
ILLUMINA PROPRIETARY
Part # 15028392 Rev. J
August 2013
MiSeq
®
Sample Sheet
Quick Reference Guide
FOR RESEARCH USE ONLY
Revision History 3
Introduction 5
Sample Sheet Parameters 6
Sample Sheet Settings for Sequencing 12
Sample Sheet Settings for Analysis 13
Sample Sheet Settings for Variant Calling 18
Naming the Sample Sheet 20
Technical Assistance
This document and its contents are proprietary to Illumina, Inc. and its affiliates ("Illumina"), and are intended solely for the
contractual use of its customer in connection with the use of the product(s) described herein and for no other purpose. This
document and its contents shall not be used or distributed for any other purpose and/or otherwise communicated, disclosed,
or reproduced in any way whatsoever without the prior written consent of Illumina. Illumina does not convey any license
under its patent, trademark, copyright, or common-law rights nor similar rights of any third parties by this document.
The instructions in this document must be strictly and explicitly followed by qualified and properly trained personnel in order
to ensure the proper and safe use of the product(s) described herein. All of the contents of this document must be fully read
and understood prior to using such product(s).
FAILURE TO COMPLETELY READ AND EXPLICITLY FOLLOW ALL OF THE INSTRUCTIONS CONTAINED HEREIN
MAY RESULT IN DAMAGE TO THE PRODUCT(S), INJURY TO PERSONS, INCLUDING TO USERS OR OTHERS, AND
DAMAGE TO OTHER PROPERTY.
ILLUMINA DOES NOT ASSUME ANY LIABILITY ARISING OUT OF THE IMPROPER USE OF THE PRODUCT(S)
DESCRIBED HEREIN (INCLUDING PARTS THEREOF OR SOFTWARE) OR ANY USE OF SUCH PRODUCT(S) OUTSIDE
THE SCOPE OF THE EXPRESS WRITTEN LICENSES OR PERMISSIONS GRANTED BY ILLUMINA IN CONNECTION
WITH CUSTOMER'S ACQUISITION OF SUCH PRODUCT(S).
FOR RESEARCH USE ONLY
© 2011–2013 Illumina, Inc. All rights reserved.
Illumina, IlluminaDx, BaseSpace, BeadArray, BeadXpress, cBot, CSPro, DASL, DesignStudio, Eco, GAIIx, Genetic
Energy, Genome Analyzer, GenomeStudio, GoldenGate, HiScan, HiSeq, Infinium, iSelect, MiSeq, Nextera, NuPCR,
SeqMonitor, Solexa, TruSeq, TruSight, VeraCode, the pumpkin orange color, and the Genetic Energy streaming bases
design are trademarks or registered trademarks of Illumina, Inc. All other brands and names contained herein are the property
of their respective owners.

Revision History
MiSeq Sample Sheet Quick Reference Guide
3
Revision History
Part # Revision Date
Description of Change
15028392 J August
2013
Added the following information:
• Sample sheet settings BaitManifestFileName,
OutputGenomeVCF, PicardHSmetrics, and StitchReads; added
description of read stitching
• Sample ID requirements for the Targeted RNA workflow
• Adapter sequences for TruSeq libraries; moved adapter
sequences to Adapter Settings section
15028392 H May 2013
Added sample sheet requirements for the Amplicon - DS
workflow.
Changed the Custom Amplicon workflow to TruSeq Amplicon
workflow.
15028392 G April 2013
Updated the Adapter sample sheet setting to include the adapter
sequence for Nextera Mate Pair libraries.
Added the ReverseComplement sample sheet setting for Nextera
Mate Pair libraries using the Resequencing or Assembly
workflows.
15028392 F March
2013
Updated the following sample sheet settings:
• CustomAmpliconAlignerMaxIndelSize—Changed default to 25
• FilterPCRDuplicates—Changed to FlagPCRDuplicates
• VariantCaller—Added Resequencing workflow
• EnrichmentMaxRegionStatisticsCount and
ExcludeRegionsManifestA—Added for the Enrichment
workflow
• Added VariantMinimumQualCutoff
• Removed VariantFilterQualityCutoff;
VariantMinimumGQCutoff is preferred
• Added requirements for Targeted RNAworkflow
• Removed Index 2 as an option for Assembly and Small RNA
workflows
• Noted that custom indices can be used for the Custom
Amplicon workflow with MCS v2.2, or later
Removed custom primer instructions. See Using Custom Primers on
the MiSeq (part # 15041638).

