
A RESIDENT’S GUIDE TO
PEDIATRIC RHEUMATOLOGY
4
th
Revised Edition - 2019

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 2
This guide is intended to provide a brief introduction to basic topics in pediatric rheumatology.
Each topic is accompanied by at least one up-to-date reference that will allow you to explore the
topic in greater depth.
In addition, a list of several excellent textbooks and other resources for you to use to expand
your knowledge is found in the Appendix.
We are interested in your feedback on the guide! If you have comments or questions, please
feel free to contact us via email at pedrheumguide@gmail.com.
Supervising Editors:
Dr. Ronald M. Laxer, SickKids Hospital, University of Toronto
Dr. Tania Cellucci, McMaster Children’s Hospital, McMaster University
Dr. Evelyn Rozenblyum, St. Michael’s Hospital, University of Toronto
Section Editors:
Dr. Michelle Batthish, McMaster Children’s Hospital, McMaster University
Dr. Roberta Berard, Children’s Hospital – London Health Sciences Centre, Western
University
Dr. Liane Heale, McMaster Children’s Hospital, McMaster University
Dr. Clare Hutchinson, North York General Hospital, University of Toronto
Dr. Mehul Jariwala, Royal University Hospital, University of Saskatchewan
Dr. Lillian Lim, Stollery Children’s Hospital, University of Alberta
Dr. Nadia Luca, Alberta Children’s Hospital, University of Calgary
Dr. Dax Rumsey, Stollery Children’s Hospital, University of Alberta
Dr. Gordon Soon, North York General Hospital and SickKids Hospital Northern Clinic in
Sudbury, University of Toronto
Dr. Rebeka Stevenson, Alberta Children’s Hospital, University of Calgary

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 3
TABLE OF CONTENTS
Section
Topic
Page #
1
Pediatric Rheumatology Clinical Assessment
4
2
Approaches and Differential Diagnoses for Common
Complaints Referred to Pediatric Rheumatology
14
3
Juvenile Idiopathic Arthritis
25
4
Systemic Lupus Erythematosus and Related Conditions
35
5
Systemic Vasculitis
42
6
Idiopathic Inflammatory Myopathies
56
7
Scleroderma and Related Syndromes
60
8
Autoinflammatory Diseases
67
9
Uveitis
76
10
Inflammatory Brain Diseases
79
11
Infection and Infection-Related Conditions
85
12
Pain Syndromes
92
13
Pediatric Rheumatology Emergencies
96
14
Medications
107
Appendix
Helpful Resources in Pediatric Rheumatology
115
Notes:
Please consider that all treatment regimens discussed in the guide are
suggestions based on evidence-based guidelines and/or common practices by
the pediatric rheumatologists who are Section Editors of the Guide. Alternative
treatment approaches may be used in other centres.
More detailed information on medications (class, action, dose, side effects, monitoring)
may be found in the Medications section.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 4
SECTION 1 – PEDIATRIC RHEUMATOLOGY CLINICAL ASSESSMENT
1A. Pediatric Rheumatologic History
An appropriate rheumatologic history for a new patient should cover the following areas:
History of presenting complaint
Onset, duration, pattern
Potential triggers, such as trauma, infection or immunizations
Severity and impact on function, including school and activities of daily living
Associated symptoms
Factors that improve or worsen symptoms
Previous investigations
Previous treatment, including effectiveness and adverse reactions
Past medical history
Chronic medical conditions
Admissions to hospital, surgeries
Eye examinations
Development
Brief review of all domains - gross motor, fine motor, speech, language, hearing, social
Immunizations
All childhood vaccinations
Varicella – Infection or vaccination?
Medications
Prescribed medications – dose, route, frequency, adherence
Over-the-counter medications, vitamins, herbal supplements
Allergies
Travel history (especially risk factors for tuberculosis or Lyme infections)
Family history
Ethnicity and consanguinuity
Rheumatologic diseases: Juvenile idiopathic arthritis (JIA), rheumatoid arthritis (RA)
Ankylosing spondylitis (AS)
Premature osteoarthritis
Inflammatory bowel disease (IBD)
Psoriasis
Systemic lupus erythematosus (SLE)
Vasculitis
Autoinflammatory diseases, including early hearing loss and early
renal failure
Other autoimmune diseases: Diabetes mellitus type I, Celiac disease, Thyroid disease
Social history
Parents marital status, occupations, care providers, drug coverage, adolescent psychosocial
assessment (e.g. HEEADSSS)
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 5
Review of systems
General: Energy level, fatigue, poor sleep, non-restful sleep
Anorexia, weight loss
Fevers → frequency, duration, pattern, associated symptoms
Functioning → home, social, school, extra-curricular activities, work
HEENT: Photophobia, blurred vision, redness, pain
Sicca symptoms (dry eyes, dry mouth)
Nasal and/or oral ulcers (painful or painless)
Epistaxis
Dysphagia
Otalgia, hearing difficulties
CVS: Chest pain, orthopnea, syncope
Peripheral acrocyanosis
Raynaud phenomenon
Respiratory: Difficulty breathing, shortness of breath
Pleuritic chest pain
Prolonged cough, productive cough, hemoptysis
GI: Recurrent abdominal pain, “heartburn”
Diarrhea, constipation, bloody stools, melena
Nausea, vomiting
Skin: Any type of skin rash on face, scalp, trunk, limbs
Petechiae, purpura
Nodules
Ulcers (includes genital/perineal)
Photosensitivity
Alopecia, hair changes
Nail changes (pits, onycholysis) and nail fold changes
Joints: Pain (day and/or night), swelling, redness, heat, decreased range of motion
Loss of function, reduced activities, pain waking from sleep
Inflammatory → morning stiffness or gelling, improves with activity or exercise
Mechanical → improves with rest, “locking”, “giving away”
Muscles: Pain
Muscle weakness (proximal vs. distal)
Loss of function, reduced activities
CNS: Headaches
Psychosis, visual distortions
Cognitive dysfunction, drop in school grades
Seizures
PNS: Motor or sensory neuropathy
GU: Dysuria, change in urine volume or colour
Irregular, missed or prolonged menstrual periods, heavy menses

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 6
1B. Pediatric Rheumatologic Examination
Vital signs (including blood pressure percentiles)
Height, weight, BMI (percentiles, recent changes)
General appearance
HEENT: Conjunctival injection or hemorrhage, pupils (shape and reaction)
Complete ophthalmoscope examination from cornea to fundus
Nasal mucosa, nasal discharge, sinus tenderness
Oropharyngeal mucosa, tongue, tonsils
Thyroid
CVS: Heart sounds, murmurs, rubs, precordial examination
Vascular bruits (if indicated)
Peripheral pulses, peripheral perfusion, capillary refill
Lungs: Respiratory excursion, percussion, breath sounds, adventitious sounds
Abdomen: Tenderness, peritoneal signs, masses, bowel sounds, bruits (if indicated)
Hepatomegaly, splenomegaly
LN: Assess all accessible lymph node groups
Skin: Any type of skin rash, including petechiae, purpura, nodules, and ulcers
Alopecia, hair abnormalities
Nails: Nail pits, clubbing, onychonychia
Nail fold capillaries – thickening, branching, drop-out, hemorrhages
Digital ulcers, splinter hemorrhages, loss of digital pulp
CNS: Mental status
Cranial nerves
Motor: muscle bulk, tone, power/strength, tenderness, deep tendon reflexes
Cerebellar
Gait (walking, running, heels, toes, and tandem)
Sensory (if indicated), allodynia borders (if indicated)
Joints: Begin with a screening exam, such as the Pediatric Gait Arms Legs Spine
(pGALS)
Assess all joints for heat, swelling, tenderness, stress pain, active and passive
range of motion, deformity
Enthesitis sites
Localized bony/joint tenderness
Leg length (functional and/or actual)
Thigh, calf circumference difference (if indicated)
Back: Range of motion, tenderness, stress pain from repetitive motion
Scoliosis
Modified Schober test (if indicated)
Other: Fibromyalgia tender points (if indicated)
References:
1. Foster HE, Jandial S. pGALS – paediatric Gait Arms Legs and Spine: a simple
examination of the musculoskeletal system. Pediatr Rheumatol Online J 2013; 11(1):44.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 7
1C. Laboratory Testing in Pediatric Rheumatology
General Principles
Interpret all laboratory results in context of specific patient
Consider the clinical rationale and potential impact of all laboratory tests that are ordered,
especially for autoantibody testing
Review all laboratory test results to guide interpretation of abnormalities
Trends in laboratory values may be more important than isolated abnormalities
Complete blood cell count and differential
Hemoglobin, red blood cell count and mean corpuscular volume
o Normocytic or microcytic anemia in chronic inflammatory disease
o Autoimmune hemolytic anemia in systemic lupus erythematosus (SLE)
o Non-immune hemolytic anemia in macrophage activation syndrome (MAS)
o Iron deficiency anemia if chronic blood loss (e.g. due to NSAIDs, inflammatory bowel
disease)
White blood cell count and differential
o High white blood cell counts may be due to infection, systemic inflammation, or side-
effect of corticosteroids
o Leukopenia with lymphopenia and/or neutropenia may be due to systemic inflammation
or medications
Platelet count
o Active inflammation may lead to increased platelet counts (e.g. subacute phase of
Kawasaki disease, systemic juvenile idiopathic arthritis (JIA), or Takayasu arteritis)
o Active disease may also lead to reduced platelet counts (e.g. SLE)
Acute phase response to systemic inflammation
Acute phase reactants are plasma proteins produced by the liver that change production
during acute phase of inflammation
Acute phase response mediated by cytokines, such as IL-1, IL-6 and TNF (which are the
target of many biologic agents used in childhood rheumatic diseases)
Substantial acute phase response may be seen in infection, trauma, burns, tissue infarction,
advanced cancer and immune-mediated disease
Mild elevation may be seen in conditions such as obesity, pregnancy, and strenuous
exercise
Overall effect of acute phase response is to protect host from damage
Excessive or prolonged acute phase response may be deleterious itself (e.g. septic shock,
MAS, malignancy)
C-reactive protein (CRP)
Direct measure of inflammation (sensitive but not specific)
Level rises rapidly in response to inflammation and falls quickly with appropriate treatment
May reflect severe disease more closely than other acute phase reactants, although this
may be patient-specific and/or disease-dependent (e.g. CRP typically rises in patients with
SLE when there is infection, serositis or MAS, but may be normal with active disease)

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 8
Erythrocyte sedimentation rate (ESR)
Indirect measure of acute phase reaction
Changes more slowly than CRP
Measure rate at which red blood cells settle in a tube of anticoagulated blood in one hour
Depends on fibrinogen, gamma globulins
Ferritin
Protein central to iron homeostasis
Serum ferritin levels increase in setting of inflammation
Very high levels suggestive of macrophage activation syndrome
May not function as a reliable measure of iron status in setting of inflammatory disease
Summary of laboratory changes in acute phase response to systemic inflammation:
Increase in acute phase response
Decrease in acute phase response
CRP, ESR
Complement proteins
Fibrinogen, coagulation proteins
Ferritin
Ceruloplasmin
Haptoglobin
G-CSF
IL-1 receptor antagonist
Serum amyloid A
Albumin
Transferrin
IGF-1
Complement
Increased levels of complement components frequently seen in inflammation
Low complement levels present in SLE, acute post-infectious glomerulonephritis,
membrano-proliferative glomerulonephritis, or liver disease
Congenital complement deficiencies predispose either to recurrent infections (mainly
encapsulated organisms) or to unusual autoimmune disease (“lupus-like” disease)
In SLE, serial measurements of C3 and C4 are useful to monitor disease activity
o Complement levels tend to fall during a flare and return to normal concentration after
appropriate therapy
o Persistently low C3 associated with lupus nephritis
Autoantibodies
Antinuclear antibodies (ANA)
Autoantibodies directed against nuclear, nucleolar or perinuclear antigens
ANA should not be used as a screening tool
o Low titres of ANA (e.g. ANA ≤ 1:80) may be present in up to 30% of normal healthy
population and may revert to negative over time
o ANA may also be present in non-rheumatologic diseases (e.g. infection, malignancy,
medications)
No need to repeat ANA regularly once positive titre established (from Choosing Wisely:
Pediatric Rheumatology Top 5 by American College of Rheumatology)

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 9
Low titres of non-specific ANA may be seen in JIA patients, but positive ANA titres ≥ 1:160
in JIA patients are associated with younger age at onset, higher risk of uveitis, asymmetric
arthritis and lower number of affected joints over time
Persistent higher titres of ANA > 1:160 suggest connective tissue diseases, such as SLE
o Negative ANA makes diagnosis of SLE unlikely
Specific antibodies (e.g. anti-double stranded DNA) should only be requested if ANA is
positive and there is evidence of rheumatic disease (highlighted in Choosing Wisely:
Pediatric Rheumatology Top 5 by American College of Rheumatology)
Anti-double stranded DNA (Anti-dsDNA)
Anti-dsDNA autoantibody targets DNA in nucleus of cell
Highly specific for SLE
Titres are affected by disease activity and may be used to monitor disease progression and
response to therapy
Autoantibodies to extractable nuclear antigen (ENA)
Specific ENA antibodies
Characteristic disease associations
Anti-Ro/SSA
SLE, Neonatal lupus erythematosus, Sjögren
Anti-La/SSB
SLE, Neonatal lupus erythematosus, Sjögren
Anti-Sm (Anti-Smith)
SLE
Anti-RNP
Mixed connective tissue disease, SLE, Systemic sclerosis
Anti-histone
Drug-induced lupus, SLE
Anti-Scl 70
Diffuse systemic sclerosis
Anti-centromere
Limited systemic sclerosis (CREST)
Anti-Jo1
Polymyositis with interstitial lung disease, juvenile
dermatomyositis (JDM)
Anti-SRP
Anti-Mi-2
JDM with profound myositis & cardiac disease
JDM with good prognosis
Rheumatoid factor (RF)
IgM autoantibody that reacts to Fc portion of IgG
Present in 85% of adults with rheumatoid arthritis
Present in only 5-10% of children with JIA
o Helpful in classification and prognosis of JIA, but should not be used as a screening test
since arthritis is a clinical diagnosis
o Children with RF-positive polyarthritis are at higher risk of aggressive joint disease with
erosions and functional disability
RF may also be detected in chronic immune-complex mediated diseases, such as SLE,
systemic sclerosis, Sjögren, mixed connective tissue disease, cryoglobulinemia and chronic
infection (subacute bacterial endocarditis, hepatitis B and C, TB)
Anti-citrullinated peptide antibodies (ACCP)
Antibodies to citrullinated peptides found in inflamed synovium
Highly specific for rheumatoid arthritis, but often positive in older children with polyarticular
Rheumatoid factor positive JIA
Indicates increased risk of aggressive disease and progressive joint damage

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 10
Antiphospholipid antibodies
Heterogeneous group of antibodies directed against cell membrane phospholipids
Include lupus anticoagulant, anticardiolipin, anti-β
2
-glycoprotein I
Associated with increased risk of arterial or venous thrombosis (but lupus anticoagulant
paradoxically prolongs laboratory PTT)
May be produced due to primary antiphospholipid antibody syndrome (APS) or secondary to
SLE, other autoimmune diseases, malignancy, infection or drugs
Antineutrophil cytoplasmic antibodies (ANCA)
Antibodies target antigens in cytoplasmic granules of neutrophils
May be pathogenic by activating neutrophils, leading to perpetuation of chronic inflammation
High sensitivity and specificity for primary small vessel systemic vasculitides
ANCA
Immunofluorescence
pattern
Antigen specificity
(ELISA)
Disease associations
c-ANCA
Cytoplasmic
Proteinase-3 (PR3)
Granulomatosis with polyangiitis
p-ANCA
Perinuclear
Myeloperoxidase
(MPO)
Microscopic polyangiitis
Eosinophilic granulomatosis with
polyangiitis
Ulcerative colitis
Primary sclerosing cholangitis
SLE
Anti-Glomerular Basement Membrange (Anti-GBM) antibodies
Antibodies target alpha-3 chain of type IV collagen, which is normally present in glomerular
and alveolar basement membranes
Antibody binding to basement membranes in lungs and kidneys activates classical
complement pathway and neutrophil-dependent inflammation, leading to small vessel
vasculitis with immune complex formation
Production of antibodies may be triggered by environmental factors (e.g. infection, cigarette
smoking)
Human Leukocyte Antigen (HLA) Genetics
Many genes of the major histocompatibility complex (especially HLA class I and II genes)
have been associated with rheumatic disorders
HLA-B27
HLA class I gene that is present in only 7-10% of the general population (may be higher in
some First Nations groups)
Found in 90-95% of Caucasians with ankylosing spondylitis and many patients with JIA
(particularly enthesitis related arthritis and psoriatic arthritis), inflammatory bowel disease,
isolated acute anterior uveitis, and reactive arthritis
HLA-B27 may play a role in the pathogenesis of inflammatory disease
HLA-B51
May be associated with Behçet disease

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 11
Additional tests:
Urinalysis
Routinely used to assess for proteinuria and hematuria associated with renal involvement in
autoimmune and autoinflammatory diseases
Fecal calprotectin
May be measured as an indicator of underlying gastrointestinal inflammation
Genetic testing
Often ordered to confirm diagnosis of genetic fever syndromes and other autoinflammatory
disorders
Cytokine profiling
May be used in research contexts to qualify the inflammatory response and guide therapy
May become more widely available in upcoming years
References:
1. Mehta J. Laboratory testing in pediatric rheumatology. Pediatr Clin N Am 2012; 59:263-
84.
2. Pilania RK, Singh S. Rheumatology panel in pediatric practice. Indian J Pediatr 2019;
56(5):407-14.
1D. Diagnostic Imaging in Pediatric Rheumatology
General Principles
Interpret all imaging results in context of specific patient
Consider the clinical rationale and potential impact of all imaging that is ordered, including
risks of sedation, radiation and contrast administration
Imaging alone is not sufficient to confirm any rheumatic disease
Repeat imaging may be helpful to assess response to therapy, disease progression and
development of damage or to screen for specific organ involvement in systemic conditions
Review of questionable or unexpected imaging findings with a pediatric radiologist who has
specific expertise (e.g. musculoskeletal or neuroradiology training) is recommended
X-ray
Bone and joint X-rays
o Often ordered as initial testing for pain and deformity
o May be used to rule out bony abnormalities or injuries, such as fracture, that may
explain symptoms
o Helpful to image both affected and non-affected sides to assess for subtle changes
o May be normal at disease onset
o Most likely to be ordered in assessment of patients with possible JIA or other
inflammatory arthritis, non-bacterial osteomyelitis, or systemic sclerosis
o Findings in JIA may include effusion, soft tissue swelling, periarticular osteopenia,
joint space narrowing, erosions, subchondral cysts, osteophytes, bone deformity,
fusion, or accelerated bone development in young children
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 12
Chest X-rays
o May be used to assess for heart and lung involvement in systemic autoimmune or
autoinflammatory diseases
Ultrasound
May be used to assess for effusions and other findings of synovitis or to facilitate joint
injections
May also be used with Doppler to assess vasculature for obstruction of blood flow, which
may be due to thrombosis or vasculitis in rheumatic diseases
Highly operator dependent and requires specific skill and experience, especially with
pediatric patients
Many rheumatologists are currently performing point-of-care ultrasonography to aid clinical
assessment of disease activity and joint injection
Computed tomography (CT)
Chest CT may be useful to identify findings of interstial lung disease and pulmonary
hemorrhage in connective tissue diseases
CT angiograms may be used to assess for findings of vasculitis when magnetic resonance
or conventional angiograms are not easily accessible
While CT may identify findings for a number of rheumatic diseases, it is often not the first
imaging of choice because of the associated radiation.
If CT is deemed necessary, radiation may be reduced with high resolution techniques
Magnetic resonance imaging
Ideal imaging modality for synovitis in specific joints (e.g. temporomandibular, sacroiliac and
cervical spine) and may identify early signs of disease and/or damage in JIA
Whole body MRI protocols have been developed to assess for enthesitis and chronic non-
bacterial osteomyelitis
Specialized protocols have been developed to identify findings in inflammatory myositis
Ideal imaging modality to identify findings of brain inflammation (especially if 3T or higher
strength magnet available) and may also be used to assess for inflammation in aorta, blood
vessels and other organs (e.g. gastrointestinal tract); MR angiography should be requested
for more accurate imaging of blood vessels
Limitations are that MRI is expensive and less accessible and longer scan times often
require sedation for younger children
Echocardiography
Typically used to assess for coronary artery aneurysms in Kawasaki disease and for carditis
or other cardiopulmonary involvement in systemic autoimmune or autoinflammatory
diseases
Antenatal use of echocardiography is important in babies at risk of neonatal lupus
erythematosus to assess for myocarditis and endocardial fibroelastosis
Some centres may require consultation with cardiologist in conjunction with imaging
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 13
Dual Energy X-ray Absorptiometry (DEXA) scan
Measures bone mineral density
Used most often in patients on chronic steroid therapy at baseline and at regular intervals to
monitor for development of osteopenia/osteoporosis
References
1. Pilania RK, Singh S. Rheumatology panel in pediatric practice. Indian J Pediatr 2019;
56(5):407-14.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 14
SECTION 2 – APPROACHES TO AND DIFFERENTIAL DIAGNOSES FOR COMMON
COMPLAINTS REFERRED TO PEDIATRIC RHEUMATOLOGY
2A. Approach to Childhood Joint Pain
Differential diagnosis for pain involving a single joint:
Traumatic
Fracture
Soft tissue injury (e.g. strains, sprains)
Foreign body synovitis
Infection-related
Septic arthritis
Osteomyelitis
Chronic infections, such as tuberculosis or Lyme disease
Reactive arthritis including post-Streptococcal reactive arthritis
Acute rheumatic fever
Inflammatory
Juvenile idiopathic arthritis (JIA)
Chronic non-bacterial osteomyelitis
Inflammatory bowel disease
Genetic autoinflammatory syndromes (e.g. familial Mediterranean fever,
pyogenic arthritis pyoderma gangrenosum and acne)
Behçet disease
Neoplastic
Musculoskeletal tumors (e.g. osteoid osteoma, osteosarcoma)
Hematologic malignancy
Hemarthrotic
Traumatic
Coagulopathy (e.g. hemophilia)
Pigmented villonodular synovitis
Arteriovenous malformation
Hematologic
Sickle cell disease (e.g. pain crisis, dactylitis)
Mechanical
Overuse or repetitive strain injury
Tendon/ligament/meniscal injury
Apophysitis
Joint damage (e.g. prior trauma, infection, congenital anomaly)
Orthopedic
Avascular necrosis (AVN)
Slipped capital femoral epiphysis (SCFE)
Osteochonditis dissecans
Pain syndrome
Complex regional pain syndromes (CRPS)
Potential investigations for pain involving a single joint:
X-rays
Joint aspiration and synovial fluid analysis and/or culture
Blood work: CBC and differential, ESR, CRP
Consider, if indicated:
o Further infectious testing (e.g. blood culture, Lyme serology, TB skin test)
o Further imaging (e.g. ultrasound, MRI)
o Autoimmune serology (e.g. ANA, HLA B27)

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 15
Differential diagnosis for pain involving multiple joints:
Inflammatory
Juvenile idiopathic arthritis (JIA)
Systemic lupus erythematosus (SLE)
Juvenile dermatomyositis
Scleroderma/mixed connective tissue disease/overlap syndromes
Systemic vasculitis (e.g. Henoch-Schönlein purpura / IgA vasculitis)
Inflammatory bowel disease (IBD)
Genetic autoinflammatory syndromes
Sarcoidosis
Chronic non-bacterial osteomyelitis / chronic recurrent multifocal
osteomyelitis
Serum sickness
Infection-related
Acute infections (e.g. parvovirus B19, EBV, Neisseria gonorrheae)
Chronic infections (e.g. tuberculosis (Poncet arthritis), Lyme disease)
Subacute bacterial endocarditis (SBE)
Reactive arthritis, including acute rheumatic fever (ARF)
Osteomyelitis and septic arthritis may rarely present with multifocal
involvement
Immunological
Immunodeficiency associated with arthritis (e.g. Wiskott-Aldrich)
Neoplastic
Leukemia, lymphoma, neuroblastoma, cancers with systemic
involvement
Mechanical
Overuse injuries, repetitive strain injuries
Apophysitis
Hypermobility – benign or due to connective tissue disease (e.g. Ehlers-
Danlos)
Skeletal dysplasias
Metabolic
Rickets
Vitamin C deficiency (scurvy)
Glycogen storage disease, mucopolysaccharidoses
Pain syndrome
Fibromyalgia
Potential investigations for pain involving multiple joints:
Blood work: CBC and differential, blood film, ESR, CRP
Infectious testing (e.g. Parvovirus B19 serology, EBV serology, throat culture, ASOT)
Consider, if indicated:
o Autoimmune serology (e.g. ANA, Rheumatoid factor, HLA B27)
o Imaging (e.g. X-rays, ultrasound, MRI)
o Urinalysis
o Bone marrow aspirate and biopsy

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 16
What do clinical features associated with joint pain tell you about underlying diagnosis?
Signs/symptoms
Associated conditions
Severe joint pain
Infection-related, malignancy, trauma, AVN, pain syndrome
Pinpoint tenderness
Osteomyelitis, trauma, AVN, malignancy, enthesitis, chronic non-
bacterial osteomyelitis
Night pain
Malignancy, osteoid osteoma, benign nocturnal limb pain
Redness
Septic arthritis, acute rheumatic fever, reactive arthritis
Migratory joint pain
Leukemia, acute rheumatic fever
Non weight bearing
Infection, malignancy, discitis, myositis, pain syndrome
Hip pain
Infection-related, AVN, SCFE, malignancy, chondrolysis, transient
synovitis, JIA (particularly enthesitis related arthritis)
Back pain
Usually benign, but consider bone or spinal cord tumour, discitis,
spondylolysis/spondylolisthesis, JIA (enthesitis related arthritis),
myositis, osteoporosis, CNO, pain syndrome
Periarticular pain
Malignancy, hypermobility, pain syndrome, CNO
Dactylitis
JIA (particularly enthesitis related arthritis and psoriatic arthritis),
sickle cell, trauma
Clubbing
Cystic fibrosis, IBD, malignancy (especially lung), familial,
hypertrophic osteoarthropathy
Weight loss
Malignancy, systemic autoimmune rheumatologic diseases, IBD
Muscle weakness
Myositis, overlap syndromes, malignancy, pain-related weakness
Rash
Systemic autoimmune rheumatologic diseases, vasculitis, JIA
(particularly systemic arthritis and psoriatic arthritis), acute
rheumatic fever, Lyme disease, serum sickness, autoinflammatory
syndromes
Oral ulcers
Vasculitis, Behçet disease, SLE, IBD, autoinflammatory
syndromes
Eye pain and redness
Reactive arthritis, enthesitis related arthritis. IBD, Behçet disease
Nail or nail fold changes
Systemic autoimmune rheumatologic diseases, psoriasis,
subacute bacterial endocarditis
Raynaud phenomenon
Systemic autoimmune rheumatologic diseases
School withdrawal
Pain syndrome, chronic fatigue
Travel
Infection-related (e.g. tuberculosis, Lyme disease, viral)
Consanguinity
Genetic or metabolic diseases (e.g. autoinflammatory diseases)
References:
1. Nannery R, Heinz P. Approach to joint pain in children. Paediatr and Child Health 2018;
28(2):43-49.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 17
2. Tse SM, Laxer RM. Approach to acute limb pain in childhood. Pediatr Rev 2006; 27:170-
80.
3. Sen ES, et al. The child with joint pain in primary care. Best Pract & Res Clin Rheumatol
2014; 28:888-906.
2B. Approach to Childhood Back Pain
Differential diagnosis for back pain in children
Inflammatory
Juvenile idiopathic arthritis (JIA)
Inflammatory bowel disease (IBD)
Chronic non-bacterial osteomyelitis, chronic recurrent multifocal
osteomyelitis
Transverse myelitis (e.g. SLE)
Infection-related
Acute infections (e.g. osteomyelitis, septic arthritis, discitis, epidural
abscess)
Chronic infections (e.g. tuberculosis (Pott disease))
Reactive arthritis
Neoplastic
Musculoskeletal tumors (e.g. osteoblastoma, osteosarcoma, spinal cord
tumors, metastases)
Leukemia, lymphoma
Mechanical
Spondylolysis, spondylolisthesis
Scoliosis
Scheuermann disease
Disc prolapse
Degenerative disc disease
Trauma
Fracture
Hematologic
Sickle cell pain crisis
Pain syndrome
Other
Fibromyalgia
Neurofibromatosis
Potential investigations for back pain in children:
Investigations may not be needed and depend on clinical assessment
Consider, if indicated:
o Imaging (e.g. X-rays, MRI)
o Autoimmune serology (e.g. ANA, Rheumatoid factor, HLA B27)
o Blood work (e.g. CBC and differential, ESR, CRP)
References:
1. Altaf F, et al. Back pain in children and adolescents. Bone Joint J 2014; 96B:717-23.
2. Nigrovic PA. Evaluation of the child with back pain. UpToDate. Updated December
2018. URL: https://www.uptodate.com/contents/evaluation-of-the-child-with-back-pain.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 18
2C. Approach to Fevers
Definition of fever of unknown origin:
Temperature > 38 degrees Celsius lasting ≥ 8 days with no clear source of fever
Differential diagnosis for fever of unknown origin in children
Infectious
Bacterial (e.g. abscess, mastoiditis, osteomyelitis, pyelonephritis,
sinusitis, typhoid fever, tuberculosis)
Viral (e.g. Adenovirus, CMV, EBV, Enterovirus, HIV)
Other infections including parasitic and fungal (e.g. malaria, Lyme
disease, Toxoplasma, Blastomycosis)
Inflammatory
Serum sickness
Systemic vasculitis (e.g. Kawasaki disease)
Systemic lupus erythematosus
Juvenile dermatomyositis
Systemic arthritis/JIA
Behçet disease
Inflammatory bowel disease
Genetic autoinflammatory syndromes
Castleman syndrome
Hemophagocytic lymphohistiocytosis (primary or secondary HLH/MAS)
Sarcoidosis
Drug-induced
Drug fevers or intoxication
Neoplastic
Leukemia, lymphoma
Langerhans cell histiocytosis
Neuroblastoma
Endocrinologic
Hyperthyroidism
Thyroiditis
Diabetes insipidus
Other
Pancreatitis
Factitious fevers
Potential investigations for fever of unknown origin in children:
Investigations will depend on clinical assessment and serial re-examination
Initial blood work: CBC and differential, blood film, electrolytes, urea, creatinine, glucose,
ESR, CRP, ferritin, liver enzymes, albumin, LDH
Urinalysis
Initial infectious work-up: blood culture, urine culture, nasopharyngeal swab for viruses
Consider, if indicated:
o Imaging (e.g. X-rays, abdominal ultrasound)
o Further infectious testing (e.g. ASOT, Monospot, cerebrospinal fluid testing)
o Testing for immunodeficiency (e.g. complement and immunoglobulin levels)

