring program
Monitoring of microorganisms and particulate matter should be routinely performed in
accordance with the monitoring program.
2. Microbiological control
The microbiological environmental monitoring program should be routinely performed to
monitor potential microbial contamination. The program should include periodic investigation
on characterization of environmental flora and isolates to assess contamination risks to
pharmaceutical products.
3. Sampling
Sampling of surfaces that come in contact with pharmaceutical products or other materials in
critical areas should be performed immediately after the completion of filling or other aseptic
processing operations.
4. Gases for manufacturing processes
Gases that may directly contact pharmaceutical products, primary containers, and surfaces that
come into direct contact with pharmaceutical products should be periodically inspected and
controlled to ensure the absence of microorganisms.
5. Routine analysis
For the adequate maintenance of the manufacturing environment, data obtained from routine
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 34 -
monitoring should be analyzed to detect any trends in changes in the environment and
establish monitoring limits for trend analysis. Even if changes in the environment do not
deviate from the specified limits (the alert level), any trends suggesting variations from normal
conditions (trend analysis level) should be predicted and the cause(s) investigated to maintain
the quality of the environment at an appropriate level. Trend analysis results should also be
utilized for the maintenance of equipment for environmental control, such as the HVAC
system, and for optimization of sterilization and disinfection procedures.
11.3 Example Assessment Criteria for Environmental Monitoring
Table 2 shows example frequencies of environmental monitoring classified by the cleanliness
level, and Table 3 shows acceptance criteria for airborne particulate matter and microorganism
counts. Since the risk of contamination varies depending on the formulation and size or volume of
pharmaceutical products, structure/function of manufacturing equipment, automation level, time of
retention of closures, and availability and performance of equipment for environmental control such
as the HVAC system, the environmental monitoring program should be prepared and implemented
by taking these factors into account:
1. The frequency of microbiological monitoring may be increased or decreased depending on the
type and time of processing activities; however, the frequency needs to be adequate for
effective monitoring of potential microbiological contamination of pharmaceutical products.
2. The frequency of microbiological sampling from the surface of personnel gown and other stuff
should be commensurate with ability and experience of individual personnel. For example,
sampling frequency should preferably be increased for operators with less aseptic processing
experience. The ability and experience of personnel should be collectively evaluated based on
the frequency of engagement in aseptic processing, microbacterial monitoring data, frequency
of participation in media fill tests, etc.
3) The monitoring frequency for Grade C and D areas should be determined by the types of
pharmaceutical products, processes, operations, etc. to be performed in the areas for
appropriate quality control and risk management. The frequency may be decreased if the risk
of contamination is low, such as when pharmaceutical products are not exposed to the
environment.
4. Monitoring should be reinforced immediately after facility operation is started (beginning of
PQ), restarted after long-term shutdown, or partially changed.
5. When personnel enter a Grade A area from a Grade B area, surface microbacterial count on
gowns and other stuff should be evaluated against stricter acceptance criteria (those for Grade
A area) depending on the level of product contamination risk.

Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 35 -
6. Sampling of particulate matter in Grade A and B areas should preferably be conducted via
continuous monitoring from equipment assembly until completion of critical operations.
7. The monitoring of particulate matter during times when no manufacturing operations are
taking place should be conducted on an as-needed basis to maintain the environment at
predetermined cleanliness levels and thereby, for example, detect malfunctions in the air
conditioning system.
8. Assessment results of particulate matter monitoring may differ depending on the amount of air
sampled and air suction capacity of monitoring devices. Air samplers and assessment method
should be appropriate for the particulate matter control system used.
Table 2. Frequency of Environmental Monitoring for Microbacterial Control
Cleanliness grade
Airborne
particulate
matter
Airborne
microorganisms
Surface microorganisms
Equipment and
walls
Gloves and
gowns
A
During
processing
Every working
shift
After completion
of processing
After completion
of processing
B
During
processing
Every working
shift
After completion
of processing
After completion
of processing
C, D
Area in which
products and
containers are
exposed to the
environment
Once a month Twice a week Twice a week ----
Other areas Once a month Once a week Once a week ----

Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 36 -
Table 3. Acceptance Criteria for Environmental Microorganism Count (during Operations)
note 1
Cleanliness
grade
Airborne microorganisms Surface microorganisms
Air
(CFU/m
3
)
Settle plate
Note 2
(CFU/plate)
Contact plate Gloves
(CFU/24−30 cm
2
)
(CFU/5 fingers)
A < 1 < 1 < 1 < 1
B 10 5 5 5
C 100 50 25
−
D 200 100 50
−
Note 1) Acceptance criteria are expressed as mean values.
Note 2) Measurement time per plate is 4 hours at maximum and the measurement is
performed during processing operation.
12. Qualification of Equipment and Utilities
12.1 General Requirements
1. In this section, the term “equipment” refers to equipment used for sterilization, filtration,
filling, capping, freeze-drying, and sealing in the manufacture of sterile pharmaceutical
products in the APA, as well as HVAC system, incubators, fermentors, and cleaning
equipment installed, as required, in indirect support areas.
2. In this section, the term “utilities” refers to systems for supplying different qualities of water,
pure steam, compressed air, and different kinds of gases in the manufacture of sterile
pharmaceutical products.
3. For the qualification of equipment and utilities, qualification protocols and SOPs should be
established to define assignment of responsibility of individual personnel and other related
matters. Equipment and utilities used for the manufacture of sterile pharmaceutical products
should be designed so as to have minimum influence on the sterility of the products. The
structure or shape and components materials of equipment and utilities should be selected to
make it easy for cleaning, disinfection, sterilization, and maintenance. Special attention should
be paid to the surface of equipment and utilities to which pharmaceutical products, component
materials, water, steam, or gases etc. may be directly exposed.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 37 -
4. Flow lines for sterile pharmaceutical products, sterile raw materials, and other sterile materials
should beadequately designed by taking into account personnel movement and airflow
patterns.
5. Personnel movement and intervention into sterile pharmaceutical products should be designed
to be minimum. In addition, operation, maintenance, repair, and adjustment of equipment
should preferably be performed from outside the critical area, whenever feasible.
6. Generation of turbulence and particles in critical areas should be controlled to a minimum.
The flow of clean air from supply vent to return or exhaust vent should be optimally designed
in direct and indirect support areas.
7. Equipment should be laid out so as to minimize the physical burden on operators.
8. Requirement specifications (user requirements specifications, URS) for equipment and utilities
should be defined in writing with regard to required quality levels, facility capacity for
amounts of use during manufacture, applicable regulatory requirements (e.g. laws, regulations,
guidelines), quality of component materials, and performance, etc., and DQ should be
conducted in accordance with the URS.
9. The duration of exposure of sterile pharmaceutical products, surface of equipment that may
contact with sterile pharmaceutical products, and containers uncapped, should be kept as short
as possible. 10. IQ should verify that the equipment and utilities have been installed as
directed in relevant design specification in accordance with written procedures.
11. OQ should verify that equipment and utilities have a capacity of performance as required by
their specifications. If the equipment and utilities are to be operated or used in APAs, it should
be verified that the required cleanliness in the APAs is maintained throughout operation or use.
12. All processes conducted in the APA that may influence the sterility of pharmaceutical
products should be scientifically evaluated and appropriately validated.
13. Operational procedures for all key equipment and control parameters, and theire acceptable
limits should be described in relevant SOPs in an appropriate manner.
14. Validation of the processes utilizing cleaning, sterilizing, incubating/fermenting, filtering,
filling, capping, freeze-drying, and sealing equipment should be conducted to assess the
sterility assurance level of pharmaceutical products in each of these processes. Sterility
assurance levels may be validated together for multiple processes using different equipment if
the processes are continuous.
15. The sterility of equipment surfaces that may come into direct contact with sterile
pharmaceutical products should be validated.
16. OQ studies should be conducted for utilities including CIP/SIP systems and equipment that
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 38 -
supply purified water, water for injection, compressed air/other gases, pure steam, etc.
17. Validated period for use of the equipment after sterilization should be established to ensure the
sterility of sterile pharmaceutical products. If critical process changes are made, the potential
impact of the changes on validated period foruse should be reevaluated.
18. Since design concepts applicable to aseptic manufacturing of sterile pharmaceutical products
vary, any additional techniques available for promoting sterility assurance should be positively
employed whenever available.
19. When sterilization equipment is a continuous system, conveyor belts should not pass through a
partition between an APA and a processing area of lower air cleanliness, unless the belts
themselves are continuously sterilized (e.g. heat sterilization tunnel). When a continuous
sterilizer is used, airflow should be monitored to ensure that air does not flow from a
non-sterile to sterile area during processing.
12.2
Equipment Maintenance
1. For the preventative maintenance of equipment and utilities, maintenance protocol and SOPs
should be prepared to define the assignment of responsibility of individual personnel and other
related matters.
2. SOPs should also be prepared for cleaning, disinfection, and sterilization procedures for
equipment and utilities and their use permission in subsequent manufacture. The procedures
for cleaning, disinfection, and sterilization should be as specific and detailed as possible to
achieve cleaning, disinfection, and sterilization of equipment in an efficient and reproducible
manner. These procedures should address the following:
(1) Assignment of responsibility of individual personnel for cleaning, disinfection, and
sterilization of equipment and utilities
(2) Cleaning, disinfection, and sterilization schedules
(3) A complete description of the procedures, instruments, apparatuses, and agents used for
cleaning, disinfection, and sterilization (including a procedure for diluting cleaning
agents) of equipment and utilities
(4) Instructions for disassembly and reassembly of pieces of equipment and utilities
necessary to ensure adequate cleaning, disinfection, and sterilization, where appropriate
(5) Instructions for the removal or deletion of description regarding previous batch
(6) Instructions for preventing contamination of cleaned equipment and utilities until next
use
(7) Inspection of equipment and utilities to confirm cleanliness level and sterility
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 39 -
immediately before use, if feasible
(8) Maximum allowable time for cleaning, disinfection, and sterilization of equipment
and utilities after completion of manufacturing, where appropriate
3. Equipment and utilities should be kept after cleaning, washing, drying, preserved, and, when
necessary, disinfected or sterilized to minimize their influence on the sterility of sterile
pharmaceutical products.
4. When successive lots of the same sterile pharmaceutical product are produced, one at a time
and in succession, using the same equipment and utilities for continuous or period (campaign)
production, the equipment and utilities should be cleaned, disinfected, and sterilized at
intervals that have been validated as effective for the prevention of microbial contamination.
5. The evaluation of cleaning, disinfection, and sterilization of equipment and utilities used in the
manufacture of sterile pharmaceutical products like vaccines containing live microorganisms
per se or other ingredients of bacterial origin should include the evaluation of efficiency of
these procedures in removing microorganisms and other ingredients of bacterial origin. The
evaluation of sterilization efficiency may be omitted when target microorganisms have been
documented to be less resistant to these processes than microorganisms specified in the
Japanese Pharmacopoeia or other official compendia.
6. The procedures for cleaning and for selecting cleaning and disinfecting agents should be
specified and justified with rationale and adequate evidence.
7. All equipment and utilities should be identified by appropriate methods based on materials or
products to be processed and cleanliness levels required.
8. If stopped for repair or inspection, equipment and utilities should be disinfected or sterilized
prior to resumption of operation, as required.
12.3 Calibration
1. For the calibration necessary for the control, measurement, and monitoring of equipment and
utilities critical in ensuring the sterility of pharmaceutical products, calibration protocol and
SOPs should be established to define assignment of responsibility of individual personnel and
other related matters. These SOPs should then be followed for calibration.
2. The calibration of equipment and utilities should be performed using certified and traceable
standards, whenever available.
3. Records of the above-mentioned calibration procedures should be maintained.
4. The current calibration status of critical equipment and utilities should be known to relevant
personnel and verifiable.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 40 -
5. Instruments that fail to meet calibration criteria should not be used.
6. When a critical instrument for ensuring the sterility of sterile pharmaceutical products shows
deviations from approved standard calibration values, investigation and assessment of the
deviations should be conducted to judge whether or not the deviations have affected sterility of
pharmaceutical product lots manufactured in an environment which has been controlled,
measured, or monitored by the instrument of concern since its last calibration.
12.4 Change Control
1. For the confirmation, verification, approval, and recording of changes in equipment, utilities
(including parameters), and procedures which may be critical in ensuring the sterility of
pharmaceutical products, change control SOPs should be established to define assignment of
responsibility of individual personnel and other related matters.
2. Changes referenced in Item 1 above should be drafted by an assigned person, reviewed by a
qualified person, and approved by the quality department, since such changes carry a risk of
altering the capacity and performance of equipment affecting the quality of pharmaceutical
products.
3. Proposed changes should be evaluated concerning their potential impact on sterility of
pharmaceutical products from the viewpoint of risk management. The impact evaluation
should be based on points of consideration referred to in Article 12.1 above.
4. Prior to the implementation of approved changes, all SOPs should have a provision ensuring
revision of all documents to be affected by approved changes.
5. Personnel responsible for operating equipment affected by approved changes should be trained
prior to the implementation of the changes.
6. The potential impact of approved changes on the valid time of use of sterilized equipment
should be assessed to ensure the sterility of sterile pharmaceutical products.
13. Sterilization Process
13.1 General Requirements
1. Containers and closures that come into direct contact with pharmaceutical products and the
surfaces of equipment that may come into direct contact with intermediate products after
sterilization should be sterilized by methods appropriate for maintaining the predetermined
sterility assurance level.
2. Equipment surfaces that may come into direct contact with containers and closures should also
be sterilized as required for maintaining the predetermined sterility assurance level.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 41 -
3. Materials to be sterilized should be handled by techniques appropriate for avoiding mix-ups of
sterilized and unsterilized materials.
4. Already-sterilized materials should be treated with appropriate preventive measures to avoid
re-contamination. As a rule, such materials should be handled in accordance with the aseptic
processing procedures recommended in this guidance, particularly when directly exposed to
the environment.
5. Sterilization processes for sterilizing pharmaceutical products and materials in critical areas
should be individually validated and also periodically evaluated at least once a year.
6. History of sterilization equipment usage should be adequately controlled and maintained by,
for example, keeping logbooks.
7. Sterilization-related procedures and control parameters for process control, routine monitoring
and control, maintenance and control, supplies, and sterility verification should be fully
documented.
13.2 Autoclaving
1. The quality of steam used for sterilization should be ensured not to adversely affect the
function and safety of materials or equipment to be sterilized. The generally recommended
procedure is to use vapor (pure steam) generated from purified water or water of high quality.
Condensate water resulting from the vapor should also meet specifications for water of higher
purity than that used for product formulation. The description of vapor should be periodically
inspected, and causes of quality deterioration should be investigated, whenever suspected, in
order to implement proper corrective measures.
2. Appropriate control procedures (e.g. visual inspection procedure, maximum allowable
frequency of steam cleaning) should be established for the sterilization of materials to be used
repeatedly (e.g. filters, utensils, aseptic gowns) to ensure maintenance of specifications, safety,
and intended functions after repeated exposure to steam at its maximum intensity. Accordingly
materials for repeated use should be properly managed by the control procedures.
13.2.1 Sterilization Process
1. Acceptable limits of sterilization-related process parameters should be established and
documented.
2. When the sterilization process includes air purging, methods and specifications should be
established for measuring and evaluating the maximum acceptable limits of air leak volume
for sterilization equipment and permissible residual volume of non-condensable gas in
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 42 -
materials to be sterilized.
3. If air or water come into direct contact with materials to be sterilized during the sterilization
process, their purity and physical characteristics (e.g. pressure, temperature) should not
adversely impact the intended performance or safety of the materials.
4. Commercial biological indicators (BIs) and chemical indicators (CIs) used in the verification
of the sterilization process should conform to international standards or other official
specifications.
5. When the validity of a certain sterilization process is tested by simulation using a dummy load,
the validity, efficacy, and time limit of use of the load should be verified and documented.
6. When the sterilization process includes a procedure or procedures other than sterilization (e.g.
drying), the assessment method for such a procedure or procedures should be established,
documented, and implemented for appropriate control.
7. Pretreatment procedures in the sterilization process (e.g. cleaning) should be defined with
appropriate conditions and controlled accordingly so as not to impair the validity of the
sterilization process.
13.2.2 Sterilization Equipment
1. Key properties of sterilization equipment including manufacturer’s name, type, size, structure,
materials of construction, functions, and capacity, should be available in writing. The user
manual should also be available with the following outlined: methods of standard operation,
default setting, emergency responses, disassembly and reassembly, maintenance control
(including calibration), etc.
2. Sterilization equipment should have basic performance requirements for sterilization such as
establishment of operational parameters and processing capacity.
3. Parts of sterilization equipment that are exposed to the stress of sterilization procedures (e.g.
inner wall surface, pipes) should be made of materials resistant to such stress. The materials
should not release any substances that may have undesirable effects (e.g. interactions,
decomposition, absorption) on the quality of sterilization processes or pharmaceutical
products.
4. Utilities such as electricity and compressed air should be constantly supplied to sterilization
equipment to ensure consistent operation throughout the sterilization process.
5. When materials to be sterilized are not hermetically sealed, gas used for aeration or pressure
recovery should be sterilized. Filters used for gas sterilization should have a structure suitable
for sterilization and be made of materials resistant to sterilization procedures. In addition,
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 43 -
filters should be tested for their integrity to ensure the sterility of gas to be supplied.
6. Cycle parameters of sterilization equipment necessary for monitoring sterility of materials or
products should be freely established in ranges suitable for the control of sterilization
processes and easily managed with excellent reproducibility. Sterilization patterns of the
equipment should also be easy to establish depending on properties and physical state of
materials to be sterilized.
7. Sterilization equipment should be equipped with functions (e.g. computerized system control)
that enable the sterilization cycle to proceed accurately. If the equipment is of the continuous
cycle type, there should be a function that enables the correct transfer of products into and out
of the sterilizer chamber.