4
Part # 15028392 Rev. J
Part # Revision Date
Description of Change
15028392 E November
2012
Added the following information:
• Description of Enrichment workflow, manifest, and data
section requirements
• Data section requirements for PCR Amplicon workflow
• Descriptions of sample sheet settings AdapterRead2 and
QualityScoreTrim
• Note about supported option for listing genome references for
multiple species in the same sample sheet when using MiSeq
Reporter v2.1
Updated the following information:
• Organized sample sheet settings into settings for sequencing,
analysis, and variant calling
• Updated Small RNA workflow to list the genome folder as
required in the sample sheet
• Updated sample sheet settings for variant calling to add
VariantMinimumGQCutoff, and to update StandBiasFilter and
MinimumCoverageDepth for the Enrichment workflow
15028392 D July 2012
Added the following information:
• Added the PCR Amplicon analysis workflow for Nextera XT
libraries and information about the manifest file
• Noted that adapter trimming is recommended for longer read
lengths up to 250 cycles
• Added descriptions of sample sheet settings for
PercentTilesToScan and StrandBiasFilter
• Changed Setup Options screen to Run Options screen per MCS
v1.2
15028392 C April 2012
Updated the following information:
• Updated name of Amplicon workflow to Custom Amplicon
• Updated name of DenovoAssembly workflow to Assembly
• Added GenerateFASTQ workflow
• Listed genome folder as required for amplicon sequencing in
Sample Sheet Parameters
15028392 B December
2011
Updated the steps in Setting Up the Sample Sheet.
Listed manifest files as required for TruSeq Custom Amplicon
libraries.
15028392 A September
2011
Initial release
Introduction
MiSeq Sample Sheet Quick Reference Guide
5
Introduction
The sample sheet is a comma-separated values (*.csv) file that stores information
required to set up, perform, and analyze a sequencing run.
Illumina recommends that you create your sample sheet before preparing your sample
libraries. You can create your sample sheet using the Illumina Experiment Manager or
create it manually using a text editor, such as Excel or Notepad.
Before starting the run, make sure that the sample sheet is accessible to the instrument.
You can either copy the sample sheet to a network location or copy the sample sheet
from a USB flash drive using the Manage Files feature in MiSeq Control Software (MCS).
When the run begins, the software copies the sample sheet from the designated sample
sheet folder to the root of the MiSeqOutput folder. At the end of the run, the sample sheet
is used for secondary analysis by the MiSeq Reporter software.
Illumina Experiment Manager
The Illumina Experiment Manager is a wizard-based application that guides you
through the steps to create your sample sheet.
Using the Illumina Experiment Manager not only reduces syntax errors, but also
provides prompts for information that applies to your sample type and analysis
workflow. It provides a feature for recording parameters for the sample plate, such as
sample ID, project name, dual indices, and barcode information. Then, the sample plate
information can be imported to the sample sheet using the Illumina Experiment
Manager.
The Illumina Experiment Manager can be run on any Windows platform. To download
the software, go to the Illumina Experiment Managersupport page on the Illumina
website (support.illumina.com/sequencing/sequencing_software/experiment_
manager/downloads.ilmn).
For more information, see the Illumina Experiment Manager User Guide (part # 15031335).
Sample Sheet Workflow
1 Create your sample sheet using one of the following methods:
• Illumina Experiment Manager—See the Illumina Experiment Manager User Guide
(part # 15031335).
• Excel or Notepad—See Sample Sheet Parameters on page 6.
2 Name your sample sheet with the reagent cartridge barcode number associated with
the sequencing run, and use a *.csv extension. For more information, see Naming the
Sample Sheet on page 20.
3 Copy the sample sheet to the sample sheet folder specified in Run Options in MCS
or other location accessible to the instrument computer.