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 19
Definition of recurrent fevers:
≥ 3 episodes of unexplained fever within 6 months separated by ≥ 7 days of good health
Differential diagnosis for recurrent fevers
Infectious
Repeated viral or bacterial infections
Viral (e.g. CMV, EBV, Parvovirus, hepatitis viruses, HIV)
Bacterial (e.g. Typhoid fever, occult dental abscess, endocarditis,
Mycobacteria)
Parasitic or fungal (e.g. malaria, Borrelia, Brucellosis, Yersinia)
Inflammatory
Genetic autoinflammatory syndromes
Periodic fever with aphthous stomatitis, pharyngitis and adenitis (PFAPA)
Systemic lupus erythematosus
Systemic arthritis/JIA
Inflammatory bowel disease
Behçet disease
Polyarteritis nodosa
Sarcoidosis
Hemophagocytic lymphohistiocytosis (primary or secondary HLH/MAS)
IgG4 disease
Hematologic
Cyclic neutropenia
Neoplastic
Leukemia, lymphoma
Immunologic
Drug-induced
DiGeorge syndrome
Chediak-Higashi
Combined immunodeficiency syndrome
Drug fevers or intoxication
Other
CNS abnormality (e.g. hypothalamic dysfunction)
Castleman disease
Factitious fevers
Potential investigations for recurrent fevers:
Clinical assessment during episode of fever and when well
Fever diary including pattern of fever and associated symptoms
Blood work during episode and when well: CBC and differential, ESR, CRP, ferritin, liver
enzymes, albumin, LDH, immunoglobulins (including IgD)
Urinalysis
Consider, if indicated:
o Infectious testing (e.g. blood culture, viral serology)
o Autoimmune serology (e.g. ANA)
o Genetic testing
References:
1. Antoon JW, et al. Pediatric fever of unknown origin. Pediatr Rev 2015; 36(9):380-91.
2. Soon GS, Laxer RM. Approach to recurrent fever in childhood. Can Fam Physician
2017; 63(10):756-62.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 20
2D. Approach to Recurrent Oral Ulcers
Differential diagnosis for recurrent oral ulcers in children
Inflammatory
Inflammatory bowel disease
Celiac disease
Behçet disease
Systemic lupus erythematosus (SLE)
Hyperimmunoglobulinemia D syndrome (HIDS)
Periodic fever with aphthous stomatitis, pharyngitis and adenitis (PFAPA)
A20 haploinsufficiency (HA20)
Sarcoidosis
Infectious
Viral (e.g. Herpes simplex, Coxsackie)
Reactive arthritis
Hematologic
Cyclic neutropenia
Drugs
Azathioprine, Methotrexate, Sulfasalazine
Other
Aphthous stomatitis
What are the characteristics of oral ulcers in different inflammatory conditions?
SLE
Painless shallow oral ulcers, typically located on roof of mouth
where hard and soft palate meet
Inflammatory bowel
disease
Painful aphthous ulcers anywhere in oropharynx, sometimes
associated with cheilitis
Behçet disease
Painful aphthous ulcers or punched-out ulcers on tongue, lips,
gingiva and/or buccal mucosa
Celiac disease
Painful recurrent aphthous ulcers
PFAPA
Painful aphthous ulcers with discrete margins, typically on
buccal mucosa, associated with febrile episodes
HIDS
Painful aphthous ulcers with discrete margins, typically on
buccal mucosa, associated with febrile episodes
Sarcoidosis
Painless well-circumscribed brownish red or violaceous lesions
(sometimes nodular), erythematous gingival enlargement,
submucosal swelling of palate
References:
1. Siu A, et al. Differential diagnosis and management of oral ulcers. Semin Cutan Med
Surg 2015; 34(4):171-7.
2. Le Doare K, et al. Fifteen-minute consultation: a structured approach to the management
of recurrent oral ulceration. Arch Dis Child Educ Pract Ed 2014; 99(3):82-6.
3. Stoopler ET, Al Zamel G. How to manage a pediatric patient with oral ulcers. J Can Dent
Assoc 2014; 80:e9.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 21
2E. Additional differential diagnoses
Differential diagnosis for lymphadenopathy in children
Inflammatory
Systemic lupus erythematosus
Systemic arthritis/JIA
Kawasaki disease
Hemophagocytic lymphohistiocytosis (primary or secondary HLH)
Kikuchi-Fujimoto disease
Castleman disease
Rosai-Dorfman disease
Monogenic autoinflammatory diseases
Periodic fever with aphthous stomatitis, pharyngitis and adenitis (PFAPA)
Serum sickness
Sarcoidosis
Infectious
Viral (e.g. EBV, CMV, HIV)
Bacterial (e.g. Bartonella, tuberculosis)
Bacterial spirochete/tick bourne (e.g. Lyme disease)
Neoplastic
Lymphoma, leukemia
Langerhans cell histiocytosis
Neuroblastoma
Other
Drug-induced
Differential diagnosis for erythema nodosum in children
Infectious
Viral (e.g. EBV, CMV, HIV)
Bacterial (e.g. Group A Streptococcus, Mycoplasma, Bartonella, Yersinia,
tuberculosis)
Inflammatory
Inflammatory bowel disease
Systemic lupus erythematosus
Behçet disease
Systemic vasculitis (e.g. polyarteritis nodosa, granulomatosis with
polyangiitis)
Sarcoidosis
Neoplastic
Lymphoma, leukemia
Hepatocellular carcinoma
Renal cell carcinoma
Drug-related
Oral contraceptives
Antibiotics (e.g. sulpha drugs, penicillins, macrolides)
Other
Idiopathic

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 22
Differential diagnosis for recurrent parotitis
Infectious
Viral: HIV (diffuse infiltrative lymphocytosis), Influenza B, mumps, EBV,
CMV, Parvovirus, Paramyxovirus, Adenonvirus
Bacterial: Streptococcal infections, Staphylococcus aureus, Bartonella,
Haemophilus
Tuberculosis
Inflammatory
Systemic lupus erythematosus
Sjögren syndrome
IgG4 related disease
Neoplastic
Parotid tumours
Lymphoma
Other
Sialolithiasis
Juvenile recurrent parotitis
Pneumoparotid
Differential diagnosis for muscle weakness
Inflammatory
Juvenile dermatomyositis
Juvenile polymyositis
Systemic lupus erythematosus
Mixed connective tissue disease
Juvenile idiopathic arthritis
Systemic sclerosis
Overlap myositis
Inclusion-body myositis
Focal myositis
Orbital myositis
Granulomatous myositis
Eosinophilic myositis
Inflammatory bowel disease
Autoinflammatory diseases (e.g. TNF-receptor associated periodic
syndrome, Chronic atypical neutrophilic dermatosis with lipodystrophy
and elevated temperature, Familial Mediterranean fever)
Infectious
Viral (e.g. Enterovirus, Influenza, Coxsackievirus, Echovirus, Parvovirus,
Hepatitis B, HTLV)
Bacterial/Spirochetal (e.g. Staphylococcus, Streptococcus, Borrelia)
Parasitic (e.g. Toxoplasmosis, Trichinosis)
Genetic
Muscular dystrophy (e.g. Duchenne, Becker)
Congenital myopathies (e.g. Spinal muscular atrophy)
Metabolic
Metabolic diseases (e.g. mitochondrial, glycogen storage)
Other
Endocrinopathies (e.g. thyroid-associated myopathies)
Trauma
Toxins
Neuromuscular transmission disorders (e.g. myasthenia gravis)
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 23
Differential diagnosis for chorea and abnormal movements in children
Inflammatory
Autoimmune encephalitis
Systemic lupus erythematosus
Antiphospholipid antibody syndrome
Behçet disease
Hashimoto encephalitis
Polyarteritis nodosa
Sjögren syndrome
Celiac disease
Sarcoidosis
Infectious
Acute rheumatic fever
Lyme disease
Malaria
Neurosyphilis
Tuberculosis
Creutzfeld-Jacob disease
Neurologic
Benign hereditary chorea
Huntington disease
Idiopathic basal ganglia calcification
Ataxia telengiectasia
Tic disorder
Neoplastic
Paraneoplastic syndromes
Tumors with basal ganglia involvement
Drug-related
Dopaminergic and other drugs
Other
Porphyria
Wilson disease
Liver failure

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 24
Differential diagnosis for stroke-like presentations in children
Inflammatory
CNS vasculitis (primary angiography-positive or secondary vasculitis)
Systemic vasculitis (e.g. polyarteritis nodosa)
Systemic lupus erythematosus
Antiphospholipid antibody syndrome
Structural
Arterial dissection
Fibromuscular dysplasia
Moyamoya disease
Hematologic
Thromboembolic disease (e.g. prothrombotic condition, atherosclerosis)
Hemoglobinopathies (e.g. sickle cell disease)
Vasospastic
Reversible vasoconstrictive syndromes
Drug-induced (e.g. cocaine)
Genetic
Deficiency of adenosine deaminase 2 (DADA2)
Channelopathies
Connective tissue disorders (e.g. Ehlers-Danlos syndrome, Marfan
syndrome)
Neurofibromatosis
Metabolic
CADASIL (cerebral autosomal dominant arteriopathy with subcortical
infarcts and leukoencephalopathy), MELAS (mitochondrial
encephalopathy, lactic acidosis, stroke-like episodes)
References:
1. Hambleton L, et al. Lymphadenopathy in children and young people. Paediatr and Child
Health 2016; 26(2):63-7.
2. Penn E, Goudy S. Pediatric inflammatory adenopathy. Otolaryngol Clin North Am 2015;
48(1):137-51.
3. Leung AKC, et al. Erythema nodosum. World J Pediatr 2018; 14(6):548-54.
4. Baszis K, et al. Recurrent parotitis as a presentation of primary pediatric Sjögren
syndrome. Pediatrics 2012; 129:179-82.
5. Huber AM. Juvenile idiopathic inflammatory myopathies. Pediatr Clin North Am 2018;
65(4):739-56.
6. Gilbert DL. Acute and chronic chorea in childhood. Semin Pediatr Neurol 2009; 16(2):71-
6.
7. Tsze DS, Valente JH. Pediatric stroke: a review. Emerg Med Int 2011; 734506.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 25
SECTION 3 – JUVENILE IDIOPATHIC ARTHRITIS
3A. Introduction to Juvenile Idiopathic Arthritis (JIA)
Arthritis is diagnosed in the presence of joint effusion OR two or more of the following:
limited range of movement with joint line tenderness or painful range of movement
Currently, the most widely-used classification criteria for JIA is by the International League of
Associations for Rheumatology (ILAR) from the late 1990’s
o Definition: JIA is arthritis of unknown etiology that begins before the 16th birthday
and persists for at least 6 weeks and in which other causes of arthritis are excluded
o Classification: Recognizes 7 distinct subtypes of JIA, based on their presentation
within the first 6 months
1. Oligoarthritis
2. Polyarthritis (Rheumatoid Factor Negative)
3. Polyarthritis (Rheumatoid Factor Positive)
4. Systemic arthritis
5. Enthesitis-related arthritis
6. Psoriatic arthritis
7. Undifferentiated arthritis
A recent re-classification of JIA was proposed by Pediatric Rheumatology International
Trials Organization (PRINTO) in 2019, but has not been validated
o Most prominent changes proposed in the new PRINTO criteria are that JIA is
considered a group of distinct clinical phenotypes (rather than a single disease) that
begin before the 18
th
birthday and are not classified by the number of joints involved
Oligoarthritis
ILAR Classification Criteria for Oligoarthritis *
Definition: Arthritis affecting 1 to 4 joints during the first 6 months of disease
Subcategories:
1. Persistent oligoarthritis: Affects not more than 4 joints throughout disease course
2. Extended oligoarthritis: Affects more than 4 joints after the first 6 months of disease
Exclusions:
o Psoriasis or a history of psoriasis in the patient or first degree relative
o Arthritis in an HLA-B27 positive male beginning after 6
th
birthday
o Ankylosing spondylitis, enthesitis related arthritis, sacroiliitis with IBD, or acute anterior uveitis,
or history of one of these disorders in 1
st
degree relative
o Presence of IgM Rheumatoid Factor on at least 2 occasions at least 3 months apart
o Presence of systemic JIA
* Classification criteria are designed to identify a homogeneous population for research studies
and are not diagnostic criteria, although they are often used for diagnosis in practice
Oligoarthritis is the most common subtype of JIA
Typical patient is a young girl with positive ANA who presents with a small number of swollen
joints
Most frequent joints to be involved are knees, ankles, wrists, or elbows

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 26
Hip involvement is distinctly uncommon, especially early in disease, unless the disease
develops into extended oligoarthritis or is really part of enthesitis-related arthritis
ANA is positive in 60-80% of patients (antigenic specificity is unknown for ANA in JIA)
Oligoarticular JIA with positive ANA is associated with a higher risk of asymptomatic uveitis
(see Section 9)
Polyarthritis (Rheumatoid Factor Negative)
ILAR Classification Criteria for Polyarthritis (Rheumatoid Factor Negative) *
Definition:
Arthritis affecting 5 or more joints during first 6 months of disease
Negative testing for RF
Exclusions:
o Psoriasis or a history of psoriasis in the patient or first degree relative
o Arthritis in an HLA-B27 positive male beginning after 6
th
birthday
o Ankylosing spondylitis, enthesitis related arthritis, sacroiliitis with IBD, or acute anterior uveitis,
or history of one of these disorders in 1
st
degree relative
o Presence of IgM Rheumatoid Factor on at least 2 occasions at least 3 months apart
o Presence of systemic JIA
* Classification criteria are designed to identify a homogeneous population for research studies
and are not diagnostic criteria, although they are often used for diagnosis in practice
Children with RF negative polyarthritis are frequently younger and have a better prognosis
than those with RF positive disease
ANA is positive in about 25% of patients
Joint involvement is frequently symmetrical, affecting large and small joints alike
Less than 50% of patients go into spontaneous remission, and long-term sequelae are
frequent, especially with hip and shoulder involvement
Polyarthritis (Rheumatoid Factor Positive)
ILAR Classification Criteria for Polyarthritis (Rheumatoid Factor Positive) *
Definition:
Arthritis affecting 5 or more joints during first 6 months of disease
2 or more positive tests for RF at least 3 months apart during first 6 months of disease
Exclusions:
o Psoriasis or a history of psoriasis in the patient or first degree relative
o Arthritis in an HLA-B27 positive male beginning after 6
th
birthday
o Ankylosing spondylitis, enthesitis related arthritis, sacroiliitis with IBD, or acute anterior uveitis,
or history of one of these disorders in 1
st
degree relative
o Presence of systemic JIA
* Classification criteria are designed to identify a homogeneous population for research studies
and are not diagnostic criteria, although they are often used for diagnosis in practice
All patients are RF positive, many are positive for anti-CCP antibodies, and ANA is positive in
40-50%

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 27
RF positive polyarthritis mostly affects adolescent girls
Patients with RF positive polyarthritis share many characteristics with adults with rheumatoid
arthritis, including symmetrical polyarthritis especially involving the PIP joints and MCP joints
Children may develop rheumatoid nodules and similar complications to adult disease,
including joint erosions and Felty syndrome (neutropenia and splenomegaly)
RF positive polyarthritis is associated with more joint erosion and damage and with worse
radiographic outcome
Remission rates (off medications) are lowest among RF positive patients
Systemic Arthritis
ILAR Classification Criteria for Systemic Arthritis *
Definition:
Arthritis affecting 1 or more joints
Associated with or preceded by fever of at least 2 weeks duration that is documented to be
daily, or “quotidian” for at least 3 days
Accompanied by 1 or more of:
Evanescent (non-fixed) erythematous rash
Generalized lymph node enlargement
Hepatomegaly and/or splenomegaly
Serositis
Exclusions:
o Psoriasis or a history of psoriasis in the patient or first degree relative
o Arthritis in an HLA B27 positive male beginning after 6
th
birthday
o Ankylosing spondylitis, enthesitis related arthritis, sacroiliitis with IBD, or acute anterior uveitis,
or history of one of these disorders in 1
st
degree relative
o Presence of IgM Rheumatoid Factor on at least 2 occasions at least 3 months apart
* Classification criteria are designed to identify a homogeneous population for research studies
and are not diagnostic criteria, although they are often used for diagnosis in practice
Typical symptoms of systemic arthritis include:
o Once or twice daily fever spikes to >38.5°C, which then return to baseline or below
o Salmon-coloured, evanescent rash accompanying the fever, occasionally pruritic, and
lesions may be elicited by scratching the skin (Koebner phenomenon)
o Lymphadenopathy (common), splenomegaly (10%) and hepatomegaly (less common)
o Arthritis may develop later (usually within first year of fever) and is usually polyarticular,
affecting knees, wrists and ankles, but cervical spine and hip involvement also occurs
An infectious work-up should be done and bone marrow aspirate to exclude malignancy
strongly considered before starting corticosteroid treatment
Systemic JIA is associated with macrophage activation syndrome, a potentially life
threatening inflammatory complication (see Section 13)
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 28
Enthesitis Related Arthritis (ERA)
ILAR Classification Criteria for Enthesitis Related Arthritis *
Definition:
Arthritis and enthesitis
Or, arthritis or enthesitis with at least 2 of the following:
Presence or history of sacroiliac joint tenderness or inflammatory back pain
Presence of HLA-B27 antigen
Onset of arthritis in a male over 6 years of age
Acute (symptomatic) anterior uveitis
History of ankylosing spondylitis, enthesitis related arthritis, sacroiliitis with
inflammatory bowel disease, or acute anterior uveitis in a first-degree relative
Exclusions:
o Psoriasis or a history of psoriasis in the patient or first degree relative
o Presence of IgM Rheumatoid Factor on at least 2 occasions at least 3 months apart
o Presence of systemic JIA
* Classification criteria are designed to identify a homogeneous population for research studies
and are not diagnostic criteria, although they are often used for diagnosis in practice
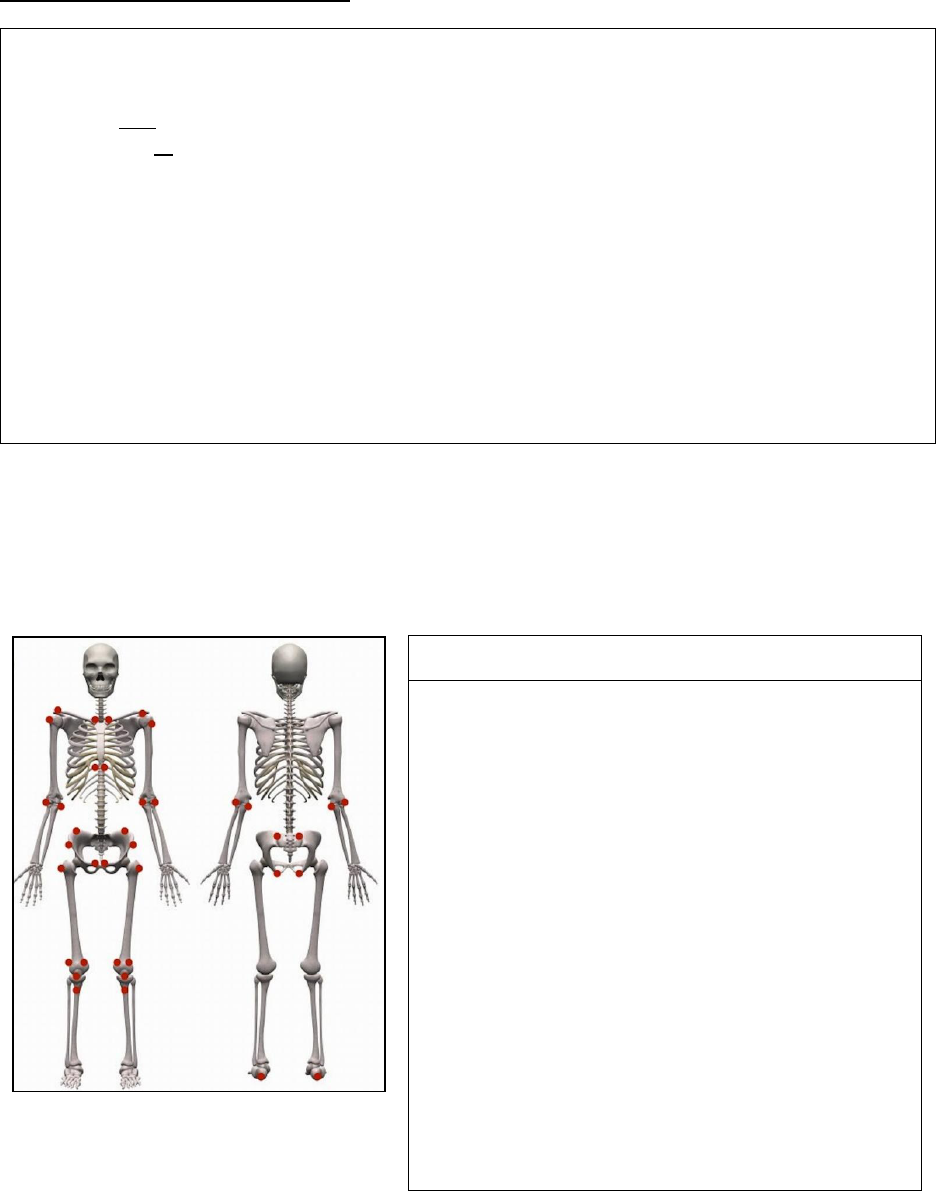
Hallmark of ERA is enthesitis (inflammation of the insertion sites of tendons, ligaments and
fascia) and asymmetrical oligoarthritis, predominantly affecting the lower extremities
Entheses by anatomic region
Region
Enthesitis exam
Chest
Costernal junctions (1
st
and 7
th
)
Shoulder
Acromioclavicular junction
Supraspinatus insertion into greater tubercle of
humerus
Elbow
Common flexor insertion into medial epicondyle
of humerus
Common extensor insertion into lateral
epicondyle of humerus
Pelvis
Abdominal muscle insertions into iliac crest
Sartorius insertion into anterior superior iliac
spine
Posterior superior iliac spine
Gracilis and adductor insertion into pubis
symphysis
Hamstring insertion into ischial tuberosity
Hip extensor insertion into greater trochanter of
femur
Knee
Quadriceps tendon insertion to patella
Infrapatellar ligament insertion to patella and
tibial tuberosity
Ankle
Achilles tendon insertion into calcaneus
Foot
Plantar fascia insertion into calcaneus,
metatarsal heads and base of 5
th
metatarsal
Image adapted with permission from: Jariwala M, Burgos-Vargas R, “Juvenile Spondyloarthropathies” in Pediatric
Rheumatology: A Clinical Viewpoint, 2017, Pages 229-46, Copyright Springer Singapore (2017).
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 29
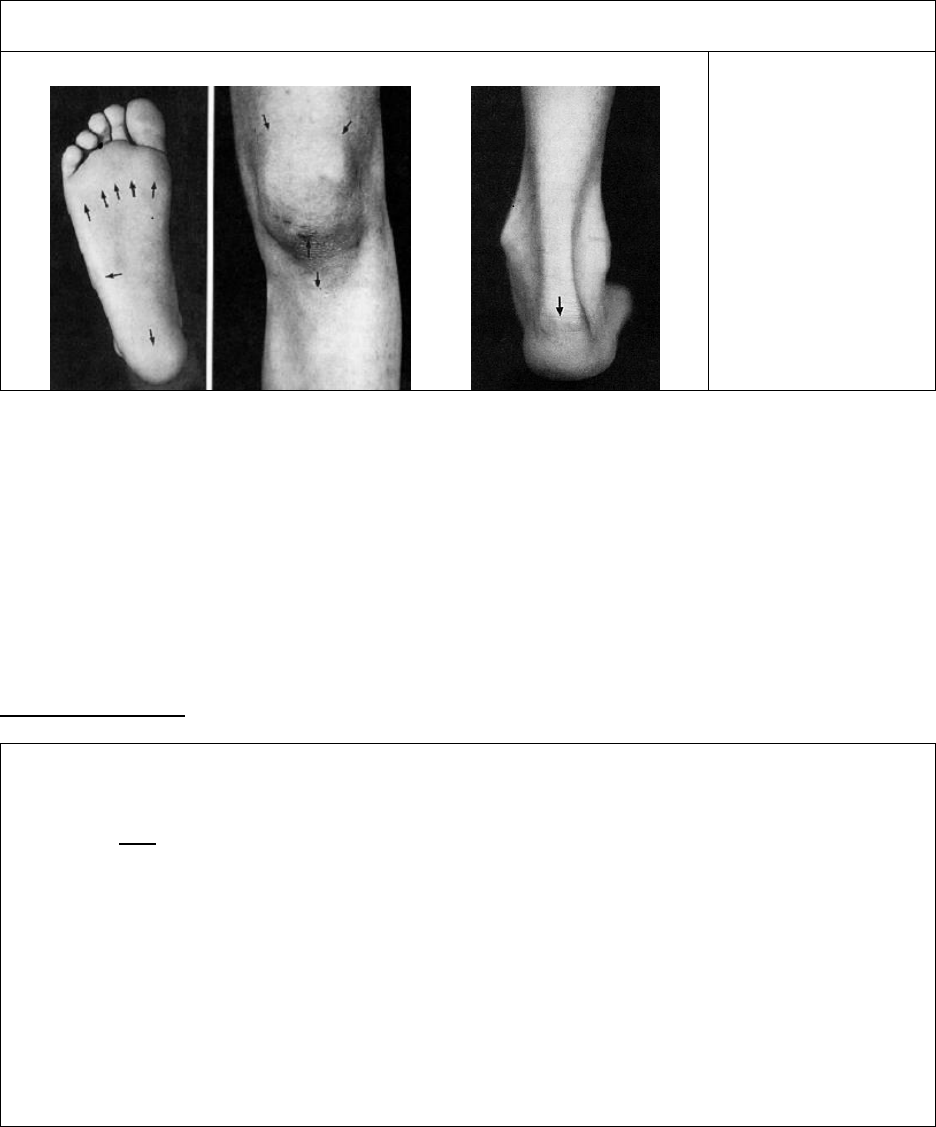
Common sites of enthesitis in the lower body
A B C
A. Insertions of
plantar fascia
B. Insertions of
quadriceps and
patellar tendons
C. Insertion of
Achilles tendon
Images were published in “Spondyloarthropathies of childhood” in Pediatrics Clinics of North America, 1995,
Volume 42, Pages 1051-1070, Copyright Elsevier (1995).
ERA typically occurs in boys, usually over 6 years of age with familial predilection
Axial involvement (involvement of the sacroiliac joints and/or spine) typically develops later
Other manifestations include tarsitis (diffuse inflammation of tarsal joints and surrounding
tendon sheaths) and dactylitis (sausage-shaped swelling of entire digit)
Symptomatic anterior uveitis may develop in children with ERA and this usually presents
with significant eye pain and redness, which may be unilateral
Gastrointestinal symptoms (e.g. chronic abdominal pain, diarrhea, hematochezia) should be
carefully evaluated for possible inflammatory bowel disease
Psoriatic Arthritis
ILAR Classification Criteria for Psoriatic Arthritis *
Definition:
Arthritis and psoriasis
Or, arthritis and at least 2 of the following:
Dactylitis
Nail-pitting or onycholysis
Psoriasis in a first-degree relative
Exclusions:
o Arthritis in an HLA B27 positive male beginning after 6
th
birthday
o Ankylosing spondylitis, enthesitis related arthritis, sacroiliitis with IBD, or acute anterior uveitis,
or history of one of these disorders in 1
st
degree relative
o Presence of IgM Rheumatoid Factor on at least 2 occasions at least 3 months apart
o Presence of systemic JIA
* Classification criteria are designed to identify a homogeneous population for research studies
and are not diagnostic criteria, although they are often used for diagnosis in practice
Psoriasis may develop after arthritis and may lead to reclassification of JIA type as psoriatic
Typically asymmetric, and involves both large and small joints