8. Sterilization equipment should be equipped with appropriate sensors and recorders for the
measurement and control of critical cycle parameters to achieve the required level of
sterilization. The specifications (e.g. type, precision, materials), location, and other
requirements for the sensor should be selected based on characteristics and required conditions
of the sterilization process to be run with the equipment.
9. Sterilization equipment should have a function to ensure maintenance of conditions
permissible for anticipated sterilization processing at all times during operation. It is
recommended that alarming and recording systems which function in response to the type and
severity of emergency be installed. It is also recommended that safety devices (e.g. safety
valves) be installed to prevent major accidents.
10. The location where sterilization equipment is installed should have sufficient space for the
operator to work and should be maintained at the required cleanliness level.
11. Sterilization equipment should be designed to facilitate easy manual operations by the operator,
such as operation of control buttons and transfer of pharmaceutical products into and out of
the chamber.
12. If the computer system for manufacturing control or other purposes is connected to and
controlled by a higher-level host computer, input/output data, control specifications, and other
processing should be precisely documented.
13. SOPs should be established and documented to ensure reflection of physical changes and
process changes made to sterilization equipment in the specifications for the equipment.
13.2.3 Validation of Sterilization Procedures
The method for validation of autoclave cycles comprises tests on heat distribution in the
sterilization chamber, heat permeability of sterilization load, and verification of sterilization capacity
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 44 -
using BIs. This validation also serves as PQ of sterilization equipment.
1. A heat permeability test should be conducted using materials to be actually sterilized. Except
for samples for temperature measurement, it is acceptable to use a reference load instead,
provided the use of the reference load can demonstrate scientific validity of its use based on
physical data obtained
2. The heat permeability test should be conducted for different patterns of loading including
maximum load, at least 3 times for each pattern. The minimum loading pattern test should be
conducted as required. Pictures or photographs showing loading patterns used in the test
should be recorded.
3. The heat permeability test may be conducted by grouping products and loading patterns if the
grouping is acceptable with regard to type and properties of materials to be sterilized and
batch sizes for sterilization.
4. Locations of verification thermometers should include cold spots of materials to be sterilized
and, as appropriate, hot spots.
5. The temperature at cold spots should be confirmed with a thermometer to verify that
predetermined sterilization conditions for materials to be sterilized have been attained with
heating.
6. The accomplishment of sterilization at cold spots should be verified using relevant BIs. For
details on available BIs, refer to ISO 14161 (Sterilization of Health Care ProductsBiological
Indicators: Guidance for the Selection, Use and Interpretation of Results).
7. When sterilization cycles are established based on bioburden determination with materials to
be sterilized, the count and resistance of BIs and assessment methods for these parameters
should be selected based on predicted or established bioburden levels.
8. The integrity of materials to be sterilized should be verified by established sterilization cycles.
9. The time for sterilization cycles should be confirmed to be compatible with the time schedule
of actual manufacturing.
10. If heat distribution is determined using a thermometer not originally provided with
sterilization equipment, the thermometer should be calibrated before and after the
determination.
11. Sterilization equipment should be validated again if the structure of the equipment is modified,
if loading conditions for materials to be sterilized are changed, or if utilities supply conditions
are changed. The scope and frequency of revalidation are dependent on risk of inadequate
sterility assurance of pharmaceutical products.
12. The sterilization of porous materials should be conducted after carefully establishing
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 45 -
sterilization cycles to achieve thorough sufficient ventilation to replace air deep in the
materials with vapor.
13. Air inside the chamber of the sterilizer should be periodically confirm to be completely
removed during sterilization cycles. This check should be added to routine monitoring and
control items, as required. The typical air removal test is the Bowie-Dick test.
13.2.4 Routine Monitoring and Control
1. Process parameters and control items necessary for routine monitoring and control of the
sterilization process should be determined and documented based on validation data. The
validity of the process parameters and control items should be verified by confirming
reproduction of specified conditions of sterility for individual materials to be sterilized.
2. Test items as well as detailed operating procedures and frequency of testing should be
documented for periodic check-ups, maintenance control, calibration, and equipment.
3. Routine management and control of the sterilization process should be performed on a
cycle-by-cycle basis.
4. Data on sterilization cycle-related parameters should be obtained and recorded to verify that
sterilization of materials has been successfully achieved. Recorded data should include
readings of the inner pressure and temperature of the sterilization chamber in each sterilization
cycle.
5. The completion of sterilization cycles within specified limits of relevant specifications should
be verified by direct measurement of selected cycle parameters, and obtained results should be
recorded. If necessary, BIs and CIs should also be monitored.
6. A leak test should be periodically performed when the sterilization process incorporates an air
elimination process for steam penetration. Any additional checks on performance other than
sterilization (e.g. drying of materials to be sterilized) that may have potential influence on
product quality should be conducted and recorded according to written procedures.
13.2.5 Handling of Sterilized Materials
1. SOPs should be prepared and implemented for handling materials following completion of the
sterilization process. The SOPs should include methods and criteria for assessing sterilized
materials to confirm that the sterilization process has been adequately conducted to comply
with relevant requirements. When additional parameters (e.g. BIs, CIs) other than process
parameters are required for the assessment of complete sterilization, specifications to be met
with such parameters should be included in the assessment criteria.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 46 -
2. The SOPs should also specify procedures for obtaining and storing various records of
sterilization processing. The records should include information on the following matters and
be reviewed and approved by the supervisor:
(1) Time started and ended and date of sterilization processing
(2) Sterilization equipment used
(3) Materials sterilized
(4) Sterilization conditions employed
(5) Assessment criteria and results of sterilization processing
(6) Records of physical process parameters (e.g. temperature, pressure, etc.)
(7) Identification of sterilized materials and their traceability
(8) Operators’ names
When sterilization is carried out by a batch process, retrospective investigations of the
sterility of sterilized materials can be easily traced by allotting batch numbers to individual
processing.
3. If any materials are judged not to have been adequately sterilized, such materials should be
handled in accordance with relevant SOPs. Causes of inadequate sterilization should be
investigated and appropriate corrective actions implemented.
4. Sterilized materials should be stored under conditions suitable for preserving and maintaining
their sterility and other properties. The location, method, environmental conditions, and
duration of storage should be predetermined and managed accordingly.
13.3 Dry Heat Sterilization
Basic requirements and control methods for dry heat sterilization should be consistent with
those specified for autoclaving. Additionally, the following dry heat sterilization-specific control
measures should be met.
1. The dry heat sterilization process should be validated via endotoxin challenge test or other
appropriate method when the process requires depyrogenation.
2. Materials to be sterilized should be periodically tested for pre-sterilization endotoxin content.
3. HEPA filters mounted on sterilization equipment should be periodically tested for leaks to
check the capacity of the filters. The test should ideally be performed once every 6 months or
at least once a year.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 47 -
13.4 Electron Beam and Gamma Ray Sterilization
Basic requirements and control procedures for electron beam and gamma ray sterilization
should be consistent with those specified above for autoclaving. Additionally, the following criteria
specific to electron beam and gamma ray sterilization should be met:
1. The dose of radiation necessary for achieving complete sterilization should be determined
based on acceptable validation data.
2. Sterilization process parameters should be established based on appropriate validation data.
Adequate records verifying that the irradiation has been performed in accordance with the
process parameters should be obtained and recorded.
3. Bioburden assay of materials to be sterilized should be performed prior to sterilization at a
predetermined frequency.
4. The loading configuration of materials to be irradiated should be evaluated and documented
based on validation data. Procedures for adequate storage and control of materials before and
after sterilization should also be documented.
5. The name, loading configuration, quantity, irradiation date, and dose absorbed should be
controlled for irradiated materials. These materials should be identified in an appropriate
manner (e.g. sterilization batch number) to ensure the traceability of individual materials.
6. Irradiated materials should be placed in the smallest packaging units available for storage and
control and labeled as “irradiated” in a readily accessible location outside the container.
7. The radiation dosage measurement system should ensure traceability of measurement results
to national standards.
8. When the irradiation sterilization process is contracted out, the consigner and consignee
should agree to at least the following matters in writing:
(1) Preservation of the sterility of consigned goods during transportation
(2) Preparation of consignee’s statement certifying that consigned goods have been
sterilized
(3) Disclosure of sterilization conditions, upon request by the consigner, for each lot of
consigned goods, as appropriate
9. The predetermined radiation dosage should be periodically checked at appropriate intervals to
ensure the efficacy of irradiation sterilization cycles (sterilization dose audit).
13.5 Other Sterilization Methods
Basic requirements and control procedures for electron beam and gamma ray sterilization
should be consistent with those specified for autoclaving. Additional control measures specific to
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 48 -
electron beam and gamma ray sterilization should be established and implemented, as appropriate.
14. Clean-In-Place System
A clean-in-place (CIP) system is a cleaning method which is designed to clean an entire system of
equipment with appropriate cleaning agents in situ without any disassembly of equipment
components, or pipes. Points to consider in designing equipment to be subjected to CI P and in
implementing the system are summarized below. Points to consider in implementing the system
should also be applied to general cleaning operations.
14.1 Design Considerations for the CIP System
When designing equipment, pipes, cleaning agent supply apparatus, etc. for the CIP system,
the following technical points should be considered:
1. Equipment and piping with smooth inner surfaces should be selected and incorporated into the
CIP system to facilitate cleaning effectiveness. The CIP system should be designed to allow
for prompt confirmation of cleanliness level after completion of the CIP process.
2. Presence of “dead legs” in piping connected to the equipment should be minimized. The
equipment, piping, and valves within the equipment should have designed to have adequate
slope to allow for draining of both cleaning agents.
3. The cleaning agent supply portion of the CIP system should be designed to maintain constant
flow rate, pressure, temperature, and concentration of cleaning agents.
4. When equipment and/or pipes to be subjected to CIP are washed by dividing them into
several segment, the segments should overlap o ensure that all portions of the system are
adequately and effectively cleaned.
5. After completion of a CIP process, equipment or systems much be able to be stored in a
manner which prevent recontamination.
14.2 Selection of Cleaning Agents
1. Cleaning agents should be selected after evaluating their ability to remove residual substances,
physicochemical properties of residual substances to be removed, and compatibility with
manufacturing equipment. All components of cleaning agents must be removed to levels
below specified detection limits before starting the final rinsing process.
2. Examples of cleaning agents include water, hot water, detergents, alkaline solutions, hot
alkaline solutions, and organic solvents.
3. The quality of water used for final rinsing of product contact equipment surfaces should be of
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 49 -
the same quality as water used for product formulation.
4. Quality control specifications of cleaning agents should be established and documented.
14.3 CIP Process Parameters
The most difficult to clean locations within a CIP system should be identified at the validation
stage, and if necessary additional cleaning operations or processes should be developed and verified
for cleaning efficacy. Based on validation results, CIP process parameters to achieve the acceptable
level of cleanliness should be specified and documented for the control of the cleaning process to
achieve the predetermined level of cleanliness. The CIP process parameters should include the
following:
1. Type and concentrations of cleaning agents
2. Flow rate of cleaning agents
3. Duration of contact between the equipment or process surfaces and cleaning agents.
4. Temperature and pressure of cleaning agents
5. Total cleaning time
6. Control parameters that indicate the acceptable residual substances after completion of CIP,
such as conductivity, pH, and total organic carbon (TOC) (to be determined based on the
composition of cleaning agent)
7. Maximum allowable time until start of CIP process after completion of the manufacturing
process (Maximum allowable time should be controlled to avoid so as not to be hard to
remove the residual substances by elapsed time until start of CIP.)
14.4 Routine Monitoring and Control
The technical performance of each CIP system should be recorded and data retained for
periodically review. CIP and related records should include, but not be limited to, the following:
1. Time and date
2. Name of equipment cleaned
3. Name and production batch number of pharmaceutical products manufactured prior to CIP
cycle
4. Name and production batch number of pharmaceutical products manufactured after cleaning
with the CIP system
5. Names of CIP operators
6. Operating conditions of the CIP system
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 50 -
7. Verification of the compatibility of CIP conditions employed
8. Allowable time between the completion of the CIP processing and the use of equipment
cleaned with the CIP system
9. Validity of calibration of instruments used to detect the completion of cleaning process and
indicate CIP process parameters such as flow rate and pressure, etc. .
14.5 Maintenance and Control
Critical equipment such as pumps, which are closely related to CIP parameters including pressure,
temperature, and flow rate, should be well controlled and subjected to maintenance at defined
intervals. When critical equipment is replaced, new replaced equipment should be selected as
equivalent performance. Replaced equipment should be assessed and documented to achieve
equivalent cleaning efficiency.
14.6 Personnel Training
The education and on job training programs for personnel engaged in CIP operations should cover
at least the following:
1. Design and functions of the CIP equipment and an operational outline of CIP process
2. Possible corrective actions to be taken in the event of deviations or OOS results in the CIP
process
3. Any other technical issues of significance in the operation of a specific CIP process.
15. Sterilization-In-Place System
A sterilization-in-place (SIP) system is a sterilization method which is designed to sterilize an
entire system of equipment in situ without disassembly of components, or piping. The most common
sterilizing agent for SIP is saturated steam (moist heat).
15.1 General Requirements
1. When equipment (e.g. tanks, filling lines, transfer lines, filtration system, water for injection
systems) that cannot to be sterilized by autoclaving due to size or shape is subjected to
sterilization-in-place (SIP), the efficacy of SIP (typical sterility assurance level [SAL]: ≤ 10
-6
)
should be demonstrated by an appropriate measuring instrument such as temperature gauge,
pressure gauge, thermocouple, and moist heat-resistant BIs. Care must be taken to ensure that
the placement of these BIs does not obstruct the flow of steam or the ability of the system to
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 51 -
drain condensate.
2. Steam used in the SIP process should be generated from purified water or water of not less
than purified water grade. Condensed water from steam should meet specifications of water
used for product formulation.
3. Locations most difficult to sterilize so-called “cold spots” in the equipment should be
identified, and the SAL achieved at these spots should be evaluated are appropriate intervals.
4. Equipment integrity should be maintained after completion of SIP. Steam and condensate
should be purged from the SIP system by either sterile compressed air or nitrogen gas, and the
system should be maintained under positive pressure until it is used for processing. For any
equipment which may be operated under negative pressure or atmospheric pressure, a
qualification test must be performed to confirm that the sterility of the entire equipment is not
compromised. The maximum allowable time between the completion of SIP and the use of the
equipment should be specified and verified.
5. When the equipment is not equipped with an automatically controlled and valve sequenced
SIP system, manual SIP procedures should be established and then strictly complied with, and
critical procedures of the system should be double-checked. Records of manual SIP operations,
when performed, should be maintained as evidence that the operations were conducted as
stipulated in the procedures.
15.2 Key Design Considerations for the SIP System
SIP equipment should be confirmed to be compatible with steam to be used and
pharmaceutical products to be sterilized and should be designed not to retain air or condensed water
within the equipment. The following matters should be taken into account:
1. Smoothness of inner surfaces of the equipment
2. The design must ensure that saturated steam to reach all surfaces to be sterilized
3. Location of the saturated steam inlet and steam distribution
4. The system design should avoid the formation of air pockets within the SIP system and ensure
that condenses water is efficiently drained from the system.
There should not be unnecessary piping branches and dead legs should be minimized
5. All piping should be properly sloped to allow for adequate draining.
6. Appropriate location for steam and steam condensate discharge
7. Heat and pressure resistance of the equipment
8. Compatibility between construction materials and steam quality
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 52 -
9. Measures necessary to maintain sterility of the systems to be sterilized during and after SIP
processing, such as the installation of appropriate vent filters and maintenance of positive
pressure
9. When equipment and/or pipes are sterilized by dividing them into several segment, the segments
should overlap o ensure that all portions of the system are adequately and effectively sterilized.
15.3 Routine Monitoring and Control
1. When equipment to be subjected to SIP is washed by certain cleaning procedures, including the
CIP system, SIP processing should also be performed promptly after the completion of CIP or
washing. Data on SIP processing should be recorded and retained for each SIP process and
periodically reviewed for completeness and correctness. It is recommended that the following
parameters should be monitored and recorded continuously from the introduction of steam until
completion of SIP for each SIP operation: temperature (e.g. supply steam, inside tanks, drain
ports), pressure (e.g. supply steam, inside tanks, inside of pipes), and duration of SIP operation.
If continuous measurement and recording are not feasible, alternate monitoring and recording
methods should be instituted to confirm that processing requirements for sterilization parameters
have been met.
2. Process operation records and other records on SIP operations should include, but not be limited
to, the following:
• Time and date
• Names of equipment subjected to SIP
• Names of operators
• Operation conditions
• Verification of compatibility of SIP conditions employed
3. An appropriate system should be established for distinguishing the status of equipment before
and after SIP processing.
4. Filters for sterilizing gas and vent filters for tanks and chambers used in the SIP process
should be periodically tested for integrity to ensure that they functioning properly.
5. Critical instruments such as thermometers should be calibrated at appropriate defined
intervals.
15.4 Maintenance and Control
Valves and steam traps should be subject to periodic maintenance checks to ensure the proper
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 53 -
injection of steam for sterilization and that condensed water forming during SIP is properly
discharged. If the shape and size of pipes to be sterilized by the SIP system or steam supply
conditions are modified, such modifications should be subject to change control, documented and
validated.
15.5 Personnel Training
Education and training programs for personnel engaged in SIP operations should cover the
following:
1. Design and functions of the SIP equipment and an operational outline of SIP process
2. Appropriate countermeasures that may be taken to correct abnormalities in the SIP process
3. Any issues that are deemed by the user to be critical in the performance and assessment of the
SIP system
16. Aseptic Filling Processes
16.1 General Requirements
Aseptic filling processes should meet the following requirements:
1. SOPs for aseptic filling processes should be established describing in detail each operation
procedure for all the steps starting from the preparatory stage including the filling machine
assembly to sterilization, stoppering, capping, washing and cleaning after filling, and further
other matters necessary for operation (e.g. control parameters for equipments, movement and
behavior in clean room, system for responsibility, permissible interventions).