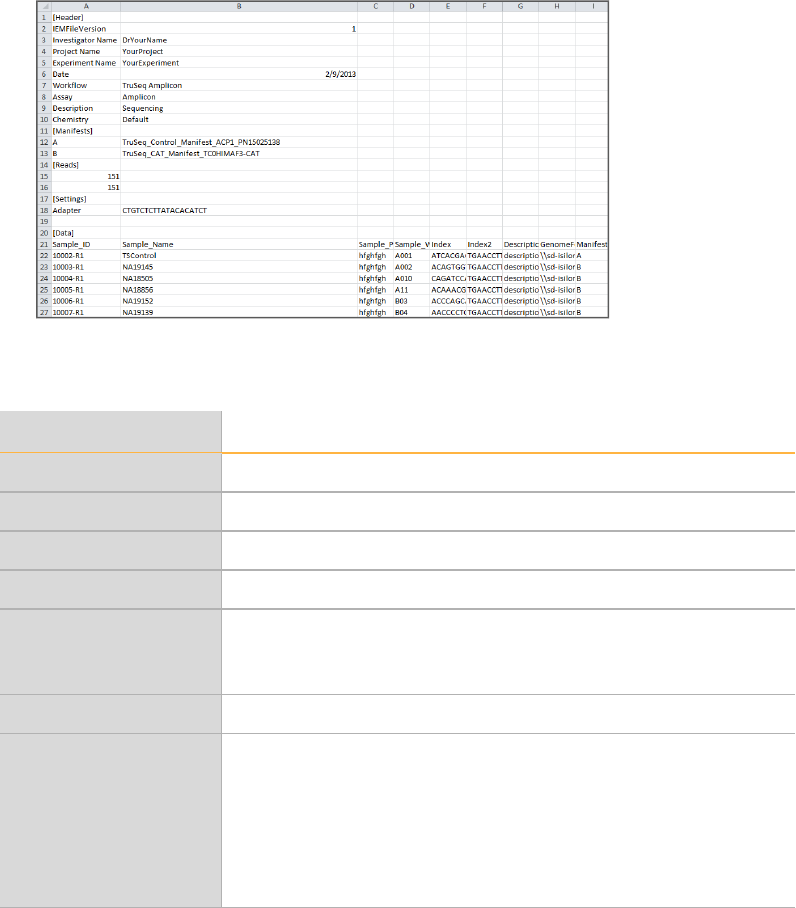
6
Part # 15028392 Rev. J
Sample Sheet Parameters
The sample sheet is organized in sections titled Header, Reads, Manifests, Data, and
Settings. Section headings are case-sensitive and shown in brackets [] in the following
example:
Figure 1 Sample Sheet Example in Excel
Header Section
Parameter Description
Investigator Name Your name.
Project Name Project name of your preference.
Experiment Name Experiment name of your preference.
Date Date of your experiment.
Workflow Required
The workflow field must list the analysis workflow name
recognized by MiSeq Reporter.
Assay The name of the assay used to prepare your samples.
Chemistry The recipe fragments used to build the run-specific recipe. If this
field is blank, the system uses the default recipe fragments.
Optional—For non-indexed or single-indexed TruSeq RNA or
TruSeq DNA libraries, leave this field blank.
Required—For any workflows that use dual indexing, such as
Nextera and TruSeq Custom Amplicon, the chemistry field is
required. Enter amplicon in this field.

Sample Sheet Parameters
MiSeq Sample Sheet Quick Reference Guide
7
Reads Section
Parameter Description
Number of cycles
in Read 1
Required
Number of cycles
in Read 2
Required for paired-end runs.
NOTE
The index sequence defined in the Data section specifies the number of cycles for
the index read.
Data Section
The following table summarizes the Data section requirements for each analysis
workflow. Column order is not important.
Workflow Required Data Columns Optional Data
Columns
Amplicon - DS Sample_ID, Sample_Name, Manifest,
GenomeFolder, Index
Index 2*
Assembly Sample_ID Sample_Name,
Index,
GenomeFolder
Enrichment Sample_ID, Manifest, GenomeFolder Sample_Name,
Index, Index 2
Generate FASTQ Sample_ID Sample_Name,
Index, Index 2
LibraryQC Sample_ID, GenomeFolder Sample_Name,
Index, Index 2
Metagenomics Sample_ID Sample_Name,
Index, Index 2
PCR Amplicon Sample_ID, Manifest, GenomeFolder Sample_Name,
Index, Index 2
Resequencing Sample_ID, GenomeFolder Sample_Name,
Index, Index 2
Small RNA Sample_ID, GenomeFolder,
Contaminants, miRNA, RNA
Sample_Name, Index
Targeted RNA Sample_ID**, Sample_Name, Manifest,
GenomeFolder, Index, Index 2
--
TruSeq Amplicon
(formerly Custom
Amplicon)
Sample_ID, Manifest, GenomeFolder,
Index
Sample_Name, Index
2*
* For the TruSeq Amplicon and Amplicon - DS workflows—Using MCS v2.2, or later, you can
optionally specify a single-index run.