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 30
Clinical hallmark is dactylitis, which is caused by simultaneous inflammation of the flexor
tendon and synovium, leading to the typical “sausage digit” appearance
Undifferentiated Arthritis
ILAR Classification Criteria for Undifferentiated Arthritis *
Definition: Arthritis that fulfils criteria in no category or in 2 or more of above categories
* Classification criteria are designed to identify a homogeneous population for research studies
and are not diagnostic criteria, although they are often used for diagnosis in practice
References:
1. Crayne CB, Beukelman T. Juvenile idiopathic arthritis: Oligoarthritis and polyarthritis.
Ped Clin North Am 2018; 65(4):657-74.
2. Weiss PF, Colbert RA. Juvenile spondyloarthritis: A distinct form of juvenile arthritis. Ped
Clin North Am 2018; 65(4):675-90.
3. Lee JJY, Schneider R. Systemic juvenile idiopathic arthritis. Ped Clin North Am 2018;
65(4):691-709.
4. Petty RE, et al. ILAR classification of juvenile idiopathic arthritis: Second revision,
Edmonton 2001. J Rheumatol 2004; 31(2):390-2.
5. Martini A, et al. Toward new classification critieria for juvenile idiopathic arthritis: First
steps, Pediatric Rheumatology International Trials Organization International
Consensus. J Rheumatol 2019; 46(2):190-7.
3B. Approach to Management of JIA
Goals of therapy
1. Eliminate inflammation with goal to achieve clinical remission
2. Prevent joint damage
3. Promote normal growth and development
4. Maintain normal function and optimize quality of life
5. Minimize medication toxicity
Timing of assessments:
o Children with suspected JIA should be reviewed by a pediatric rheumatologist in 4-6
weeks and those with possible systemic JIA within 7 days
o Follow-up is recommended at intervals of 3-4 months in patients with controlled
disease and more often in those with uncontrolled disease
Disease monitoring:
o Assessments of disease activity by a pediatric rheumatologist and multidisciplinary
team are essential for disease monitoring
o Laboratory monitoring is often an essential part of management, especially during
disease flares and medication changes (escalation and weaning)
o Surveillance joint X-rays should not be ordered routinely to monitor disease activity,
but may be used as needed to assess for joint damage (highlighted in Choosing
Wisely: Pediatric Rheumatology Top 5 by American College of Rheumatology)
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 31
o Ultrasound and/or MRI could be considered to detect early or subclinical disease
activity or damage and MRI is usually indicated for monitoring of temporomandibular,
sacroiliac, hip, and subtalar joints
o Careful monitoring by an eye care provider is essential to assess for chronic anterior
uveitis, especially in patients with oligoarthritis and positive ANA
o Screening for asymptomatic uveitis should take place within 4 weeks of diagnosis
Multidisciplinary approach:
o Multidisciplinary team is part of comprehensive JIA management
o Occupational and physical therapists play an important role in treating JIA
o Psychosocial aspects of disease must be recognized and addressed
Treat-to-Target Strategy for JIA:
o Target of treatment is complete remission, which means absence of signs and
symptoms of inflammatory disease activity, including extra-articular manifestations
(e.g. uveitis)
o Minimal or low disease activity may be an alternative target, particularly in patients
with longstanding or difficult-to-treat disease
o Setting the target and therapeutic decisions should be based on individual patient
characteristics and agreed on with the patient/parents
o Rapid escalation and changes in therapy may be required until target is achieved
o In all patients, at least a 50% improvement in disease activity should be reached
within 3 months and the target within 6 months; however, patients with systemic JIA
should be fever-free within 1 week
Medications:
o Initial therapy with an NSAID may be started by a patient’s primary care physician;
however, further therapy should be directed by a pediatric rheumatologist
o Intra-articular corticosteroids and methotrexate remain key medications for JIA
o Potential algorithms for treatment of oligoarthritis, polyarthritis and systemic JIA are
included in the following pages
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 32
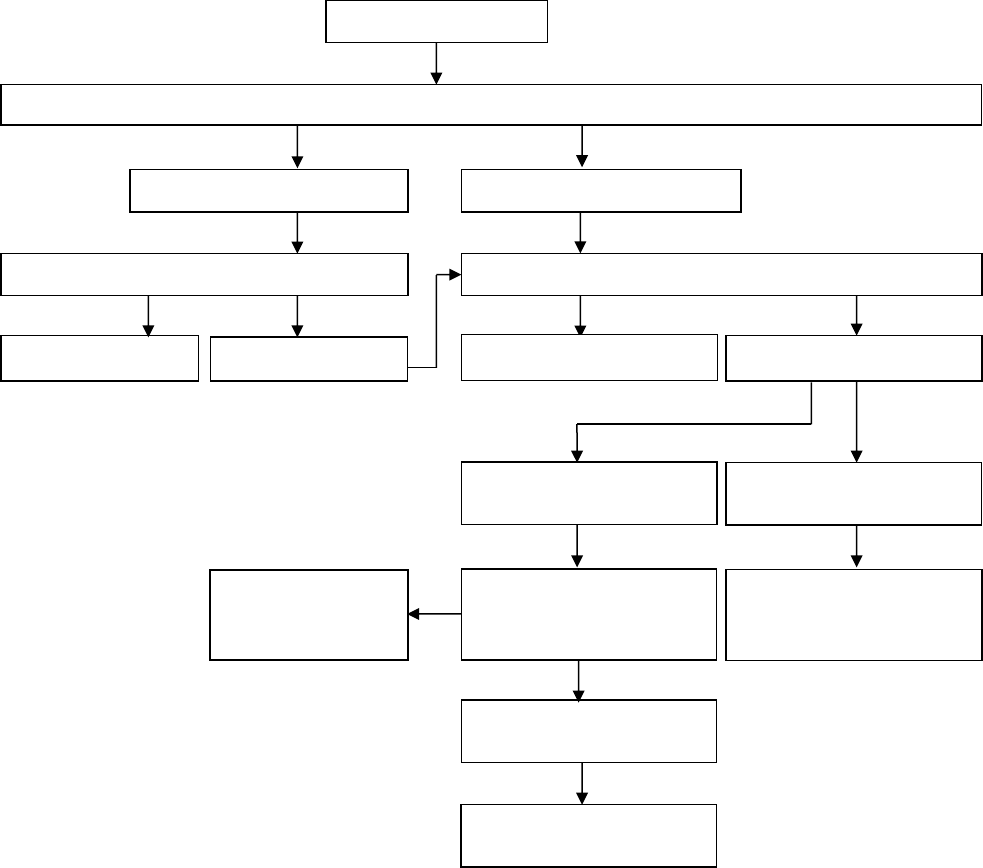
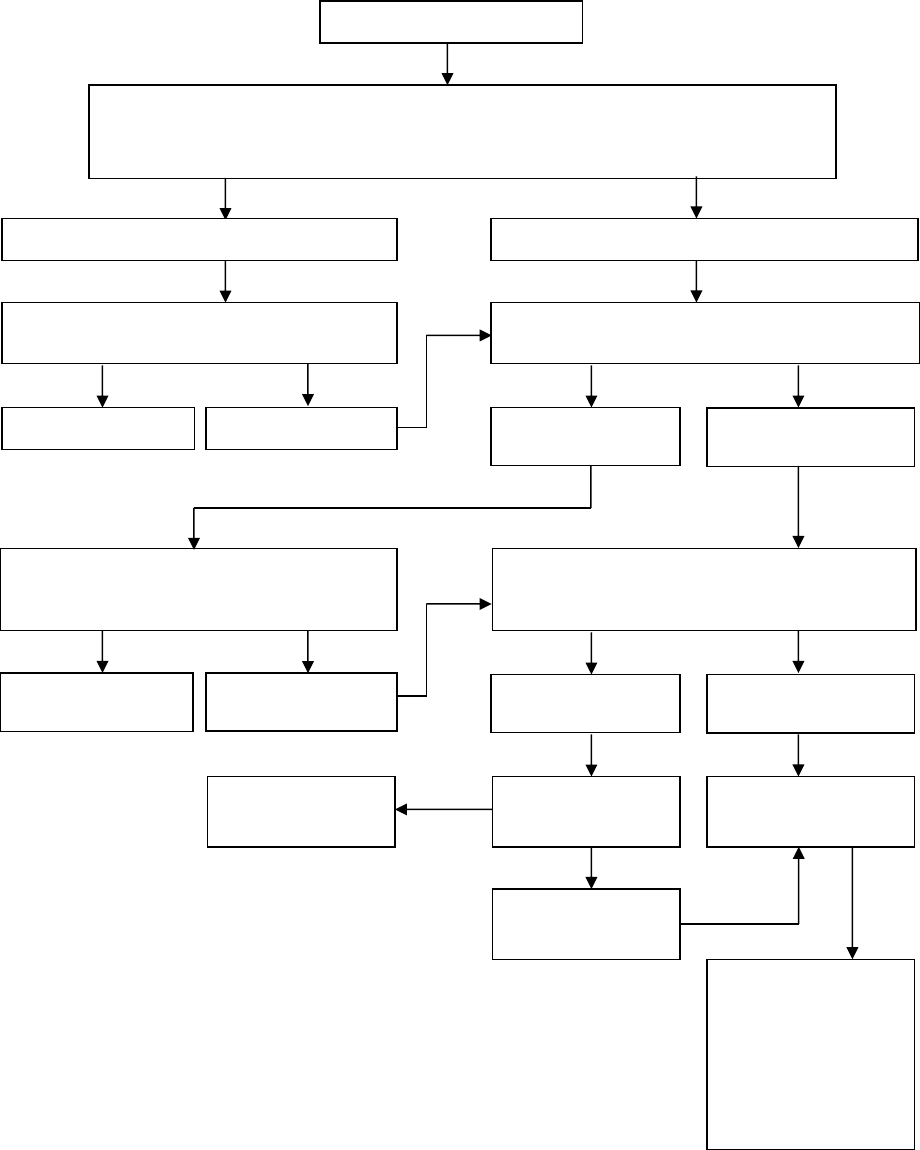
An Algorithm for Treatment of Oligoarthritis
Persistent oligoarticular
arthritis
Inadequate response
Oligoarticular arthritis
NSAID or Intra-Articular Corticosteroid injection (IAC) withTriamcinolone hexacetonide
Improvement
Inadequate response
Follow and, if no IAC, continue NSAID
Recurrence
Remission
Evolves into polyarticular
arthritis
Intermittent IAC, or
consider adding disease-
modifying drug
Management same as
polyarticular JIA
(see next algorithm)
Inadequate response
Remission
Consider biologic agent
Repeat or first IAC
Remission
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 33
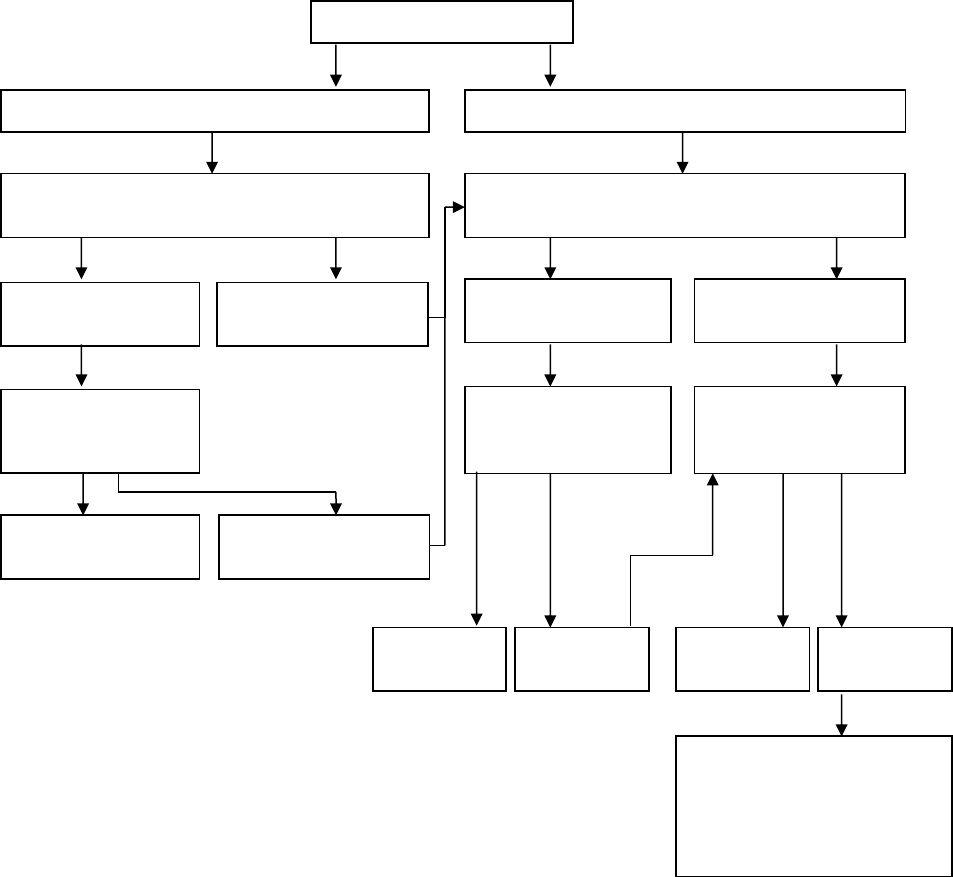
An Algorithm for Treatment of Polyarthritis
Inadequate
response
If inadequate
response, consider
switch to different
anti-TNF agent or
other biologic agent
(e.g. abatacept,
tocilizumab)
Consider adding
anti-TNF therapy
Continue therapy
and follow
Remission
Recurrence
Optimise disease-modifying drug
Consider IAC or low dose oral
corticosteroids as bridging therapy
Continue therapy and follow
Polyarthritis
NSAID with or without Intra-Articular Corticosteroid injections (IAC)
Consider using disease-modifying drug, such as methotrexate, as part of initial
therapy for moderate to severe polyarthritis
Improvement
Inadequate response
Add disease-modifying drug, such as
methotrexate or leflunomide
Continue NSAID and follow
Recurrence
Remission
Improvement
Recurrence
Remission
Improvement
Inadequate
response
N.B. A limited course of Corticosteroids
(< 3 months) during initiation or
escalation of therapy may be added in
patients with high-moderate disease
activity
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 34
An Algorithm for Treatment of Systemic JIA
References
1. Giancane G, et al. Recent therapeutic advances in juvenile idiopathic arthritis. Best Pract
Res Clin Rheumatol 2017; 31(4):476-87.
2. Ravelli A, et al. Treating juvenile idiopathic arthritis to target: recommendations of an
international task force. Ann Rheum Dis 2018; 77(6): 819-28.
3. Beukelman T, et al. 2011 American College of Rheumatology recommendations for the
treatment of juvenile idiopathic arthritis... Arthritis Care Res 2011; 63(4):465-82.
4. Ringold S, et al. 2019 American College of Rheumatology/Arthritis Foundation Guideline
fot the Treatment of Juvenile Idiopathic Arthritis: Therapeutic Approaches for Non-
Systemic Polyarthritis, Sacroilitis, and Enthesitis. Arthritis Care Res 2019; 71(6):717-34.
5. Cellucci T, et al. Management of Juvenile Idiopathic Arthritis 2015: A Position Statement
from the Pediatric Committee of the Canadian Rheumatology Association. J Rheumatol
2016; 43(10):1773-6.
Systemic arthritis
Mild to moderate disease
Moderate to severe disease
NSAID and/or Corticosteroidsand/or
Anakinra
Corticosteroids and/or biologic agent,
such as anti-IL-1 or anti-IL-6 therapy
Improvement
Inadequate
response
Improvement
Inadequate
response
Continue therapy
and follow
Continue therapy
and follow
Add or change
biologic anti-IL-1 or
anti-IL-6 agent
Remission Recurrence
Recurrence
Remission
Recurrence
Remission
Change biologic therapy or
consider methotrexate,
leflunomide, or anti-TNF
agent, or abatacept if
prominent arthritis

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 35
SECTION 4. SYSTEMIC LUPUS ERYTHEMATOSUS & RELATED CONDITIONS
4A. Systemic Lupus Erythematosus (SLE)
Multi-system inflammatory disease characterized by autoantibody and immune-complex
mediated inflammation of blood vessels and connective tissues
Pediatric-onset SLE accounts for 10-20% of all cases of SLE
Female predominance, especially in adolescence and adulthood
Ethnic predilection in Blacks, Hispanics, and Asians
Positive family history of SLE in 10%
1997 American College of Rheumatology (ACR) Classification Criteria for SLE *
Patients are classified as having SLE if they have ≥ 4/11 of following criteria:
Malar rash (butterfly rash sparing nasolabial folds)
Discoid lupus rash **
Photosensitivity
Oral or nasal mucocutaneous ulcerations (typically painless)
Non-erosive arthritis involving two or more peripheral joints
Nephritis (characterized by proteinuria and/or cellular casts)
CNS involvement (characterized by seizures and/or psychosis)
Serositis (pleuritis or pericarditis)
Cytopenia (thrombocytopenia, lymphopenia, leukopenia, hemolytic anemia with
reticulocytosis)
Positive ANA
Positive immunoserology (anti-dsDNA, anti-Sm (anti-Smith), antiphospholipid antibodies)
* Classification criteria are designed to identify a homogeneous population for research studies
and are not diagnostic criteria, although they are often used for diagnosis in practice
** Uncommon in children
1997 ACR classification criteria are not diagnostic criteria and were designed to identify a
homogeneous population of SLE patients for research studies; however, the presence of ≥ 4
criteria is specific for SLE (>93%) and so the criteria have been widely used for diagnosis
In 2012, newer Systemic Lupus International Collaborating Clinics (SLICC) classification
criteria for SLE were developed that incorporated more immunologic criteria and were more
sensitive (>95%) but less specific (83%) than the 1997 ACR criteria
New EULAR/ACR classification criteria for SLE have been developed in 2019 and may be
adopted in the future since they are both sensitive (>95%) and specific (>93%) for SLE

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 36
2019 Proposed European League Against Rheumatism (EULAR) and American College
of Rheumatology (ACR) Classification Criteria for SLE *
Clinical domains
Points**
Constitutional domain
Fever
2
Cutaneous domain
Non-scarring alopecia
Oral ulcers
Subacute cutaneous or discoid lupus
Acute cutaneous lupus
2
2
4
6
Arthritis domain
Synovitis or tenderness in at least 2 joints
6
Neurologic domain
Delirium
Psychosis
Seizure
2
3
5
Serositis domain
Pleural or pericardial effusion
Acute pericarditis
5
6
Hematologic domain
Leukopenia
Thrombocytopenia
Autoimmune hemolysis
3
4
4
Renal domain
Proteinuria >0.5 g/24 hours
Class II or V lupus nephritis
Class III or IV lupus nephritis
4
8
10
Immunological domains
Points**
Antiphospholipid antibody domain
Anti-cardiolipin IgG >40 GPL
Or anti-β
2
-glycoprotein I IgG >40 units
Or lupus anticoagulant
2
Complement proteins domain
Low C3 or low C4
Low C3 and low C4
3
4
Highly specific antibodies domain
Anti-dsDNA antibody
Anti-Sm (Anti-Smith) antibody
6
6
* In order to be classified as having SLE, patients must have all of the following: (a) ANA
≥1:80 (entry criterion); (b) ≥10 points in total; and (c) at least one clinical criterion
N.B. Classification criteria are designed to identify a homogeneous population for research
studies and are not diagnostic criteria, although they are often used for diagnosis in practice
** Only the highest criterion in a given domain is counted toward total number of points
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 37
Other clinical features of SLE not included in any of the above classification criteria:
o Additional constitutional symptoms (i.e. fatigue, weight loss, anorexia)
o Other rashes (e.g. annular erythema, maculopapular or linear (nonspecific) rash, bullous
lupus (rare), palmar/plantar/periungual erythema, livedo reticularis, or vasculitic rash)
o Myalgia, and/ or myositis
o Raynaud phenomenon (see Section 7E)
o Lymphadenopathy
o Hepatomegaly, splenomegaly
o Decreased concentration and cognitive dysfunction, stroke, mood disorder, headache
o Pneumonitis, pulmonary hemorrhage
o Myocarditis, Libman-Sacks endocarditis
Other common laboratory features of SLE:
o Elevated ESR with normal CRP (except high CRP in infection and/or serositis)
o Elevated IgG levels
o Other autoantibodies: anti-Ro, anti-La, anti-RNP, Rheumatoid factor
May be accompanied by macrophage activation syndrome (MAS) at onset or anytime during
course
Treatment
o Use minimum required treatment to maintain clinical and laboratory quiescence
o More aggressive treatment used for more severe organ involvement
o Hydroxychloroquine (Plaquenil™)
Considered standard therapy for SLE
Proven efficacy in decreasing frequency and severity of disease flares
Improves serum lipid profile
May be helpful in lowering antiphospholipid antibody titres and preventing thrombotic
recurrences in patients with SLE
o Corticosteroids
Often used in initial therapy for SLE with dose depending on severity and organ
involvement
Pulse (very high dose) therapy is used for severe lupus nephritis, hematologic crisis,
CNS disease or other life or organ-threatening manifestations
o Azathioprine
Typically used for hematologic and renal manifestations
o Mycophenolate mofetil
Used for hematologic, renal and CNS manifestations
o Cyclophosphamide
Used for severe renal and CNS manifestations
o Rituximab
Used for resistant thrombocytopenia and in other specific scenarios, such as when a
patient is unresponse to other therapies
o Belimumab
Adjunctive therapy for mild/moderate SLE (trials excluded those with severe CNS
and renal involvement)
Course and Outcomes
o Relapsing and remitting course of disease
o 10-year survival >90%
o Most deaths related to infection, renal, CNS, cardiac, and pulmonary disease

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 38
o Additional morbidity related to disease and/or treatment:
Early-onset coronary artery disease
Bone disease → osteopenia, osteoporosis, avascular necrosis
Malignancy
Infection
Childhood-onset SLE vs. adult-onset SLE
Children have more active disease at presentation and over time
Children more likely to have active renal disease (~70% vs. 30-60% in adults)
Children receive more intensive drug therapy and sustain more long-term damage
References:
1. Harry O, et al. Childhood-onset systemic lupus erythematosus: a review and update. J
Pediatr 2018; 196:22-30.
2. Tarvin SE, O’Neil KM. Systemic lupus erythematosus, Sjögren syndrome, and mixed
connective tissue disease in children and adolescents. Ped Clin North Am 2018;
65(4):711-37.
3. Weiss JE. Pediatric systemic lupus erythematosus: More than a positive antinuclear
antibody. Pediatr Rev 2012; 33(2):62-73.
4. Tedeschi SK, et al. Multicriteria decision analysis process to develop new classification
criteria for systemic lupus erythematosus. Ann Rheum Dis 2019; 78(5):634-40.
4B. Neonatal Lupus Erythematosus (NLE)
Disease of developing fetus and newborn characterized by transplacental passage of
maternal autoantibodies
Pathogenesis linked to maternal anti-Ro and anti-La antibodies
Presence of autoantibodies is necessary but not sufficient to cause NLE since many
mothers with autoantibodies deliver healthy, unaffected infants
Mothers of infants with NLE may have SLE, Sjögren syndrome, or another autoimmune
disease; however, many mothers are healthy with no known autoimmune disease
Incidence of NLE is 1-2% in children of mothers with anti-Ro and/or anti-La antibodies
Higher risk for subsequent children once one child has been affected (e.g. 16% of
subsequent siblings of child with congenital heart block)
Clinical features
o Cardiac
Most important and severe manifestation is complete congenital atrioventricular (AV)
heart block
Complete heart block is associated with significant morbidity and mortality
(congestive heart failure, fetal hydrops, intrauterine death)
Other manifestations include less severe conduction abnormalities, carditis and risk
of endocardial fibroelastosis
o Skin
Classic NLE rash is annular, erythematous papulosquamous rash with fine scale and
central clearing
Predilection for face and scalp (not malar distribution)
Typically photosensitive

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 39
Dermatitis may be present at birth, but commonly develops in first 6 weeks of life
New lesions appear for several months, but rarely develop after 6 months and
typically heal without scarring
Telangiectasias may develop starting at 6-12 months of age, may not be in areas
affected by previous rash
o Hematologic
Thrombocytopenia is most common
Neutropenia and anemia are less common
Usually resolve without sequelae and rarely require treatment
Neutropenia is not typically associated with increased risk of infection
o Hepatic
Asymptomatic cholestatic hepatitis with mildly to moderately elevated liver enzymes
Hepatomegaly and less commonly splenomegaly
May be the only manifestation(s) of NLE
Typically resolves before 6 months without treatment
o Neurologic
Reported CNS manifestations include macrocephaly, hydrocephalus, spastic
paraparesis, asymptomatic neuroimaging abnormalities, and vasculopathy
Clinical significance still unclear
Important to monitor head circumference
Treatment
o If fetal bradycardia found during pregnancy, require fetal echocardiography to assess for
heart block and endocardial fibroelastosis (EFE) and may require treatment with
Dexamethasone/Betamethasone ± sympathomimetics
o Pacemaker may be required soon after birth for neonates with complete heart block
o Classic NLE rash does not require treatment (although sun avoidance and sunscreen
are recommended) since rash will completely resolve; Corticosteroids may hasten
healing, but may increase risk of telengiectasias
o Severe cytopenias may require treatment with IVIG
o Future pregnancies require expectant management with fetal heart rate monitoring and
mothers with autoantibodies may be treated with Hydroxychloroquine (Plaquenil)
References:
1. Vanoni F, et al. Neonatal systemic lupus erythematosus syndrome: a comprehensive
review. Clin Rev Allergy Immunol 2017; 53(3):469-76.
2. Johnson B. Overview of neonatal lupus. J Pediatr Health Care 2014; 28(4):331-41.
4C. Drug-Induced Lupus
Development of lupus-like symptoms that is temporally related to continuous drug exposure
(>1 month) and that resolves with cessation of the offending drug
Usually accompanied by serologic findings of positive ANA as well as anti-histone
antibodies (in approximately 90% of patients)
Variable time from drug exposure to onset of symptoms
Onset generally insidious
Patients commonly present with fever, arthralgias or arthritis, myalgias and serositis
Usually mild, although life threatening disease has been reported
Rarely involve classic malar or discoid rash, oral ulcers or major organ involvement