2. Processing of sterile pharmaceutical products (e.g. filling, capping, freeze-drying) and
operations where sterile containers (including stoppers) that directly contact with aseptically
filtered products will be exposed to the environment, should be done in a critical area (Grade
A). If vial capping is undertaken outside the critical area, vials should be protected with Grade
A air supply after leaving the aseptic processing area and until the cap has been crimped
completely. Crimping of vial cap should be undertaken in areas of at least Grade C taking into
consideration of contamination risks due to container-closure integrity, and if necessary,
additional supplementary measures should be implemented to prevent or minimize the risks of
contamination during crimping by microbial and non-viable particles. The distance of the
location and the duration of time between stoppering and capping should be as short as
possible.
3. In aseptic filling processes, environmental monitoring should be undertaken for the full
duration of critical processing, starting from preparatory stage such as assembly of filling
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 54 -
machines and container supply machines that would directly contact drug products, and then
monitoring data obtained should be duly evaluated. The information on how to undertake
environmental monitoring, such as frequency, should be referred to the Environmental
Monitoring Section of this guidance.
4. Equipment surfaces that come into direct or indirect contact with sterile pharmaceutical
products should be decontaminated or sterilized prior to manufacturing according to validated
sterilization procedures.
5. Sterilized equipment should be preserved in validated procedures to keep sterile condition
until use.
6. Connecting area of a reserve tank of sterile bulk product with filling equipment (including
filling lines) should be sterilized by the SIP system in a critical area (Grade A). If connecting
area cannot be sterilized by the SIP system, the following alternative method to secure sterility
assurance, may be employed :
● Containers and filling equipment should be aseptically connected in a critical area.
● If the connection is performed in Grade B or lower environment, the connecting area
and any downstream thereafter should be sterilized using the SIP system.
These procedures may not be applied to connecting system which is proved to ensure a higher
sterile assurance (e.g. commercial available sterile connectors).
7. The transfer or supply of sterile materials such as sterilized rubber closures through indirect
support areas should be conducted by validated procedures that ensure to maintain the sterility
of such materials. The frequency of such transfer or supply should be as minimum as possible.
8. The sterility assurance level of aseptic filling process should be verified by process simulation.
9. If the active ingredients of sterile pharmaceutical products have high potent physiologic
activity or are bacteria which may carry a risk of infection, the premise and equipment must be
in compliaqnce with requirements and rules stipulated in the Regulations for Buildings and
Facilities of Pharmacies and the Standards for Manufacturing Control and Quality Control of
Drugs, Quasi-drugs (known as “GMP Regulations”). Further, the equipment and processing
areas should be inactivated and cleaned after completion of processing, if necessary . If an air
circulating the filling area is to discharge outside, the air should be pre-treated by an
appropriate cleaning procedure prior to discharge.
10. The maximum allowable time for filling process should be established and validated for the
adequateness.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 55 -
16.2 Filling of Liquid Products
Aseptic liquid filling processes should meet the following requirements:
1. Sterile bulk products should be prepared using sterile containers equipped with gas filtration
filters. The filters should be tested for integrity after use.
2. The maximum allowable time should be established for preparation of sterile bulk products
and the process during the preparation to the filling. The maximum allowable storage period
should be specified for sterile bulk products. If a solution of bulk products is prepared in a
non-sterile area and subsequently sterilized by filtration during filling processing, the steril
filtration should be undertaken promptly after preparation of a bulk solution to prevent or
minimize the growth of bacteria or endotoxins in the bulk solution.
3. The integrity of containers used for the preparation of sterile bulk solution and connection of
the containers with filling equipment should be periodically assessed and confirmed, and the
procedures for the assessment should be established. Appropriate period for replacement of
the gaskets should also be established.
16.3 Powder Filling Processes
Aseptic powder filling processes should meet the following requirements:
1. Bulk powder to be filled should be stored in hermetic containers, unless an alternate method
has been verified to be equivalent or more effective in keeping the powder free from
contamination with foreign matter or microorganisms.
2. SOPs for assessing the integrity of hermetic containers used to store bulk powder should be
established and verified. The frequency and procedures for replacing gaskets should also be
established.
3. Control criteria for airborne particulates should be established f during powder filling
processes in APA filling areas by taking into account the potential influence of dust on
counting particulate matter. The criteria should be based on the following data obtained
through validation conducted under operating conditions with the HVAC system running.
• Particulate count determined with the powder filling machine halted
• Particulate count determined with the powder filling machine idle
• Particulate count determined during operation of powder filling machine (monitoring
during periodic validation of process control)
4. If the outer surface of the product container is cleaned with compressed air following filling of
bulk powder, the dispersion of powder into the surrounding environment should be minimized
by appropriate preventive measures.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 56 -
17. Filtration Sterilization Processes
17.1 Liquid Filtration Sterilization Processes
17.1.1 Selection of Filters for Sterile Liquid Filtration
Filters for sterile liquid filtration should be selected based on their physicochemical properties,
biological safety profile, bacterial retentionperformance, andextractable profile, followed by the
assessment of compatibility with pharmaceutical products and process characteristics such as
required membrane surface areas in accordance with the assessment protocol or procedure.
Generally, the nominal pore size for sterilizing filters suitable for sterile liquid filtration is less than
0.2/0.22 μm.
17.1.2 Implementation of Sterile Liquid Filtration and Process Control
Process parameters necessary for sterile liquid filtration should be established based on
characteristics of filters and pharmaceutical products, and then be validated for these parameters.
1. Cleaning procedures
The filtration system (including secondary fluid path [e.g. pipes and holding tanks set after the
filter]) should be assessed for efficiency in removing extracts, insoluble particulate matter,
oxidisable substances, etc.
2. Filter sterilization procedures
A sequence should be established for filtration sterilization procedures, and these procedures
should be verified to be efficient in cleaning and sterilizing without damaging the filters. The
maximum cumulative time allowed for use of an individual filter under applicable sterilization
conditions should be specified under conditions of repeated use. Common procedures for filter
sterilization are autoclaving, gas sterilization, and radiation sterilization.
3. Filter integrity test method
Filters used in the manufacturing process should be tested for integrity by a non-destructive
method experimentally demonstrated to provide data that correlate well with data on the
filter’s bacterial retention capacity. Methods of testing filter integrity include the diffusion
flow (forward flow) and bubble point test. “Demonstration of correlation” means verification
that a filter satisfying the integrity test limit value can maintain bacterial retention capacity (if
the limit value is not exceeded, the filter is not guaranteed to have sufficient bacterial retention
capacity). Basic data on filter integrity should be obtained from the filter’s manufacturer.
(1) Filters should be wetted with suitable wetting solutions recommended by the filter’s
manufacturer or products actually used for filtration sterilization.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 57 -
(2) SOPs for the integrity test should include, but not be limited to, the following:
• Procedures for filter wetting
• Environmental conditions for integrity testing
• Confirmation of testing processes
• Evaluation of filter test failure and trouble shooting
• Recording of test results
• Conditions for filtration sterilization process
4. The filtration sterilization process should be validated under operating conditions, assuming
the worst-case scenario, by taking into account the points listed below. Potential risks
associated with aseptic processing should be assessed, and introduction of multistage filtration
should be evaluated as needed. If a multistage filtration system is employed, the sterilizing
filter should be placed as close as possible to the filling valve.
(1) Compatibility of filters with pharmaceutical products (e.g. chemical resistance)
(2) Maximum filtration time or maximum time of contact with pharmaceutical products
(3) Maximum filtration volume
(4) Maximum flow rate
(5) Temperature
(6) Maximum differential pressure
17.1.3 Filter Validation of Product-Specific Bacterial Retention Performance
1. Bacterial challenge test
Filters should be validated for ability to capture bacteria potentially present in individual
pharmaceutical products under operating conditions, assuming the worst-case scenario, e.g.
maximum filtration volume or maximum differential pressure. Filters may be validated in
groups classified by properties of sterilization solution or process conditions.
2. Challenge solutions and challenge bacteria
(1) Challenge solution
The solution used in the bacterial challenge test should be a solution of pharmaceutical
product which is sterilized by filtration in actual manufacturing production. If the
challenge test procedures need to be modified for some reason, such as because the
pharmaceutical product has bactericidal prpperty, filtration processing should first be
conducted with the drug solution to be sterilized under simulated worst-case scenario
conditions for actual manufacturing production in order to verify the compatibility of a
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 58 -
drug substance with the filter, and then the challenge test should be performed under
modified test conditions.
(2) Challenge bacteria
The challenge test should be conducted with Brevundimonas diminuta (ATCC 19146) or
other scientifically selected bacteria to confirm that the filtration process generates a
sterile filtrate. The challenge level should be at least 10
7
colony-forming units (cfu) of
test organism per cm
2
sample surface.
17.1.4 Routine Procedures
1. Cleaning of filtration system
Filter housing and pipes of filtration system should be cleaned by appropriate procedures
established during the process development phase of the filtration system. As a rule, filters are
not generally cleaned or reused; however, if used again, filters should be cleaned via
established appropriate procedures.
2. Sterilization of filtration system
Filtration system should be sterilized promptly after completing the cleaning process by
appropriate procedures established during the process development phase of the filtration
system to prevent microbiological proliferation.
3. Filter integrity test
Filters should be verified for integrity after filtration processing (after use of filters) without
disassembling the entire filter. Integrity should also be confirmed prior to the filtration process
(before use of filters), as appropriate, by evaluating potential risks inherent to the process.
4. Bioburden control
Bioburden level of pharmaceutical products prior to filtration should be checked with
appropriate frequency.
5. Maintenance and change control
Appropriate procedures should be established and implemented to maintain and control filters
and filtration system equipment including testing and inspection equipment. Procedures
should also be established for confirmation and recording of changes to be made to the
conditions for filter use and maintenance control.
6. Personnel training
Personnel involved in filtration sterilization during manufacture should be adequately trained.
Training program should include, but not be limited to, operation procedures for integrity
testing, procedures and implementation of investigation into reasons for integrity test failure,
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 59 -
loading and unloading of filters, and cleaning and sterilization of filters.
7. Manufacturing records
Manufacturing records should include, but not be limited to, the following filtration
sterilization-related information:
(1) Procedures for filtration sterilization
(2) Name and batch number of pharmaceutical products filtered
(3) Names and signatures (or seals) of operators in charge of filtration sterilization
(4) Name of filter manufacturer and types, lot numbers, and/or serial numbers of filters
(5) Cleaning and sterilization conditions for filters and filtration system
(6) Conditions of the filtration sterilization process (e.g. differential pressure, primary and
secondary side pressure, flowrate, operating temperature, duration of filtration,
processing volume)
(7) Conduct and outcome of filter integrity test
17.2 Air and Other Gases
17.2.1 Selection of Filters for Gas Filtration Sterilization
Filters for gas filtration sterilization should be selected from those made of hydrophobic
materials based on their physicochemical properties, biological safety profile, and bacterial
retentionperformance. The membrane surface area necessary for efficient filtration should be
calculated based on flow rate and differential pressure specific to individual processes. Generally,
the nominal pore size for sterilizing filters suitable for sterile air filtration is less than 0.2/0.22 μm.
17.2.2 Implementation and Control of Air Filtration Sterilization
1. Procedures for air filtration sterilization
Gas filters are generally used repeatedly. The maximum allowable cumulative time of
filtration under applicable sterilization conditions should be established before use. Common
procedures for filter sterilization include SIP system, steam sterilization in an autoclave, and
radiation sterilization. With steam sterilization, water may be retained in the filter to possibly
reduce filtration flow rate; therefore, the filter needs to be dried well but as quickly as possible
to prevent bacteria proliferation.
2. Filter integrity test procedures
(1) The filter integrity test should be non-destructive and suitable for determining a filter’s
bacterial retention capacity (refer to Section 17.1.2 3).
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 60 -
(2) Processes in which filtered gas comes into direct contact with sterilized products
Air filters used in processes in which filtered gas comes into direct contact with sterile
pharmaceutical products (e.g. processes using aseptic filling equipment, vent filters of
sterile bulk holding tanks, freeze-drying equipment, vacuum-break filters of autoclave)
should be subjected to an integrity test which provides data positively correlated to data
from the bacteria challenge test performed with liquids used in testing filters for sterile
liquid filtration (in general, the challenge test is conducted by the filter manufacturer
using liquid [water, routinely]). Details of the filter integrity test should be confirmed
with the filter manufacturer (refer to Section 17.1.3).
(3) Air filters used in processes in which filtered gas does not come into direct contact with
sterilized products (e.g. air supply during bulk intermediate product manufacturing
process and fermentation process) should be controlled by establishing appropriate
control procedures based on risk analysis.
3. Conditions of filtration sterilization process
Gas filters are generally used repeatedly for a significant period of time. The materials of the
filter should be examined for durability, including resistance to oxidation and degradation. In
addition, the following parameters (1) to (5) for gas filtration sterilization should be
established prior to processing. Unlike with filters for sterile liquid filtration, process
parameters for gas filters cannot be realistically established by assuming the worst-case
scenario; therefore, it is not absolutely required for the manufacturer to validate the bacterial
retention capacity of each process.
(1) Temperature
(2) Maximum pressure differential
(3) Gas flow direction
(4) Duration of use
(5) Frequency of filter sterilization
17.2.3 Confirmation of Bacterial Retention Capacity
Bacterial retention capacity of gas filters should be confirmed by evaluating the methodology
and results of the retention test documented in the filter manufacturer’s product warranty certificate
and validation support data.
17.2.4 Design of Filtration System
Condensation tends to build up on filters of filtration system, leading to reduction in the flow
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 61 -
of filtrate and inviting proliferation of bacteria. Filtration system should be designed to remove
condensed water from filters and their housings promptly upon generation. If such generation is
inevitable as with the WFI tank, certain preventive measures such as heating of filter housing should
be instituted (refer to Section 17.1.4).
17.2.5 Routine Procedures and Validation
If particles and fibers of gas filters may become detached and affect the quality of
pharmaceutical products in processes during which filtered gas comes into direct contact with
sterilized products, the detachment can be evaluated using liquids. Generally, the need for filter
cleaning validation (e.g. CIP, cleaning prior to sterilization) by the drug manufacturer should be
determined based on data provided by the filter manufacturer (refer to Section 17.1.4).
18. Freeze-Drying Process
18.1 General Requirements
1. Vials must remain unstoppered and ampoules unsealed in the freeze-drying process to be
exposed to the environment. Appropriate measures should be established to prevent microbial
contamination of pharmaceutical products during transfer from the filling area to a
freeze-drying chamber, while being held in a freeze-drying chamber, or while being processed
from freeze-drying to sealing.
2. Transfer of materials and products into the freeze-drying chamber should be carried out in a
working area maintained at the critical area cleanliness level (Grade A). If possible, the
transfer method should be one which does not require human intervention, such as tunnel-type
automatic transfer lines, transportation vehicles equipped with a unidirectional airflow device,
and isolators.
3. Vials freeze-dried but yet to be capped and ampoules to be heat-sealed or have caps screwed
on should be processed in a pathway or working area maintained at the critical area cleanliness
level (Grade A).
4. Containers and closures should be designed so as to maintain suitable air-tightness between
time from capping in a freeze-drying chamber to having caps screwed on. If the screwing
process is conducted in a non-aseptic processing area, the cleanliness of capped vials should
be maintained by applying Grade A air until completion of cap-screwing after transfer from a
critical area (Grade A). Cap-screwing should be performed in an area of Grade C or higher
cleanliness, depending on the level of contamination risk anticipated given container-closure
tightness requirements, and additional preventive measures should be taken depending on the
level of contamination risk with microorganisms or particulate matter generated during
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 62 -
cap-screwing. The distance between points of cap application and cap-screwing and time
between these processes should be as short as possible.
5. Microbial cleanliness level should be supervised in the areas where the operations described in
Items 2), 3), and 4) above are conducted.
6. Sterility of pharmaceutical products with screwed-on caps should be ensured via the validated
container/closure system integrity test and in-process control tests. All ampoules and other
containers sealed by fusion should be subjected to a leak test or other test that ensures the
integrity of the products after the fusion process. Vials mounted with closures for screwing
should be checked for completeness of closure placement to eliminate vials with missing
closures or those inappropriately stoppered. Recommended procedures to confirm the
tightness of closures include close torque control and press pressure control.
7. Entry of air into processing chambers (e.g. machinery room) under reduced pressure relative
to the outside environment should be strictly kept to a minimum to ensure the sterility of
pharmaceutical products during the freeze-drying process. Procedures necessary for ensuring
the reliability of leak tests to supervise air entry and integrity tests of vacuum break filters and
leak filters to control vacuum level should be established and implemented
.
18.2 Validation
1. The sterility of the freeze-drying process should be ensured by developing and validating
microbiological and physical monitoring programs for the process itself and processes
immediately before and after. The microbiological monitoring program is usually comprised
of a media fill test, process simulation test, assessment of bioburden during sterilization
(including for freeze-drying equipment for general use), and bioburden control features. The
physical monitoring program is comprised of a leak test and integrity test for vacuum break
filters and leak filters. Routine validation of the sterilization process, bioburden control, and
filter integrity test should be conducted in a manner similar to that employed with equipment
actually used in the manufacture of sterile pharmaceutical products.
2. Process simulation should be conducted in accordance with provisions stipulated in Chapter
20 as one of the critical control programs for the freeze-drying process. It is important to select
appropriate conditions by referring to actual manufacturing processes in order not to inhibit
the growth of bacteria nor impair the viability of culture media.