8
Part # 15028392 Rev. J
** For the Targeted RNA workflow, the sample ID and sample name must be specified in the sample
sheet in such a way to specify replicates. For more information, see Data Columns for the Targeted
RNA Workflow on page 10.
Column
Heading
Description
SampleID Required
Every sample must have a unique sample ID.
At least one sample must be listed. List one sample per line.
Sample_Name Optional
The sample name is used in reporting and file naming.
Index Required for multi-sample assays with single or dual indexing.
Nucleotide sequence—Valid characters are A, C, G, T, and N, where
N matches any base. Enter the index sequence of the i7 index.
Index2 Required for multi-sample assays with dual indexing.
Nucleotide sequence—Valid characters are A, C, G, T, and N, where
N matches any base. Enter the index sequence of the i5 index.
NOTE
For the appropriate index sequences, see the user guide for your sample preparation kit.
Genome Folder Path
The GenomeFolder contains the reference genome in FASTAfile format. For optimal
results, store reference genomes on the local drive or use BaseSpace.
Enter the full path (UNC path) to the GenomeFolder in the sample sheet. Do not enter the
path using a mapped drive.
NOTE
Introduced in MiSeq Reporter v2.1, you can specify genome references for
multiple species in the same sample sheet for all workflows except the Small RNA
workflow.
Data Section Requirements by Workflow
Not all columns used in the Data section of the sample sheet apply to every analysis
workflow. For example, the Manifests column is required for some workflows, and not
required for other workflows.
Data Columns for the Amplicon - DSWorkflow
Column Heading Description
Sample Name Required
Group sample names in pairs. List exactly two samples with
identical sample names, one sample for the forward direction and
one sample for the reverse direction.

Sample Sheet Parameters
MiSeq Sample Sheet Quick Reference Guide
9
Column Heading Description
GenomeFolder Required
Specify the path to the reference genome folder, which contains the
FASTA files to be used in the alignment step. It is the same
reference genome specified in the manifest file. Specifying the
genome folder provides coordinates and chromosome mapping.
For example, the genome folder for human is hg19 (Homo_
sapiens\UCSC\hg19\Sequence\WholeGenomeFASTA).
Manifest Required
Specify the manifest key for this sample, which is located in the first
column of the Manifests section. Two manifests must be specified,
one manifest containing the reverse complement of the probes
listed in the other manifest.
Data Columns for the Assembly Workflow
Column Heading Description
GenomeFolder Optional
If provided, MiSeq Reporter compares the de novo assembly against
the reference genome, and generates a dot-plot that graphically
summarizes the results.
• If the specified folder does not exist, Illumina Experiment
Manager combines the GenomePath configuration setting with
the genome string.
• If the path does not exist, MiSeq Reporter stops processing.
Data Columns for the Enrichment Workflow
Column Heading Description
GenomeFolder Required
Specify the path to the reference genome folder, which contains the
FASTA files to be used in the alignment step.
Manifest Required
Specify the manifest key for this sample, which is located in the first
column of the Manifests section.
Data Columns for the Library QC Workflow
Column Heading Description
GenomeFolder Required
Specify the path to the reference genome folder, which contains the
FASTA files to be used in the alignment step.
10
Part # 15028392 Rev. J
Data Columns for the PCR Amplicon Workflow
Column Heading Description
GenomeFolder Required
Specify the path to the reference genome folder, which contains the
FASTA files to be used in the alignment step.
Manifest Required
Specify the manifest key for this sample, which is located in the first
column of the Manifests section.
Data Columns for the Resequencing Workflow
Column Heading Description
GenomeFolder Required
Specify the path to the reference genome folder, which contains the
FASTA files to be used in the alignment step.
Data Columns for the Small RNA Workflow
Column Heading Description
GenomeFolder Optional
If provided, reads are aligned against the full reference genome.