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 40
Laboratory findings may include mild cytopenias, high ESR
Drugs that have been implicated in drug-induced lupus include: Minocycline,
anticonvulsants, Hydralazine, and biologic agents that target tumor necrosis factor (TNF)
Treatment
o Stop the offending drug
o Corticosteroids and/or Hydroxychloroquine (Plaquenil™) may be used for moderate to
severe manifestations (e.g. cardiac tamponade)
References:
1. Vaglio A, et al. Drug-induced lupus: Traditional and new concepts. Autoimmun Rev
2018; 17(9):912-8.
4D. Antiphospholipid Syndrome (APS)
Systemic autoimmune disorder characterized by recurrent arterial and/or venous thrombosis
and elevated levels of one or more antiphospholipid antibodies
Primary APS if occurs without apparent underlying disease
Secondary APS due to SLE, other autoimmune diseases, drugs or viral infections (e.g. HIV)
Venous thrombosis in ~60%, arterial thrombosis in ~30%, small vessel thrombosis in ~5%,
and mixed thrombosis in ~2%
Thrombotic manifestations are most common, followed by hematologic, skin and non-
thrombotic neurologic manifestations
Adaptation of the Updated Sapporo Classification Criteria for Pediatric APS Patients *
Definite APS is considered to be present if the clinical criterion and at least 1 of the laboratory
criteria are met
Clinical criterion:
Vascular thrombosis: ≥1 clinical episode(s) of arterial, venous, or small vessel thrombosis
in any tissue or organ confirmed objectively by validated criteria
Laboratory criteria:
Lupus anticoagulant on ≥ 2 occasions at least 12 weeks apart
Anticardiolipin antibody (IgG and/or IgM isotype) in medium or high titre (>40 GPL or MPL,
or >99
th
percentile) on ≥ 2 occasions at least 12 weeks apart
Antibodies to β
2
-glycoprotein I (IgG and/or IgM isotype) in medium or high titre (>99
th
percentile) on ≥ 2 occasions at least 12 weeks apart
* Classification criteria are designed to identify a homogeneous population for research
studies and are not diagnostic criteria, although they are often used for diagnosis in practice
Higher risk of thrombosis associated with higher antibody titres, IgG isotype and specific
antibodies (e.g. anti-cardiolipin and lupus anticoagulant)
Deep venous thrombosis is the most common type of venous thrombosis, while stroke is the
most common type of arterial thrombosis (see Section 2: Differential Diagnosis of stroke-like
presentations in children)
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 41
Additional clinical features of APS:
o Livedo reticularis, Raynaud phenomenon, and skin ulcers
o Cardiac valve disease (Libman-Sachs endocarditis)
o Chorea
o Seizures
o Transient cerebral ischemia
o Transverse myelopathy
Additional laboratory features of APS:
o Thrombocytopenia
o Hemolytic anemia
o Additional antibodies to prothrombin, annexin, and/or other phospholipids
o False positive VDRL
Treatment
o If primary, treat as a disorder of coagulation
o If secondary (most commonly due to SLE), treat underlying disorder (often using
Corticosteroids and/or Hydroxychloroquine)
o Anticoagulation using heparin (e.g. low molecular weight heparin (LMWH)) is usually
required at least initially, but patients could require LMWH or warfarin therapy lifelong
o Consider anti-platelet agents (e.g. ASA)
o May consider Rituximab as direct therapy to target pathogenic autoantibodies in APS
References:
1. Rumsey DG, et al. Diagnosis and treatment of antiphospholipid syndrome in childhood:
A review. Blood Cells Mol Dis 2017; 67:34-40.
2. Aguiar C, et al. Pediatric antiphospholipid syndrome. Curr Rheumatol Rep 2015;
17(4):27.
3. Avcin T, et al. Pediatric antiphospholipid syndrome: Clinical and immunologic features of
121 patients in an international registry. Pediatrics 2008; 122(5):e1100-7.
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 42
SECTION 5 – SYSTEMIC VASCULITIS
5A. Introduction to Vasculitis
Group of multi-system inflammatory diseases characterized by inflammation and necrosis of
blood vessels, resulting in vessel occlusion and tissue ischemia
Consider vasculitis when:
o Unexplained prolonged constitutional symptoms (fever, weight loss, fatigue)
o Multiple organ system involvement – see table below:
Organ System
Clinical features that suggest possible systemic vasculitis
Head and Neck
Chronic sinusitis
Epistaxis
Chronic otitis
Hearing loss
Chondritis
Ophthalmologic
Episcleritis
Iritis
Panuveitis
Retinitis
Central nervous system
Headaches
Seizures
Strokes
Cardiac
Pericarditis
Myocarditis
Myocardial infarction
Pulmonary
Hemorrhage
Nodules
Cavities
Infiltrates
Renal
Nephritis
Nephrotic syndrome
Hypertension
Rapidly progressive renal failure
Gastrointestinal
Ischemic abdominal pain
Dermatologic
Palpable purpura
Nodules
Livedo reticularis
Ulcers
Vascular
Chronic vascular insufficiency
Vascular bruits
Claudication
Peripheral nervous system
Mononeuritis
Musculoskeletal
Arthritis, arthralgia
Myalgia, calf pain

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 43
Classification of vasculitis based on size of vessel (predominantly) involved
Large vessel vasculitis
Takayasu arteritis
Giant cell arteritis (older adults)
Medium vessel vasculitis
Kawasaki disease
Polyarteritis nodosa (systemic, cutaneous)
Small vessel vasculitis
Immune complex vasculitis
IgA vasculitis (Henoch-Schönlein purpura)
Cryoglobulinemia
Hypocomplementemic urticarial vasculitis
ANCA-associated vasculitis
Microscopic polyangiitis
Granulomatosis with polyangiitis (previously Wegener
granulomatosis)
Eosinophilic granulomatosis with polyangiitis (previously
Churg-Strauss Syndrome)
Variable vessel vasculitis
Behçet disease
Cogan syndrome
Other vasculitis
Primary CNS vasculitis (see Section 10)
Primary cutaneous vasculitis
Vasculitis secondary to drugs, infection (e.g. hepatitis B virus,
Parvovirus), or malignancy
Monogenic disease causing vasculitis (e.g. Deficiency of
Adenosine Deaminase 2)
Investigations
o Look for end-organ damage (eyes, skin, heart, lungs, kidneys, nervous system)
o Look for triggers or underlying disease (drugs, malignancy, infection, CTD)
o Inflammatory markers (CRP, ESR)
o Immune serology (ANA, ANCA)
o Tissue biopsy (histopathology & immunofluorescence)
o Angiography (conventional; magnetic resonance; computed tomography); vessel wall
itself may be assessed using magnetic resonance or computed tomography
Treatment
o Depends on specific disease, organ involvement, severity
o Immunosuppressive agents plus supportive therapy
Potential complications
o Acute: organ failure (renal, pulmonary, cardiac), hemorrhage (pulmonary, GI), thrombus
(renal, pulmonary, coronary, cerebral, GI vessels), infection (often treatment-related)
o Chronic: hypertension, renal failure, pulmonary insufficiency, hearing loss, saddle nose
deformity, subglottic stenosis, hemiplegia, neuropathy
References:
1. Barut K, et al. Pediatric Vasculitis. Curr Opin Rheumatol 2016; 28:29-38.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 44
2. Jennette JC, et al. 2012 Revised International Chapel Hill Consensus Conference
Nomenclature of Vasculitides. Arthritis Rheum 2013; 65(1):1-11.
3. Weiss P. Pediatric Vasculitis. Pediatr Clin North Am 2012; 59(2):407-23.
5B. Takayasu arteritis
2008 EULAR/PRINTO/PRES Classification Criteria for Childhood Takayasu arteritis *
Angiographic abnormalities of the aorta or its main branches and pulmonary arteries showing
aneurysm/dilatation, narrowing, or thickened arterial wall (mandatory criterion)
Plus ≥ 1/5 of the following:
Peripheral pulse deficit or claudication (focal muscle pain induced by physical activity)
Discrepancy of four limb systolic BP >10 mm Hg in any limb
Bruits or thrills over the aorta and/or its major branches
Hypertension (>95
th
percentile for height)
Acute phase reactants (ESR >20 or increased CRP)
* Classification criteria are designed to identify a homogeneous population for research studies
and are not diagnostic criteria, although they are often used for diagnosis in practice
Large vessel vasculitis involving the aorta and its branches (thoracic, abdominal, carotid)
Chronic, relapsing disease
Initially can present as non-specific inflammatory illness with fever
Evolution to chronic, fibrotic phase with signs and symptoms of chronic vascular
insufficiency (pulse deficit, claudication, BP discrepancy, bruits)
Investigations
o Magnetic resonance angiography useful to show extension of disease and vessel wall
inflammation; often used to follow disease (less invasive than conventional angiography)
o Rule out associated TB infection (PPD, chest X-ray)
Treatment
o Depends on degree of inflammation
o If “active” disease (by acute phase reactants +/- wall enhancement on MRA):
Corticosteroids plus second line agent
Second line agents include Methotrexate, Mycophenolate mofetil, Infliximab or
Adalimumab
May also use Tocilizumab, Cyclophosphamide, or Rituximab if refractory disease
o If “inactive” disease:
Manage end-organ manifestations (medical therapy +/- vascular surgery)
References:
1. Ozen S, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood
polyarteritis nodosa, childhood Wegener’s granulomatosis and childhood Takayasu
arteritis: Ankara 2008. Ann Rheum Dis 2010; 69(5):798-806.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 45
2. Brunner J, et al. Takayasu arteritis in children and adolescents. Rheumatology 2010;
49:1806-14.
3. de Ranieri D. Great vessels of children: Takayasu’s arteritis. Pediatr Ann 2015;
44(6):e148-52.
5C. Kawasaki disease (KD)
Diagnostic Criteria for Kawasaki disease *
Fever persisting for ≥5 days
Plus ≥4/5 of the following:
Changes in peripheral extremities (edema/erythema) or perineal area (erythema/peeling)
Polymorphous exanthem
Bilateral conjunctival injection, non-exudative
Changes of lips and oral cavity (injection of oral and pharyngeal mucosa, fissured lips,
strawberry tongue)
Cervical lymphadenopathy (frequently unilateral, ≥1.5 cm)
* Other ways to make diagnosis of KD:
a) In presence of fever and coronary artery involvement on echo, <4/5 criteria sufficient
b) Incomplete KD diagnosed if ≥5 days of fever with 2 or 3 features (common in infants,
who are at higher risk of coronary artery involvement)
c) Atypical KD diagnosed if KD with an unusual manifestation (e.g. renal failure)
Medium vessel vasculitis, with predilection for coronary arteries
Most common between 1 and 5 years of age
Most common cause of acquired heart disease in children in developed countries
May be triggered by infectious agent (viral and/or bacterial super-antigen implicated)
Polygenic with genes identified that influence risk of KD and coronary artery involvement
Clinical features
o Common: irritability (aseptic meningitis), arthritis (at onset or delayed), sterile pyuria
(urethritis), gastroenteritis (abdominal pain, vomiting, diarrhea), uveitis, periungual
desquamantion in weeks 2 or 3 (subacute phase)
o Uncommon: gallbladder hydrops, GI ischemia, jaundice
o Cardiac involvement: myocarditis, pericarditis, cardiac failure, valvular regurgitation
Complications
o Coronary artery disease
Major concern is the development of coronary artery aneurysms, which most
commonly occurs at 4-6 weeks after the acute illness
Risk factors: males, infants <1 year or >9 years of age, prolonged fever, Asian or
Hispanic ethnicity, thrombocytopenia, hyponatremia
o KD shock syndrome (characterized by hypotension or poor perfusion)
o Macrophage activation syndrome (MAS)
o Dissemeniated intravascular coagulation
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 46
Investigations
o Leukocytosis with left shift, normocytic anemia, elevated ESR/CRP, hypoalbuminemia,
hyponatremia, may have elevated transaminases
o Thrombocytosis in second week of illness with return to normal by 4-8 weeks
o Echoardiogram required at the time of diagnosis and 6 weeks later
Treatment
o See treatment algorithm on next page
o Target treatment within 10 days of fever onset
o IVIG
o Unequivocally reduces the occurrence of coronary artery aneurysms
Recommended dose 2 g/kg
If still febrile 24-36 hours after IVIG → second dose of IVIG
Consider monitoring for IVIG-related hemolysis with CBC, blood film, reticulocytes
and direct antiglobulin test initially at 24-92 hours and then at 5-7 days after IVIG
o ASA
Historically, started with high-dose ASA 80-100 mg/kg/day (anti-inflammatory) until
afebrile x 24 hours, then switched to low-dose 3-5 mg/kg/day (anti-platelet)
Many centres now start with low-dose ASA 3-5 mg/kg/day
Low-dose ASA (and/or other antiplatelet agents) may be continued longer than 6
weeks in patients with coronary artery aneurysms
o Corticosteroids
May be used with first dose of IVIG in patients at high risk of coronary artery disease
(see risk factors above) or may be reserved for patients with refractory fever after
two doses of IVIG
Additional indications: myocarditis, MAS, Kawasaki shock-syndrome
Common treatment regimens include IV Methylprednisolone for 1-3 days or
Prednisone PO 2 mg/kg/day for 3 days or for a longer course with slow wean in high-
risk patients
o If large coronary aneurysm → Abciximab (glycoprotein IIb/IIIa receptor inhibitor) in acute
or subacute phase; long-term antiplatelet (+ Heparin or Warfarin if giant aneurysm)
Prognosis
o In-hospital mortality 0.17% (all cardiac-related)
o ~ 2% risk of recurrent KD
o Without treatment, coronary artery aneurysms occur in ~25% of patients → reduced to
~4% if IVIG treatment within 10 days
o If coronary artery aneurysm → risk for thrombosis, obstruction and stenosis, ventricular
dysfunction/arrhythmia, early atherosclerosis, myocardial infarction (highest if ≥8 mm)
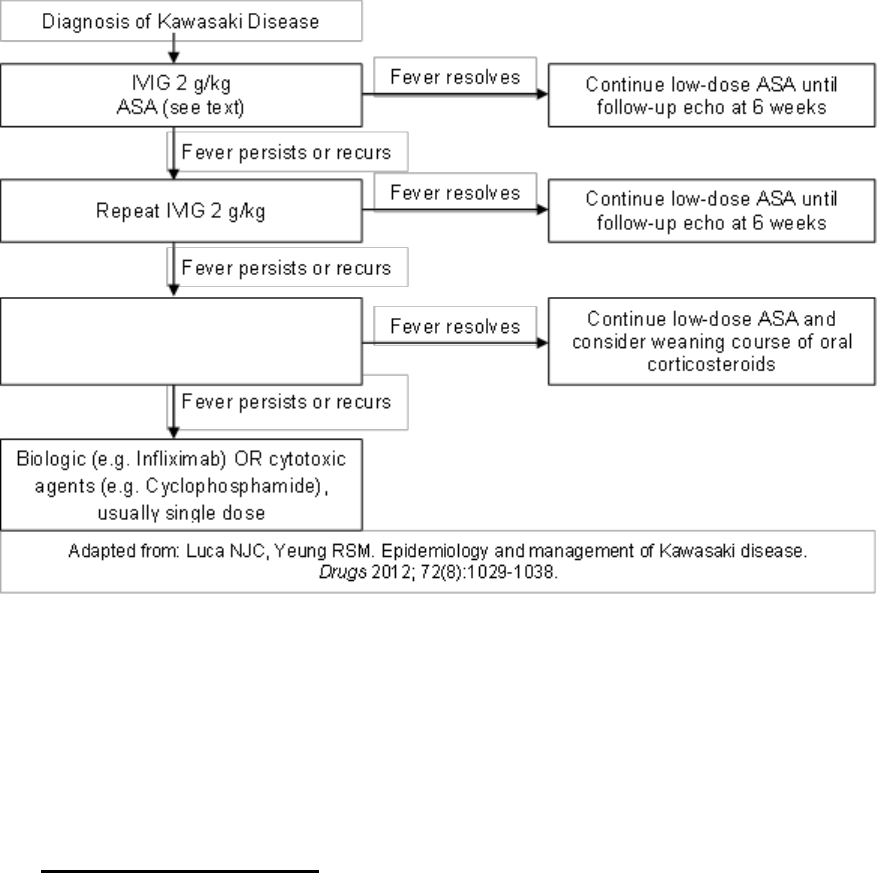
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 47
An algorithm for treatment of Kawasaki disease
References:
1. McCrindle BW, et al. Diagnosis, treatment, and long-term management of Kawasaki
disease: A scientific statement for health professionals from the American Heart
Association. Circulation 2017; 135:e927-99.
2. Yellen ES, et al. Performance of 2004 American Heart Association recommendations for
treatment of Kawasaki disease. Pediatrics 2010; 125(2): e234-e241.
3. Scuccimarri R. Kawasaki disease. Ped Clin North Am 2012; 59(2):425-445.
5D. Polyarteritis nodosa (PAN)
Necrotizing vasculitis in medium or small size muscular arteries
Very rare in childhood
Corticosteroids (see text)

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 48
2008 EULAR/PRINTO/PRES Classification Criteria for Childhood PAN *
Systemic illness characterized by:
Histological findings of necrotizing vasculitis in medium or small sized muscular arteries,
or
Angiography showing aneurysm, stenosis or occlusion of medium or small sized arteries
Plus ≥1/5 of the following:
Skin involvement (livedo reticularis, tender subcutaneous nodules, superficial skin
infarctions, or deep skin infarctions)
Myalgia or muscle tenderness
Hypertension (>95
th
percentile for height)
Peripheral neuropathy (motor mononeuritis multiplex, sensory peripheral neuropathy)
Renal involvement (proteinuria >0.3 g in 24 hrs, hematuria, red blood cell casts, impaired
renal function)
* Classification criteria are designed to identify a homogeneous population for research studies
and are not diagnostic criteria, although they are often used for diagnosis in practice
Systemic PAN
Additional clinical features
o Constitutional symptoms, including prolonged fever
o Testicular pain or tenderness
o Stroke or coronary artery disease
o Bruits
o Ischemic abdominal pain
Laboratory features
o Leukocytosis, thrombocytosis, and elevation of ESR and CRP
o Positive hepatitis B serology can occur although it is unusual
Treatment
o Prednisone plus second line agent (e.g. Methotrexate, Cyclophosphamide, Azathioprine,
Mycophenolate mofetil)
o Plasma exchange may be considered in acute life-threatening disease
Cutaneous PAN
Clinical syndrome characterized by absence of major organ involvement, but may involve
skin, joints, muscles and peripheral nervous system
Skin findings (tender subcutaneous nodules, livedo reticularis, superficial or deep ulcers)
Additional clinical features
o Constitutional features
o Myalgia, arthralgia, non-erosive arthritis
o Peripheral neuropathy

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 49
Investigations
o Diagnosis requires deep skin biopsy to get small muscular arteries showing necrotizing,
non-granulomatous vasculitis
o Negative testing for ANCA
o May be associated with serological (ASOT) or culture evidence of Streptococcal
infection
Treatment
o Corticosteroids with rapid wean or another second line agent (e.g. IVIG, Methotrexate)
o Penicillin treatment (if proven associated Streptococcal infection) and prophylaxis
Deficiency of Adenosine Deaminase 2 (DADA2)
Consider in differential diagnosis of polyarteritis nodosa
Recently identified monogenic autoimmune recessive disease (mutations in ADA2 gene)
leading to vasculitis
Clinical features are variable, depending on mutation type and number of affected alleles
Common features:
o Recurrent lacunar stroke (ischemic or hemorrhagic) with onset at young age
o Fever
o Vasculitis, including polyarteritis nodosa
o Livedo reticularis
o Hepatosplenomegaly
o Various ophthalmologic manifestations
Investigations
o Elevated inflammatory markers
o Diagnosis confirmed through genetic testing
o MRI brain to assess for stroke
Treatment
o TNF-inhibitors have been successful in suppressing fever, vasculopathy and strokes
o Hematopoetic stem cell transplantation may offer definitive treatment
o Future therapies may include recombinant ADA2 protein or gene therapy
References:
1. Ozen S, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood
polyarteritis nodosa, childhood Wegener’s granulomatosis and childhood Takayasu
arteritis: Ankara 2008. Ann Rheum Dis 2010; 69(5):798-806.
2. Beckum KM, et al. Polyarteritis nodosa in childhood: recognition of early dermatologic
signs may prevent morbidity. Pediatr Dermatol 2014; 31(1):e6-9.
3. Falcini F, et al. Clinical overview and outcome in a cohort of children with polyarteritis
nodosa. Clin Exp Rheumatol 2014; 32(3 Suppl 82):S134-7.
4. Ombrello AK, et al. Treatment strategies for deficiency of adenosine deaminase 2. N
Engl J Med. 2019; 380(16):1582-84.
5E. IgA Vasculitis (also known as Henoch-Schönlein Purpura, or HSP)
Most common vasculitis in children

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 50
Often follows a respiratory infection, most commonly Group A Streptococcus
Predominantly small vessel vasculitis, characterized by IgA deposition and leukocytoclastic
vasculitis
2008 EULAR/PRINTO/PRES Classification Criteria for Childhood HSP *
Purpura (commonly palpable and in crops) or petechiae with lower limb predominance **
Plus ≥1/4 of the following:
Diffuse abdominal colicky pain with acute onset (may include intussusceptions and
gastrointestinal bleeding)
Skin biopsy showing leukocytoclastic vasculitis with predominant IgA deposits, or kidney
biopsy showing proliferative glomerulonephritis with predominant IgA deposits
Arthritis or arthralgias of acute onset
Renal involvement (proteinuria >0.3 g in 24 hrs, hematuria, or red blood cell casts,
impaired renal function)
* Classification criteria are designed to identify a homogeneous population for research
studies and are not diagnostic criteria, although they are often used for diagnosis in practice
** If purpura in atypical distribution, demonstration of IgA deposition is required
Clinical features
o Cutaneous purpura (100% of patients) with palpable lesions 2-10 mm in diameter,
usually concentrated on lower extremities
o Arthritis (75%) usually affecting knees and ankles, associated with painful oedema
o GI involvement (50-75%), including abdominal pain and intussusception
o Renal involvement (40-50%)
Most commonly microscopic hematuria
Proteinuria accompanies hematuria in 25%
Nephrotic syndrome in 5%
Renal abnormalities may not manifest initially, thus must regularly monitor blood
pressure and urinalysis x 6 mos after acute illness
o Orchitis (10-20% of males) associated with pain and swelling
Investigations
o No distinctive or diagnostic laboratory abnormalities
o May have elevated WBC, platelets, ESR and/or CRP
o Coagulation profile must be normal and thrombocytopenia should be absent
o Serum IgA increased in 50% of patients
o Biopsy of skin or kidneys may be needed (see classification criteria above)
Treatment
o Largely supportive
o NSAIDs may be used for joint pain (caution required due to potential renal involvement)
o Prednisone in select patients
May decrease severity and duration of GI symptoms and may help bullous lesions
Unclear impact on risk of persistent renal disease (controversial)
No definite benefit for prevention of HSP recurrence

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 51
o If severe nephritis (e.g. nephrotic syndrome, decreased renal function, crescentic
nephritis): pulse IV Methylprednisolone ± second line agent (e.g. Azathioprine,
Mycophenolate mofetil, Cyclophosphamide)
Prognosis
o Usually a self-limited condition that resolves within 4 weeks (average)
o Recurrence occurs in about 1/3 of patients
o Long-term prognosis depends on severity of nephritis (poorer prognosis with nephrotic
syndrome or if >50% crescent formation on biopsy)
o End-stage renal disease occurs in 1-3% of patients; in ~20% of those with nephritic or
nephrotic syndrome (N.B. % varies among different studies)
References:
1. Gonzalez-Gay MA, et al. IgA vasculitis: Genetics and clinical and therapeutic
management. Curr Rheumatol Rep 2018; 20(5):24.
2. Ozen S, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood
polyarteritis nodosa, childhood Wegener’s granulomatosis and childhood Takayasu
arteritis: Ankara 2008. Ann Rheum Dis 2010; 69(5):798-806.
3. Hahn D, et al. Interventions for preventing and treating kidney disease in Henoch-
Schönlein Pupura (HSP). Cochrane Database of Systematic Reviews 2015;
8:CD005128.
4. Weiss PK, et al. Corticosteroids may improve clinical outcomes during hospitalization for
Henoch-Schönlein purpura. Pediatrics 2010; 126(4):674-81.
5F. Granulomatosis with Polyangiitis (GPA, formerly Wegener Granulomatosis)
Predominantly small vessel vasculitis, characterized by granulomatous inflammation
Generally occurs in the second decade of life, with a female preponderance
Hallmark of GPA is triad of upper and lower respiratory tract inflammation and renal disease
2008 EULAR/PRINTO/PRES Classification Criteria for Childhood GPA *
At least 3 of the 6 following criteria:
Histopathology showing granulomatous inflammation within wall of artery or in
perivascular or extravascular area
Upper airway involvement (chronic purulent or bloody nasal discharge, recurrent
epistaxis, nasal septum perforation, saddle nose deformity, chronic or recurrent sinus
inflammation)
Laryngo-tracheo-bronchial involvement (subglottic, tracheal or bronchial stenoses)
Pulmonary involvement (nodules, cavities, or fixed pulmonary infiltrates)
ANCA positive by immunofluorescence or ELISA
Renal involvement (proteinuria >0.3 g in 24 hrs, hematuria, or red blood cell casts,
impaired renal function)
* Classification criteria are designed to identify a homogeneous population for research studies
and are not diagnostic criteria, although they are often used for diagnosis in practice

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 52
Clinical features (in order of decreasing frequency)
o Constitutional: fatigue, malaise, fever, weight loss
o Pulmonary: dyspnea, chronic cough, hemoptysis/alveolar hemorrhage, lung
nodules/cavitations/fixed infiltrates
o Ear, nose and throat: nasal involvement (epistaxis, ulcers), sinusitis, otitis/mastoiditis,
hearing loss, subglottic involvement
o Renal: abnormal urinalysis, biopsy-proven glomerulonephritis, elevated creatinine, acute
renal failure
o Musculoskeletal: arthralgia/myalgia, arthritis
o Gastrointestinal: nonspecific abdominal pain, chronic nausea
o Eye: nonspecific red eye, conjunctivitis, scleritis
o Cutaneous: palpable purpura/petechiae
o Neurological: severe headache, dizziness
Investigations
o ANCA positive in ~90% of patients (~80% are c-ANCA positive with anti-PR3 positivity)
Treatment
o Initial therapy involves combination of Corticosteroids and a second-line agent, such as
Cyclophosphamide, Rituximab or Methotrexate (choice depends on disease severity)
o Plasma exchange may be used as part of induction therapy for children with life-
threatening disease
o Maintenance therapy with Methotrexate, Azathioprine, Rituximab, plus tapering doses of
Corticosteroids
o May consider endoscopic intervention for subglottic stenosis and endobronchial disease
Prognosis
o Significant morbidity associated with disease and medications
o Rare complications include mechanical ventilation and dialysis in around 10% of patients
References:
1. Jariwala MP, Laxer RM. Primary vasculitis in childhood: GPA and MPA in childhood. Front
Pediatr 2018; 16(6):226.
2. Ozen S, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood
polyarteritis nodosa, childhood Wegener’s granulomatosis and childhood Takayasu
arteritis: Ankara 2008. Ann Rheum Dis 2010; 69(5):798-806.
3. Villa-Forte A. EULAR /European vasculitis study group recommendations for the
management of vasculitis. Curr Opin Rheumatol 2010; 22:49-53.
4. McGeoch L, et al. CanVasc recommendations for the management of antineutrophil
cytoplasm antibody (ANCA)-associated vasculitides. Can J Kidney Health Dis 2015;
2(43):1-6.
5G. Microscopic Polyangiitis (MPA)
Pauci-immune, necrotizing, non-granulomatous small vessel vasculitis
Rare in childhood
No classification criteria have been developed