(1) Temperature and time for cooling purpose should be appropriately specified.
(2) Pressure reduction should be gradual so as not to cause explosive boiling or spontaneous
freezing.
(3) The freeze-drying program (particularly, drying time) should be carefully established to
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 63 -
avoid drying the culture media or impairing the viability of culture media.
(4) With regard to the freeze-drying processes, those processes which cause turbulence at
the time of starting pressure reduction, vacuum break, or loading of materials or
products to be freeze-dried, and processes with the highest risk of microbial
contaminationsuch as a process with human interventionshould be simulated and
evaluated repeatedly several times under the worst-case scenario.
(5) Some freeze-dried products need to be prepared with containers filled with inert gas
such as nitrogen to ensure product stability. If growth conditions for aerobic bacteria
need to be secured, air should be used instead of inert gas. If anaerobic bacteria are
identified or suspected to be present in the preparation, inert gas and growth media for
anaerobic bacteria should be used.
3. If the capacity of the freeze-dryer is equal to or smaller than the standard equipment with
respect to accommodating the standard unit number of media, the equipment concerned should
be loaded with a unit number of media suitable to the size. If the capacity of the freeze-dryer is
larger than the standard (5,000 units of media), containers filled with media should be placed
at appropriate locations within the freeze-drying chamber: medium containers should be
randomly placed or decimated in sequential order to be evenly placed for unbiased evaluation.
If the evaluation assumes the worst-case scenarioincluding incomplete integrity of vacuum
break filters, leakage from doors or ice-condensers, or back-diffusion of gas or air from
vacuum pumpmedium containers should be placed in locations where the risk of
contamination associated with any one of these abnormalities is particularly high.
4. The integrity of containers and closures should be validated to ensure the sterility of
pharmaceutical products.
5. The validity of the leak test and the integrity of vacuum break filters and leak filters for
vacuum control should be validated for the control of air entry into the freeze-dryer chamber
under negative pressure. The judgment criteria for the leak test should be strictly established
by taking the following factors into account so as to minimize the risk of microbial
contamination within the chamber of freeze-drying equipment: volume of the freeze drying
chamber, retention time under reduced pressure in the freeze-drying process, and environment
surrounding the freeze-drying equipment.
18.3 Cleaning and Sterilization of Freeze-Drying Equipment
1. Cleaning of freeze-drying equipment should be scheduled after taking the following factors
into account:
(1) Cleaning procedures for freeze-drying equipment should be established with due
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 64 -
awareness of the difficulties involved in cleaning the complex inner structure of the
freeze-drying chamber.
(2) Sampling procedures for verifying cleaning efficiency should include sampling of
drained rinse water and combine the swab method to sample materials at the nearmost
surfaces of shelves and areas around drains. The transfer method using clean sticky tape
is also an effective sampling procedure. For verification of cleaning efficiency using the
rinse-water sampling and swab methods, a pharmaceutical product (actual or simulated)
employed as an indicator of cleaning efficiency should be selected based on ease of
cleaning and pharmacological activity of the pharmaceutical product.
(3) When a detergent is used in cleaning, toxicity and other relevant data for the agent
should be obtained from the supplier for evaluation, and appropriate assessment
procedures for the swab and rinse-water sampling methods should be established to
assess potential effects of residual agents on pharmaceutical products to be freeze-dried.
2. Appropriate sterilization procedures should be established and validated to ensure the
sterilization of freeze-drying equipment.
(1) Freeze-drying equipment has a complex inner structure and is composed of materials
varying in type and size. Sterilization procedures for the equipment should therefore be
sufficiently comprehensive to secure complete sterilization after taking into account
possible cold spots and diffusion of sterilization gas throughout the complex chamber.
In particular, with regard to sterilization procedures which use gas, temperature and
humidity inevitably vary within the chamber, and therefore sterilization should be
conducted over sufficient time to allow for permeation. Circulation and diffusion
patterns of gas should be evaluated in detail for optimization of sterilization procedures.
(2) With regard to sterilization procedures which use steam, given the complex inner
structure of the chamber, due care should be exercised to achieve efficient displacement
of stagnant air and removal of condensed water.
(3) Steam sterilization should be conducted for every freeze-drying cycle, as a rule. When
the interval of sterilization is changed in accordance with properties of pharmaceutical
products or for other reasons, the validity of sterilization between intervals should be
ensured by microbiological validation.
18.4 Routine Monitoring and Control and Maintenance of Freeze-Drying Equipment
1. The leakage of gas from freeze-drying equipment should be measured at the time points
described below. Caution should be taken when identifying or measuring the volume of
pseudo-leaks of gas generated inside the freeze-drying chamber.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 65 -
(1) Leak test for every batch of pharmaceutical products at the completion of freeze-drying
Records of leak test data should be obtained in brief at the completion of freeze-drying.
(2) Leak test at the completion of steam sterilization
Records of leak test data should be obtained after cooling of freeze-drying equipment
since steam sterilization exerts significant stress on the chamber.
(3) Leak test during periodic revalidation
Freeze-drying equipment should be run through an empty cycle overnight to measure
leakage of air or gas from the equipment during periodic revalidation.
(4) An additional leak test should be conducted upon detecting actual or signs of abnormal
leakage in the leak test in Items (1) or (2) above.
2. The program for periodic equipment function testing should include, but not be limited to, the
functional diagnosis of the heat transfer/circulation system for shelves, the cooling system for
refrigerating machines, and the vacuum/exhaust system.
3. Vacuum break filters, leak filters, gaskets for vacuum sealing, and other parts should be
periodically replaced depending on their cumulative duration and frequency of use.
4. Critical instruments for monitoring and controlling freeze-drying equipment such as
temperature regulators and vacuum gauges should be calibrated periodically and have their
calibration results documented for records. A calibration interval of approximately 6 months is
recommended unless previous calibration results suggest a need to change the interval.
5. Vacuum gauges are highly sensitive meters used to measure very minute changes in pressure,
and on-site calibration via the method confirmed to be traceable is practically impossible. As
such, it may be acceptable to contract out calibration to an authorized testing facility.
19. Isolator System, Barrier System, and Blow-Fill Seal
19.1 Isolator System
A properly designed isolator system provides an extremely aseptic environment but does not
provide a hermetically sealed enclosure. Although highly potent pharmaceutical products with high
pharmacological activities are occasionally manufactured in an isolator system with a cabinet
maintained under negative pressure, sterile pharmaceutical products are usually manufactured using
an isolator system operated under positive pressure. In addition, ensuring product sterility requires
the establishment and the implementation of a comprehensive preventive maintenance program
including maintenance and control procedures for HEPA filters, gloves, half suits, and any other
design features that are intended to provide enclosure integrity.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 66 -
19.1.1 General Requirements
1. The air cleanliness level of an environment where an isolator system is installed for the
manufacture of sterile pharmaceutical products should be Grade D or higher.
2. Connectors combining two isolators and connection ports for input and output of aseptic
materials or bulk product should be structurally designed to be suitable for maintaining the
integrity of the isolator enclosures.
3. Numbers of half suits, gloves, transfer ports, and connection ports for output should be kept at
a minimum in order to reduce the risk of contamination.
4. The ports on isolators such as those used to transport finished products outside the isolator,
should be suitable to prevent contamination from entering the enclosure. These port openings
should have adequate air flow moving from the enclosure to the surrounding environment.
Generally, this is achieved by maintaining the pressure difference at an appropriate level.
5. The efficacy of decontamination process applied to the isolator enclosure and associated
equipment should be verified biologically by confirming a 4 to 6 log reduction on biological
indicators known to be resistant to antimicrobial agent utilized. The level of decontamination
should be established based on the intended use of the isolator and bioburden requirements.
Process for decontaminating materials to be transferred into the isolator should also be
validated to be capable of achieving a 4 to 6 log reduction of suitable biological indicator.
6. Product contact equipment/surface should reduce surface bioburden to the possible level prior
to decontamination. Procedures for decontamination product contact equipment/surfaces
within the isolator will generally require 6 log or greater spore log reduction established
through the use of appropriate biological indicators.
7. A periodic leak test should be performed based on the criteria predetermined for leak detection
sensitivity.
8. Decontamination frequency should be established based on the level of potential risks of
contamination, verified by appropriate validation studies, and reviewed at regular intervals.
19.1.2 Design of Isolator System
The isolator system should be designed after appropriate consideration of technical
requirement. Design should consider isolator structural requirements, operational conditions, and
risks associated with manufacturing operations performed inside the isolator.
19.1.3 HVAC System
1. The air cleanliness inside the isolator system should be Grade A.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 67 -
2. The rate of air exchange in the isolator should be sufficient to prevent retention of particulate
matter or contaminants and conditioning of air should enable temperature to be maintained
within the predeterminedrange.
3. Air velocity and airflow patterns should be sufficient to maintain a clean environment
necessary for operations within the isolator system.
4. Air in the isolator system should be circulated through a HEPA or higher-grade filter. The
supply of outside air to the HVAC inlet and the isolator exhaust should also take place through
a HEPA or higher-grade filter.
5. The pressure differential between the isolator and isolator room should be maintained at a
minimum of 17.5 Pascals. A greater differential may be necessary depending on the type of
operation, such as when half suits and gloves are used during operation. The pressure
differential should be continuously monitored and recorded throughout operation, and an
alarm system should be installed to warn operators of an abnormal drop in pressure.
19.1.4 Decontamination
1. The decontamination process should be established by taking the following points into
account:
(1) Cleaning and drying of the internal surface of isolator system, as required, prior to
decontamination of the enclosure and equipment contained therein
(2) Amounts of decontaminant injected into the isolator
(3) Biological indicators (BIs)
(4) Chemical indicators (CIs)
(5) Temperature distribution within the isolator and in the surrounding environment
(6) Humidity
(7) Duration of decontamination process
(8) Concentrations of decontaminants when using gas decontaminants
(9) Pressure differential
(10) Verification of a relatively uniform distribution of decontaminants within the isolator
(11) Bioburden
2. Decontaminants should be selected based on evaluation of compatibility with the isolator’s
materials of construction, type of operations to be performed inside the isolator, volume and
configuration of materials brought into the isolator, and bioburden in the isolator. Possible
agents for isolator decontamination include peracetic acid, ozone, chlorine dioxide, and
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 68 -
hydrogen peroxide.
3. Decontamination should be conducted by personnel with sufficient knowledge and
understanding of the characteristics of the decontamination mist, vapor, or gas and with
familiarity with the operation of the decontamination employed.
4. The residual level of the decontaminant should be confirmed to have been reduced to equal to
or less than acceptance criterion value after completion of decontamination. The value should
be established by considering not only the safety of factory personnel but also potential
influence on product quality and subsequent processes.
5. Every batch of decontamination agent used should be analyzed before use to confirm that they
meet their predetermined composition and identity.
19.1.5 Personnel Training
The education and training program on the use of the isolator system should include but not be
limited to the following:
1. General requirements on aseptic processing
2. Proper utilization of gloves and half suits
3. The isolator decontamination process
4. Integrity testing for isolator equipment
5. Procedures for loading of materials and unloading of finished products
6. Operation, monitoring, and maintenance/control of isolator equipment
7. Safety handling and use of decontamination agents based on the relevant Material Safety Data
Sheet (MSDS) and the known compatibility of decontaminants with the isolator equipment
8. Process-specific SOPs
19.1.6 Routine Monitoring and Control
Routine control requirements for the isolator system should include, but not be limited to, the
following:
1. SOPs for the operation of the isolator system should be developed based onvalidated
processing conditions.
2. While the isolator system is assumed to be maintained at a high level of integrity, its
maintenance is not absolutely perfect. Therefore, a leak test should be performed at periodic
intervals and also prior to each decontamination. Methods to be used in the leak test should
include but not be limited to the following:
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 69 -
(1) Pressure hold test
(2) Gas detection method
3. Glove material should be resistant to all chemicals and decontaminants to be used.
4. Gloves should be checked visually for damage such as puncture or tears before each use.
5. In operations performed with gloves, gloves should preferably be supplemented by inner
gloves worn beneath the isolator gloves.
6. Gloves should preferably be tested for physical leaks and subjected to microbiological
monitoring using swabbing or other methods at regular intervals.
7. A maintenance and control program should be developed for consumable materials and items
to specify a suitable time interval for replacement.
8. Prior to decontamination, parameters that may affect decontamination efficacy such as
temperature, humidity, gas concentration, etc.should be monitored at predetermined
locations and the results should be recorded for each decontamination cycle.
9. The total particulate count in the isolator should be monitored at predetermined locations at
suitable time intervals.
10. Microbiological monitoring should be conducted at suitable time intervals at locations
predetermined based on potential contamination risk in relation to structural characteristics of
isolator system and properties of operations to be performed in the system. Typical example
locations for monitoring include the inner surface of the isolator, glove surface, materials
carried into the isolator, and material contact surfaces. The validity of the locations and
frequency of monitoring must be verified at regular intervals.
19.2 Restricted Access Barrier System (RABS)
A restricted access barrier system (RABS) is a means to produce sterile pharmaceutical
products by separating personnel from critical areas and minimizing direct human intervention in
critical areas during aseptic processing.
The RABS is an integrated aseptic processing system to be implemented in aseptic processing
areas (critical areas) comprising both hardware and software components, such as physical partitions,
air supplied through HEPA filters, and appropriate operational procedures.
The structure and composition of the RABS are varied from a hard wall to a structure with
tight barriers and a high degree of isolation like isolator. The HVAC system accompanying the
RABS is also not uniform: the HVAC system either utilizes the air conditioning system originally
available with the RABS or is an independent HVAC system. This chapter describes basic
requirements specific to the design and operations of the RABS.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 70 -
19.2.1 General Requirements
The RABS should be designed by taking into account the following matters as general
requirements:
1. The inner environment and HVAC system for the RABS should meet the following
requirements for the critical area, as stated in Chapter 7 of this document.
2. The area or room where the RABS is installed should be defined as a direct support area and
the air cleanliness level in the area or room should be Grade B or higher.
3. Interventions by factory personnel, if required, should be conducted through sealed gloves or
half suits. Appropriate procedures for disinfection, inspection, and replacement of gloves or
half suits should be instituted and implemented to minimize risks of product contamination.
Refer to “Isolator System” in Section 19.1 for detailed requirements regarding glove use.
4. The inner surface of the RABS that comes into direct contact with pharmaceutical products
should preferably be disinfected with the SIP system. Parts of the surface that cannot be
treated by the SIP system should be disassembled and disinfected by autoclaving or other
methods and then assembled. Further, if equipment such as an isolator can be decontaminated,
the surfaces of such equipment that come into direct contact with pharmaceutical products can
then achieve still higher levels of microbacterial cleanliness.
5. The inner surfaces of the RABS that do not come into direct contact with pharmaceutical
products should be disinfected via appropriate methods of disinfection.
6. When sterile materials need to be carried into the RABS, a transfer system resistant to
contamination should be employed. If sterile materials in containers are carried into the system,
the surface of the containers should be adequately decontaminated prior to the transfer.
7. When human intervention is required during processing while keeping the RABS door open,
the following points to consider should be taken into account to reduce risk of product
contamination:
(1) The inside of the RABS should be appropriately disinfected after intervention to
eliminate potential product contamination.
(2) SOPs for handling containers present in the RABS at door opening should be
established in advance based on potential risks of product contamination. If the door
needs to be opened for an unexpected reason, all containers present in the system should
be removed from the system, as a rule.
(3) Intervention procedures should be thoroughly recorded.
8. If the door might be opened during aseptic processing, an ISO 5 (at least, no-load level)
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 71 -
protection booth should be installed outside the system. Air should be ensured to run from
inside the RABS to the protection booth upon opening the door.
19.2.2 Personnel Training
The education and training program on the use of the RABS should cover but not be limited to the
following:
1. General requirements on aseptic processing
2. Proper handling procedures for gloves and half suits
3. Procedures for decontaminating the inside of the RABS
4. Procedures for loading and unloading materials and intermediate products
5. Details on operation, monitoring, measurement, and maintenance/control of the RABS
6. Procedures for intervention while the door is kept open and relevant points to consider
19.3 Blow-Fill-Seal (BFS) System
The blow-fill-seal (BFS) system is a specialized aseptic packaging technology used in the
manufacture of sterile pharmaceutical products which uses in-line forming, filling, and sealing of
sterile plastic containers. Since plastic containers are molded from plastic pellets, filled, and sealed
by fusion in a continuous production run under a closed and sterile environment without human
intervention during the filling process, the sterility of pharmaceutical products can be ensured
without terminal sterilization processing (e.g. autoclaving) after sealing. As operations are carried
out as a closed, automatic, and continuous process, the BFS system features a relatively low chance
of contamination during production. However, the system type varies: one type permits filling and
closure in a perfectly closed system, while another requires caps and inner plugs to be supplied from
outside. A sterility control program should therefore be established based on features of each system.
19.3.1 Scope of Blow-Fill-Seal System and Processes to be Addressed
Among the manufacturing processes for sterile pharmaceutical products to be manufactured
using the BFS system, this guidance document is applicable to those processes not requiring
sterilization (e.g. autoclaving) following filling and fusion-sealing processes. In the manufacture of
liquid pharmaceutical products, the BFS system is applied to processes involving sterilized filtration
of drug solution, loading of plastic pellets, molding of containers, filling of drug solution into
containers, and sealing of containers. In the manufacture of powder pharmaceutical products, the
system is applied to processes involving loading of sterile bulk powder, loading of plastic pellets,
molding of containers, filling of the sterile powder into containers, and sealing of containers.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 72 -
Particular attention should be paid to the following:
1. Elution of plasticizers, additives, and unpolimerized monomers, etc. from plastic containers
2. Pyrogenicity of plastic pellets
3. Environmental conditions for plastic container molding
4. Sterilization of drug solution (preparation of drug solution by filtration sterilization)
5. Compatibility of containers with drug solution
6. Cleanliness level of the filling environment and environment where the equipment is installed
7. Fusion-sealing operations
It is critical to establish strict criteria for the above items 2), 3), 6), 7), and “assessment of
sterility” from a sterility control point of view.