Contaminants Required
Specify the path to the folder containing the FASTA files of
contaminants.
miRNA Required
Specify the path to the folder containing FASTA files of mature
miRNAs.
RNA Required
Specify the path to the folder containing FASTA files of small RNAs.
Data Columns for the Targeted RNA Workflow
Column Heading Description
GenomeFolder Required
Specify the path to the reference genome folder, which contains the
FASTA files to be used in the alignment step.
Manifest Required
Specify the manifest key for this sample, which is located in the first
column of the Manifests section of the sample sheet.

Sample Sheet Parameters
MiSeq Sample Sheet Quick Reference Guide
11
Data Columns for the TruSeq Amplicon Workflow
Column Heading Description
GenomeFolder Required
Specify the path to the reference genome folder, which contains the
FASTA files to be used in the alignment step.
The reference genome must be the same genome used to generate
the manifest file. The genome folder is used to provide variant
annotations and set the chromosome sizes in the BAM file output.
Manifest Required
Specify the manifest key for this sample, which is located in the first
column of the Manifests section of the sample sheet.
Sample Sheet Settings
The Settings section of the sample sheet is optional for all workflows. Settings control
various sequencing and analysis parameters. Each line in the Settings section contains a
setting name in the first column and a value in the second column.
12
Part # 15028392 Rev. J
Sample Sheet Settings for Sequencing
Parameter Description
CustomRead1PrimerMix
CustomIndexPrimerMix
CustomRead2PrimerMix
Create one line for each custom primer used. Indicate C1 for the
Read 1 primer, C2 for the Index primer, or C3 for the Read 2
primer. Custom primers are supported for Read 1, Index 1 Read,
and Read 2 only.
For more information, see Using Custom Primers on the MiSeq (part
# 15041638).
PercentTilesToScan If set to the default value of 1, 100% of the tiles are scanned. Valid
values are 0 through 1.
• If set to 0, the software rounds up to one tile.
• For all other settings, the software rounds down. For example, a
value of 0.99 results in one less tile than the maximum number
of tiles possible on the flow cell.
For more information about dual-surface scanning, see the MiSeq
System User Guide (part # 15027617).
Sample Sheet Settings for Analysis
MiSeq Sample Sheet Quick Reference Guide
13
Sample Sheet Settings for Analysis
Parameter Description
Adapter For all workflows
Specify the 5' portion of the adapter sequence to prevent
reporting sequence beyond the sample DNA.
Illumina recommends adapter trimming for Nextera
libraries and Nextera Mate Pair libraries.
To specify two or more adapter sequences, separate the
sequences by a plus (+) sign. For example:
CTGTCTCTTATACACATCT+AGATGTGTATAAGAGA
CAG
For more information, see Adapter Settings on page 15
and Adapter Sequences on page 16.
AdapterRead2 For all workflows
Specify the 5' portion of the Read 2 adapter sequence to
prevent reporting sequence beyond the sample DNA.
Use this setting to specify a different adapter other than
the one specified in the Adapter setting.
For more information, see Read 1 and Read 2 Adapters on
page 16.
Aligner For the Resequencing workflow and Library QC
workflow
As of MiSeq Reporter v2.2, Eland has been deprecated,
but not removed. For backward compatibility, use the
Aligner setting to specify Eland.
When using the default aligner for any workflow, you
do not need to specify the alignment method in the
sample sheet.
BaitManifestFileName For the Enrichment workflow
Specify the full path to the bait file. This setting is used
only if the PicardHSmetrics setting is used and set to
true.
CustomAmpliconAlignerMax
IndelSize
For the TruSeq Amplicon workflow
By default, the maximum detectable indel size is 25.
A larger value increases sensitivity to larger indels, but
requires more time to complete alignment.
EnrichmentMaxRegion
StatisticsCount
For the Enrichment workflow
Default is 40000. Sets the maximum number of rows
shown in the Targets table and recorded
EnrichmentStatistics.xml.
14
Part # 15028392 Rev. J
Parameter Description
ExcludeRegionsManifestA For the Enrichment workflow
This setting excludes one or more region groups
(separated by plus signs) from consideration. For
example, if this setting specifies ABC+DEF, any region
that has either ABC or DEF specified in the Group
column of the manifest is ignored when parsing the
manifest. No variant calling is performed for this region
or reported in enrichment statistics.