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 53
Clinical features
o Rapidly progressive, necrotizing, crescentic glomerulonephritis (90% of patients)
o Pulmonary capillaritis leading to hemorrhage (30-60%)
o Pulmonary-renal syndrome (30-50%)
o Hypertension (50-60%)
o Palpable purpura (common)
o May have refractory anemia
Diagnosis
o Serology: 50-75% p-ANCA positive with anti-MPO on ELISA
o Renal biopsy with immunofluorescence: pauci-immune glomerulonephritis
Treatment
o Induction: Corticosteroids + Cyclophosphamide, Methotrexate or Rituximab
o Maintenance: Azathioprine, Methotrexate, or Rituximab
References:
1. Jariwala MP, Laxer RM. Primary vasculitis in childhood: GPA and MPA in childhood. Front
Pediatr 2018; 16(6):226.
2. Cabral D, et al. Comparing presenting clinical features in 48 children with microscopic
polyangiitis to 183 children who have granulomatosis with polyangiitis (Wegener's): An
ARChiVe Cohort Study. Arthritis Rheumatol 2016; 68(10):2514-26.
3. Sun L, et al. Clinical and pathological features of microscopic polyangiitis in 20 children.
J Rheumatol 2014; 41(8):1712-9.
5H. Eosinophilic Granulomatosis with Polyangiitis (formerly Churg-Strauss Syndrome)
Granulomatous small vessel vasculitis
No EULAR/PRINTO/PRES classification criteria – very rare in children
Characterized by:
o Preceding history of “difficult to control” chronic asthma
o Paranasal sinus abnormalities
o Peripheral eosinophilia (≥10%) + eosinophilic infiltration on biopsy
o Non-fixed pulmonary infiltrates
o Peripheral neuropathy
Additional clinical features
o Cardiovascular (50%): myocardial ischemia, pericarditis, cardiac failure
o Ischemic abdominal pain
o Cutaneous nodules
Diagnosis
o Biopsy (lung, skin) showing eosinophilic infiltrates and granulomas
o Peripheral eosinophilia and increased IgE levels
o ANCA, usually anti-MPO, present in less than 50% patients
Treatment
o Prednisone plus second line agent
o Cyclophosphamide or Rituximab if cardiac, GI or neurologic involvement

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 54
References;
1. Gendelman S, et al. Childhood-onset eosinophilic granulomatosis with polyangiitis
(formerly Churg-Strauss syndrome) : a contemporary single-center cohort. J Rheumatol
2013; 40(6):929-35.
2. Boyer D, et al. Churg-Strauss syndrome in children: A clinical and pathologic review.
Pediatrics 2006; 118:e914-20.
5I. Behçet Disease
Systemic vasculitis with characteristic oral and genital ulcers, vasculopathy and uveitis
Among the systemic vasculitides, Behçet disease is remarkable for its ability to involve
arteries and veins of all sizes (small, medium, large)
More common in certain ethnic groups along the “Silk Road” (Turks, Greeks)
Uncommon in children
1990 International Study Group for Behçet Disease Criteria for Diagnosis
Recurrent oral ulcers (major or minor aphthous ulcers, or herpetiform ulceration recurring
at least 3 times in 12 months)
Plus ≥ 2 of the following criteria:
Recurrent genital ulcers (aphthous ulceration or scarring)
Eye lesions (including anterior or posterior uveitis, cells in vitreous on slit lamp
examination, or retinal vasculitis, observed by an ophthalmologist)
Skin lesions (including erythema nodosum, pseudofolliculitis, papulopustular lesions, or
acneiform nodules consistent with Behçet)
Pathergy (skin papule 2 mm or more in size developing 24 to 48 hours after oblique
insertion of a 20-25 gauge needle 5 mm into the skin, generally of the forearm)
2014 International Classification Criteria for Behçet Disease *
Ocular lesions (anterior or posterior uveitis or retinal vasculitis)
Genital aphthosis
Oral aphthosis
Skin lesions (erythema nodosus, pseudofolliculitis, skin aphthosis)
Neurologic manifestations (peripheral and central)
Vascular manifestations (arterial and/or venous thrombosis, phlebitis)
Positive pathergy test
2 points
2 points
2 points
1 point
1 point
1 point
Bonus point
* Patients with total score ≥4 are classified as having Behçet disease
N.B. Classification criteria are designed to identify a homogeneous population for research
studies and are not diagnostic criteria, although they are often used for diagnosis in practice
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 55
Other clinical features include:
o CNS: aseptic meningitis, encephalitis, cerebral venous sinus thrombosis, or
pseudotumour cerebri
o MSK: oligoarthritis or polyarthritis
o GI: abdominal pain, diarrhea, colitis
o Vascular: arterial and/or venous thrombosis
Diagnosis
o Currently made clinically
o Several sets of diagnostic criteria have been proposed (see above)
o No pathognomonic clinical finding or laboratory test to provide definitive diagnosis
o Genetics may be helpful – HLA-B51 (more prevalent in Mediterranean and Far East) and
HLA-B51 (more prevalent in East Asian)
Treatment
o No controlled studies have been performed on children
o Corticosteroids, Colchicine, Thalidomide, and Anti-TNF agents (e.g. Infliximab) have
been shown to be helpful
o May treat isolated oral and/or genital ulcers with topical therapy, including analgesics
and/or steroids
o Apremilast (an oral phosphodiesterase 4 inhibitor) has recently been demonstrated to be
effective for treatment of oral ulcers in Behçet disease
References:
1. Demir S, et al. Vasculitis in systemic autoinflammatory diseases. Front Pediatr 2018;
6:377.
2. Koné-Paut I. Behçet’s disease in children, an overview. Pediatr Rheumatol Online J
2016; 14(1):10.
3. Ozen S, Eroglu FK. Pediatric-onset Behçet disease. Curr Opin Rheumatol 2013;
25(5):636-42.
4. Hatemi G, et al. EULAR recommendations for the management of Behçet disease. Ann
Rheum Dis 2008; 67: 1656-1662.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 56
SECTION 6 – IDIOPATHIC INFLAMMATORY MYOPATHIES
6A. Juvenile Dermatomyositis (JDM)
JDM is an autoimmune myopathy characterized by capillary vasculopathy primarily affecting
skin and muscle
Not associated with cancer in children, unlike dermatomyositis in adults
Bohan and Peter Criteria for Diagnosis of Juvenile Dermatomyositis
Symmetrical proximal muscle weakness
Characteristic skin changes, including Gottron papules on the dorsal surface of the
knuckles and heliotrope rash over the eyelids
Elevated muscle enzymes, including CK, AST, LDH, aldolase
Abnormal EMG demonstrating denervation and myopathy
Abnormal muscle biopsy demonstrating necrosis and inflammation
Recently, MRI has become an important diagnostic tool to look for muscle inflammation and
to direct a site for biopsy (if needed)
Please see Section 2E for other diagnoses to consider when assessing a child with muscle
weakness
Clinical features
o Proximal muscle weakness (which is present in 95% of patients) may be described on
history as difficulty getting up from sitting or lying, difficulty climbing stairs, and frequent
falls; also, children may demonstrate a Gower sign on physical exam (see
https://www.youtube.com/watch?v=IpoT46EAuCU)
o It is important to assess for 3D’s – dysphagia, dysphonia and dyspnea – that indicate
severe disease
o Nasal voice, difficulty swallowing and choking on foods (18-44%) may indicate weakness
of the palate and cricopharyngeal muscles
o Characteristic skin rashes include:
Gottron papules (57-100%, but may be confused with psoriasis given location on
extensor surfaces, see https://www.nejm.org/doi/full/10.1056/NEJMicm1002816)
Heliotrope rash (66-100%)
Malar rash (42-73%)
Photosensitive rashes
Skin ulceration in severe cases.
o Capillary vasculopathy can be seen using capillaroscopy to look at changes in the nail
fold capillaries (91%) such as tortuosity, dilatation, and dropout
o Other organ systems may also be involved:
Arthritis (23-58%)
GI tract symptoms (22-37%), including dysphagia, GI ulceration, perforation
Lungs (interstitial lung disease)
Heart (cardiomyopathy) – very rare
o Constitutional features, such as fever and fatigue, are common
o Anasarca can be a rare initial manifestation and is associated with treatment resistance
and poor prognosis
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 57
o Amyopathic JDM (skin features without muscle involvement) is rare in children and may
represent JDM with mild muscle involvement that has not yet been identified; however,
treatment to prevent future complications (e.g. calcinosis) is recommended
Investigations
o Muscle enzymes (CK, AST, ALT, LDH, aldolase) and inflammatory markers (ESR, CRP)
likely to be elevated
o Positive ANA is common (up to 70% of patients) but not specific
o Myositis-specific antibodies (MSA) are identified in up to 2/3 of children with JDM, but
are not routinely available in all laboratories
MSA
Frequency
Associated Clinical Characteristics
Anti-p155/p140
60%
Rash (Gottron papules, malar rash, “shawl-sign” rash)
Photosensitivity
Low CK levels
Chronic illness course
Generalized lipodystrophy
Anti-MJ
20%
Muscles cramps
Dysphonia
High rate of hospitalization
Monocyclic disease course
Anti-synthetase
5-10%
Interstitial lung disease
“Mechanic’s hands”
Arthralgia
Older age at diganosis
Anti-Mi2
5%
Hispanic ethnicity
Rash (Gottron papules, heliotrope rash, malar rash)
High CK
Low mortality
Anti-SRP
**In patients with
polymyositis
25%
Black race
Severe onset
Distal weakness
Raynaud phenomenon
Cardiac involvement
High CK levels
Chronic disease course, may require wheelchair use
Anti-MDA5
7.4%
Japanese and East Asian patients
Skin ulceration
Milder muscle disease
Arthritis
May have progressive interstitial lung disease
Complications
o Long delays in diagnosis or insufficiently aggressive treatment may put patients at higher
risk for complications and poor outcome
o Muscle weakness and pain can lead to joint contractures

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 58
o Soft tissue calcification, or calcinosis, can develop within a few years of diagnosis or
may be seen at presentation of longstanding disease (see
https://www.dermnetnz.org/topics/calcinosis-cutis/)
o Lipoatrophy may occur accompanied by hyperinsulinism, hypertriglyceridemia, liver
dysfunction, acanthosis nigricans, and type 2 diabetes
o Medication-related side effects from Corticosteroid toxicity (see Section 14) can include
infection, osteoporosis, growth delay, cataracts and glaucoma, type 2 diabetes, and
hypertension
Monitoring disease activity
o Clinical: skin rash; periungual capillaroscopy; muscle strength and function as measured
by the Childhood Myositis Activity Scale (CMAS)
o Laboratory: muscle enzymes (CK, AST, ALT, LDH, aldolase), inflammatory markers
(ESR), lipid abnormalities & organ involvement
Treatment
o Supportive: adequate nutrition, physiotherapy, exercise, sunscreen for photosensitive
rash
o Medications:
Induction therapy using Corticosteroids starting from 1-2 mg/kg/day with slow taper
and subcutaneous Methotrexate
Cyclophosphamide may be used for interstitial lung disease, gastrointestinal disease
and vasculitis
IVIG, Cyclosporine, Mycophenolate mofetil or Rituximab if resistant or refractory
Topical therapies may also be considered for resistant skin disease
Course and Outcomes
o 40-60% of patients have a chronic course, 40-60% have a monophasic course, and <5%
have a polyphasic course
o Ongoing rash and nail fold abnormalities in first 6 months are best predictors of longer
time to remission
o Persistent skin and nail fold changes may represent ongoing inflammatory disease and
should be treated accordingly
o Outcomes are favourable, since most children have no functional disability and <10%
have moderate-to-severe disability
References:
1. Huber AM. Juvenile idiopathic inflammatory myopathies. Pediatr Clin North Am 2018;
65(4):739-56.
2. Huber A, Feldman BM. An update on inflammatory myositis in children. Curr Opin
Rheumatol 2013; 25(5):630-5.
3. Rider LG, et al. Developments in the classification and treatment of the juvenile
idiopathic inflammatory myopathies. Rheum Dis Clin North Am 2013; 39(4):10.
4. Rider LG, et al. The myositis autoantibody phenotypes of the juvenile idiopathic
inflammatory myopathies. Medicine (Baltimore) 2013; 92(4):223.
5. Huber AM, et al. Validation and clinical significance of the Childhood Myositis
Assessment Scale for assessment of muscle function in the juvenile idiopathic
inflammatory myopathies. Arthritis Rheum 2004; 50(5):1595-603.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 59
6B. Juvenile Polymyositis
Uncommon in children
Characterized by proximal and distal muscle weakness
No associated skin findings and normal nail fold capillaries
Myositis is typically more severe than in juvenile dermatomyositis or in other connective
tissue diseases
Resistant to treatment
Anti-signal recognition particle (SRP) autoantibodies are seen in children with polymyositis
and are associated with black race, severe onset, distal weakness, Raynaud phenomenon,
cardiac involvement, high CK levels, chronic disease course and wheelchair use
References:
1. Huber AM. Juvenile idiopathic inflammatory myopathies. Pediatr Clin North Am 2018;
65(4):739-56.
2. Huber A, Feldman BM. An update on inflammatory myositis in children. Curr Opin
Rheumatol 2013; 25(5):630-5.
3. Rider LG, et al. The myositis autoantibody phenotypes of the juvenile idiopathic
inflammatory myopathies. Medicine (Baltimore) 2013; 92(4):223.
6C. Myositis in other connective tissue diseases
Myositis may be present in other connective tissue diseases, such as systemic lupus
erythematosus, systemic sclerosis, mixed connective tissue diseases and overlap
syndromes
Please see Section 2E for other diagnoses to consider when assessing a child with muscle
weakness
Typically accompanied by other features of the various connective tissue diseases, such as
arthralgia, malar rash, Raynaud phenomenon, interstitial lung disease
Laboratory findings include high titres of ANA and myositis-associated antibodies
o Anti-PM-Scl and anti-Ku associated with scleroderma-myositis overlap syndrome
o Anti-U1-RNP associated with mixed connective tissue disease and overlap
syndromes
Associated with higher mortality than other categories of myositis
References:
1. Huber AM. Juvenile idiopathic inflammatory myopathies. Pediatr Clin North Am 2018;
65(4):739-56.
2. Shah M, et al. The clinical phenotypes of the juvenile idiopathic inflammatory
myopathies. Medicine 2013; 92(1):25-41.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 60
SECTION 7 – SCLERODERMA & RELATED SYNDROMES
7A. Classification of Scleroderma and Scleroderma-like Disorders
Morphea/ Localized scleroderma
(See Section 7B)
Circumscribed morphea
Linear scleroderma
Generalized morphea
Pansclerotic morphea
Mixed morphea
Systemic sclerosis
(See Section 7C)
Diffuse cutaneous *
Limited cutaneous **
Overlap syndromes
Scleroderma-like disorders
Graft versus host disease
Drug or toxin induced (e.g. L-tryptophan,
vinyl chloride, bleomycin)
Diabetic cheiroarthropathy
Phenylketonuria
Eosinophilia-myalgia syndrome
Eosinophilic fasciitis
Nephrogenic systemic fibrosis
Premature aging syndromes
*Diffuse cutaneous systemic sclerosis characterized by skin sclerosis extending proximal to
wrists and ankles and involving trunk and face; associated with internal organ involvement and
earlier organ dysfunction
**
Limited cutaneous systemic sclerosis (formerly known as CREST syndrome – calcinosis,
Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) progresses
more slowly, but has higher risk for later development of pulmonary hypertension
7B. Localized Scleroderma or Morphea
Morphea refers to a group of autoimmune disorders with sclerotic skin and subdermal
connective tissue changes due to excessive accumulation of collagen
25% of children can have extracutaneous manifestations: arthritis, uveitis, neurologic
findings (e.g. seizures, headache), Raynaud phenomenon
Circumscribed morphea
o Includes superficial lesions sometimes referred to as “plaque” morphea
o May involve superficial and deep dermis as well as subcutaneous tissues
o Early lesions are firm, ivory-coloured oval lesions surrounded by reddish-lilac coloured
ring suggesting active inflammation
o Later, there is atrophy, hyper- ( or rarely hypo-) pigmentation and softening of lesions
o More common on trunk than extremities

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 61
Generalized morphea
o When ≥4 individual circumscribed lesions become confluent and affect ≥2 anatomic sites
o Often rapid onset over months
Linear scleroderma
o Most common form in children and adolescents
o Characterized by ≥ 1 linear streaks (often following dermatomal distribution) extending
over face, head, trunk and/or extremities
o Unilateral in greater than 85% cases
o Complications include joint flexion contractures, limb atrophy, leg length discrepancy
o Facial Linear Variants: may be associated with intracranial lesions, seizures, uveitis, and
dental abnormalities.
En coup de sabre: involves face or scalp, usually forehead; often with alopecia
Parry-Romberg syndrome: progressive hemi-facial atrophy; often involves face
below the forehead; more disfiguring; no epidermal involvement
Pansclerotic morphea
o Least common subtype, but most disabling
o Circumferential changes (often affecting a limb) that extend into tissues below dermis
including muscle, tendon and bone; frequently spares the fingers and toes
Mixed morphea
o Morphea of ≥ 2 subtypes in an individual patient
Diagnosis
o Clinical, although skin biopsy may be performed (to exclude other disorders or to assess
if lesion actively inflamed or “burnt out”)
o MRI may be useful to determine extent of deep lesions
Treatment
o Topical: Corticosteroids, Calcipotriene (vitamin D), Imiquimod 5%, Tacrolimus
o Systemic: Corticosteroids, Methotrexate, Mycophenolate mofetil, Cyclosporine, Biologic
therapy may be considered in recalcitrant disease
o Other: Phototherapy with Ultraviolet A rays
o Supportive: physiotherapy, occupational therapy, psychosocial support
o Surgery for facial lesions, tendo-achilles lengthening
References
1. Li SC. Scleroderma in children and adolescents. Ped Clin North Am 2018:757-781.
2. Zulian F. Scleroderma in children. Best Pract Res Clin Rheumatol 2017; 31:576-595.
7C. Systemic Sclerosis (SSc)
Rare autoimmune disease in children, characterized by symmetrical skin thickening or
hardening and fibrosis of internal organs
Sexes equally affected until onset age of 8 years, followed by 3:1 ratio of females:males
90% of pediatric patients have diffuse cutaneous subtype and 10% have limited cutaneous
subtype (formerly known as CREST syndrome – calcinosis, Raynaud phenomenon,
esophageal dysmotility, sclerodactyly, and telangiectasia)

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 62
2013 ACR/EULAR Classification Criteria for SSc *
Items
Score
Skin thickening **
Skin thickening of fingers of both hands extending proximal to
metacarpophalangeal (MCP) joints
9
Skin thickening of whole finger distal to MCP joints
4
Puffy fingers
2
Finger tip lesions **
Digital tip ulcers
2
Pitting scars in fingertips
3
Telengiectasia
2
Abnormal nailfold capillaries (enlarged capillaries and/or capillary loss with
or without peri-capillary hemorrhages)
2
Pulmonary arterial hypertension and/or interstitial lung disease
2
Raynaud phenomenon
3
Scleroderma related antibodies (any of anti-centromere, anti-Scl70 (also
known as anti-topoisomerase I), or anti-RNA polymerase III)
3
* Patients with total score of ≥ 9 are classified as having definite systemic sclerosis
N.B. Classification criteria are designed to identify a homogeneous population for research
studies and are not diagnostic criteria, although they are often used for diagnosis in practice
** Only include highest score from these categories in calculation of total score
2007 PRES/ACR/EULAR Provisional Classification Criteria for Juvenile SSc *
Major criterion (mandatory): Sclerosis/induration of skin proximal to MCP joints
Plus ≥ 2 of the following minor criteria:
Cutaneous (sclerodactyly)
Peripheral vascular (Raynaud phenomenon, nail fold capillary abnormalities, digital tip
ulcers)
Gastrointestinal (dysphagia, gastroesophageal reflux)
Cardiac (arrhythmias, heart failure)
Renal (renal crisis, new-onset arterial hypertension)
Respiratory (pulmonary fibrosis, decreased DLCO, pulmonary arterial hypertension)
Neurologic (neuropathy, carpal tunnel syndrome)
Musculoskeletal (tendon friction rubs, arthritis, myositis)
Serologic (antinuclear antibodies, SSc-selective autoantibodies including anti-
centromere and anti-Scl70 (also known as anti-topoisomerase I))
* Classification criteria are designed to identify a homogeneous population for research studies
and are not diagnostic criteria, although they are often used for diagnosis in practice
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 63
Common clinical features of SSc:
Raynaud
Phenomenon
Common in children with SSc
Associated with abnormal nail fold capillaries
Can lead to digital pitting and gangrene
Dermatologic
Non-pitting edema and/or induration of skin resulting in restricted range
of motion, usually in fingers; later evolves to skin thickening causing
joint contractures (sclerodactyly)
Calcium deposits under the skin, often develop over bridge of nose and
extensor surfaces
Telangiectasias
Abnormal nail fold capillaries
Musculoskeletal
Arthralgias
Polyarthritis with minimal joint effusion
Joint contractures often secondary to skin changes
Subclinical myositis with mild weakness and slight elevation in muscle
enzymes
Gastrointestinal
Major cause of morbidity
Severe gastroesophageal reflux disease (GERD) due to dysfunction of
lower esophageal sphincter
Dysmotility leads to stasis, bacterial overgrowth and malabsorption with
diarrhea; may also result in severe constipation and megacolon
Respiratory
Major cause of mortality
Pulmonary arterial hypertension (most severe)
Interstitial lung disease (most common, usually bibasilar)
Inflammatory alveolitis (precedes fibrosis)
Cardiac
Pericarditis (small pericardial effusions are very common)
Micro-infarction of cardiac vasculature leads later to cardiomyopathy
Arrhythmias (from fibrosis of conducting system)
Renal
Major cause of morbidity prior to development of ACE inhibitors
Renal vasculopathy leads to renal hypertension (may be life-
threatening)
Proteinuria (may precede hypertension)
Glomerular disease is unusual
Neurologic
Rare (e.g. trigeminal neuropathy, carpal tunnel syndrome)
Investigations
o Blood work to assess for evidence of systemic inflammation and organ involvement
o Serology helpful for diagnosis and classification: ANA (common), Rheumatoid factor
(rare), anti-Scl 70 (also known as anti-topoisomerase1, usually associated with diffuse
cutaneous SSc), anti-centromere (usually associated with limited cutaneous SSc)
o Blood pressure and urinalysis to evaluate renal involvement
o ECG and echocardiogram to evaluate possible cardiac involvement and screen for
pulmonary arterial hypertension; consider cardiac MRI
o Chest X-ray, pulmonary function tests with DLCO and high resolution CT chest to
assess for lung disease, especially alveolitis and interstitial pulmonary fibrosis
o Upper GI series to look for dysmotility and GERD

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 64
Treatment
o Primarily supportive care
Avoid cold, stress, caffeine and nicotine (to prevent Raynaud phenomenon)
Eat small meals, avoid foods that exacerbate gastric acidity, remain upright after
eating and elevate head of bed (for dysmotility and GERD)
Physiotherapy and occupational therapy
o Symptomatic treatment
GERD: Proton pump inhibitors (e.g. Omeprazole)
Raynaud phenomenon: peripheral vasodilators (e.g. Nifedipine)
Hypertension, renal disease: ACE Inhibitors (e.g. Enalapril)
Pulmonary hypertension: endothelin-1 receptor antagonists (e.g. Bosentan),
prostacyclin analogs (Epoprostenol)
o Systemic therapy
Methotrexate or Mycophenolate mofetil (MMF) for active skin disease
Cyclophosphamide, MMF and Corticosteroids for alveolitis and interstitial lung
disease
Other immunomodulatory agents have unclear efficacy in treatment of SSc
Autologous stem cell transplantation has been demonstrated to be efficacious and
best cadidates appear to be patients with severe disease of short duration before
irreversible damage has occurred
Prognosis and outcome
o Prognosis depends on degree of organ dysfunction, which either later stabilizes or
progresses to significant morbidity and mortality
o Survival much better in children (5 year survival approximately 90%) compared to adults
References
1. Li SC. Scleroderma in children and adolescents. Ped Clin North Am 2018:757-781.
2. Zulian F. Scleroderma in children. Best Pract Res Clin Rheumatol 2017; 31:576-595.
3. Zulian F, et al. The Pediatric Rheumatology European Society/American College of
Rheumatology/European League against Rheumatism provisional classification criteria
for juvenile systemic sclerosis. Arthritis Rheum 2007; 57(2):203-12.
7D. Mixed Connective Tissue Disease (MCTD)
Autoimmune disorder characterized by several clinical and laboratory features:
o High titre anti-U1 RNP antibodies
o Swollen hands
o Raynaud phenomenon
o Arthritis
o Myositis
o Skin rashes (may include malar rash, Gottron-like papules, sclerosis)
Children may also develop over time GI manifestations (similar to SSc), interstitial lung and
renal diseases
Multiple different diagnostic criteria for MCTD exist (e.g. Sharp, Alarcon-Segovia, Kasukawa,
Kahn), but no single set of criteria is validated in children
Investigations should be directed to assess for multi-organ involvement
Treatment depends on severity of clinical manifestations and organ involvement