19.3.2 Process Flow and Environments for Container Molding and Filling
1. Critical processes of the BFS system
(1) Preparation of drug solution
(2) Filtration sterilization
(3) Temporary storage of filtered drug solution
(4) Molding (including clean air supplied into the molding environment)
(5) Filling
(6) Fusion sealing
2. Characteristics of BFS processes
(1) All processes of the BFS system from molding of plastic containers to filling and
fusion-sealing should be performed in a continuous automatic operation.
(2) Filling and fusion-sealing operations are carried out in a restricted, isolated area;
therefore, the working space is not necessarily an aseptic area with air cleanliness grade
of “A” which is routinely required for the manufacture of sterile pharmaceutical
products. As such, it is acceptable to maintain Grade A cleanliness level only for the
restricted processing areas of molding and filling. For this reason, however, accurately
controlling the cleanliness level of the molding and filling areas is quite important for
protecting pharmaceutical products from contamination by foreign and particulate
matter.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 73 -
19.3.3 Sterility Assurance of Plastic Containers
The inner surfaces of plastic containers molded by the BFS system must be sterile. Sterility
assurance for these surfaces requires the following actions be implemented:
1. The quality of plastic pellets should be adequately controlled throughout the storage period to
prevent excessive microbial contamination that may affect sterility and pyrogenicity of
pharmaceutical products.
2. Temperature and time are key factors for not only efficiently melting and molding of plastic
pellets but also eradicating microorganisms present on the pellets. Temperature and time
established for processes from melting to molding need to be verified and controlled to be
suitable for dry heat sterilization (plastic pellet melting and molding processes are conducted
under dry-heat conditions, free from moisture).
Note: The Japanese Pharmacopoeia recommends the use of Bacillus atrophaeus as an
indicator for dry heat sterilization. The D160 value for B. atrophaeus ATCC 9372 has been
reported to be 0.89 – 1.22 on glass plates and 1.22 – 2.07 on plastic plates.
3. The sterility of the molding and filling processes should be verified by process simulation.
19.3.4 Critical Control Parameters of the BFS Process
The following are critical control parameters of the BFS process:
1. Bioburden of plastics
Bioburden (in particular, fungi) of plastic materials and additives thereof should be determined
prior to use. If the plastic supplier fails to provide sufficient information on these materials,
the cleanliness level of the plastic should be closely supervised throughout the manufacturing
process to maintain adequate bioburden control.
2. The temperature for melting plastic pellets and the time from melting to extrusion for blow
molding should be monitored and controlled at respective predetermined optimal levels.
3. The equipment for the preparation and transportation of the drug solution should preferably be
designed to be adaptable to the CIP and SIP systems to ensure proper disinfection of drug
solution preparation processes and sterility of pharmaceutical products. If the equipment is not
adaptable to these systems, certain off-line control systems should be in place to ensure
cleanliness and sterility levels similar to those achieved by the CIP and SIP systems.
4. Quality of environment air
In the BFS system, pharmaceutical products are exposed to environment air only during
molding and filling processes. Local environments for these processes and air supplied to
these processing areas should therefore be monitored to maintain a Grade A air cleanliness
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 74 -
level, and the quality of air surrounding the equipment should be of Grade C or higher
cleanliness. Operators should wear gowns suitable for these cleanliness levels.
5. Quality of air for extrusion blow molding and integrity of air filters
Air contacting the inner surface of the container should be supplied after passing through
sterilizing filters. When compressed air is used instead, the oil and water content in the air
must be strictly controlled. The cleanliness level based on viable bacterial and particulate
matter counts should be Grade A equivalent.
6. Air supply to local spaces for filling operations
Air supplied to local spaces for filling operations, which are air-shower rooms equipped with a
filling nozzle and parisons where melted plastic resin is inflated to form the container, is
generally passed through sterilizing filters. Environmental monitoring of these local spaces is
usually performed by measuring airborne particle matter. If the spaces are purged with sterile
air supplied through sterilizing filters, the filter integrity test may be employed instead of
particulate matter monitoring as a means to ensure air sterility. Additional environmental
monitoring is necessary if filling operations in these spaces require human intervention to
prepare materials for filling, adjust materials and equipment during operation, or clean up
equipment.
7. Heat transfer medium and product quality
Although the heat transfer medium is unlikely to come into direct contact with the molded
plastic containers, the possibility of leakage or contamination of the medium into melted
plastic should not be ruled out.
8. Integrity of fusion-sealing performance
The integrity of sealing performance is a highly critical process parameter for the BFS system,
and a number of methodsincluding rare gas leak and high voltage leak detectionhave
been developed to test the integrity. Seal integrity should be ensured via an appropriate
method, and validity of the method should be verified after employment.
9. The CIP and SIP of the BFS process (temperature, time, and F
0
)
10. The integrity of the SIP in the filling process and maintenance of sterility
11. Challenge test of fusion-sealing process
12. Media fill process simulation test of filling process
13. Continuous operation (acceptability of continuous operation and verification of the maximum
allowable time of operation)
The BFS process is often operated continuously with no break; therefore, the maximum time
allowed for continuous operation should be established depending on stability of the drug
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 75 -
solution in question and microbial contamination risk over the entire process. Procedures and
control parameters should be specified for resuming processing after interruption or
discontinuation of operation.
20. Process Simulation
20.1 Outline and Scope
Process simulation is a technique of applying the “media fill test” concept to all aseptic
processes. Sterile pharmaceutical products are manufactured by complex processing requiring
handling of sterile bulk materials and other raw materials as well as multiple aseptic processes.
Aseptic filling is only one of these processes involved in sterile pharmaceutical product
manufacturing. The validity and reliability of the method employed for sterility assurance of
pharmaceutical products manufactured by aseptic processing should be verified by validating all
processes involved in aseptic processing. Process simulation is a validation method using
microbiological growth media or a substance that supports microbiological growth in place of active
pharmaceutical ingredients to assess not only the performance of aseptic filling process but also that
of the overall aseptic manufacturing process for sterile pharmaceutical products. This process
simulation is applied to the assessment of manufacturing processes for sterile API including
filtration, crystallization, drying, milling, mixing, and freeze-drying on trays to obtain a powder and
also overall manufacturing processes for finished sterile pharmaceutical products such as filling and
sealing. Process simulation should be designed to emulate the routine production process as closely
as possible, including personnel movement, working environment, and manufacturing activities and
operations, under “practical” but worst-case conditions. Before proceeding with actual
implementation of process simulation, the “Media Fill Test (Process Simulation)” in the General
Information section of the Japanese Pharmacopoeia should be referenced.
20.2 Process Simulation Procedures
20.2.1 Frequency of Process Simulation
1. Initial process simulation
Materials subject to initial process simulation are equipment, instruments, processes,
containers of different design (except for containers different in size but same in design), etc.
to be used for the first time in manufacture. Based on reference information contained in the
Japanese Pharmacopoeia, media fill test should be conducted using a sufficient number of
liquid products filled in containers that adequately reflect the actual filling line of one
production run, with at least three replicate runs on separate days. If the product is bulk
product, the test should be conducted using one production unit amount of the bulk product.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 76 -
2. Repeated process simulation
Based on reference information contained in the Japanese Pharmacopoeia, process simulation
should be repeated at periodical intervals of at least 6 months on each working shift for every
aseptic process and every filling line, using liquid products in containers in a sufficient
number that adequately represents the actual aseptic and filling operations. If the product is
bulk powder, the test should be conducted using one unit production amount of the bulk
product. Personnel assigned to critical aseptic processing should be trained on aseptic
processing operations and take part in process simulations at least once a year.
When an aseptic process or filling line has not been used for over 6 months, process
simulation should be conducted with a frequency similar to that of the initial process
simulation prior to resumption of use.
Process simulation should also be conducted with a frequency similar to that of the initial
process simulation prior to reuse, as appropriate, if any process, facility, or equipment is
significantly modified to affect the level of sterility assurance, if personnel assigned to critical
aseptic processing are changed, if results of environmental bacterial tests are not acceptable, or
if sterility testing of finished products identifies contaminated products.
20.2.2 Medium Selection and Performance Testing
Process simulation should use soybean-casein digest medium or other media suitable for
testing bacterial growth. If the product is bulk powder, surrogate media (e.g. lactose, D-mannitol,
polyethylene glycol, powder medium) sterilized by radiation should be used instead. Details on
growth promotion tests of media to be used and bacterial growth inhibitory activity assays of
surrogate media should be referenced in the Japanese Pharmacopoeia.
20.3 Points to Consider for Process Simulation
Process simulation should be performed for all manufacturing processes, equipment, and
operational activities that may be correlated with sterility assurance of pharmaceutical products. The
key points to consider are as follows:
1. Cleaning of facilities and equipment and cleaning and disinfection of manufacturing
equipment, containers, closures, and trays should be conducted in accordance with SOPs.
2. Process simulation should be performed for all routine activities at different manufacturing
stages and temporal processing interventions.
3. Process simulation for temporal processing interventions which are known to occur on a
routine basis (e.g. weight adjustment and supply of sterile materials, containers, closures,
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 77 -
environmental monitoring) and anticipated but non-programmed interventions (e.g.
modification of manufacturing line, adjustment of equipment conditions, repair or replacement
of equipment parts) should be performed under practical operating conditions that simulate the
worst possible intervention conditions.
4. Process simulation should be performed under equipment operating conditions (e.g. lines
speed) that would most likely cause contamination.
5. Process simulation should be performed over the time period determined by taking the longest
possible time of actual operations into account.
6. All personnel engaged in aseptic processing are required to participate in process simulation.
The simulation test should be designed by simulating the largest possible number of
participating personnel and working shifts.
7. Enough medium should be filled in the container to allow the medium to contact the entire
inside surface of the container on rotation or inversion, thereby rendering a reliable judgment
of bacterial growth.
8. Even if inert gas is not routinely used in manufacturing, process simulation should be
performed by replacing inert gas with air unless the simulation test is not intended for
anaerobic growth.
20.4 Incubation and Inspection of Media Fill Units
1. If there is leakage of contents from the container or damage to the container prior to
incubation, these findings should be recorded and the media fill units of concern removed
from the simulation test.
2. The medium should contact all container surfaces on rotation or inversion of the container.
3. Media should be incubated for at least 14 days at a predetermined temperature within a
preferred range of 20 to 35°C. If the test temperature does not fall within this range, the
temperature should be justified and be within ± 2.5°C of the predetermined temperature.
4. If two different temperatures are employed in the test, the media should be incubated for at
least 7 days at the lower temperature and then for the same duration at the higher temperature.
5. Growth of microorganisms should be observed on the last day of incubation when determining
the absence or presence of growth.
6. If microorganisms are found to have contaminated test materials, they should be identified and
characterized to clarify the causes of contamination.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 78 -
20.5 Acceptance Criteria for Process Simulation
The acceptance criteria for process simulation should be consistent with those stipulated in the
“Media Fill Test” listed in the General Information section of the Japanese Pharmacopoeia. If
process simulation provides positive results indicating contamination, necessary actions should be
taken in accordance with the Japanese Pharmacopoeia.
20.6 Process Simulation of Manufacturing Lines Equipped with Isolator System
Process simulation of a manufacturing line equipped with an isolator system may be
performed with reduced frequency compared with that for a non-isolator system, provided the line
has passed the initial performance qualification test, meets the conditions described below, and can
be verified to have a low risk of bacterial contamination.
1. Manufacturing lines are structurally designed to completely separate the environment where
personnel engage in operations from the environment where pharmaceutical products are
exposed, and personnel can directly intervene only through barriers and gloves.
2. Risk management of the isolator system is performed using reliable technologies and with
frequency suitable to individual control parameters (e.g. gloves, pressure difference, reverse
current from the opening, loading and unloading operations, effects of cool zones in the
sterilization tunnel, decontamination).
3. HVAC system, surfaces of equipment and devices that come to contact with drug solutions,
filter performance, and other factors that comprise the sterility of pharmaceutical products are
separately evaluated and verified to be sterile by appropriate validation and periodic
reevaluation.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 79 -
Annexes
A1. Active Pharmaceutical Ingredients (APIs) Manufactured via Cell
Culture/Fermentation
A1.1 General Requirements
This section addresses the specific control of active pharmaceutical ingredients (APIs)
manufactured by cell culture or fermentation to supplement API-related regulations, guidelines,
guidance, and the main text of this guidance document.
1. The term “classical process” refers to manufacturing processes using microorganisms and
cells which exist in nature or those modified by conventional methods employed from the old
days. The term “biotechnological process” refers to manufacturing processes using cells and
microorganisms derived or modified by recombinant DNA, hybridoma, or other
biotechnological manipulation. The degree of control for microorganism in biotechnological
process is usually greater than that for classical process to produce proteins or polypeptides. If
natural human or animal cells are used for classical process, special precautions should be
taken against potential contamination of such cells with microorganisms and viruses derived
from the original human or animal cells.
2. Raw materials (e.g. (culture) media, buffer components) used in the production of APIs by cell
culture or fermentation may serve as good nutrients for microbes contaminated, so that
adequate control parameters, such as control of bioburden levels should be developed and
implemented, taking into account the supplier information and its preparation method for raw
materials, and type and characteristics of the final sterile pharmaceutical products and its
manufacturing processes. Media or other materials used in cell culture should also be managed
to control Mycoplasma and other microorganism levels, as appropriate.
3. The cleanliness level of cell culture/fermentation processing areas should be designed and
controlled depending on the type of operations performed in the areas. The processing area
may not necessarily be designated as a critical area, if the equipment installed in the area is a
closed system. However, an adequate cleanliness level should be established and maintained
to prevent contamination.
4. Preventive and safety measures against potential viral contamination in the manufacture of
APIs should be implemented pursuant to ICH guideline Q5A “Quality of
Biotechnological/Biological Products: Viral Safety Evaluation of Biotechnology Products
Derived from Cell Lines of Human or Animal Origin.”
5. With regard to in-process control and quality control (including monitoring of critical
processes), the records for sterilization of the equipment, environmental microorganisms
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 80 -
monitoring, and all deviations thereof should be prepared and maintained to adequately
control environmental microorganism level.
6. For appropriate environment control of cell culture or fermentation processing areas, the
criteria for hygiene control for restriction of personnel entry, gowning, and operators health
should be established and reduced to training, as appropriate.
A1.2 Cell Culture and Fermentation
In order to prevent viral or microbial contamination, working cell banks and other starting
materials used in the cell culture or fermentation should be subjected to characteristic analyses
and evaluation for viral and microbial safety every time they are prepared. Based on such data,
contamination preventive measurements or procedures should be established and implemented
to use them in the manufacturing.
2. Where aseptic addition of cell substrates, media, buffer, and gases is need in a closed or open
system, the equipment should be selected to achieve sterility assurance and containment
condition. If inoculation into the culturing vessels, transfer of the cultures thereafter and/or
additions of media or buffer is to be necessary, an adequate operating control and procedures
should be established to minimize the contamination risk.
3. Critical process control parameters (e.g. pH, temperature, dissolved oxygen) and productivity
(yield) should be monitored. If any unusual results are noted in these parameters, the
possibility of contamination with bacterial, fungal, or Mycoplasma should be assessed.
4. If the cell culture or fermentation is performed with continuous culturing apparatuses like a
perfusion system, which is designed to continuously feed media and discharge culture solution
from the vessel, an appropriate operating procedure should be established to ensure that cell
culture can be continuously performed throughout the culturing period, without any unwanted
influence to product quality, like contamination.
5. Equipment used in cell culture and fermentation should be cleaned and sterilized after every
use. Fermentation equipment used in classical process should be cleaned and disinfected
appropriately. When genetically engineered microorganisms or cells are to be transferred (or
disposed) outside from the biological safety management areas, the transfer should be started
only after inactivation of such microorganism or cells is performed by a validated procedure,
as stipulated in the Law Concerning the Conservation and Sustainable Use of Biological
Diversity Through Regulations on the Use of Living Modified Organisms (the Cartagena Law),
and completion of inactivation should be confirmed every time before transfer. Washing of
cell culture and fermentation equipment should be performed by a validated cleaning
procedures established by taking into account the characteristics of the materials to be washed
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 81 -
off. In addition to CIP and SIP, other cleaning methods such as cleaning with complete
disassembly and manual cleaning should be conducted, as appropriate, depending on the
structural characteristics of the equipment.
6. Culture media should be sterilized before use via a method suitable for protecting the quality
of culture solution or fermentation solution.
7. Standard procedures for counter-measurements in the event of contamination with bacteria or
others (e.g. decontamination, disposal, washing feasibility check, potential impact on finished
products) should be established.
8. The process for seeding and additive addition should be basically performed in a closed
system. If these processes are to be performed using open vessels, it should be conducted in a
biosafety cabinet or similarly well-controlled environment to prevent contamination. These
measures should be controlled to prevent contamination from personnel, environment, and
production process.
A1.3 Harvesting, Isolation, and Purification
1. Cell harvesting to either remove cells or cellular components or recover cellular components
after cell disruption should be performed in an area and with equipment that are designed to
minimize the risk of microbial contamination of the harvested material, as well as the risk of
environment and personnel.
2. If an open system is used in the purification process, purification should be performed under
well-controlled, clean environment conditions suitable for maintaining the quality of
intermediates purified.
3. Removal or inactivation of microorganisms, cells, cellular debris, and media components
should be conducted under conditions suitable for minimizing risk of API quality deficiency
due to degradation, contamination, or other causes.