If the sample sheet contains more than one manifest, use
multiple lines, such as ExcludeRegionsManifestB,
ExcludeRegionsManifestC.
FlagPCRDuplicates For the Enrichment workflow, Library QC workflow,
PCR Amplicon workflow, and Resequencing
workflow
Settings are 0 or 1. Default is 1, filtering.
If set to 1, PCR duplicates are flagged in the BAM files
and not used for variant calling. PCRduplicates are
defined as two clusters from a paired-end run where
both clusters have the exact same alignment positions
for each read.
(Formerly FilterPCRDuplicates. FilterPCRDuplicates is
acceptable for backward compatibility.)
Kmer For the Assembly workflow
This setting overrides the k-mer size used by Velvet.
Default is 31; odd-numbered values up to 255 are
supported.
OutputGenomeVCF For the Enrichment workflow, PCR Amplicon
workflow, and TruSeq Amplicon workflow
Settings are 0 or 1.
If set to true (1), this setting turns on genome VCF
(gVCF) output for single sample variant calling.
This setting requires MiSeq Reporter v2.3.
QualityScoreTrim For all workflows
If set to a value > 0, then the 3' ends of non-indexed
reads with low quality scores are trimmed. Trimming is
automatically applied by default at a value of 15 when
using BWA for alignment.
PicardHSmetrics For the Enrichment workflow
Settings are 0 or 1. Default is 0.
If set to true (1), this setting generates Picard HS metrics
for the given bait and manifest file. If the bait file is not
explicitly identified, the manifest file is used as the bait
file.
Use the BaitManifestFileName setting to specify the bait
file.
Sample Sheet Settings for Analysis
MiSeq Sample Sheet Quick Reference Guide
15
Parameter Description
ReverseComplement For the Assembly workflow, Library QC workflow,
and Resequencing workflow
Settings are 0 or 1. Default is 1, reads are reverse-
complemented.
If set to true (1), all reads are reverse-complemented as
they are written to FASTQfiles. This setting is necessary
in certain cases, such as processing of mate pair data
using BWA, which expects paired-end data. Per-cycle
metrics might be disrupted by this setting.
Set this setting to 1 when using the Resequencing
workflow or Assembly workflow with Nextera Mate
Pair libraries.
Set this setting to 1 when using the Workflow workflow
with Nextera Mate Pair libraries.
StitchReads For the Amplicon - DS workflow, Generate FASTQ
workflow, and TruSeq Amplicon workflow
Settings are 0 or 1. Default is 0, paired-end reads are not
stitched.
If set to true (1), paired end reads that overlap are
stitched to form a single read. To be stitched, a minimum
of 10 bases must overlap between Read 1 and Read 2.
Paired-end reads that cannot be stitched are converted
to two single reads.
This setting requires MiSeq Reporter v2.3.
For more information, see Read Stitching on page 17.
TaxonomyFile For the Metagenomics workflow
This setting overrides the taxonomy database; default is
taxonomy.dat.
As of MiSeq Reporter v2.3, species-level classification is
enabled, by default. For faster, but less granular genus-
level classification, specify gg_13_5_genus_32bp.dat.
VariantCaller For the Enrichment workflow, PCRAmplicon
workflow, Resequencing workflow, and TruSeq
Amplicon workflow
Specify one of the following variant caller settings:
• GATK (default)
• Somatic (recommended for tumor samples)
• Starling (legacy variant caller)
• None (no variant calling)
When using the default variant caller for the workflow,
it is not necessary to specify the variant calling method
in the sample sheet.
Adapter Settings
Some clusters can sequence beyond the sample DNA and read bases from a sequencing
adapter, particularly with longer read lengths up to 250 cycles.
The Adapter sample sheet setting prevents reporting sequence beyond the sample DNA
by trimming the specified sequence in FASTQ files. Trimming the adapter sequence
avoids reporting of spurious mismatches with the reference sequence, and improves
performance in accuracy and speed of alignment.

16
Part # 15028392 Rev. J
For workflows using BWA, the Adapter setting trims reads from the start of the adapter
sequence. When using ELAND (deprecated in MiSeq Reporter v2.2), reads are N-masked
or replaced with Ns (no-call) from the start of the adapter sequence.