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 65
Reference
1. Berard RA, Laxer RM. Pediatric mixed connective tissue disease. Curr Rheumatol Rep
2016; 18(5):28.
2. Mier RJ, et al. Pediatric-onset mixed connective tissue disease. Rheum Dis Clin North
Am 2005; 31(3):483-96.
7E. Raynaud Phenomenon
Vascular spasm in extremities leading to triphasic colour sequence: white (blanching due to
ischemia), blue (cyanosis, related to desaturation), then red (erythema due to reperfusion)
Well-demarcated areas of colour change
Usually affects fingers and toes, but may also involve other areas (lips, tongue, tip of nose,
earlobes)
Precipitated by cold, physical or emotional stress, caffeine, medications or smoking
Raynaud phenomenon may be primary or secondary
o Primary
No underlying etiology, but often positive family history
No peripheral ulcerations
o Secondary
Due to underlying autoimmune disease (scleroderma, overlap syndromes, MCTD,
SLE, JDM), mechanical obstruction (thoracic outlet syndrome, cervical rib),
hyperviscosity (polycythemia), cryoglobulinemia, drugs/toxins, or vibration-induced
phenomenon
In isolated Raynaud phenomenon, two best predictive factors for future development of
autoimmune diseases are:
1. Positive ANA
2. Abnormal nail fold capillaries
Investigations
o Blood work – complete blood count and differential, inflammatory markers, complement
levels, serology (ANA, specific autoantibodies, RF)
o Urinalysis
Treatment
o Preventive (avoid triggers; warm mittens, socks and boots in winter etc)
o Systemic therapy may be used to prevent ischemic tissue injury
Peripheral vasodilator, such as Nifedipine, may be titrated to alleviate the Raynaud
episodes; avoid medication-related hypotension, headaches or dizziness
If severe, may require IV prostaglandins
o Topical therapy (e.g. nitroglycerin 2% ointment) may be used for digital ulcers
References
1. Nigrovic PA, et al. Raynaud’s phenomenon in children: a retrospective review of 123
patients. Pediatrics 2003; 111(4):715-721.
2. Gargh K, et al. A retrospective clinical analysis of pharmacological modalities used for
symptomatic relief of Raynaud’s phenomenon in children treated in a UK paediatric
rheumatology centre. Rheumatology 2010; 49(1):193-4.
3. Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Engl J Med 2016; 375(6):556-65.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 66
7F. Sjögren Syndrome
Multisystem autoimmune disease characterized by decreased secretion of lacrimal and
salivary glands leading to dry eyes (keratoconjunctivitis sicca) and xerostomia (dry mouth)
Most common presentation in children is parotid swelling or parotitis
Clinical diagnosis, given lack of validated pediatric criteria
Diagnosis in adults based on weighted scoring system, including the following:
o Presence of focal lymphocytic sialadenitis in labial salivary gland biopsy (higher weight)
o Positivity for anti-SSA/Ro (higher weight)
o Ocular staining score (see below)
o Schirmer’s test (see below)
o Unstimulated whole saliva flow
Sjögren syndrome may be primary or secondary
o Primary (idiopathic) has no underlying etiology
o Secondary occurs in the context of an autoimmune disease, such as systemic lupus
erythematosus
Investigations
o Ocular: Schirmer’s test (tear production ≤ 5 mm in 5 minutes is abnormal), tear break-up
time, Rose Bengal staining of devitalized areas
o Salivary glands: scintigraphy, biopsy
o Blood work: complete blood count and differential, inflammatory markers,
immunoglobulin levels, serology (ANA, anti-Ro/SSA, anti-La/SSB, specific
autoantibodies, RF)
Treatment
o Supportive (artificial tears for dry eyes; increase fluid intake, chewing gum for dry mouth)
o Hydroxychloroquine (Plaquenil) may be helpful
Complications
o Increased risk of eye irritation and conjunctivitis
o Oral problems (dental caries, gingivitis, and infections such as Candida)
o Increased risk of non-Hodgkin lymphoma
References
1. Baszis K, et al. Recurrent parotitis as a presentation of primary pediatric Sjögren
syndrome. Pediatrics 2012; 129(1):e179-82.
2. Shiboski SC, et al. 2016 American College of Rheumatology/European League Against
Rheumatism classification criteria for primary Sjogren’s syndrome. Arthritis Rheumatol
2017; 69(1):35-45.
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 67
SECTION 8 – AUTOINFLAMMATORY DISEASES
8A. Periodic Fever/Autoinflammatory Syndromes
The recurrent or periodic fever syndromes are defined by ≥3 episodes of unexplained fever
in a 6-month period, occurring at least 7 days apart, separated by at least one week of good
health
Fevers are typically associated with a constellation of symptoms, including ocular,
oropharyngeal, gastrointestinal, dermatologic, musculoskeletal, and neurologic
manifestations
Interval between attacks of fever may be irregular or regular
Patients typically feel well between episodes
Characteristic Features of the Periodic Fever Syndromes
Features
FMF
TRAPS
HIDS
CAPS
PFAPA
FCAS
MWS
NOMID
Age of
onset
< 20 yrs
< 20 yrs
< 1 yr
< 1 yr
Often < 1yr
At birth or
within first
months
< 5 yrs
Duration of
attack
1-3 days
1-4 weeks
3-7 days
1-3 days
1-3 days to
continuous
Hours or
continuous
3-6 days
Interval of
attacks
Weeks to
months
Weeks to
months
Weeks to
months
Variable;
cold-induced
Variable
Days
3-6 weeks
Skin rash
Erysipelas-
like in ~40%
Migratory
rash; may be
painful
Maculopapular
in 90%
Cold-induced;
urticarial
Urticarial
Urticarial
No
Adenopathy
No
Not typical
Common; may
be generalized
Not typical
Not typical
Not typical
Yes
Oral ulcers
No
No
May occur
No
No
No
Yes
Abdominal
pain
In ~95%;
pain,
peritonitis,
constipation
Common;
colicky
Often present;
can be severe
with diarrhea
May occur
May occur
May occur
May occur
MSK
Arthralgia;
oligoarthritis;
myalgia
Localized
myalgia;
arthralgia;
arthritis
Symmetric
oligoarthritis of
large joints;
arthralgia
Arthralgia
Arthralgia;
arthritis;
clubbing
Arthralgia;
osseous
overgrowth;
clubbing
Arthralgia
Serositis
Peritonitis;
pleuritis;
pericarditis
Pleuritis;
peritonitis
No
No
Pericarditis
(uncommon)
Not typical
No
Amyloidosis
Occurs in
60% if
untreated
Occurs in
~25% if
untreated
Uncommon,
<5-10%
May occur
Occurs in
~30% if
untreated
May occur
No
Other
Scrotal
swelling and
pain
Periorbital
edema;
conjunctivitis;
headache;
testicular
pain
Headache
Conjunctivitis
Conjunctivitis;
episcleritis;
sensorineural
hearing loss
Conjunctivitis;
episcleritis;
papilledema;
chronic
meningitis;
sensorineural
hearing loss
Inheritance
AR
AD
AR
AD
AD
AD / de novo
None
Mutation
Chromosome
Gene
Protein
16p13
MEFV
Pyrin
12p13
TNFRSF1A
TNF receptor
P55
12q24
MVK
Mevalonate
kinase
1q44
NLRP3
Cryopyrin
1q44
NLRP3
Cryopyrin
1q44
NLRP3
Cryopyrin
No gene
identified
AD: autosomal dominant, AR: autosomal recessive
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 68
Familial Mediterranean fever (FMF)
Most common hereditary autoinflammatory disease
Typically autosomal recessive inheritance but occasionally autosomal dominant
transmission; linked to genetic mutation in MEFV gene encoding pyrin
Ethnic predilection among Sephardi and Ashkenazi Jewish, Arab, Armenian, and Turkish
populations with carrier rates as high as 1:3 to 1:5
Usually presents in childhood with 60% of patients presenting prior to 10 years of age
Clinical features
o Fever episodes last for 1-3 days and occur at irregular intervals
o Clinical hallmark is serositis (e.g. peritonitis, pleuritis, pericarditis)
o Skin: Erysipelas-like rash on shins and dorsum of feet
o MSK: Monoarthritis or oligoarthritis, arthralgia, myalgia
Morbidity is associated with amyloidosis, especially renal amyloidosis
Treatment
o Colchicine is highly effective therapy for most patients with FMF
o Anti-IL-1 therapy with Anakinra, Canakinumab or Rilonacept is effective in Colchicine-
resistant or intolerant FMF
TNF-Receptor Associated Periodic Syndrome (TRAPS)
Rare recurrent fever syndrome
Originally known as Familial Hibernian Fever
Autosomal dominant inheritance; linked to genetic mutation in TNFRSF1A gene that
encodes TNF receptor
Age of onset ranges from early childhood to later adulthood
Clinical features
o Distinguishing feature is relatively long duration of most attacks, which can last 3-4
weeks and occur at irregular intervals
o Skin: Migrating erythematous, maculopapular rash that spreads from trunk to extremities
o MSK: Severe migratory myalgias associated with rash, arthralgias, arthritis
o Ocular: Conjunctivitis, periorbital edema
o GI: Severe abdominal pain
o Other: oral ulcers, lymphadenopathy
Treatment
o Corticosteroids provide symptomatic relief but do not diminish frequency of attacks
o Anti-TNF agents (e.g. Etanercept) thought to be promising, but results of studies
disappointing
o Anti-IL-1 therapy may be beneficial
Mevalonate kinase deficiency- Hyperimmunoglobulinemia D Syndrome (HIDS)
Rare recurrent fever syndrome
Autosomal recessive inheritance; linked to genetic mutations in MVK gene encoding
mevalonate kinase
More than 90% of patients show symptoms within first year of life
Clinical features
o Fever episodes lasting 3-7 days that recur every 4-8 weeks
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 69
o Fever typically associated with abdominal pain, vomiting, diarrhea and a diffuse
maculopapular or urticarial rash
o Other common features include tender cervical lymphadenopathy, oral ulcers,
headaches, arthralgias, and large joint symmetric arthritis
o May have a striking elevation of serum IgD and IgA during fever episodes
o Elevation of urinary mevalonic acid during episodes
o Often triggers are identified, especially immunizations
Treatment
o NSAIDs and corticosteroids often limit symptoms
o Biologic agents (Anti-IL-1 and Anti-TNF) may be more effective
Cryopyrin Associated Periodic Syndrome (CAPS)
Group of autoinflammatory syndromes that are associated with genetic mutations involving
NLRP3 gene encoding cryopyrin
FCAS and NOMID characterized by disease onset in infancy; MWS may develop later
Spectrum of 3 diseases on a continuum of increasing disease severity
1. Familial Cold Autoinflammatory Syndrome (FCAS)
Autosomal dominant inheritance of NLRP3 mutations
Children develop fever, chills and generalized, non-pruritic urticarial skin lesions
within 30 minutes to 6 hours of exposure to cold
Symptoms persist up to 24 hours
Associated symptoms during attacks include conjunctivitis and arthralgias
Amyloidosis extremely rare
2. Muckle Wells Syndrome (MWS)
Autosomal dominant inheritance of NLRP3 mutations
Frequent episodes of fever lasting 24-48 hours
Characterized by generalized urticarial-like rash, arthralgias, myalgias, arthritis, and
conjunctivitis
Progressive sensorineural hearing loss emerges in adolescence
Higher risk of amyloidosis (25%)
3. Neonatal Onset Multisystem Inflammatory Disease (NOMID)
Spontaneous NLRP3 mutations
Nearly continuous clinical features that develop shortly after birth
Frequent fever episodes lasting 24-48 hours several times per week
Distinguishing features from other autoinflammatory syndromes are poor growth or
failure to thrive, severe neuroinflammation and deforming arthropathy
Skin: Nearly-constant generalized urticarial-like rash
CNS: Aseptic meningitis, intellectual disability, neurosensory hearing loss, optic
nerve atrophy
MSK: Deforming arthropathy with epiphyseal overgrowth, clubbing
Ocular: Conjunctivitis, episcleritis, uveitis, papilledema, visual loss

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 70
Hepatomegaly, splenomegaly
Poor long-term prognosis with high morbidity and mortality
Treatment
o Anti-IL-1 therapy with Anakinra, Canakinumab, or Rilonacept are highly effective
o Early treatment may reduce risk of amyloidosis and improve functional outcome
Periodic Fever, Aphthous Stomatitis, Pharyngitis and Adenitis (PFAPA)
Most common recurrent fever syndrome in children in North America
No known genetic association or inheritance pattern
Typically starts before 5 years and is self-limited (usually resolves within 5 years)
Clinical features
o Episodes of high fever that occur with regular periodicity every 3-6 weeks
o Fever episodes generally last up to 3-6 days
o Characteristic findings of small non-scarring aphthous ulcers, non-exudative pharyngitis,
and cervical adenitis
o May have associated nausea, vomiting, abdominal pain and headache
o Throat cultures are consistently negative
Treatment
o No consensus regarding treatment
o Single dose of prednisone at onset of symptoms and, if necessary, the following day can
abort the attack; however, interval between fever attacks may shorten
o Other options include cimetidine and tonsillectomy +/- adenoidectomy
References:
1. Ostring GT, Singh‐Grewal D. Periodic fevers and autoinflammatory syndromes in
childhood. J Paediatr Child Health 2016; 52:865-71.
2. Soon GS, Laxer RM. Approach to recurrent fever in childhood. Can Fam Physician
2017; 63(10):756-62
3. Tsoukas P, Laxer RM. Follow the complex bread crumbs: A review of autoinflammation
for the general paediatrician. Paediatr and Child Health [published online ahead of print,
July 2019].
4. Federici S, Gattorno M. A practical approach to the diagnosis of autoinflammatory
diseases in childhood. Best Pract Res Clin Rheumatol 2014; 28(2):263-76.
5. Goldsmith DP. Periodic fever syndromes. Pediatr Rev 2009; 30(5):e34-41.
8B. Other Inherited Autoinflammatory Diseases
The term ‘autoinflammatory’ has been used to distinguish disorders of innate immune
system characterized by recurrent, seemingly unprovoked episodes of inflammation from
more common ‘autoimmune’ diseases characterized by dysregulation of the adaptive
immune system (with high-titre autoantibodies and proliferation of antigen-specific T cells)
Hereditary periodic fever syndromes (described above) were first group of monogenic
disorders to be classified as autoinflammatory
New monogenic autoinflammatory diseases continue to be discovered (described below)
Spectrum of autoinflammatory diseases is now thought to include disease, such as systemic
juvenile idiopathic arthritis, Behçet disease, and chronic non-bacterial osteomyelitis (CNO),
which may prove to be polygenic in origin
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 71
Pyogenic Arthritis, Pyoderma Gangrenosum, and Acne (PAPA) Syndrome
Rare autosomal dominant autoinflammatory syndrome
Clinical features
o Recurrent episodes of sterile, erosive arthritis in early childhood
o As patients progress to puberty, skin involvement may predominate
o Characterized by cystic acne, recurrent and often debilitating aggressive ulcerative skin
lesions of the lower extremities indistinguishable from pyoderma gangrenosum
Treatment
o Arthritis may respond to corticosteroids, but adverse effects often limit their use
o Reports of successful treatment with Anti-IL-1 and Anti-TNF therapy
Deficiency of the Interleukin-1 Receptor Antagonist (DIRA)
Rare autosomal recessive autoinflammatory syndrome
Clinical features
o Systemic inflammation in the perinatal period
o Bone pain with characteristic radiographic findings of multifocal sterile osteolytic bone
lesions, widening of multiple anterior ribs, and periostitis
o Pustular skin lesions
Treatment
o Patients treated with Anakinra have shown rapid clinical and immunological responses
Deficiency of the Interleukin-36 Receptor Antagonist (DITRA)
Rare life-threatening multisystem disease with repeated flares of sudden onset
Clinical features
o High-grade fever, malaise
o Generalized pustular psoriasis
Treatment
o Treatment with anakinra has been described
Deficiency of Adenosine Deaminase 2 (DADA2) (also see Section 5D)
Newly recognized autosomal recessive syndrome with presentation very early in life
Clinical features
o Recurrent fevers, livedoid skin rash and vascular involvement
o Vascular involvement may include recurrent lacunar strokes, cerebral haemorrhage,
polyarteritis nodosa
o May have hypertension, hepatosplenomegaly and cutaneous vasculitis
Treatment
o Reports of successful treatment with Anti-TNF therapy
o Hematopoeitic stem cell transplantation considered for severe phenotypes
Type 1 Interferonopathies
Rare diseases characterized by mutations in interferons, which are molecules that represent
the cell’s first lines of defence again pathogens, mainly viruses
Type I interferon (INFαß) signalling defects can phenotypically manifest as a group of
heterogeneous autoinflammatory diseases

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 72
Common clinical features in type 1 Interferonopathies
o Lupus-like symptoms during infancy or prepubertal age
o Signs of vasculopathy such as chilblains or strokes
Specific conditions
o Aicardi-Goutières syndrome
Prototypic type 1 interferonopathy
Characterized by neonatal onset
Clinical features include progressive congenital encephalopathy, intracranial
calcification, white matter disease, chilblain-like skin lesions, glaucoma,
hypothyroidism, cardiomyopathy, demyelinating peripheral neuropathy
o Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature
(CANDLE) syndrome, also known as Nakajo-Nishimura syndrome (NNS)
Diagnosis
o Gene studies are the most accurate diagnostic test
o Interferon gene signature test
Treatment
o Unlike other fever syndromes, general immunosuppression with Corticosteroids,
Methotrexate, or anti-IL1 (e.g. Anakinra or Canakinumab which are effective in other
periodic fever syndromes) are not effective
o Anti-IL6 and Jak inhibitors have been effective in these conditions
References:
1. Federici S, Gattorno M. A practical approach to the diagnosis of autoinflammatory
diseases in childhood. Best Pract Res Clin Rheumatol. 2014; 28(2):263-76.
2. Hashkes PJ, Toker O. Autoinflammatory syndromes. Pediatr Clin North Am 2012;
59(2):447-70.
3. Davidson S, et al. An Update on Autoinflammatory Diseases: Interferonopathies. Curr
Rheumatol Rep 2018; 20(7):38
8C. Role of Genetic Testing in Suspected Autoinflammatory DIsease
Genetic testing may be used to confirm a diagnosis when the clinical pattern fits with one of
the autoinflammatory diseases
A genetic diagnosis should be pursued in a logical manner recognizing the cost and
limitations of testing, although panels of genetic mutations associated with these conditions
are now more accessible and cost-effective
While genetic testing may help to confirm a diagnosis, it is important to consider the
differential diagnosis and potential investigations for recurrent fevers outlined in Section 2C
A simple interactive tool is available online (https://www.printo.it/eurofever/scoreCriteria.asp)
to guide ordering of genetic tests for recurrent fever syndromes
References:
1. Gattorno M, et al. A diagnostic score for molecular analysis of hereditary
autoinflammatory syndromes with periodic fever in children. Arthritis Rheum 2008;
58(6):1823-32.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 73
8D. Chronic Non-Bacterial Osteomyelitis (CNO)
A non-infectious, autoinflammatory disease involving bone
Pathophysiology poorly understood, neutrophil mediated
CNO affects females > males and is more common in children and adolescents
Known as chronic recurrent multifocal osteomyelitis (CRMO) if multiple bony sites
Some cases (20-30%) are unifocal at diagnosis and many have non-recurrent disease
Clinical and radiographic findings initially mimic septic osteomyelitis; however, no abscess
formation is noted, cultures are negative, and there is a poor response to antibiotic therapy
Must consider bone malignancy, infection, and histiocytosis in work-up as CNO is a
diagnosis of exclusion
There are no validated diagnostic criteria for CNO/CRMO, but Jansson et al proposed a
clinical score (see table below) that may aid in differentiating non-bacterial osteitis from
other bone lesions and may guide the diagnostic approach
Proposed clinical score for nonbacterial osteitis (Jansson et al)
Risk factor
Score
Normal blood count
Symmetrical bone lesions
Lesions with marginal sclerosis
Normal body temperature
Vertebral, clavicular or sternal lesions
Radiographically proven lesions ≥ 2
CRP ≥ 1 mg/dL (10 mg/L)
13
10
10
9
8
7
6
Total clinical score *
63
* Total clinical score
Recommended diagnostic approach
1 lesion on whole body imaging
>1 lesion on whole body imaging
≤ 28
Bone biopsy with culture
Clinical monitoring
28-38
Clinical monitoring
Clinical monitoring
≥ 39
Consistent with CNO diagnosis
Consistent with CNO diagnosis
Adapted from Jansson et al, Arthritis Rheum 2009.
Clinical features
o Presents with acute or insidious onset of bone pain often associated with localized
swelling, tenderness and warmth; some patients also have fever and malaise
o Typical sites of involvement include the clavicles, pelvis, vertebral bodies and
metaphyses of long bones
o CNO is associated with inflammatory disorders of skin (e.g. palmoplantar pustulosis,
psoriasis, generalized pustulosis, severe acne, pyoderma gangrenosum), disorders of
the gastrointestinal tract (e.g. inflammatory bowel disease), and arthritis adjacent to
active bone lesions and (less commonly) distant to the osteitis

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 74
o The term SAPHO (synovitis, acne, pustulosis, hyperostosis, osteitis) syndrome is often
used in adults -- SAPHO may represent a later presentation of childhood CNO or may
be a distinct disorder within the same disease spectrum
o Clinical course characterized by periods of exacerbation with symptom-free intervals
Imaging
o X-Rays:
Mixed osteolytic and sclerotic bone lesions localized in the metaphyses close to the
growth plate
Periosteal reaction may be present
Cortical thickening may occur later in disease course
o MRI (whole body, if available): sensitive to assess extent and activity of lesions, as well
as asymptomatic lesions
o Bone scan: may be helpful to assess the extent of lesions, but is associated with
significant radiation exposure and may not distinguish inflammatory lesions from
metabolically active growth plates
Treatment
o Most lesions resolve without significant sequelae and spontaneous remission can occur;
however severe pain, recurrences, and functional limitations may necessitate therapy
o First-line therapy: NSAIDs provide symptomatic relief in up to 80% of patients
o Corticosteroids may sometimes be used for brief periods of time to provide symptomatic
relief or as a bridge to second-line therapy
o Second-line agents include Bisphosphonates (e.g. Pamidronate, Zolendronate),
Sulfasalazine, Methotrexate, anti-TNF agent (e.g. Infliximab)
References:
1. Zhao Y, Ferguson PJ. Chronic nonbacterial osteomyelitis and chronic recurrent
multifocal osteomyelitis in children. Pediatr Clin North Am 2018; 65(4):783-800.
2. Twilt M, Laxer RM. Clinical care of children with sterile bone inflammation. Curr Opin
Rheumatol 2012; 23(5):424-31.
3. Jansson AF, et al. Clinical score for nonbacterial osteitis in children and adults. Arthritis
Rheum 2009; 60(4):1152-9.
4. Zhao Y, et al. Consensus treatment plans for chronic nonbacterial osteomyelitis
refractory to Nonsteroid anti-inflammatory drugs and/or with active spinal lesions.
Arthritis Care Res 2018; 70(8):1228-37.
8E. Relapsing Polychondritis
A rare immune-mediated condition associated with inflammation in cartilage and other
tissues (particularly ears, nose, eyes, joints, respiratory tract, and heart valves)
Children have similar clinical features to adults, but are more likely to have family history of
autoimmunity and less likely to have associated inflammatory diseases
Early manifestations often remain unrecognized until emergence of classic features, such as
auricular inflammation and saddle-nose deformity
Associated with high morbidity and mortality
Screening for complications (e.g. aortic dilatation, cardiac lesions) is mandatory
Diagnosis based on clinical criteria – all 3 sets of criteria were established based on single-
centre cohort studies and none have been validated in an independent cohort

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 75
1976 McAdam et al Criteria for Relapsing Polychondritis
≥ 3 of the following clinical features:
Bilateral auricular chondritis
Non-erosive, seronegative inflammatory polyarthritis
Nasal chondritis
Ocular inflammation (conjunctivitis, keratitis, scleritis/episcleritis, uveitis)
Respiratory tract chondritis (laryngeal and/or tracheal cartilages)
Cochlear and/or vestibular dysfunction (neurosensory hearing loss, tinnitus, vertigo)
1979 Damiani and Levine Criteria for Relapsing Polychondritis
≥ 1 of the clinical features proposed by McAdam plus positive histologic confirmation
or
≥ 2 of the clinical criteria proposed by McAdam plus response to Corticosteroids or Dapsone
1989 Modified Michet et al Criteria for Relapsing Polychondritis
Proven chondtritis in at least 2/3 cartilage sites of:
Auricular cartilage
Nasal cartilage
Laryngotracheal cartilage
or
Proven inflammation in at least 1/3 of the above cartilage sites (auricular, nasal,
laryngotracheal) plus 2 other minor criteria:
Occular inflammation
Vestibular dysfunction
Seronegative inflammatory arthritis
Hearing loss
Treatment
o No evidence-based guidelines for treatment
o In adults, largely empiric and based on disease severity
o Options include NSAIDs, corticosteroids, Methotrexate, Dapsone, Azathioprine and
anti-TNF therapy
References:
1. Belot A, et al. Pediatric-onset relapsing polychondritis: case series and systematic
review. J Pediatr 2010;156(3):484-9.
2. Kemta LF, Chevalier X. Refractory relapsing polychondritis: Challenges and solutions.
Open Access Rheumatology: Research and Reviews 2018; 1-11.
3. Borgia F, et al. Relapsing polychondritis: An updated review. Biomedicines 2018;
6(3):E84.
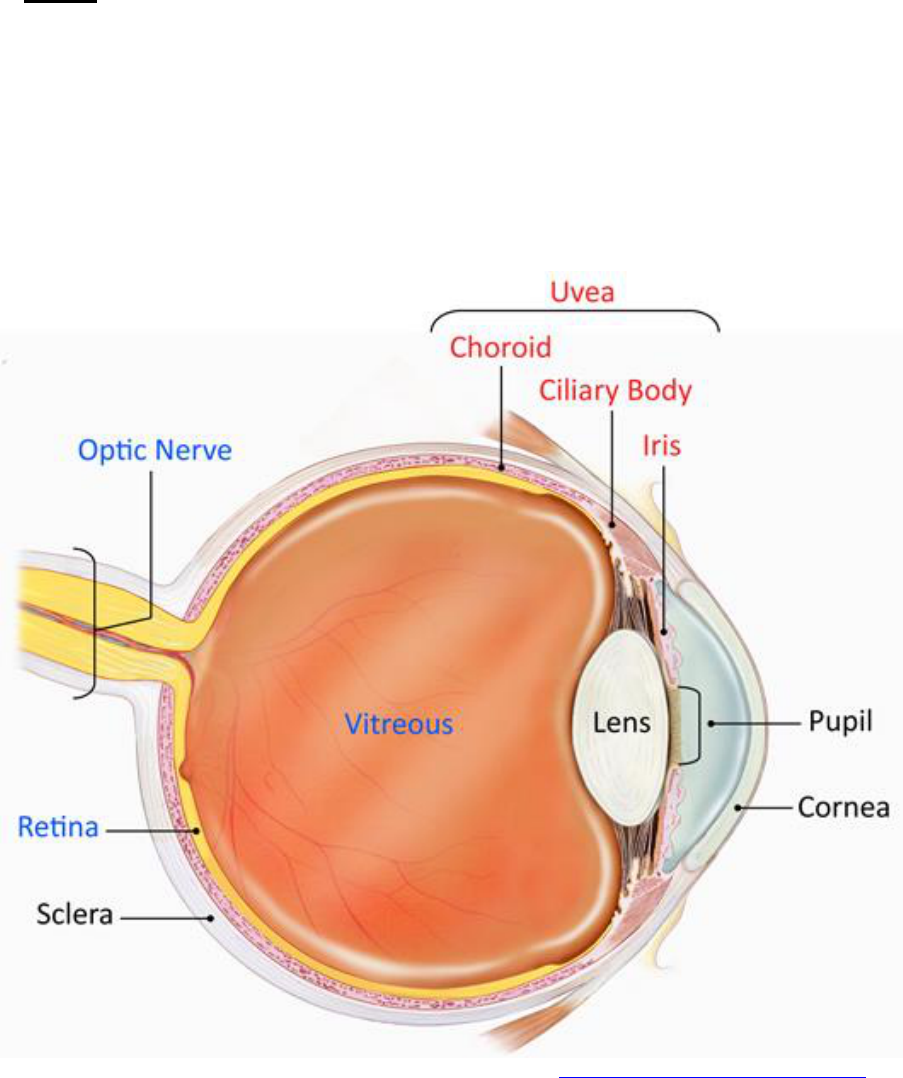
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 76
SECTION 9 – UVEITIS
9A. Uveitis
Inflammation of the uvea, which is the middle layer of the eye
May be asymptomatic or symptomatic
Classification based on anatomic location of inflammation:
o Anterior uveitis involves the iris and/or ciliary body
o Intermediate uveitis involves the pars plana between the ciliary body and retina
o Posterior uveitis involves the choroid and/or retina
o Panuveitis describes the presence of inflammation in all three anatomic locations in
which there is no predominant site of inflammation
* Image published in “Facts About Uveitis” by the National Eye Institute: https://nei.nih.gov/health/uveitis/uveitis
Complications of uncontrolled uveitis include:
o Cataracts
o Glaucoma
o Band keratopathy
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 77
o Synechiae (adhesion of iris to lens)
o Cystoid macular edema
o Vision loss
Treatment
o Prompt and aggressive treatment to prevent or minimize visual complications
o Minimize chronic use of topical corticosteroids (due to side effects such as cataract
formation and glaucoma)
o Close collaboration between rheumatologists and ophthalmologists is essential
o Options include topical (corticosteroids, cycloplegics, mydriatics, anti-glaucoma agents)
and systemic (Methotrexate, Infliximab, Adalimumab, other) therapies
9B. Systemic Inflammatory Diseases Associated with Uveitis
Disease
Acute/Chronic
Location
Associated clinical
features
Investigations
Juvenile
idiopathic arthritis
(except enthesitis
related arthritis)
Chronic, recurrent,
asymptomatic
Anterior >
Posterior
Oligoarthritis >>
Polyarthritis
ANA
Enthesitis related
arthritis
Acute symptomatic,
often recurrent
Anterior
Enthesitis, sacroiliitis;
often associated with
reactive arthritis, IBD, or
a family history of these
conditions
HLA-B27
Behçet disease
Acute or chronic
Posterior
Recurrent oral and/or
genital ulcers, arthritis,
skin rash
Pathergy
Blau syndrome
(Infantile
sarcoidosis)
Chronic
Posterior,
Anterior,
Panuveitis
Skin rash, arthritis
Consider genetic
testing (NOD2/
CARD15
mutations)
Kawasaki disease
Acute,
asymptomatic
Anterior
Consider if patient
presents with severe
conjunctivitis and
photophobia
Echocardiogram
Sarcoidosis
Chronic
Posterior,
Anterior,
Panuveitis
Skin rash, arthritis, lung
involvement,
lymphadenopathy
Biopsy, consider
genetic testing
(NOD2/CARD15
mutations)
Tubulo-interstitial
nephritis and
uveitis (TINU)
Acute
Anterior
Fever, arthralgias,
fatigue, abdominal pain,
and nephritis; uveitis may
present before or after
renal disease
U/A, renal function
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 78
9C. Infectious Causes of Uveitis
Disease
Acute/Chronic
Location
Associated clinical
features
Investigations
Cat scratch
(Bartonella
henselae)
Chronic
Anterior,
Posterior
Fever of unknown origin,
regional lymphadenopathy,
abdominal pain, weight
loss, hepatosplenomegaly
Cat exposure
Serology
Cytomegalovirus
Chronic
Posterior
Congenital; fever, malaise
Immunocompromised host
Serology, viral PCR
Herpes virus
Acute or chronic
Anterior,
posterior
Keratouveitis, fever,
gingivostomatitis
Serology, viral
culture and/or PCR
Lyme disease
Chronic
Anterior,
Posterior
Erythema migrans, arthritis,
CNS symptoms
Tick bites in endemic areas
Serology
Toxoplasmosis
Chronic, acute
recurrences
Posterior
Congenital exposure
(chorioretinitis,
hydrocephalus, intracranial
calcifications); bilateral
symmetric non-tender
cervical lymphadenopathy,
constitutional symptoms,
headaches, myalgias and
hepatosplenomegaly
Immunocompromised host;
cat exposure
Serology
Tuberculosis
Chronic
Anterior
Chronic cough, fever,
weight loss, multi-organ
manifestations
Travel/exposure history
PPD,
Chest X-ray
References:
1. Krishna U, et al. Uveitis: a sight-threatening disease which can impact all systems.
Postgrad Med J 2017; 93(1106);766-73.
2. Heilingenhaus A, et al. Update of the evidence based, interdisciplinary guideline for anti-
inflammatory treatment of uveitis associated with juvenile idiopathic arthritis. Semin
Arthritis Rheum 2018; Epub ahead of print.
3. Sen ES, et al. Uveitis associated with juvenile idiopathic arthritis. Nat Rev Rheumatol
2015; 11(6):338-48.
4. Angeles-Han ST, et al. 2019 American College of Rheumatology/Arthritis Foundation
Guideline for the Screening, Monitoring, and Treatment of Juvenile Idiopathic Arthritis-
Associated Uveitis. Arthritis Care Res 2019; 71(6):703-16.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 79
SECTION 10 – INFLAMMATORY BRAIN DISEASES
10A. Introduction to Inflammatory Brain Diseases
Inflammatory brain disease encompasses a wide range of disorders
Clinical and diagnostic features vary depending on the underlying disease
A broad differential diagnosis should be considered when a child presents with newly
acquired neurological or psychiatric deficits
Types of inflammatory brain diseases in children:
Vasculitis
Primary Angiitis of the Central Nervous System in childhood (cPACNS)
Angiography-positive cPACNS: progressive and non-progressive
Angiography-negative cPACNS
Secondary CNS vasculitis
Non-vasocentric
neuroinflammatory
disorders
Demyelinating disorders
Multiple sclerosis, acute demyelinating encephalomyelitis (ADEM),
optic neuritis, transverse myelitis
Antibody mediated inflammatory brain disease
Autoimmune encephalitis, neuromyelitis optica, Hashimoto
encephalopathy
Systemic inflammatory diseases with CNS involvement
Systemic lupus erythematosus, antiphospholipid syndrome, celiac
disease, Beçhet disease, sarcoidosis
Post-infectious or infection-associated inflammatory encephalopathy
Post-Streptococcal neuropsychiatric disorders (including acute
rheumatic fever, Pediatric Acute-onset Neuropsychiatric Syndrome
(PANS) and Pediatric Autoimmune Neuropsychiatric Disorders
Associated with Streptococcal infections (PANDAS)), post-
Mycoplasma basal ganglia encephalitis, post-Herpes Simplex Virus
encephalitis, Febrile infection-related epilepsy syndrome (FIRES)
Other neuroinflammatory disorders
Rasmussen encephalitis
10B. Childhood Primary Angiitis of the Central Nervous System (cPACNS)
Currently defined by modified Calabrese criteria:
o Clinical evidence of a newly-acquired focal or diffuse neurologic and/or psychiatric deficit
in child <18 years of age, plus
o Angiographic or histologic evidence of CNS vasculitis, plus
o Absence of an underlying systemic condition
2 clinically and radiologically distinct types of cPACNS
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 80
1. Angiography positive cPACNS (Large-medium vessel CNS vasculitis)
o Clinical features: stroke presentation with headache, acute hemiparesis, hemisensory
deficits, and/or fine motor deficits
o Inflammatory markers: often normal
o CSF: often normal
o MRI: unilateral focal areas of acute ischemia in a vascular distribution
o Evidence of vasculitis on angiography (conventional angiography or MRA)
o Brain biopsy not required
o Further divided into progressive and non-progressive subtypes
o Non-progressive cPACNS
Defined by absence of progression on imaging 3 months after initial angiography (i.e.
monophasic disease)
More common than progressive cPACNS
o Progressive cPACNS
Defined by progression on neuroimaging 3 months after initial angiography
Presents with both focal and diffuse neurologic deficits
Multifocal T2 lesions on MRI, proximal and distal stenosis on angiography
2. Angiography negative cPACNS (Small vessel CNS vasculitis)
o Clinical features: systemic symptoms (fever, malaise), headache, seizures, ataxia,
cognitive decline and/or behaviour changes
o Inflammatory markers: may be elevated
o CSF: more likely to have pleocytosis, elevated protein and/or elevated opening pressure
compared to angiography-positive disease; oligoclonal bands may also be present
o MRI: multifocal T2 hyperintensities in both white and grey matter, lesions do not conform
to large-vessel vascular territory
o By definition, angiography is negative
o Brain biopsy (ideally lesional): non-granulomatous, intramural and perivascular T
lymphocytes in small arteries, arterioles, capillaries or venules
Treatment
o Based on type of cPACNS
o Angiography positive cPACNS
Anti-coagulation with or without anti-platelet agent
Corticosteroids in non-progressive cPACNS may improve outcome
Progressive cPACNS treated with same protocol as for angiography negative
cPACNS
o Angiography negative cPACNS
Induction (first 6 months) using Cyclophosphamide and Corticosteroids
Maintenance (up to 24 months) using Mycophenolate mofetil and Corticosteroids
o Rehabilitation addressing cognitive, behavioural, physical and psychological deficits
o Adjunctive therapy: PJP prophylaxis while on Cyclophosphamide; Vitamin D
supplementation and ensure adequate calcium intake while on steroids
Prognosis
o Complications: persistent neurological deficits, seizures, cognitive disability