4. Buffers, column chromatography apparatus, and other materials used for the purification
process are not necessarily required to be sterile; however, the microorganisms level should be
controlled not to make any influence to the product quality. If purification and column
chromatography equipment cannot be sterilized, it should be decontaminated with a suitable
organic or alkaline solution. As bioburden level may vary depending on the type of process,
operation time, buffer solution, temperature, pH, etc., the control level should be established
for individual processes and conditions involved in manufacturing. If the process cannot be
sterilized, the level of endotoxins should be measured as a part of in process control, and
appropriate endotoxin control levels should be established to detect the increase of endotoxins
beyond the control level.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 82 -
5. All equipment should be properly cleaned after use and, if appropriate, disinfected, , or
sterilized, whenever feasible.
6. Purified API and intermediate products should be stored under predetermined appropriate
conditions, such as sterilization by filtration or other appropriate methods.
A2 Pharmaceutical Water
Basic concepts applicable to the manufacturing control and quality control of pharmaceutical water
are indicated as follows.
A2.1 Considerable Points Essential tor Basic Design of Pharmaceutical Water Equipment
The basic design of the equipment, and other subsystems s applicable to pharmaceutical water
production should be developed after establishing the procedures necessary for the efficient
manufacturing and quality control of pharmaceutical water so well as to maintain a
constant supply
of pharmaceutical water in required quality. Critical points to consider on designing the water
systems should include, but not limited to, the following:
1. All of the grades, specifications, quantities, and control methods for pharmaceutical water(s)
should clearly be defined.
2. The variant quality of source water including seasonal changes y should be thoroughly be
ascertained.
3. The principal water system design should be predetermined on maximum momentary water
flow rate, application time and frequency of water to be used, and such conditions demanded at
the points of use as temperature, number of ports, and piping specifications,
including branches
and pipes’ diameters.
4. Pharmaceutical water equipment should have such a reliable sterilization or sanitization
system as to ensure microbial control provided.
5. The locational adequacy of water sampling ports for water quality control should be evaluated
so well as to ensure stable supply of pharmaceutical water that fulfill required quality
specifications. Water samples should be collected from the locations not only of points of use
but also other critical points for the pharmaceutical water process. Locations necessary for water
quality assessment should be provided with certain structural features that facilitate the sampling
for quality analysis. If no sampling ports can structurally be set up at the expected locations, the
ports should preferably be located as close to the points of use as possible.
6. Although the water supply to the points of use should in principle be made through a loop
system, any appropriate alternative means for maintaining water quality should be employed,

Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 83 -
when water circulation is inapplicable s. No filters should be placed at any downstream
locations in the water purification system from the viewpoint of potential risk free from
microbial growth. However, filters may be placed at some upstream points of the water
purification system to eliminate impurities (e.g. with a protective filter attached to the outlet of
activated carbon filtration system).
7. Any backflow of water from the points of use should be prevented in consideration of such
appropriate procedural and mechanical steps as to adjust the pressure differences and to regulate
the valves.
8. Member materials used for pharmaceutical water equipment should be selected so suitably as
to maintain and control water quality at the required level. In particular, such corrosion-resistant
materials as AISI stainless steel 316 grade should be selected and have smooth surfaces given
especially at the locations contacting water for injection (WFI). The surfaces subject to
sterilization with pure steam or high-temperature water circulation should preferably be finished
by means of passivation.
9. The entire piping in the high-purity quality water system should be installed at such an angle
as to allow complete and easy drainage of water from the system.
10. As water will readily stagnate at “dead legs” occurring in T-shaped branches from the main
piping leading thence to a closure mechanism such as valves, the distance between the
diametrical axial center of the main piping and the closure mechanism in use should not be
longer than six times to the inner diameter of the branch, but desirably not longer than three
times if possible.
11. Measuring instruments should be a sanitary type free of water stagnation.
12. The directions and contents of fluids running through the various installed pipes should be
displayed on the outer surfaces of the piping at locations accessible to operators at an
appropriate interval.
A2.1.1 Pretreatment Equipment
Pretreatment equipment should be selected in consideration of the capacity suitable for maintaining
invariable water quality within the specifications required and for maximizing water treatment
efficiency and system life on the basis of elaborate investigation of the amounts of heavy metals,
free chlorine, organic matter, microorganisms, and colloidal particles, etc. present in the source
water.
A2.1.2 Equipment for Producing Water for Injection
The manufacturing equipment for water for injection should be designed so as to facilitate periodic

Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 84 -
sterilization with pure steam. If steam sterilization is inapplicable to the equipment because of its
low heat resistance, an alternative system should be allowed to perform sterilization or sanitization
using hot water or chemical agents. Some points to consider in designing the equipment for
producing WFI are summarized below.
1. Distillation Equipment (Water Stills)
Commonly applied types of distillation are single-effect, multi-effects, and vapor-compressors.
The latter two types are so highly efficient in producing high-quality water and highly
energy-saving as to be recommended for a large-scale of pharmaceutical water production.
Since each of these three methods has different attributes, it is important to select a proper
distillation method based on the intended use,
and the estimated f consumption in order to
fully utilize the advantages of each method.
The design of the water distillation system should include such adequate and practical
considerations as to satisfy the specifications required for the still combined with the
procedures of such feed water pretreatment as ion-exchange, reverse osmosis, ultrafiltration,
and any other subsystem used and to prevent any entrainment of impurities carried over with
vapor and to determine a blow-down flow appropriate for prevention from scaling due to
condensation.
2. Reverse Osmosis (RO)
The reverse osmosis (RO) is used to improve various factors in water quality by allowing
water to flow through permeable and semipermeable membranes based on osmotic pressure
differentials to remove small molecular solutes similar in size to inorganic salts as well as
solvent molecules, microorganisms, endotoxins, etc. depending on their respective
concentrations in source water. Although RO can be treated at an ambient temperature and its
performance is highly cost effective in energy-saving compared with distillation, stricter
control than that of distillation is required to prevent any leaks due to pinholes into the
downstream and microbial contamination. Points to consider in designing RO membrane units
are shown below:
(1) As no gaseous carbon dioxide and ammonia can be removed from feed water by RO,
such prior pretreatment as deaeration, neutralization, and/or ion-exchange should be
required on occasional demand.
(2) Appropriate equipment for the microbiological control and monitoring should be
included in place in the pretreatment system for feed water to meet the predetermined
control criteria.
(3) As RO generally operated at an ambient temperature may cause some concern about

Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 85 -
downstream contamination due to leaks through pinholes developed in the membrane, the
structural system composed of two ROs in series should preferentially be designed to provide
enhanced reliability and better control. Additionally, UV sterilization, heat-sterilization, and other
appropriate treatments in the downstream should be performed to inhibit microbial growth in the
system
3. Ultrafiltration (UF)
Ultrafiltration (UF) is a hyper-filtration method capable of removing endotoxins from feed water.
Unlike RO systems, UF units can generally be operated at a far lower pressure than RO and are
excellent in heat-resistance. Some UF membranes are made of materials resistant to steam
sterilization, and hence the membranes can be
relatively allow their surfaces to be easily sterilized
with high-temperature water or chemical agents. It is recommended to select high-grade UF
membranes capable of removing organic substances with a molecular weight of fraction exceeding
6,000 Daltons and suitable for the intended use. Although the purification performance of UF
modules is dependent on the upstream water quality and the modules’ grade as that of RO, routine
maintenance and control of the UF system should be made in order to exercise no ill effects on the
purification performance and water quality developed by microbial growth due to the fouling of
particulate matter and microorganisms.
4. Storage Tanks for Water for Injection (WFI) and Other High-purity Waters
WFI should preferably be used so immediately after production as to avoid any intermixture
contamination with microorganisms and other chemical substances. The following factors
should be taken into account in storing WFI and other high-purity pharmaceutical waters in
tanks.
(1) Storage tanks should be a closed-type with smooth inner surfaces. The nozzles of a level
indicator attached to the tank should be minimum in number and as short as possible.
(2) Storage tanks should be structured to allow no water stagnation and easier cleaning of
the inner surfaces and to facilitates
complete drainage.
(3) The appropriate capacity of a storage tank should be determined to provide a water
turnover at the highest possible rate. However, wherever feasible, a longer storage of
pharmaceutical water in the tank should be avoided. The maximum storage time should be
established by validation.
(4) The storage tank should be provided with such a hydrophobic ventfilter with micropores
of 0.2 or 0.22 μm as to prevent the tank from any invasion of microorganisms and foreign

Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 86 -
matter. The integrity of the ventfilter should be ensured prior to installation and at regular
intervals thereafter.
(5) Where hot water is supplied into a storage tank, a heater should preferably be set up
around a vent filter to prevent the filter from obstruction due to condensation of the hot
water.
(6) When the tank is disinfected with hot water, the tank should be equipped with such an
additional mechanism as to have heat spread over the whole inner surface of the tank
including its upper part.
(7) As common safety valves are difficult to disinfect or sterilize because of their structural
complexity, a sanitary type of safety valves should be employed or combined with a
rupture disc type of valve to ensure the water quality in the tank. When a rupture disc
valve is used, an alarm system should desirably be in place employed to give an alarm
signal on the rupture.
(8) As the gas-liquid interface in a tank is a part ready to induce microbial growth and
develop corrosion, water should preferably be spread over throughout the entire tank,
including the tank top with constant water fluidity kept.
5. Piping Structure
Pharmaceutical water stored in a tank is transported to the points of use through the piping
with relatively small diameters and structured as a closed system so that the inner situation of
the piping, once installed, is difficult to examine and inspect . Therefore, thorough review of
control methods and preventive measures for troubles in the piping system should be made at
the design phase. Key points to consider in the piping system are described below:
(1) It is basically preferable that the piping system should not be provided as allowable as
possible with any bypass or branch through which water is not constantly running.
(2) WFI should preferably be circulated constantly at a temperature not lower than 80°C
and at a turbulent flow rate enough to prevent microorganisms and organic matter from
anchoring on the surfaces. Where no water circulation is given, the unused water should
be drained and refilled with new water.
(3) Any loop circulated at an ambient temperatur should be considered on some preventive
measures for microbial growth. One example is the employment of UV lamps (Germicidal
Ultraviolet Lamp) placed at an appropriate location along the piping route.
(4) If The piping system is designed as a closed-loop should be provided with some
preventive measures in the loop to maintain a positive inner pressure against any backflow

Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 87 -
from points of use.
(5) Where no circulation system is adopted, the system should be provided with such
preventive measures as hot water flushing or steam sterilization for microbial
contamination prior to water supply.
(6) Every horizontally arranged piping should have an inclination of at least 1/100 given to
prevent water from stagnation in the piping in drainage and steam or hot water
sterilization .
(7) The piping system should be provided with an ejection port allowing for adequate and
easy drainage and designed to prevent backflow.
(8) Special considerations should be taken to eliminate any risks of cross contamination at
the shutdown, in abnormality, in maintenance and check of the system relating to the
piping of supply water used for manufacturing pharmaceutical products which quickly
disperse into the air to induce hypersensitive reactions in minute amounts or such products
that may have significant effect on the attributes of other products upon cross
contamination. The separate piping route for different products should be arranged in the
other exclusive system. If a single system is inevitably used in the manufacture of
different products, such efficient measures should be implemented as to prevent or
minimize cross contamination.
6. Heat Exchangers
Any contamination of feed water due to the leakage of heat medium contained in the heat
exchanger should be prevented. A double tube type or a double tube-sheet type (shell-and-tube
type) of heat exchanger is generally used. Any plate-type of heat exchangers should not be
used for manufacturing WFI. When If
a heat exchanger other than the first two types is used, a
heat exchanger allowing no contamination of feed water due to heat medium should be
selected. If any potential risk of contamination of feed water is supposed, a positive pressure
on the feed water side should be maintained at a level higher than that on the heat medium side,
and an appropriate monitoring
system and alarm for the pressure differential should be
attached to the heat exchanger.
7. Points of Use and Sampling Points
Adequate design and control of the points of use and sampling points should be made in
consideration of the following:
(1) No sterilizing filters should in principle not be placed at any points of use, since the

Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 88 -
filters may hamper adequate monitoring of microbiological contamination in the water
system and endotoxins could be released from microorganisms retained by the filters or
from dead microorganisms in the filters. If the attachment of sterilizing filters is
unavoidable, the frequency of disinfection/sterilization and replacement should be
determined based on validation. No sterilizing filters should preferably not
be placed in
the loop for water supply.
(2) When no water samples can be collected at the points of use, sampling ports should be
installed as close to the points of use as possible, except for the cases where the sampling
and/or the installation of such sampling ports are regarded to be obviously
disadvantageous.
(3) Sampling locations should be structured to have neither influence of initial blow-down
prior to sampling nor limitation of sampling containers.
8. Valves and Instruments
Diaphragm valves, instruments, detectors, etc. mounted on the pharmaceutical water system
should be free from water stagnation or dead spaces. Valves should be designed to achieve
sanitary application. Electric conductivity meters and total organic carbon (TOC) meters
should preferably be installed in the in-line in order to monitor chemical quality of water in a
timely manner. The locations of these instruments should be selected at the points that best
represent a local water quality in the piping system.
9. Pumps
Pumps should be a sanitary type in structure of a sealed casing protected against
contamination and be capable of withstanding hot water sanitization and/or pure steam
sterilization. Although centrifugal pumps made with stainless steel are mostly used, some
appropriate pumps should preferably be selected from the viewpoint of such key functions as a
head, discharge capacity, contact surface smoothness, and mechanical seal integrity in
consideration of various essential factors such as maximum momentary water consumption, an
average flowrate, the piping system from a water tank to the points of use, and some other
points relating to the piping.
10. Ultraviolet Radiation (UV) Lamps for Disinfection
Although UV lamps may be placed in the running water pipes for disinfection against
microorganisms grown, the bactericidal effectiveness of any UV lamps is so limited that

Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 89 -
mastering of the principles in UV lamps should be premised on the application. Points to
consider in proper design of UV lamp installation are indicated below:
(1) A UV lamp with a wavelength of 254nm is commonly used for disinfection, and hence
careful attention should be paid to the limited bactericidal effect, which is exercised only
within narrow emitted spectra close to the irradiated wavelength. Further, the disinfection
efficacy will vary with water temperature, flow rate, the intensity and duration of
irradiation, and the types of target microorganisms. It should also be noted that
microorganisms cannot be completely eliminated by UV irradiation, as some UV
irradiated microorganisms may assume such phenomena as photo-reactivation and/or
dark-reactivation.
(2) When If UV lamps are placed in the n loop for the purpose of disinfection, the locations of the
lamps should appropriately be selected in view of their inherent advantageous efficacy.
A2.2 Validation of Pharmaceutical Water
A validation program for pharmaceutical water equipment is to establish the water quality
monitoring program and the operation and maintenance program for equipment control including the
sampling plan as well as the qualification of equipment design, installation, operation and
performance.
1. Determination of quality characteristics of pharmaceutical water to be validated
2. Determination of equipment appropriate for manufacturing water of intended quality from
source water
3. Selection of equipment, process control, and a monitoring program
4. Design Qualification (DQ)
5. Installation qualification (IQ)
IQ of equipment should include the following:
(1) Calibration of instruments
(2) Qualification in that all pharmaceutical water equipment as shown in the specifications is
installed on their
drawings, and ready for operation as well as verification of instruments.
6. Operational Qualification (OQ)
OQ of equipment should include the following:
(1) Qualification of the reliable operation performance of equipment, alert system, and control
system
(2) Qualification of the appropriateness of established alert and action levels

Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 90 -
7. Performance Qualification (PQ)
In the early stage (phase 1) of PQ, g the capacity of equipment enough for the stable
manufacture and supply of pharmaceutical water in the required quality should be qualified.
In the subsequent stage (phase 2), alert and action levels for the required water quality should
be established, and SOPs for routine control should also be established for the purpose of
effecting this stage. However, at the start of this qualification, the capacity of equipment to
produce water meeting predetermined specifications should be ascertained for at least 3
consecutive days to 1 week at all points of use in critical processes for the systems of WFI and
purified water, and at points of use in the sub-loops and critical processes for the equipment
applied to source water and feed water. The same ascertainment of the above items should be
followed to conduct PQ at the Phase 2 over 1 year on documented procedures and control
criteria to obtain the practical data of seasonal variations in water quality and to confirm
secure water system performance. During this phase-2 period, the frequency of the
replacement for parts and instruments concerned should be examined, and potential problems
with routine control should be extracted to take effectual measures for solution.
In the final phase (phase 3), constant and stable pharmaceutical water production should be
ensured in the required quality within the alarm level on the trend analyses of the data on
treated water given under the influence of seasonal variations in the quality of source water, so
as to make a full report on the results,
and to evaluate the reliability of the entire equipment
control program.
8. Equipment maintenance program
The procedural or other related documents on the periodical review of the process control of
validated equipment for pharmaceutical water should appropriately be drawn up to be actually
conducted, and also the implementation plan with a schedule and procedures for the validation
on periodical revalidation including recalibration should adequately be made into practice so
that some preventive action or corrective action including proper change control should be
taken into account of the results on occasional demand.
A2.3 Routine Control of Pharmaceutical Water
A2.3.1 Outline
The demonstrated process qualification fully implemented at the initial validation stage should be
premised on the routine and periodic control programs of pharmaceutical water. The routine control
should include conductivity and total organic carbon (TOC) essential as critical control parameters,
and the periodic control should include these two parameters as well as additional control parameters
such as the counts of viable microorganisms, endotoxins, and particulate matter, depending on the

Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 91 -
intended use of the water. The frequency of measuring these parameters should be determined based
on the stability of water quality. For more details on the r
routine control of pharmaceutical water,
refer to the “Quality Control of Water for Pharmaceutical Use” in the General Information
section of
the Japanese Pharmacopoeia 16.