Read 1 and Read 2 Adapters
If you specify an adapter sequence for the Adapter setting in the sample sheet, the same
adapter sequence is trimmed for Read 1 and Read 2. To trim a different adapter sequence
in Read 2, use the AdapterRead2 setting in the sample sheet.
To trim two or more adapters, separate the sequences by a plus (+) sign.
How Adapter Trimming Works
MiSeq Reporter considers each potential adapter start position (n) within the sequence,
starting at the first base (n=0). The process continues to count matches and mismatches
between sequence (n) and adapter (0), sequence (n + 1), and adapter (1), and so on. This
loop terminates if the following occurs:
MismatchCount > 1 and MismatchCount > MatchCount
Otherwise, the count continues until the end of the sequence or end of the adapter is
reached, whichever comes first. The sequence is trimmed starting at position n, if:
MatchCount / (MatchCount + MismatchCount) > Cutoff
By default, the cutoff is 0.9 or < 10% mismatch rate. This default setting can be modified
using the configurable setting AdapterTrimmingStringency.
Masking Short Reads
MiSeq Reporter includes the NMaskShortAdapterReads configuration setting, which
prevents reads that have been almost entirely trimmed or masked from confounding
downstream analysis. This setting is applied under the following conditions:
} If the adapter is encountered within the first 32 bases of the read, then the adapter
sequence is N-masked.
} If the adapter is identified in the first 32 bases and the read includes ten or more
bases from the start of the adapter, the entire read is N-masked. The configuration
setting NMaskShortAdapterReads controls the ten-base limit.
Adapter Sequences
Library Type Adapter Sequence
Nextera libraries Illumina recommends adapter trimming for Nextera
libraries.
CTGTCTCTTATACACATCT
Nextera Mate Pair libraries Illumina recommends multiple adapter trimming for
Nextera Mate Pair libraries.
Read 1:
CTGTCTCTTATACACATCT
Read 2:
AGATGTGTATAAGAGACAG

Sample Sheet Settings for Analysis
MiSeq Sample Sheet Quick Reference Guide
17
Library Type Adapter Sequence
Small RNA libraries By default, adapter trimming is performed in the Small
RNA workflow using the standard adapter sequence:
TGGAATTCTCGGGTGCCAAGGC
Use the Adapter setting to specify an adapter sequence
other than the standard sequence.
TruSeq libraries Read 1:
AGATCGGAAGAGCACACGTCTGAACTCCAGTC
A
Read 2:
AGATCGGAAGAGCGTCGTGTAGGGAAAGAGT
GT
Read Stitching
MiSeq Reporter v2.3, or later, is required to use the optional StitchReads setting. Read
stitching is compatible only with the Amplicon - DS workflow, GenerateFASTQ
workflow, and TruSeq Amplicon workflow. Read stitching is not possible with any other
Illumina alignment method or analysis workflows, but might be allowable input with
some third-party analysis tools.
When set to true (1), paired-end reads that overlap are stitched to form a single read in
the FASTQfile. At each overlap position, the consensus stitched read has the base call
and quality score of the read with higher Q-score.
When the StitchReads setting is applied, the stitched read and each individual read are
aligned and the alignment information is used in variant calling. A BAM file is written
for the stitched read, Read 1, and Read 2. In some cases, this setting can improve
accuracy of variant calling.
For each paired read, a minimum of 10 bases must overlap between Read 1 and Read 2
to be a candidate for read stitching. The minimum threshold of 10 bases minimizes the
number of reads that are stitched incorrectly due to a chance match. Candidates for read
stitching are scored as follows:
} For each possible overlap of 10 base pairs or more, a score of 1 – MismatchRate is
calculated.
} Perfectly matched overlaps have a MismatchRate of 0, resulting in a score of 1.
} Random sequences have an expected score of 0.25.
} If the best overlap has a score of ≥ 0.9 and the score is ≥ 0.1 higher than any other
candidate, then the reads are stitched together at this overlap.
Paired-end reads that cannot be stitched are converted to two single reads in the FASTQ
file.
18
Part # 15028392 Rev. J
Sample Sheet Settings for Variant Calling
Setting Name Description
FilterOutSingleStrandVariants For the PCRAmplicon workflow and Resequencing
workflow
This setting filters variants if they are found in one
read-direction only.