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 81
References:
1. Gowdie P, et al. Primary and secondary CNS vasculitis. J Child Neurol. 2012;
27(11):1448-59.
2. Van Mater H. Pediatric inflammatory brain disease: a diagnostic approach. Curr Opin
Rheumatol 2014; 26(5):553-61.
10C. Secondary Central Nervous System Vasculitis
Occurs in context of an underlying systemic illness
Can occur in context of infections, as well as other systemic inflammatory and autoimmune
diseases
Treatment may involve Corticosteroids as well as medications directed to underlying cause
Causes of secondary CNS vasculitis:
Infections
Bacteria: Mycobacterium tuberculosis, Mycoplasma pneumonia,
Streptococcus pneumonia
Virus: Epstein-Barr virus, Cytomegalovirus, Enterovirus, Varicella zoster
virus, Hepatitis C virus, Parvovirus B19, West Nile virus
Fungus: Candida albicans, Actinomycosis, Aspergillus
Spirochete: Borrelia burgdorferi, Treponema pallidum
Inflammatory
diseases
Systemic vasculitis: granulomatosis with polyangiitis, microscopic
polyangiitis, Kawasaki disease, polyarteritis nodosa, Behçet disease
Systemic lupus erythematosus
Juvenile dermatomyositis
Morphea
Autoinflammatory syndromes
Inflammatory bowel disease
Hemophagocytic lymphohistiocytosis
Other
Drug-induced vasculitis
Malignancy-associated vasculitis
References:
1. Gowdie P, et al. Primary and secondary CNS vasculitis. J Child Neurol. 2012;
27(11):1448-59.
10D. Pediatric autoimmune encephalitis (AE)
Brain inflammation caused by antibodies directed against intracellular neuronal antigens,
synaptic receptors, ion channels and other neuronal proteins
Most common autoantibodies in children target the N-methyl-D-aspartate (NMDA) receptor,
myelin oligodendrocyte glycoprotein (MOG) and glutamic acid decarboxylase 65 (GAD65)
Addional antibody targets identified in children include aquaporin 4, dopamine-2 receptor,
gamma-aminobutyric acid (GABA) (A) receptor, GABA(B) receptor, and glycine receptor
Better prognosis if antibody target is extracellular or synaptic protein
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 82
Clinical features of pediatric AE include seizures, memory deficits, behaviour changes,
psychiatric symptoms, altered mental state, and focal neurological deficits
Investigations
o MRI may be normal or abnormal (findings often depend on antibody)
o Serum testing may show inflammatory changes or may be normal
o CSF may show increased white blood cell counts
o EEG is often abnormal with seizures, epileptiform discharges and/or slowing
o Psychoeducational testing often shows cognitive dysfunction, including impaired
memory and slow cognitive processing speeds
Diagnosis confirmed by identification of anti-neuronal antibodies in CSF or serum
Management in collaboration with pediatric neurologist is recommended
Treatment typically involves Corticosteroids, IVIG and other immunosuppressants, such as
Rituximab
Anti-NMDA receptor encephalitis
Most common neuronal antibody mediated encephalitis syndrome in children
Clinical features
o Typically evolves in stages
o Prodrome of fever and headache
o Subsequent development of psychiatric or behavioral manifestations, speech changes,
decreased consciousness, seizures, choreoathetoid movements and autonomic
instability (tachycardia, fever, hypertension, hypoventilation)
Investigations
o Diagnosed by presence of anti-NMDA receptor antibodies in CSF or serum (testing more
sensitive in CSF)
o MRI: frequently normal
o CSF: usually abnormal (lymphocytic pleocytosis, increased protein, or oligoclonal bands)
o EEG: often abnormal with diffuse slowing in children and more focal findings in
teenagers and adults
o Consider imaging for ovarian or testicular teratoma (association between anti-NMDA
receptor encephalitis and tumor in adults)
Treatment
o First line therapy includes Corticosteroids, IVIG and/or plasma exchange
o Rituximab may also be considered
Outcome
o Over 80% of patients have full recovery
o Recovery may be slow with continued improvement seen up to 2 years after onset of
symptoms
Autoimmune encephalitis associated antibodies to MOG
Most common antibody associated with autoimmune demyelination
Antibodies more common in children than adults
Clinical features
o Typically patients have symptoms consistent with ADEM, including encephalopathy,
weakness, ataxia, sensory changes and/or seizures
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 83
o Also associated with optic neuritis (especially bilateral) and transverse myelitis
Investigations
o Diagnosed by presence of anti-MOG antibodies in CSF or serum (testing more sensitive
in serum)
o MRI: focal or multifocal white matter lesions, longitudinally extensive myelitis and/or optic
neuritis
o CSF: neutrophilic pleocytosis may be present
o EEG: non-specific slowing
Treatment
o First line therapy with Corticosteroids
o IVIG and/or plasma exchange added for severe disease
o Chronic immunotherapy, including IVIG, Azathioprine and//or Mycophenolate mofetil,
may be considered for relapsing disease
Outcome
o Significant improvement expected within three months of initiating therapy
o Disappearance of antibodies associated with monophasic course, whereas persistent
antibodies are associated with relapsing course
Neuromyelitis Optica (NMO)
Neuroinflammation due to antibodies to aquaporin-4
Inflammation and demyelination mostly affecting the spinal cord and optic nerves
Clinical features
o Commonly present with acute optic neuritis and transverse myelitis
o Other reported clinical features: encephalopathy, ophthalmoparesis, vertigo, nausea and
vomiting, hyponatremia, inappropriate diuresis, intractable hiccups
o Reported in association with Sjögren syndrome
Investigations
o Diagnosis requires identification of antibodies to aquaporin-4 in serum or CSF
o CSF: pleocytosis and elevated protein
o MRI: lesions in the periventricular regions of the third and fourth ventricles and in the
periaqueductal grey matter
Treatment
o Initial therapy: Corticosteroids, IVIG and/or plasma exchange
o Maintenance with second line agent should be considered (e.g. Azathioprine, Rituximab)
Outcome
o Frequent relapsing course with accumulation of neurological deficits
References:
1. Dalmau J, Graus F. Antibody-mediated encephalitis. N Eng J Med 2018; 378:840-51.
2. Armangue T, et al. Autoimmune encephalitis in children. J Child Neurol 2012; 27(11):
1460-9.
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 84
3. Van Mater H. Pediatric inflammatory brain disease: a diagnostic approach. Curr Opin
Rheumatol 2014; 26(5):553-61.
4. Co DO, et al. Immune-mediated diseases of the central nervous system. Pediatr Clin
North Am 2017; 64(1):57-90.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 85
SECTION 11 – INFECTION & INFECTION-RELATED CONDITIONS
11A. Bone and Joint Infections
Osteomyelitis
Intraosseous infection with bacteria or rarely, fungi
Classified as acute, subacute, or chronic
o Acute osteomyelitis is of recent onset and short duration
Most often hematogenous in origin but may result from trauma such as a compound
fracture or puncture wound
Can be metaphyseal, epiphyseal, or diaphyseal in location
o Subacute osteomyelitis is of longer duration and is usually caused by less virulent
organisms
o Chronic osteomyelitis results from ineffective treatment of acute osteomyelitis and is
characterized by necrosis and sequestration of bone
Source may be (1) hematogenous (2) local invasion from contiguous source (3) direct
invasion of bone
Usually blood-borne to metaphysis, slow blood flow allows organisms to pass through
fenestrations in vessel wall, migrate through haversian canal to sub-periosteal space
Unique features:
o Neonates may present with pseudoparalysis or sepsis; fever is common; organisms
frequently cross the physis and cause growth arrest
o Patients with hemoglobinopathy frequently have Salmonella and other gram-negative
organisms
Key symptoms:
o Fever, severe bone pain, and tenderness with or without local swelling should suggest
the possibility of acute osteomyelitis
Bones involved:
o Femur, tibia, humerus, fibula, calcaneus, pelvis
Organisms:
o Staphylococcus most common
o Group A Streptococcus, MRSA, atypical Gram negative bacteria and Salmonella
Investigations
o Blood work: Elevated WBC, ESR, CRP are non-specific
o Blood cultures (sensitivity 60%), bone biopsy culture (sensitivity 80%)
o Imaging:
X-rays important for exclusion of other diagnoses
X-ray signs include soft-tissue swelling, soft tissue edema, subperiosteal changes
and bone destruction (diagnostic findings may not be clear until days 10 to 21)
Bone scan has positive predictive value of 83% (MRI 85%) and allows detection of
other sites
Treatment
o For the treatment of uncomplicated osteomyelitis, in which fever and symptoms resolve
rapidly, 2 to 4 days of intravenous antibiotics can be followed by high dose oral
antibiotics, for a total antibiotic course of 3 weeks.
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 86
Septic Arthritis
Intra-articular infection with bacteria or rarely, fungi
Medical emergency (surgical emergency if hip or shoulder involved)
Key symptoms:
o Usually accompanied by systemic signs of illness (e.g., fever, vomiting, headache)
o May be a component of a more generalized infection that may include meningitis,
cellulitis, osteomyelitis, or pharyngitis
o Joint pain is usually severe, and the infected joint and periarticular tissues are swollen,
hot, and sometimes erythematous; often difficulty weight bearing if lower extremities are
involved
Joints involved:
o Joints of lower extremity are most commonly the sites of infection
o Knees, hips, ankles, and elbows account for 90% of infected joints in children
Organisms:
o Staphylococcus aureus and non–Group A β Streptococcus are most common overall
o Streptococcus pneumoniae is common in children younger than 2 years
o Neisseria gonorrheae in sexually active adolescents
o Salmonella is commonly associated with sickle cell disease
o Mycobacterium tuberculosis is an unusual cause of septic monarthritis in childhood
o Kingella kingae is emerging as an important pathogen in children with septic arthritis
and
may also account for a significant portion of culture negative cases
Investigations
o Joint aspiration - need to aspirate joint prior to antibiotics
Synovial fluid usually appears cloudy
Very high WBC count (50,000-300,000, > 75% neutrophils)
Gram stain positive
o Blood work
Elevated WBC with neutrophilia, CRP and ESR are non-specific
o Cultures
Synovial fluid culture (sensitivity 80%), blood culture (sensitivity 10%)
Require special handling if suspect Neisseria or Mycobacterium tuberculosis
Kingella kingae may require cultures for 7 days to isolate the organism
o Imaging
Plain X-rays are not diagnostic, but may be helpful in excluding other disorders; may
show an underlying osteomyelitis as the etiology of the septic arthritis; may
demonstrate only increased soft tissue and capsular swelling
Ultrasound may be helpful in identifying/quantifying joint effusions and in joint
aspiration for diagnostic purposes
MRI superior to CT in delineation of soft tissue structures and MRI changes may be
seen as soon as 24 hours following infection; synovial enhancement detected in
virtually all patients
Bone scans are not used for diagnositic evaluation
Treatment
o Antibiotics
Choice of antibiotics depends on presence of predisposing factors, age of child and
suspected organism
Cefazolin often first line antibiotic or Clindamycin for penicillin allergic patients

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 87
Course of antibiotics typically 2 to 7 days of intravenous therapy (depending on
organism) followed by high dose oral antibiotics for a total duration of 2 weeks
o Surgical
May require surgical debridement, joint irrigation, drainage or recurrent aspiration for
infections in deep joints (e.g. hips)
References:
1. Paakkonen M, Peltola H. Bone and Joint Infections. Ped Clinic North Am 2013;
60(2);425-436.
2. Castellazzi L, et al. Update on the management of pediatric acute osteomyelitis and
septic arthritis. Int J Mol Sci 2016; 17(6):855-63.
3. Montgomery NI, Rosenfeld S. Pediatric osteoarticular infection update. J Pediatr Orthop
2015; 35(1):74-81.
4. Kocher MS, et al. A clinical practice guideline for treatment of septic arthritis in children:
efficacy in improving process of care and effect on outcome of septic arthritis of the hip.
J bone Joint Surg Am 2003; 85(6):994-9.
11B. Reactive Arthritis
A form of non-septic arthritis developing after an extra-articular infection
Arthritogenic bacteria:
o GI: Salmonella, Shigella, Yersinia, Campylobacter
o GU: Chlamydia, Ureaplasma
Clinical manifestations
o Several stages involved:
1. Clinical infection precedes appearance of arthritis and/or enthesitis by 1 to 4 weeks
2. Active period of weeks to months
3. Sustained remission or recurrent episodes which may evolve to ERA, especially in
patients that are positive for HLA B27
o Acute arthritis (marked pain, sometimes erythema over affected joint) and/or enthesitis
o May see tenosynovitis, bursitis, dactylitis
o Patients may continue to have fever, weight loss, fatigue and muscle weakness
o Painless, shallow mucosal ulcers are common
o Urethritis and cervicitis are rare
o Conjuctivitis occurs in about two thirds of children at onset
o Skin lesions include erythema nodosum, circinate balanitis and keratoderma
blennorrhagicum
Investigations
o Mild decrease in hemoglobin, mild leukocytosis with neutrophilia
o Elevated inflammatory markers (platelets, immunoglobulins, ESR and CRP)
o Autoantibodies (RF and ANA) are usually absent, but reactive arthritis is more common
in HLA-B27 positive individuals
o Synovial fluid is sterile
o Cultures (blood, urine, stool) obtained at the time of infection may be positive
Treatment:
o NSAIDs

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 88
o No clear evidence that antibiotics during inflammatory phase alter course of disease
o Rarely, Corticosteroids (oral or intra-articular) may be required
o Sulfasalazine is recommended in the management of resistant arthritis and enthesitis
References:
1. Carter JD, Hudson AP. Reactive arthritis: clinical aspects and medical management.
Rheum Dis Clin North Am 2009; 35(1):21-44.
2. Rihl M, et al. Infection and musculoskeletal conditions: Reactive arthritis. Best Pract Res
Clin Rheumatol 2006; 20(6):1119-37.
11C. Acute Rheumatic Fever (ARF)
Inflammatory illness following Group A Streptococcus (GAS) infection
Diagnosis of ARF using Jones Criteria
o Most recent revision of Jones Criteria in 2015 developed distinct criteria for low and
moderate-high risk populations to increase sensitivity for patients at higher risk
2015 Revised Jones Criteria for Diagnosis of Acute Rheumatic Fever
For all patient populations with evidence of preceding GAS infection, diagnosis of initial ARF
requires 2 major criteria or 1 major plus 2 minor criteria
Population
risk
Low risk populations
Cases of ARF ≤ 2/100,000 in school-
aged children or rheumatic heart
disease in ≤ 1/1000 at any age
Moderate- and high-risk populations
Cases of ARF > 2/100,000 in school-
aged children or rheumatic heart
disease in > 1/1000 at any age
Major
criteria
1. Carditis (clinical and/or subclinical)
2. Polyarthritis
3. Chorea
4. Erythema marginatum
5. Subcutaneous nodules
1. Carditis (clinical and/or subclinical)
2. Monoarthritis, polyarthritis or
polyarthralgia
3. Chorea
4. Erythema marginatum
5. Subcutaneous nodules
Minor
criteria
1. Polyarthralgia
2. Fever ≥ 38.5 degrees Celsius
3. ESR ≥ 60 and/or CRP ≥ 3.0 mg/dL
(30 mg/L)
4. Prolonged PR interval (unless
carditis is a major criterion)
1. Monoarthralgia
2. Fever ≥ 38 degrees Celsius
3. ESR ≥ 30 and/or CRP ≥ 3.0 mg/dL
(30 mg/L)
4. Prolonged PR interval (unless
carditis is a major criterion)
Adapted from Gewitz et al, Revision of Jones Criteria for the diagnosis of ARF, Circulation 2015.
Clinical features
Arthritis in ARF has characteristics that help differentiate it from other causes
Characteristically migratory and additive starting with monoarthritis of large joints
Short duration of arthritis (hours to days)
Dramatic response to ASA/NSAIDs

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 89
Chorea often occurs as a late manifestation; may be diagnosed as being due to ARF
without accompanying evidence of GAS infection if other causes (e.g. tic disorder,
encephalitis, familial chorea, etc…) have been excluded
Subcutaneous nodules not often present as sole major manifestation
Investigations
Infectious testing
Diagnosis of ARF requires supporting evidence of antecedent GAS infection with
positive throat culture or elevated/rising streptococcal antibody titres
Echocardiography
Echo should be performed in all suspected cases of ARF
Common cardiac findings of ARF include pathological mitral valve regurgitation,
pathologic aortic regurgitation, acute/chronic mitral or aortic valve changes
Treatment
o Antibiotic therapy: 10 days oral antibiotics, usually Penicillin
o Anti-inflammatory therapy:
ASA 100 mg/kg/day divided QID PO for 3–5 days, then 75 mg/kg/day divided QID
PO for 4 weeks (or may consider Naproxen instead)
Prednisone may be used for carditis/cardiomegaly and heart failure +/- Digoxin
o Carbamazepine, Phenobarbital, Haloperidol, or Chlorpromazine for chorea
o Prophylaxis for recurrence:
Without carditis: Up to age 21 or 5 years post initial attack, whichever is later
With carditis, but without residual heart disease: Up to age 21 or 10 years post initial
attack, whichever is later
With carditis and residual heart disease: Up to age 40 or 10 years post initial attack,
whichever is later
References:
1. Karthikeyan G, Guilherme L. Acute rheumatic fever. Lancet 2018; 392(10142):161-74.
2. Gewitz MH, et al. Revision of the Jones Criteria for the diagnosis of acute rheumatic
fever in the era of doppler echocardiography: a scientific statement from the American
Heart Association. Circulation 2015;131(20):1806-18.
3. Gerber MA, et al. Prevention of rheumatic fever and diagnosis and treatment of acute
streptococcal pharyngitis. Circulation 2009;119:1541-51.
11D. Post-Streptococcal Reactive Arthritis (PSRA)
PSRA defined as inflammatory arthritis of ≥1 joint (with poor response to NSAID) associated
with a recent Group A Streptococcus (GAS) infection, but not meeting the Jones criteria to
diagnose acute rheumatic fever (ARF) (see Section 12C)
Characteristics that help distinguish PSRA from ARF include the following:
PSRA
ARF
Age distribution
Bimodal: 8-14 years and 21-
37 years
5-15 years with peak around
12 years
Timing of disease onset
following GAS infection
7-10 days
10-28 days

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 90
Pattern of joint involvement
Additive and persistent, non-
migratory arthritis involving
large, small and axial joints
Migratory, transient arthritis
involving mainly large joints
Response to ASA/NSAID
Poor to moderate
Dramatic improvement
Carditis
Uncommon
Occurs in 60-70% of ARF
patients
Treatment
o Antibiotic therapy: 10 days oral antibiotics, usually Penicillin
o Anti-inflammatory therapy:
ASA or NSAID
Corticosteroids may be used in refractory cases
o Prophylaxis for recurrence:
Controversial
Antibiotic prophylaxis may be given for up to 1 year after onset of symptoms with
close monitoring for development of carditis; if clinically well after one year and
echocardiogram remains normal, then may discontinue prophylaxis
Prognosis
o Most cases resolve spontaneously within a few weeks, but some recurrent or prolonged
Reference:
1. Bawazir Y, et al. Post-streptococcal reactive arthritis (PSRA). Curr Rheumatol Rev 2019
[Epub ahead of print].
2. Van der Helm-van Mil AH. Acute rheumatic fever and poststreptococcal reactive arthritis
reconsidered. Curr Opin Rheumatol 2010; 22(4):437-42.
11E. Lyme Disease
Complex tic-borne disease with multiple organ involvement (skin, joint, neurologic)
Most common vector-borne infection in North America and Europe
o Borrelia burgdorferi spirochete transmitted by hard-bodied ticks of the genus Ixodes
o Found in the temperate zones of the northern hemisphere
Incidence continues to rise
Clinical manifestations divided into early and late manifestations
o Early manifestations develop within weeks or few months of tick bite
o Late manifestations begin several months or even years later
Organ system
Early Lyme disease
Late Lyme disease
Skin
Nervous system
Musculoskeletal system
Cardiovascular system
Erythema migrans
Cranial nerve palsy
Lymphocytic meningitis
Arthralgia or arthritis
Carditis*
Acrodermatitis chronic
atrophicans*
Chronic encephalomyelitis
Arthritis
*Rare in childhood
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 91
o Erythema migrans usually begins as a round, erythematous macule or papule that
rapidly expands, often with central clearing, to a diameter of at least 5 cm and resolves
within four weeks if untreated
o Arthritis is typically monoarthritis, but may sometimes be polyarthritis
Investigations
o Elevated ESR
o CSF lymphocytic pleocytosis
o Serologic confirmation (initial screening performed with ELISA, then positive or
equivocal tests confirmed with Western blot)
o Do not test for Lyme disease as a cause of musculoskeletal symptoms without an
exposure history and appropriate examination findings (highlighted in Choosing Wisely:
Pediatric Rheumatology Top 5 by American College of Rheumatology)
Treatment
o Varies according to disease manifestations
o Erythema migrans only:
Amoxicillin or Doxycycline (only if >10 years of age) PO x 14-21 days
o Early Lyme disease (except isolated rash) or Late Lyme disease:
Ceftriaxone or Cefotaxime IV x 2-4 weeks, or
Amoxicillin or Doxycycline (only if >10 years of age) PO x 4 weeks
o Post-exposure prophylaxis:
If tick removed (while in endemic area) and was engorged, may benefit from
Doxycycline 200 mg (or 4.4 mg/kg) PO as a single dose within 72 hours of removing
tick (however, not enough data to recommend amoxicillin prophylaxis)
Prevention
o Appropriate clothing (e.g. long pants and sleeves)
o Tick repellents (e.g. DEET, permethrim) applied to clothing
o Search for and remove ticks promptly with tweezers
References:
1. Shapiro ED. Lyme disease. New Engl J Med 2014; 370(18):1724-31.
2. Sood SK. Lyme disease in children. Infect Dis Clin North Am 2015: 29(2);281-94.
3. Eldin C, et al. Review of European and American guidelines for the diagnosis of Lyme
borreliosis. Med Mal Infect 2019;49(2):121-32.
4. Onyett H, et al. Lyme disease in Canada: Focus on children. Canadian Pediatric Society
position statement. Paediatr Child Health 2019. [Epub ahead of print]