A2.3.2 Sanitization
The water sanitization system should be validated to demonstrate the capability effective in
reducing microbial contamination to such an acceptably low level as to be maintained. For the heat
sanitization system a heat distribution test should be made to demonstrate that heat is distributed
throughout the entire water system. The chemical sanitization system needs the demonstration that a
selected chemical agent is spread throughout the entire system at an optimal bactericidal
concentration. It is also required to demonstrate y
that any residual chemical agent can effectively be
removed after sanitization. In general, the frequency of sanitization should be determined based on
monitoring results for the water equipment in order to adequately operate the equipment under
microbiologically well-controlled conditions and to maintain the bacterial count below the alert
level.
A2.3.3 Water Sampling
A monitoring program for pharmaceutical water equipment should be implemented at an
appropriate frequency to ensure that the equipment is well managed and controlled and allows water
of required quality to be continuously produced. Water should be sampled at the points that the
representative water quality in the process or the distribution system can be expected to be shown,
and the sampling frequency should be established based on validation data so that sampling locations
should be determined to cover all critical regions in the systems
. A sampling schedule should be laid
out in view of required quality characteristics of water to be collected. For example, as equipment
for WFI must meet microbiologically strict control requirements, the sampling frequency for WFI
needs to be higher than that for other types of pharmaceutical water.
Any water sample for microbiological testing should be tested immediately after collected, or the
sample should appropriately be stored for the following analysis. When the sample stored for a
certain period, the records of n
storage conditions and time should be kept.
Microbiological analysis of sampled running water only indicates that any presence or situation of
microorganisms in the pharmaceutical water will be suspected or supposed. Therefore, airborne
microbial counts are used as an indicator of contamination for the pharmaceutical water equipment
and also serve as a basis for establishing the alert level for the equipment. If a high level of airborne
microbial counts is consistently found, this phenomenon will indicate some growth on the biofilm so

Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 92 -
that appropriate control measures should be used.
A2.3.4 Alarm Level and Action Level
A quality monitoring program should be developed and implemented to ensure that pharmaceutical
water is constantly produced at acceptable quality, when pharmaceutical water equipment is
continuously operated within the design specifications. Monitoring data obtained should be
compared with established process parameter limits or product water specifications. In addition,
appropriate alert and action levels should be separately established on referencing the “Quality
Control of Water for Pharmaceutical Use” in the General Information of the Japanese
Pharmacopoeia 16 and used for process control as well as in judging the adequacy of operation
conditions of equipment.
A2.3.5 Microbiological Monitoring Program
The primary purpose of the microbiological monitoring program for controlling pharmaceutical
water equipment is to predict deterioration of microbiological quality of water and to ensure water
quality by maintaining constant function of the pharmaceutical water system to prevent undesirable
effects of microbiological deterioration on product quality. The microbiological quality of generated
water should be controlled at an appropriate level by not only counting but also identifying the type
of microorganisms present in the water system with the trend analysis approach.
Although it is not unnecessary to detect all types of microorganisms present in water; a monitoring
approach capable of detecting the widest possible range of microorganisms including any
microorganisms of slow growth should be adopted. The microbiological limits for pharmaceutical
water should be appropriately established by referencing the limits specified in “Microbial Limit
Test” in the General Tests, Processes, and Apparatus section of the Japanese Pharmacopoeia 16. If
the microbial count exceeds the specified action level during validation or routine control,
microorganisms should be identified or have their properties examined. If specific microorganisms
are detected in large quantities, possible biofilm formation in the water system should be suspected,
and appropriate sterilization or sanitization should be implemented to decontaminate the system.
A2.3.6 Monitoring of Conductivity and Total Organic Carbon (TOC) in Pharmaceutical
Water
The procedures of the action levels or alert levels for conductivity and TOC should conform to
those shown in the Microbiological Limits.
When water quality is controlled based on conductivity and TOC, no tests of individual metals or

Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 93 -
inorganic ions may be made. The tests of these metals and ions, however, should preferably be made
to clear up the causes whereby these parameters exceed the action or alert level.
A2.4 Training of Personnel Engaged in Pharmaceutical Water Equipment
An appropriate education and training program should be prepared concerning the production and
quality control of pharmaceutical water and be conducted periodically and as needed for personnel
engaged in the operation, maintenance, and control of the facility and equipment as well as water
quality control in order to produce and control pharmaceutical water in the required quality level.
Education and training results should be recorded in writing and retained in an archive. Main
education and training items included in the program are listed below. These items may be
performed at one time or successively over scheduled courses.
1. Relationship between pharmaceutical water quality and pharmaceutical product groups
classified by the types of water used
2. Relationship between variations in source water quality, pharmaceutical water equipment, and
pharmaceutical water quality
3. Control methods for pharmaceutical water equipment (including methods for sanitization ,
sterilization, and disinfection)
4. Test methods and control limits for pharmaceutical water
5. Ecological activities of microorganisms present in pharmaceutical water equipment (e.g. inner
surface condition of pipes, influence of water flow, formation of biofilms and endotoxins, etc.)
6. Sampling procedures and precautions for sampling
7. Validation, change control, and deviation control for pharmaceutical water equipment
A2.5 Maintenance and Control of Pharmaceutical Water Equipment
A preventative maintenance program should be established and implemented to maintain
pharmaceutical water equipment under well-controlled conditions. The program should include, but
not be
limited to, the following items:
1. Procedures for operating water equipment
2. Programs for monitoring critical water quality characteristics and equipment-operating
conditions
3. Schedules for periodic sanitization
4. Preventative maintenance and calibration of each component of water equipment
5. Change control of water equipment and operating conditions
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 94 -
6. Procedures for temporary stoppage and resumption of water equipment
In particular, procedures for the stoppage and resumption of ultrafiltration membrane
treatment equipment should be carefully evaluated by taking into account risk of leakage due
to functional degradation of membrane surfaces
6. Understanding of water equipment and its maintenance
A2.6 Change Control
When water equipment is remodeled or expanded or system operation procedures are modified,
potential impact of these changes on the quality of pharmaceutical water should be evaluated. If the
reevaluation of the equipment is judged to be necessary, equipment properties concerned should be
validated again, as appropriate. Procedures for change control should be established as part of an
equipment maintenance program. These Change control procedures should also be established in the
equipment maintenance program as part of validation and maintenance and included.
A2.7 Deviation Control
When parameters for the pharmaceutical water equipment may exceed the predetermined alert or
action levels, procedures to be taken should be documented in advance. If any deviations from the
action level occur, at least the following items should be recorded:
1. Procedures for resampling and retesting
2. Reporting procedures
3. Procedures for the action of pharmaceutical water and pharmaceutical products manufactured
using pharmaceutical water
4. Preventive measures
5. Corrective measures
6. Reevaluation of the monitoring program and established alert and action levels
A3 Pest Control of Aseptic Manufacturing Facilities
A3.1 General Requirements
Pest control of facilities for manufacturing sterile pharmaceutical products (as well as
pharmaceutical manufacturing facilities in general) is critical in maintaining a clean manufacturing
environment. Identification of insect species found within aseptic processing areas is also critical for
maintaining cleanliness of facilities, since arthropods serve as an indicator of the presence of
mold-induced food chains and are indicative of potential problems in overall biological cleanliness.
Arthropods have a rich bioburden of microorganisms and their spores on their bodies. Thus, proper
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 95 -
pest control is important from the view point of microbiological control of sterile pharmaceutical
products.
Arthropods captured in pharmaceutical manufacturing facilities may include members of
Insecta, Arachnida (spiders, mites), Chilopoda (centipedes), and Isopoda (sow bugs) classes. In this
document, these arthropods are collectively referred to as “arthropods.”
Although quite rare, arthropods may be found in APAs. An appropriate sampling method for
arthropods should be developed for estimating the population of minute arthropods in very low
density. Separately, a suitable pest control program should be established and implemented to
monitor and remove arthropods (especially fungivorous arthropods) found inside facilities, because
arthropods are rarely carried into or invade APAs from the outside.
A3.2 Pest Control Program
1. Pest control programs applicable to each clean area should be established and implemented,
and control practice records should be produced and retained.
2. Pest control programs should preferably include the following procedures:
(1) Procedures to be taken from pest monitoring activities to the implementation of
corrective actions
(2) Pest control procedures that are to be implemented after pest monitoring results which
deviate from established control criteria
(3) Follow-up survey procedures for pest monitoring data which deviates from control
criteria
(4) Survey procedures for identifying the source of fungal contamination, if fungivorous
arthropods are detected
(5) Reevaluation of cleaning procedures if arthropods that feed on organic matter in dust are
detected
3. Scope of pest monitoring
Arthropods should primarily be monitored in indirect support areas in manufacturing facilities
for sterile pharmaceutical products, and then, as monitoring results may require, in direct
support areas to assess potential influence of arthropods on the quality of pharmaceutical
products. The scope of pest monitoring should preferably be examined at each instance of
newly constructing or remodeling facilities and equipment.
4. Sampling method and size
(1) Equipment used for pest monitoring should be carried into the APA while exercising
suitable precautions to avoid contamination of the production environment.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 96 -
(2) Sampling methods should be established by taking into account the ecology of target
arthropods that may inhabit manufacturing facilities.
5. Control standards
(1) Pest control acceptance criteria should be established.
(2) In a practical manner, pest control should be conducted based on the maximum number
of arthropods, not the mean, as most arthropods are not uniformly distributed but
congregate in clumps.
(3) Arthropods emerging inside processing/manufacturing areas should be counted
separately from those invading from outside.
(4) Not only should the insect population be evaluated, but also the growth and distribution
pattern.
(5) Insect monitoring results should be classified and evaluated by the area and type of
arthropods.
6. Corrective and preventive actions
(1) Corrective and preventive actions should be promptly implemented based on monitoring
results, and the effectiveness of these actions should be confirmed.
(2) Historical records regarding the growth and distribution patterns of arthropods should be
analyzed, and appropriate preventive measures should be implemented based on results
of trend analysis.
A3.3 Preventive Measures against Arthropods
Appropriate and effective species-specific pest control measures should be developed and
implemented, targeting arthropods identified through the monitoring program.
1. Species-specific insect control
Different species of arthropods have different food habits, life history, ecology, and behaviors.
The pest control program should be tailored for the treatment of individual target species.
For example, an existing cleaning program should be reviewed to strengthen the control of
arthropods that consume organic matter in dust, and a fungus treatment program should be
implemented for fungivorous species.
2. Fungus control
In most cases, the appearance of arthropods in clean areas may be associated with fungi
growing in a facility with inadequate design and operational features. If fungivorous
arthropods are detected, the design, construction and operation of the facility should be
reevaluated, and fungus control measures should be implemented or reinforced.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 97 -
3. Supervision of facility and its design for the prevention against insects
If there are signs or evidences of insect invasion from outside or if abnormal insect population
growth is observed inside the facility, if there are insects invading from outside or the
population of insects breeding inside, facility and its design should be reevaluated to prevent
the insects.
4. Precautions in the use of insecticides
(1) Insecticides should not be used in clean areas, as a general rule.
(2) When the use of insecticides is necessary to control abnormal population growth of
insect pests, appropriate preventive measures should be instituted to prevent the
contamination of pharmaceutical products. When insecticides are used in controlled but
unclassified areas, due care should be exercised to prevent the insecticides from
dispersing into the surrounding areas.
(3) When insecticides are used in clean environments, the surfaces of these areas should be
cleaned following application using suitable method to remove the insecticides.
Following cleaning all surfaces should be confirmed to be free from insecticide
residues.
(4) For all insecticides used in pharmaceutical manufacturing facilities, the Material Safety
Data Sheets (MSDS) and use records of the insecticides should be retained for archival
purposes.
A4 Biosafety and Biosecurity Measures
Biotechnological processing using microorganisms and toxins for manufacturing
pharmaceutical products must be managed by physical containment facilities, equipment, and/or
procedures in addition to the procedures for sterility assurance (Guidance on Biosafety Practices in
Manufacturing Facilities for Biopharmaceutical Products, Notification No. 14 of the Inspection and
Guidance Division, PMSB dated February 14, 2000, and Laboratory Biosafety Manual Version 3
issued by WHO).
Biological and pharmaceutical products manufactured using genetic engineering technologies
should be manufactured in compliance with the Guidance on Regulations for the Transport of
Infectious Substances 2009-2010 issued by WHO, and biohazardous raw materials that require
biosecurity measurements should be in compliance with the Act on Prevention of Infectious Diseases
and Medical Care for Patients Suffering Infectious Diseases and Biorisk Management: Laboratory
Biosecurity Guidance issued by WHO.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 98 -
A4.1 Biosafety Levels
The manufacture of pharmaceutical products using microorganisms (the term
“microorganisms” denotes both “viruses” and “bacteria” in this chapter) should be conducted at
biosafety levels (BSLs) suitable for safe handling of materials depending on the level of risks from
individual pathogenic microorganisms. Microorganisms used in the manufacture are classified into
the three risk groups defined below, and BSL for manufacturing facilities are designated as BSL 1 to
3 consisting of the combined elements of physical capability of microorganism containment,
availability of safety instruments and protective products against infection, and operational
procedures to be implemented. As an exception, the procedures for inactivation or removal of
microorganisms may be conducted in the manner typically applied to non-biological products.
1. Risk group 1 (BSL: 1): No known or minimal risk of exposure to pathogenic agents (e.g.
pathogenic isolates of viruses for vaccine production, such as measles, rubella, mumps,
chickenpox, BCG, etc.) for operators engaged in microorganism handling or in the
surrounding area
2. Risk group 2 (BSL: 2): Moderate risk of exposure to pathogenic agents (e.g. Bordetella
pertussis, Corynebacterium diphtheriae, Clostridium tetani, Vibrio cholerae, etc.) for
operators engaged in microorganism handling and low risk for other operators in the
surrounding area
3. Risk group 3 (BSL: 3): High risk of exposure to pathogenic agents for operators engaged in
microorganism handling and low risk for other operators in the surrounding area.
Person-to-person transmission of infectious diseases does not occur under routine working
conditions. Effective therapeutic and preventive measures are available.
A4.2 Biosecurity Measures
Microorganisms and certain types of bacterial toxins have a great potential to damage regional
societies surrounding laboratory facilities when dispersed or intentionally released. Therefore,
biological security measures are usually required in biopharmaceutical sectors. Stringent control
measures should be implemented and followed at the very least for registration or designation of
operators engaged in operations, entry and exit from facilities, and storage and transfer of
biomaterials, as described below:
1. Registration or designation of operators engaged in handling of microorganisms and bacterial
toxins
2. Preparation of SOPs for storage, transfer, and transport of microorganisms and bacterial toxins
and record keeping of storage, transfer, and transport practices
In addition, microorganisms and toxins designated as specific pathogens by the Act on
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 99 -
Prevention of Infectious Diseases and Medical Care for Patients Suffering Infectious Diseases
should be classified as Types 2, 3, or 4, in accordance with the Act, and facility construction, import,
transfer, transportation, use, storage, and sterilization of such materials should be handled in
compliance with governing laws and regulations.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 100 -
A4.3 Biosafety Management Areas (Controlled Areas)
Biosafety management areas (“controlled areas”) should be established which meet the levels
of containment required for proper handling of microorganisms based on pathogenic potential of
microorganisms to be handled. If the BSL of the controlled area is 2 or higher, international
biohazard symbol containing the supervisor’s name and emergency contact information should be
displayed on entry and exit doors of the area.
The entry of non-registered personnel into controlled areas should be restricted by instituting
appropriate measures or systems at the entrances/exits and in storage rooms of microorganisms and
toxins, and entry and exit records of registered personnel should be retained as required by
applicable regulations and guidelines established for handling microorganisms and toxins.
A4.4 General Requirements for BSL1
1. There are no specific biosafety requirements necessary for BSL1 for facilities and equipment.
2. Waste materials carrying risk of infection (the term “waste materials” includes carcasses and
denotes materials contaminated with microorganisms in this document) should be sterilized
with chemical agents or disinfected with heat and transferred out of the controlled area. The
materials may be placed in leakproof containers and, after disinfection of their surfaces,
transferred out of the controlled area. Then, the materials should be incinerated. It is
acceptable to contract out the incineration, provided the materials are sterilized beforehand.
A4.5 General Requirements for BSL2
1. Any operations that may generate aerosolized microorganisms should be conducted using
closed-system equipment provided with HEPA filters, safety cabinets (Class IIA or higher), or
other equipment having a similar capacity for microorganism containment. In addition, air
exhausted from such equipment or systems should be cleaned so as to completely eliminate
aerosolized microorganisms.
2. Waste materials carrying risk of infection should be disposed of using one of the procedures
below. The incineration of the sterilized materials may be contracted out.
(1) Chemically disinfect or heat-sterilize waste materials, transfer them out of the controlled
area, and burn them using an incinerator within the facility.
(2) Place waste materials in leakproof containers to prevent dispersion during transportation,
and after disinfection of their surfaces, transfer them out of the controlled area for
incineration within the facility.
(3) Transfer waste materials from the controlled area directly to either an incinerator or
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 101 -
sterilizer using an appropriately managed closed-system procedure, and then incinerate
the materials within the facility.
3. Waste fluids containing microorganisms and fluids that come into direct contact with
microorganisms should be disposed of after appropriate treatment with chemical disinfection
or heat sterilization in a tank or other closed system placed inside or outside the controlled
area.
4. The disposal of toxins and other wastes should be conducted while taking their properties into
account.
A4.6 General Requirements for BSL3
1. Controlled areas designated for handling microorganisms that require BSL3 containment
should be structurally separated from other areas.
2. Personnel entry into BSL3 facility should be controlled by displaying “restriction notices” and
establishing procedures required to obtain permission to enter. Additionally, physical entry
restrictions, such as a security door, should be installed.
3. Ceilings, walls, and floors of controlled areas should be smooth-surfaced, crack-free,
non-dust- or debris-shedding, and resistant to chemicals or other types of disinfectants in order
to maintain a closed system in these areas.