Default value:
• 1 (on)—Somatic variant caller (0 for TruSeq
Amplicon workflow)
This setting does not apply to the TruSeq Amplicon
workflow.
IndelRepeatFilterCutoff This setting filters indels if the reference has a 1-base
or 2-base motif over eight times (by default) next to
the variant.
Default value:
• 8—GATK
• 8—Somatic variant caller
• 8—Starling
MinimumCoverageDepth For the Enrichment workflow
The variant caller filters variants if the coverage depth
at that location is less than the specified threshold.
Decreasing this value increases variant calling
sensitivity, but raises the risk of false positives.
Default value:
• 20—GATK (Enrichment workflow only; 0 for any
other workflow)
MinQScore This setting specifies the minimum base call Q-score to
use as input to variant calling.
Default value and variant caller:
• 20—Somatic variant caller
• 0—Starling
StrandBiasFilter For the Enrichment workflow
This setting filters variants with a significant bias in
read-direction. Variants filtered in this way have SB in
the filter column of the VCF file, instead of PASS.
Default value:
• -10—GATK (Enrichment workflow only; no filter
for any other workflow)
• 0.5—Somatic variant caller
VariantFrequencyEmitCutoff This variant caller does not report variants with a
frequency less than the specified threshold.
Default value:
• 0.01—Somatic Variant Caller

Sample Sheet Settings for Variant Calling
MiSeq Sample Sheet Quick Reference Guide
19
Setting Name Description
VariantFrequencyFilterCutoff This setting filters variants with a frequency less than
the specified threshold.
Default value:
• 0.01—Somatic variant caller
• 0.20—GATK
• 0.20—Starling
VariantMinimumGQCutoff This setting filters variants if the genotype quality
(GQ) is less than the threshold. GQ is a measure of the
quality of the genotype call and has a maximum value
of 99.
(Formerly, VariantFilterQualityCutoff, which is acceptable
for backward compatibility.)
Default value:
• 30—GATK
• 30—Somatic variant caller
• 20—Starling
VariantMinimumQualCutoff This setting filters variants if the quality (QUAL) is less
than the threshold. QUAL indicates the confidence
that the variant is genuine.
Default value:
• 30—GATK
• 30—Somatic variant caller
• 20—Starling

20
Part # 15028392 Rev. J
Naming the Sample Sheet
During the run setup steps, MCS looks for a sample sheet with a name matching the
barcode number of the reagent cartridge. Therefore, Illumina recommends that you name
your sample sheet with the barcode number of the reagent cartridge followed by *.csv
extension. The barcode number is on the reagent cartridge label directly below the
barcode.
In the following example, the sample sheet name is MS2000006-500.csv. You do not
need to include the kit version in the sample sheet name.
Figure 2 Reagent Cartridge Label
If the barcode number is not known, use a preferred name for the sample sheet followed
by *.csv. When the software cannot locate the sample sheet during the run setup steps,
browse to the appropriate sample sheet.

Technical Assistance
MiSeq Sample Sheet Quick Reference Guide
Technical Assistance
For technical assistance, contact Illumina Technical Support.
Illumina Website
www.illumina.com
Email
techsupport@illumina.com
Table 1 Illumina General Contact Information
Region Contact Number Region Contact Number
North America 1.800.809.4566 Italy 800.874909
Austria 0800.296575 Netherlands 0800.0223859
Belgium 0800.81102 Norway 800.16836
Denmark 80882346 Spain 900.812168
Finland 0800.918363 Sweden 020790181
France 0800.911850 Switzerland 0800.563118
Germany 0800.180.8994 United Kingdom 0800.917.0041
Ireland 1.800.812949 Other countries +44.1799.534000
Table 2 Illumina Customer Support Telephone Numbers
MSDSs
Material safety data sheets (MSDSs) are available on the Illumina website at
www.illumina.com/msds.
Product Documentation
Product documentation in PDF is available for download from the Illumina website. Go
to www.illumina.com/support, select a product, then click Documentation & Literature.

Illumina
San Diego, California 92122 U.S.A.
+1.800.809.ILMN (4566)
+1.858.202.4566 (outside North America)
techsupport@illumina.com
www.illumina.com