4. If air current is controlled in working areas of a controlled area, inward current should be
secured to minimize leakage of microorganisms from the area. The direction of the current
should be monitored by, for example, measuring and recording pressure differences against
adjacent areas. Entrances to areas with pressure difference should be equipped with airlocks,
and substantial pressure differences should be maintained in order to prevent inversion of
pressure differences or air currents with the surroundings.
5. Effective disinfecting apparatuses or devices should be available in BSL3 areas to implement
appropriate disinfection measures against pathogenic contamination.
6. Faucets in restrooms, washing sinks, and other places should be touchless, elbow-handled, or
pedal operated to prevent cross-contamination.
7. Work areas in the controlled area should be sufficiently spacious to prevent contamination
during operations.
8. The HVAC system (e.g. ducts) should be structurally designed to facilitate sterilization with
gases, as appropriate.
9. Any operation which may carry a risk of generating aerosolized microorganisms should be
conducted in a safety cabinet (Class II or
III) equipped with HEPA filters or other closed and
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 102 -
contained systems of an equivalent or higher safety level. Additionally, air from such a work
environment should be evacuated outside the facility after passing through HEPA filters.
10. Air in controlled areas should be filtered and evacuated through an independent HVAC system
equipped with HEPA filters.
11. BSL3 facilities should be designed to be capable of physically containing microorganisms
within the controlled area under any contingent circumstances, such as shut-down of the
HVAC system.
12. An emergency power supply should be available to maintain continuous operation of the
HVAC system in the event of power failure.
13. The drain system should be mounted with devices to prevent backflow.
14. Waste fluids containing microorganisms or those that come into direct contact with
microorganisms should be chemically disinfected or heat sterilized in a tank or other closed
system in either controlled or non-controlled areas and then disposed of.
15. Waste materials carrying risk of infection should be disposed of using one of the following
procedures:
(1) Chemically disinfect or heat sterilize waste materials, transfer them out of the controlled
area, and burn them using an incinerator within the facility.
(2) Place waste materials in leakproof containers to prevent dispersion during transportation,
and after disinfection of their surfaces, transfer them out of the controlled area for
incineration within the facility.
(3) Transfer waste materials from the controlled area directly to either an incinerator or
sterilizer using an appropriately-managed closed-system procedure, and then incinerate
the materials within the facility.
16. Personnel should use infection protection supplies (e.g. clothes, mask, gloves). Gowning and
degowning procedures should be appropriate. Infection protection-reinforced clothes such as
positive-pressure protective suit should be used if the situation requires it.
A4.7 Emergency Procedures
A4.7.1 Emergency Safety Measures
The following emergency procedures should be established and documented in preparation for
emergency situations such as leakage of aerosolized microorganisms or culture media, exposure to
microorganisms, fire outbreak, or natural disaster:
1. Rescue of personnel exposed to microorganisms, emergency attention and first aid for injured
personnel, and therapy for personnel infected with microorganisms
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 103 -
2. Wastes material containment methods
3. Procedures for decontamination
4. Emergency SOPs and communication networks
A4.7.2 Preventive and Counter Measures for Accidents Due to Specific Pathogens
The term “accident” in this document means a loss, theft, disappearance, intentional release,
etc. of retained specific pathogens and other microorganisms. The following procedures should be
established and documented in preparation for accidents:
1. Establishment of measures to both prevent and respond to accidents
2. Emergency communication networks
3. Reporting and notification procedures for accidents (as required depending on accident
classification)
A4.8 Personnel Training
Personnel who engage in operations in controlled areas should undergo biosafety and
biosecurity training programs prior to initial engagement and at periodic intervals thereafter. The
education and training programs should cover the following:
1. Characteristics of microorganisms to be handled in the controlled area (e.g. BSL, mode of
infection)
2. Procedures for entering and exiting the controlled area
3. Procedures for handling and operating equipment and devices installed in the controlled area
4. Procedures for safely handling microorganisms
5. Records of microorganism retention and supply
6. Containers and procedures for transporting infectious substances
7. Procedures for disposal of infectious waste materials
8. Emergency safety measures
9. Procedures for the use, storage, control, transportation, and disposal of microorganisms and
for reporting/notification in facilities where specific pathogens are handled
A5 Chemical Hazard Control
A5.1 Principles
Control procedures for hazardous materials should be evaluated and established based on the
assessment of potential health risks to patientsi.e. cross-contamination risks as well as potential
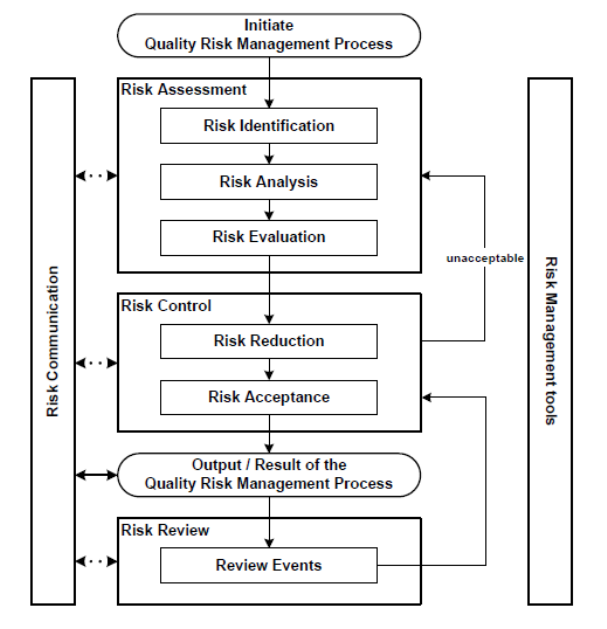
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 104 -
health risks to factory personnel. The ICH Q9 guideline “Quality Risk Management” states: “Risk is
defined as the combination of the probability of occurrence of harm and the severity of that harm.”
Health risks for patients and factory personnel may be defined as the combination of the probability
of contact with or extent of exposure to chemical substances (or drug substances) and a substance’s
potential (or hazard) to cause health damage. Chemical hazard control should be planned and control
measures implemented in accordance with the risk management process (Figure 1) proposed by the
ICH Q9 guideline “Quality Risk Management.” It is important to develop and implement efficient
measures to reduce the risks to permissible levels at every step of production process and throughout
the lifecycle of a product.
Under no circumstances should hazard prevention be used to justify compromising or
obfuscating the sterility assurance level of products.
Figure 1. Overview of a Typical Quality Risk Management Process
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 105 -
A5.2 Risk Management Processes
A5.2.1 Identification of Risks (Hazards)
It is important to establish permissible exposure limits of individual chemical substances (or
drug substances) based on scientific rationale, thereby potentially eliminating health hazards to
patients and factory personnel. These limits are obtained by extrapolating data from animals
(preclinical) or humans (clinical) to manufacturing plant setting. Such limits are defined as
“permitted daily exposure (PDE),” “acceptable daily exposure (ADE)”, or “acceptable daily intake
(ADI)” in various guidelines.
The exposure limits thus established are based on the risk of damage to human health and are
then used as reasonable references for determining safe exposure limits for work environments (e.g.
acceptable exposure concentrations and residual amounts), permissible cross-contamination limits,
and permissible residue levels on product contact surfaces. These limits provide the basis for
establishing manufacturing conditions adequate for ensuring that health risk to patients and factory
personnel are consistently below the maximum acceptable exposure level.
A5.2.2 Risk Analysis (Exposure Analysis)
The risk level for patients and factory personnel exposed to chemical substances depends on
the nature or properties of chemical substances (e.g. powder, liquids, or aerosol form; particle size;
specific gravity; solubility; etc.) over the production system life-cycle (all manufacturing steps,
including production, changeover cleaning, and parts replacement) in each manufacturing process as
well as the properties of the manufacturing processes themselves (e.g. open/closed systems,
containment equipment, protectors, dust collection, clean-in-place systems, wet-in-place, etc.).
Exposure analysis should be performed at both the steady- and non-steady-state production system’s
life-cycle, examining potential risk modalities such as the loss of containment equipment integrity.
A5.2.3 Risk Assessment
Outcomes of risk analysis regarding human exposure to hazards should be evaluated in
comparison with established exposure limits to assess potential health risks to patients and factory
personnel. Potential health risks to patients are only acceptable if the cross-contamination of one
pharmaceutical product with another product is known not to exceed defined cross-contamination
limits during the manufacturing process of a pharmaceutical product. However, health risks to
factory personnel may be acceptable if exposure to chemical substances is known to be controlled to
quantities equal to or less than the ADE via exposure routes such as inhalation, direct contact, and
oral.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 106 -
A5.2.4 Risk Control
If risk assessment results are judged to be unacceptable, appropriate risk mitigation measures
should be implemented in line commensurate with the magnitude of the risks. While risk control
measures vary, they should always address the following issue according to the prioritized list shown
below:
• Removal of target chemical substances
• Use of alternative chemical substances
• Use of containment equipment (as a physical risk mitigation measure)
• Protection of personnel working in an open system
The basis of risk control is to prevent dispersion of highly active (hazardous) chemical
substances into areas where residue control is not feasible or where decontamination is difficult.
The construction of a dedicated facility or the use of dedicated equipment should be
considered in the course of developing control measures if validation-based cleaning procedures
cannot be securely established or if risks of transferring chemical substances to non-product contact
areas or risks of contaminating products with dispersed chemical powder are not acceptable.
A5.2.5 Risk Review
Once chemical hazard preventive measures have been implemented based on a systematic risk
assessment and the implementation of well-defined control activities, the, results of the risk
management activities should be reviewed to confirm that the expected outcome has been achieved.
Measuring amounts of chemical dispersed and chemical residuerecovered from equipment
surface is a valuable and effective means of risk review.
A5.2.5 Risk Communication
Risk management processes included in the chemical hazard control program should be
systemically implemented by an organization with well-defined decision-making authority and
responsibility. The implementation should preferably be undertaken by interdisciplinary teams
comprised experts, as recommended in the ICH Q9 guideline. When teams are formed, they should
include experts from appropriate functional disciplines (e.g. quality unit, business development,
engineering, regulatory affairs, production operations, sales and marketing, legal, statistics, and
clinical), in addition to individuals knowledgeable on the quality risk management process.
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 107 -
A5.3 Personnel Training
1. The education and training programs for personnel engaged in chemical hazard control should
cover the following:
(1) Properties of chemical or drug substances to be handled
(2) Procedures for entering and exiting the controlled area
(3) Procedures for handling and operating equipment and devices within the controlled area
(4) Procedures for disposal of active waste materials
(5) Emergency safety measures
2. Education and training programs for personnel engaged in emergency operations should
include the following topics:
(1) Emergency treatment and first-aid care for factory personnel
(2) Decontamination procedures
(3) Emergency communication networks
A6 Tests and Inspections
A6.1 Endotoxins
A6.1.1 General Requirements
1. The possibility of endotoxin contamination should be considered and appropriate preventive
measures implemented for raw materials, containers, closures, pharmaceutical water used to
manufacture parenteral pharmaceutical products, and surfaces of manufacturing equipment
contacting products.
2. Surfaces of manufacturing equipment contacting products should be properly cleaned, dried,
and maintained in a clean condition to prevent an increase in bioburden and associated
endotoxin levels.
3. The final rinse of surfaces of manufacturing equipment contacting products as well as
containers and closures should use water for injection to prevent contamination with
endotoxins. The manufacturing equipment after washing should be kept dry unless
immediately followed by sterilization.
4. The efficiency of endotoxin removal from containers and closures should be validated when
the removal is performed by heat inactivation, surface washing, or adsorption or membrane
filtration of prepared drug solution.
5. Endotoxin testing procedures should generally comply with the Endotoxins Test in the
Japanese Pharmacopoeia. Prior to testing, a test for interfering factors should be performed to
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 108 -
identify the maximum valid dilution to confirm the absence of enhancing or inhibiting factors
for the reaction in sample solutions.
A6.1.2 Validation
1. When endotoxin inactivation or removal is performed by heating, washing, membrane
filtration, or adsorption, the post-processing residual endotoxin level should be verified to fall
within specified control limits by determining the post-processing removal rate after loading a
known amount of endotoxin.
2. Endotoxin testing should be supplemented by appropriate method validation.
3. Lysate and other reagents necessary for the endotoxin test should be appropriately controlled
with regard to storage temperature and expiration date.
A6.2 Insoluble Particulate Matter
A6.2.1 General Requirements
1. The amount of insoluble particulate matter remaining on washed containers, closures, and
drug solution-contact surfaces of manufacturing equipment after filtration sterilization as well
as in filtered drug solution should be controlled within certain acceptance limits.
2. Insoluble particulate matter occurring with time after production by interaction between
containers or closures and drug solution or by aggregation of high-molecular-weight
substances such as proteins should be closely monitored to control the levels below maximum
acceptable limits. The control efficiency should be verified by long-term stability tests.
3. Insoluble particulate matter testing procedures should generally comply with the Insoluble
Particulate Matter Test in the Japanese Pharmacopoeia.
A6.2.2 Validation
The Insoluble Particulate Matter Test should be supplemented by appropriate method
validation.
A6.3 Container Integrity
A6.3.1 General Requirements
1. Containers for sterile pharmaceutical products should be closed by an appropriately validated
method. The integrity-related parameters should be adequately controlled, since the integrity
of the container-closure system may be compromised, if operation conditions of
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 109 -
manufacturing equipment are not optimal.
2. Flawed containers or closures may become a contributory factor to the compromise of the
integrity. The integrity should be ensured by routine monitoring or by 100 percent inspection
of containers or closures, and necessary safety measures be implemented to prevent the supply
of pharmaceutical products with a risk of non-sterility.
3. Containers closed by fusion such as glass or plastic ampoules should be subjected to 100
percent integrity testing. Samples of other containers should be checked for integrity sccording
to appropriate procedures.
4. Container integrity and hence product sterility should be verified to be maintained until use.
5. Procedures for the integrity test should be established in a manner suitable for properties of
individual containers and closures.
A6.3.2 Validation
1. The container integrity test to be employed should be validated by appropriate methods.
2. The container integrity test should take at least the following test conditions into consideration,
wherever feasible: temperature variations during product storage, packaging forms, vibrations
and shocks during transportation, variations in atmospheric pressure during air transportation.
Rationale for these conditions should be documented.
A6.4 Visual Inspection
A6.4.1 General Requirements
1. If the sterility of pharmaceutical products is ensured by eliminating products that exhibit
observable container integrity failure, the relationship between container integrity and visual
characteristics should be appropriately defined for use as visual inspection criteria.
2. The visual inspection criteria should be optimized for each formulation of each pharmaceutical
product based on product characteristics.
3. Standards of foreign matter in pharmaceutical products should be established based on the
Foreign Insoluble Matter Test for Injectables in the Japanese Pharmacopoeia. Standards for
“readily detectable foreign insoluble matter” and “clearly detectable foreign insoluble matter”
should be specified for features of pharmaceutical products and types of foreign matter.
4. Pharmaceutical products should be subjected to the inspection of all products (“100%
inspection”) during the manufacturing process to remove products containing “readily
detectable foreign insoluble matter” or “clearly detectable foreign insoluble matter.” This
100% inspection should be followed by sampling inspection, as appropriate. The sample size
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 110 -
for sampling inspection should be sufficient for statistical analysis with respect to batch size
(refer to AOL sampling plan, as an example).
5. The test method of visual inspection should be specified in SOPs. For example, visual
inspection by humans should include, but not be limited to, the following test conditions:
(1) Inspection procedures, inspection pitch, time required for inspection per unit of
inspection, and intervals of inspectors’ rest breaks
(2) Inspection benches, inspection conveyers, inspection lamps, inspection magnifiers, and
inspection posture (e.g. seated on chair)
(3) Light intensity ininspectionposition, light intensity in inspection area or room, and color
of background plate
If all pharmaceutical products are required or planned to be visually inspected during the
manufacturing process, the conditions for inspection such as the duration of observation and
light intensity, should be specified and optimized for individual products to completely
remove “readily detectable foreign insoluble matter” and “clearly detectable foreign insoluble
matter.” The light intensity over the area of inspection should range from 2000 to 3750 lux if
sampling inspection or quality test is performed after 100 percent visual inspection. The time
required for visual inspection should be 5 seconds per unit of inspection per background color
of white and black each. The light intensity and duration of observation may be increased, as
appropriate.
6. If visual inspection is conducted using automatic inspection equipment, at least the following
matters should be determined and specified:
(1) Model of automatic inspection equipment, speed of work, and time required for
inspection per unit of inspection
(2) Assessment methods for the performance of the equipment at the beginning and end of
inspection operation as well as periodic verification of the performance using reference
standards
(3) Calibration
7. When a boundary sample is to be prepared for use in the interpretation of inspection results,
the quality of the sample should be evaluated and approved by the quality control department.
Since boundary samples deteriorate or decompose over time, an expiration date should be set
or sample quality should be periodically evaluated.
A6.4.2 Validation
1. When visual inspection is conducted manually by the inspector, the ability of individual
Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing
- 111 -
inspectors should be evaluated using boundary samples to ensure that they have the
predetermined competency and satisfy qualification requirements on visual inspection. Their
ability as well as eyesight should be assessed periodically.
2. The capacity of automatic inspection equipment should be periodically validated using
boundary samples to ensure that the equipment has the required capacity for inspecting and
eliminating foreign insoluble matter.
3. Samples of foreign insoluble matter to be used to validate the inspection process should
preferably be obtained through actual production. The use of validation samples should be
approved by the quality control department.
B Revision Records
Initially issued by the Compliance and Narcotics Division, PMSB, MHLW as an office
communication in July 2006.
Totally revised and issued by the Compliance and Narcotics Division, PMSB, MHLW as an
office communication in April 2011.