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 92
SECTION 12 – PAIN SYNDROMES
12A. Chronic Pain Syndromes
Primary pain syndromes may have a greater impact on patients’ and families’ quality of life
than inflammatory disease
Many children with chronic musculoskeletal (MSK) pain do not have an identified cause
Potential role of psychosocial stress in development of chronic pain syndromes
Growing Pains
Onset usually between 4 and 10 years of age
Typical history is deep aching cramping pain in bilateral thighs or calves, usually at night
and intermittently waking the patient from sleep
Improve with gentle massage, heat and/or analgesia
Symptoms disappear by morning
Normal physical examination
Investigations not necessary for diagnosis
Fibromyalgia (aka Generalized Amplified Musculoskeletal Pain)
Chronic generalized pain syndrome
May be triggered by change in physical activity due to injury or chronic illness
Clinical features
o Widespread pain with gradual onset and chronic course lasting at least 3 months
o Associated with fatigue, poor sleep and waking feeling unrefreshed
o Pain may be affected by anxiety, stress, activities and weather
o Symptoms may wax and wane over time
o Absence of physical findings that indicate another condition (caveat – fibromyalgia may
occur in context of another medical condition (e.g. JIA) but would be disproportionate
for that condition and would involve pain at sites that are not affected by the disease)
Diagnosis
o No confirmatory blood or imaging investigations, as these are typically normal
o Tender points are no longer included in most recent diagnostic criteria due to
inconsistencies in examination
o Most recent criteria for juvenile fibromyalgia were adapted from adult 2010 ACR criteria
for fibromyalgia (sensitivity 83.8%, specificity 89.4%) and published in 2016
Adapted Juvenile Fibromyalgia Diagnostic Criteria
Diagnosis requires all 3 of the following criteria:
1. Widspread pain index (WPI) ≥7 points and symptom severity (SS) scale ≥5 points or WPI
3-6 points and SS scale ≥9 points (see next page for WPI and SS scale)
2. Symptoms have been present at a similar level for at least 3 months
3. Patient does not have a disorder that would otherwise explain the pain
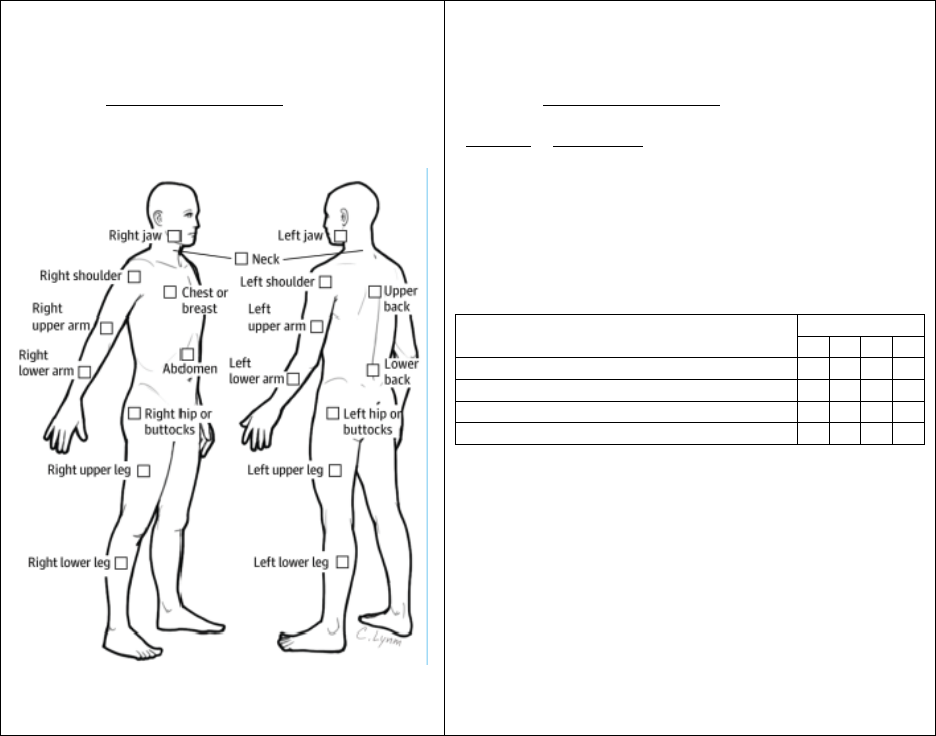
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 93
Widespread Pain Index
(1 point per check box; score range 0-19 points)
Patient instructed to indicate if they have had pain or
tenderness during the past 7 days in the areas
shown below and to check the boxes in the diagram
for each area in which they have had pain or
tenderness.
Symptom Severity Scale
(Score range 0-12 points)
Patient instructed to indicate the severity of the following
symptoms during the past 7 days using the scale below:
Severity
Description
0
No problem
1
Slight or mild problem: generally mild or
intermittent
2
Moderate problem: often present and/or at
moderate level of intensity
3
Severe problem: continuous, life-disturbing
symptoms
Symptoms
Severity
0
1
2
3
Fatigue
Trouble thinking or remembering
Waking up tired (unrefreshed)
Somatic symptoms* in general
* Somatic symptoms include: muscle pain, irritable bowel
syndrome, fatigue, thinking or remembering problem,
weakness, headache, abdominal pain or cramps, nausea,
loss of appetite, numbness or tingling, dizziness, insomnia,
depression, constipation, chest pain, shortness of breath,
blurred vision, dry mouth, dry eyes, itching, wheezing,
ringing in ears, heartburn, hair loss, easy bruising, frequent
or painful urination, bladder spasms.
Final SS score is sum of severity of first 3 symptoms
(fatigue, waking unrefreshed and cognitive symptoms
each on a scale of 0-3 points) plus the score of
severity of somatic symptoms in general (0-3 points).
Criteria and scales adapted from:
1. Zemel L, Blier PR. Juvenile fibromyalgia: A primary pain or pain processing disorder. Semin Pediatr
Neurol 2016; 23:231-41.
2. Clauw, DJ. Fibromyalgia: A clinical review. JAMA 2014. 311(15):1547-55.
Treatment strategies for chronic pain in children and adolescents that are supported by
research evidence include:
o Education about chronic pain
o Progressively increasing aerobic physical activity over time to a target of 60 minutes
daily
o Improving sleep hygiene, including consistent bed and waking times, and eliminating
long naps during the day
o Learning coping strategies for chronic pain
o Counselling, cognitive behavioural therapy (CBT), other psychotherapy and/or
biopsychosocial approaches to manage anxiety, low mood, and other consequences
and contributors to pain
o Intensive interdisciplinary pain treatment, including physiotherapy, recommended for
more severe pain-related disability
Medications less effective in childhood fibromyalgia
Better outcomes in children compared to adults

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 94
Complex Regional Pain Syndrome (CRPS) Type I (previously known as Reflex
Sympathetic Dystrophy)
Chronic pain often involving peripheral extremity (lower extremities more common in kids)
Initiating mild injury or cause of immobilization can lead to CRPS Type I
Continuing pain, allodynia, and/or hyperalgesia in which pain is disproportionate to inciting
event
Associated autonomic signs, including swelling, changes in skin blood flow leading to
discolouration, and/or abnormal sweating in the region of pain
Diagnosis using Budapest clinical criteria (see below) involves exclusion, therefore no other
condition should account for the degree of pain and dysfunction
Treatment involves intense physiotherapy with manipulation of extremity with goal to restore
function; another potential treatment option is desensitization
Complex Regional Pain Syndrome Type II
Pain caused by nerve injury, but not limited to distribution of injured nerve
Similar to type I in symptoms and treatment
References:
1. Weiss JE, Stinson JN. Pediatric pain syndromes and non-inflammatory musculoskeletal
pain. Pediatr Clin North Am 2018; 65(4):801-26.
2. Zemel L, Blier PR. Juvenile fibromyalgia: A primary pain or pain processing disorder.
Semin Pediatr Neurol 2016; 23:231-41.
3. Sherry DD, et al. The treatment of juvenile fibromyalgia with an intensive physical and
psychosocial program. J Pediatr 2015; 167(3):731-7.
4. Hechler TH, et al. Systematic review on intensive interdisciplinary pain treatment of
children with chronic pain. Pediatrics 2015; 136(1):115-27.
5. Kashikar-Zuck S, et al. Longitudinal evaluation of patient-reported outcomes
measurement information systems measures in pediatric chronic pain. Pain 2016;
157(2):339-47.
6. Weissmann R, Uziel Y. Pediatric complex regional pain syndrome: a review. Pediatr
Rheumatol 2016; 14(29):1-10.
Budapest Clinical Criteria for Complex Regional Pain Syndrome
Diagnosis requires all 4 of the following critieria:
1. Continuing pain, disproportionate to inciting event
2. At least 1 symptom (reported) in 3 or more categories*
3. At least 1 sign (at evaluation) in 2 or more categories*
4. No other diagnosis can better explain the patient’s signs and symptoms
* Categories
Sensory: hyperesthesia, hyperalgesia or allodynia
Vasomotor: temperature or skin colour asymmetry
Sudomotor/Edema: edema or sweating asymmetry
Motor/Trophic: decreased range of motion, motor dysfunction, trophic changes in skin,
hair and nails

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 95
12B. Hypermobile Joint Syndrome
Joint pain caused by idiopathic increased flexibility – may be generalized or local
Pain typically occurs after activity
More common in females
Need to consider and exclude syndromes associated with generalized joint hypermobility
(e.g. Ehlers-Danlos, Marfan, Down, Turner, Stickler and osteogenesis imperfecta
syndromes)
Several different sets of criteria for diagnosis
Beighton Criteria for Hypermobile Joint Syndrome
Able to touch thumb to volar surface of forearm (1 point each for left and right)
Able to hyperextend 5
th
finger MCP joint to 90 degrees (1 point each for left and right)
Able to hyperextend elbows >10 degrees (1 point each for left and right)
Able to hyperextend knees >10 degrees (1 point each for left and right)
Able to touch palms to floor with knees extended (1 point)
Diagnosis requires ≥6/9 points prior to puberty and ≥5/9 points after puberty
Additional features consistent with hypermobility include:
o Flat feet
o Able to sit in “W” position
o Able to touch elbows behind back
o Able to put heel behind head
Treatment
o Education and reassurance
o Activity modification (avoid exacerbating activity)
o Physiotherapy to strengthen muscles around affected joints
o Orthotics and supportive footwear
o Cognitive behavioural therapy for more severely affected individuals
Course
o Can predispose to injuries in sports
o Does not seem to increase prevalence of joint dislocations in early teens
o In general, quality of life may be lower due to frequent joint pain
References:
1. Weiss JE, Stinson JN. Pediatric pain syndromes and non-inflammatory musculoskeletal
pain. Pediatr Clin North Am 2018; 65(4):801-26.
2. Cattalini M, et al. When flexibility is not necessarily a virtue: a review of hypermobility
syndromes and chronic or recurrent musculoskeletal pain in children. Pediatr Rheumatol
Online J 2015; 13(1):40.
3. Pacey V, et al. Joint hypermobility syndrome: A review for clinicians. J Paediatr Child
Health 2015; 51(4):373-80.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 96
SECTION 13 – PEDIATRIC RHEUMATOLOGY EMERGENCIES
13A. Introduction to Pediatric Rheumatologic Emergencies
Can present with a wide spectrum of clinical illness, affecting virtually any organ
Prompt recognition and treatment may be organ and even life saving
May occur in the context of a pre-existing rheumatic disease or may be the initial
presentation
13B. Neonatal Lupus Erythematosus with Complete Heart Block (CHB)
85% of neonates with CHB have transplacentally acquired maternal antibodies to Ro/SSA or
La/SSB
Prevalence of CHB is 0.65-2% in infants of anti-Ro/SSA women; in affected mother,
likelihood of recurrence is 19%
1 year mortality up to 54% if untreated
Rheumatology consultation may be requested urgently for complete heart block with signs
of active inflammation (such as pericardial effusion or carditis), congestive heart failure or
antenatal fetal hydrops
Clinical Presentation
o Bradycardia with potential congestive heart failure (CHF)
o May already have been diagnosed antenatally
o May manifest other findings typical of NLE such as rash, hepatitis and cytopenias
Diagnostic Investigations
o Confirm CHB with electrocardiogram
o Cardiology assessment with echocardiogram to assess for active inflammation or
endocardial fibroelastosis (EFE)
o Presence of antinuclear antibodies, specifically those against Ro/SSA and La/SSB in
maternal and neonatal serum
o Elevated troponin levels may indicate secondary myocardial ischemia
Treatment
o Infants with complete heart block may need pacemaker soon after birth
o If active inflammation is seen on echocardiogram, may consider steroids +/- IVIG
(treatment will depend on presence of CHF/myocarditis and EFE)
References:
1. Izmirly PM, et al. Progress in the pathogenesis and treatment of cardiac manifestations
of neonatal lupus. Curr Opin Rheumatol 2017: 29(5):467-72.
2. Hornberger LK, Al Rajaa N. Spectrum of cardiac involvement in neonatal lupus. Scan J
Immunol 2010;72(3):189-97.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 97
13C. Macrophage Activation Syndrome
Macrophage activation syndrome (MAS) is a potentially life-threatening multisystem
inflammatory condition
Consider in the broad differential of an unexplained persistently febrile child, especially in
the presence of pancytopenia – a high index of suspicion is required
MAS may complicate a number of autoimmune diseases (e.g. systemic arthritis/JIA, SLE,
Kawasaki disease most commonly)
May occur at any time during the disease course (especially following a change in therapy)
or may be part of the initial presentation
Classified as a form of secondary hemophagocytic lymphohistiocytosis (HLH)
o Primary HLH is an inherited multi-system inflammatory disease caused by genetic
abnormalities affecting natural killer cell, macrophage and T cell function
o Similar abnormalities have recently been identified in patients with systemic JIA
o Secondary HLH in children can also be associated with malignancy or infection,
especially EBV
o Primary and secondary HLH share similar clinical and biochemical features
o Recent development of an MAS/HLH score to discriminate between primary HLH and
MAS includes age, splenomegaly, neutrophil count, platelet count, hemoglobin and
fibrinogen
Diagnostic clinical and laboratory features of MAS
o Fever (continuous/persistent)
o Splenomegaly
o Cytopenias (anemia, thrombocytopenia, neutropenia) or, in systemic JIA, may see
decrease in previously elevated cell counts
o Elevated triglycerides
o Decreased fibrinogen
o Elevated ferritin
o Hemophagocytosis on bone marrow, lymph node, liver or spleen biopsy
Other important clinical and laboratory features
o Bleeding, bruising, petechiae, due to DIC-like picture with prolonged INR/PTT, elevated
D-dimers
o Hepatic dysfunction with hepatomegaly, elevated bilirubin and liver enzymes
o Elevated LDH
o Persistently raised CRP, but decreasing ESR (due to consumption of fibrinogen)
o CNS dysfunction, including headache, confusion, seizures, and coma
o Respiratory distress including ARDS, pulmonary dysfunction
o Lymphadenopathy
o Changes in blood pressure and heart rate
o MAS may be life-threatening and can result in death
o Critically important to monitor clinical features and trends in laboratory investigations
Diagnostic criteria
o No single universally-accepted diagnostic criteria for MAS
o Different criteria using a range of abnormal laboratory values have been proposed for
various diseases
o Most criteria involve a combination of the features listed above
o A high index of suspicion is needed to make the diagnosis

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 98
Additional urgent investigations prior to starting treatment (especially prior to IVIG)
o Cultures of blood, urine and throat should be ordered to rule out an underlying bacterial
infection since it will take time to receive results
o Infectious serology and PCR (e.g. EBV, CMV, Parvovirus B19, Herpes viruses) may
be helpful to diagnose an underlying viral infection in primary or secondary HLH
o If the child does not have an established diagnosis and a systemic rheumatologic
condition is suspected, autoantibodies (e.g. ANA, ENA panel, rheumatoid factor,
ANCA) and direct antiglobulin test should be ordered
o Soluble CD163, soluble IL-2 receptor, NK cell function and lymphocyte typing may be
helpful to identify underlying immune dysfunction and/or monitor inflammation
Treatment
o Very close monitoring of labs, vital signs, and fluid input/output
o All patients require supportive management
Fluids for hypotension
Blood products (platelets, red blood cells)
Respiratory support
o Consider informing and/or involving the pediatric intensive care unit early – if site does
not have ability to provide critical care, consider transfer to a different institution
o If patient is critically ill and complete evaluation is not possible, additional treatment
should be commenced without delay
o If infection suspected, concurrent treatment with appropriate antimicrobial therapy
should be started
o Immunosuppressive therapy
IVIG often used initially during diagnostic work-up, but is rarely sufficient
Current HLH protocol involves a step-wise algorithm starting with high-dose or
pulse IV Corticosteroids (may use Dexamethasone or Methylprednisolone) and
followed by addition of Cyclosporine and then Etoposide if there is no improvement
Plasmapheresis has been used in life-threatening disease
Case series suggest that biologic agents, in particular Anakinra (anti-IL-1), may be
effective treatments for MAS
In children with primary HLH or refractory HLH, bone marrow transplant is definitive
treatment
References:
1. Sen ES, et al. Macrophage activation syndrome. Indian J Pediatr 2016; 83(3):248-53.
2. Minoia F, et al. Development and initial validation of the macrophage activation
syndrome/primary hemophagocytic lymphohistiocytosis score, a diagnostic tool that
differentiates primary hemophagocytic lymphohistiocytosis from macrophage activation
syndrome. J Pediatr 2017;187:72-8.
3. Borgia RE, et al. Features, treatment, and outcomes of macrophage activation syndrome
in childhood-onset systemic lupus erythematosus. Arthritis Rheumatol 2018;70(4):616-
624.
4. Ravelli A, et al. 2016 Classification criteria for macrophage activation syndrome
complicating systemic juvenile idiopathic arthritis: a European league against
rheumatism/American College of rheumatology/paediatric rheumatology international
trials organization collaborative initiative. Ann Rheum Dis 2016;75(3):481-9.
5. Wang W, et al. Macrophage activation syndrome in Kawasaki disease: more common
than we thought? Semin Arthritis Rheum 2015; 44(4):405-10.

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 99
13D. Pulmonary Renal Syndrome
Should be considered in any child presenting with respiratory distress and renal involvement
Clinical presentation of diffuse alveolar hemorrhage in combination with rapidly progressive
glomerulonephritis
May be rapidly fatal from devastating pulmonary hemorrhage or progressive renal failure
Causes of pulmonary renal syndrome
Specific
Systemic lupus erythematosus (SLE)
Granulomatosis with polyangiitis (GPA, formerly Wegener granulomatosis)
Microscopic polyangiitis (MPA)
Eosinophilic granulomatosis with polyangiitis (EGPA, formerly Churg-
Strauss)
Henoch-Schönlein purpura (HSP, now also known as IgA vasculitis)
Goodpasture syndrome
Non-
specific
Pulmonary edema
Pulmonary embolism in a child with renal disease
Pulmonary infection
Renal disease in a child with pulmonary disease, usually infection
Hemolytic uremic syndrome
IgA nephropathy
Clinical Presentation
o Dyspnea and cough associated with hypoxemia in room air
o Frank hemoptysis may not be present in all cases
o Renal involvement/failure: oliguria, hypertension, nephritic syndrome, nephrotic
syndrome
Diagnostic Investigations
o Tests to assess presence of pulmonary hemorrhage or vasculitis
Complete blood count showing anemia (often microcytic) or decreasing hemoglobin,
elevated reticulocyte count
Chest X-ray may show diffuse alveolar infiltrates
Chest CT may show patchy ground glass opacities or nodules
Pulmonary function tests may show an increase in DLCO consistent with intra-
alveolar bleeding
Bronchoscopy may demonstrate fresh blood and bronchalveolar lavage (BAL)
demonstrates presence of red blood cells and hemosiderin-laden macrophages; fluid
should also be sent for culture to exclude infection
o Tests to assess presence of renal involvement
Urinalysis demonstrating proteinuria, hematuria, cellular casts and/or elevated urine
protein:creatinine ratio
Increases in creatinine and/or urea

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 100
o Tests to determine underlying cause of pulmonary renal syndrome
Autoantibodies and immune markers:
Positive ANCA in GPA, MPA, EGPA (see Section 5)
ANA, anti-dsDNA, antibodies to extractable nuclear antigens, and
antiphospholipid antibodies may be positive in SLE
Anti-glomerular basement membrane (GBM) antibodies seen in Goodpasture
syndrome
Complement levels decreased in SLE
Renal biopsy:
ANCA-associated vasculitis: pauci-immune necrotizing crescentic
glomerulonephritis
SLE: glomerular immune deposits with histologic changes of lupus nephritis
Goodpasture syndrome: IgG deposition along glomerular basement membrane
with crescentric changes
HSP: deposition of IgA-containing immune complexes in glomeruli with
mesangial cell proliferation, glomerular sclerosis and crescent formation
Skin biopsy:
HSP: leukocytoclastic vasculitis with IgA deposits
SLE: immunofluorescence demonstrates immunoglobulins and complement at
the dermal-epidermal junction; may see damage of keratinocytes, follicular
plugging, basal layer vacuolation, perivascular infiltrates and dermal mucin
deposition
Treatment
o Early recognition and management of pulmonary renal syndrome is critical
o Initial therapy is identical for any underlying cause of pulmonary renal syndrome and
should be started promptly
o Supportive therapy may include oxygen, intubation, ventilation and/or dialysis
o Initial immunomodulatory therapy with pulse IV methylprednisolone followed by high
dose prednisone (1-2 mg/kg/day)
o Cyclophosphamide or Rituximab may be used depending on the underlying disease
o Addition of plasmapheresis may be considered; commonly done for anti-GBM disease
but benefits less clear for ANCA-associated vasculitis (PEXIVAS trial)
o If concurrent infection cannot be excluded, appropriate anti-microbial coverage should
be considered
References:
1. West SC, et al. Pulmonary Renal Syndrome: a life threatening but treatable condition.
Postgrad Med J 2013: 89(1051):274-83.
13E. Catastrophic Antiphospholipid Syndrome (APS)
A severe variant of the classic APS, characterized by:
o Multiple organ dysfunction and failure developing over short period of time
o Histopathological evidence of multiple small vessel occlusions, although the patient may
not have obvious thrombosis
o Laboratory confirmation of the presence of antiphospholipid antibodies (aPL), usually in
high titre

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 101
Multisystem microvascular thrombosis with a secondary systemic inflammatory response
syndrome (SIRS) due to tissue damage
2/3 of patients have an underlying trigger (infection, surgery, trauma, malignancy, and flares
of SLE) and children are more likely to have infectious trigger compared to adults
Catastrophic APS more likely to be first manifestation of APS in children compared to adults
Majority of catastrophic APS patients do not have underlying rheumatic disease
Occurs in <1% of patients with APS; mortality rate of 33-50%
Clinical Presentation
o May be mistaken for overwhelming sepsis
o Cardiopulmonary manifestations are the most frequent at presentation
May look like acute respiratory distress syndrome
Pulmonary embolus or alveolar hemorrhage may occur
o CNS features are next most common
Cerebral infarction, seizures, and encephalopathy
Cerebral venous sinus thrombosis
o Renal and abdominal involvement is common
Renal failure, proteinuria, significant abdominal pain
80% of patients experience an intra-abdominal thrombotic event over the course of an
episode
o Clinical signs of systemic inflammation and lab features of DIC
Diagnostic Criteria for Catastrophic Antiphospholipid syndrome
Definite diagnosis requires all of the following criteria:
Evidence of vessel occlusion, or effect of vessel occlusion, in ≥3 organs or tissues
Occurrence of diagnostic features simultaneously or in <1 week
Histopathologic evidence of small vessel occlusion in at least one affected organ or tissue
Presence of antiphospholipid antibodies (lupus anticoagulant, anti-cardiolipin) persistent
over at least 6 weeks
Probable diagnosis if:
Only 2 organ systems affected, or
Occurrence of two diagnostic features in <1 week and another within 4 weeks, or
Histopathologic demonstration of small vessel occlusion not possible
Unable to demonstrate persistence of antibodies due to death
Diagnostic Investigations
o Tests to confirm presence of thrombotic disorder
Look for organ infarction (kidney, spleen, or bowel) on imaging or organ failure
(cardiac or renal) with markers of DIC, coagulation dysfunction, and/or peripheral
destruction of blood elements
May require tissue sample
o Tests to confirm presence of aPL antibodies
Lupus anticoagulant, anti-cardiolipin, anti-β2-glycoprotein I

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 102
Novel aPL antibodies (e.g. anti-phosphatidylserine-prothrombin antibodies) also
reported in catastrophic APS cases but testing not widely available
o Investigate underlying triggers for the episode
Cultures and infectious serology to assess for infection (respiratory, skin, urinary tract)
Bone marrow biopsy or imaging may be needed to assess for an underlying
malignancy
Investigations for a systemic inflammatory condition, such as SLE, may be indicated if
the child does not have a previous diagnosis
Treatment
o Patients are often critically ill
o ICU support should be available and anticipated
o May need acute measures such as mechanical ventilation or dialysis
o Treatment aimed at removing triggering factor, if known, eliminating existing thrombus,
and controlling SIRS
o Empiric antibiotics until infection ruled out
o Targeting two main pathologic processes may reduce mortality
Thrombosis: treated acutely with Heparin; may also need vasodilators, fibrinolytics,
and embolectomy; long-term anticoagulation with either Low Molecular Weight
Heparin or Warfarin
SIRS: treated with systemic corticosteroids, therapeutic plasma exchange (TPE) and
IVIG (should be given after TPE)
o Other agents for severe or refractory cases include Rituximab and Eculizumab (anti-C5
agent)
References:
1. Go EJL, O'Neil KM. The catastrophic antiphospholipid syndrome in children. Curr Opin
Rheumatol 2017; 29(5):516-22.
2. Rodríguez-Pintó I, et al. CAPS Registry Project Group (European Forum on
Antiphospholipid Antibodies). Catastrophic antiphospholipid syndrome (CAPS):
descriptive analysis of 500 patients from the International CAPS Registry. Autoimmun
Rev 2016; 15:1120-24.
3. Asherson RA, et al. Catastrophic antiphospholipid syndrome: international consensus
statement on classification criteria and treatment guidelines. Lupus 2003; 12(7):530-4.
4. Litvinova E, et al. Prevalence and signification of non-conventional antiphospholipid
antibodies in patients with clinical APS criteria. Front immunol 2018;9:2971
13F. Cardiac Tamponade
Uncommon but life-threatening complication of pericarditis with effusion
Autoimmune cause identified in 13-30% of children with tamponade
May occur in children with known rheumatologic disease or as part of initial presentation
Clinical Presentation
o Typically presents with dyspnea, tachypnea and chest pain
o May have elevated jugular venous pressure, facial edema or plethora, tachycardia,
pulsus paradoxus, muffled heart sounds, and if advanced, hypotension
o Fever is common

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 103
o May see clinical features suggestive of associated rheumatic disease (e.g. SLE,
systemic JIA)
Diagnostic Investigations
o ECG typically shows sinus tachycardia and may also show low voltage QRS complexes,
ST elevation/PR segment depression, and electrical alternans
o Chest X-ray may show a large cardiac silhouette
o Echocardiography may demonstrate a moderate to large pericardial effusion, findings of
chamber collapse, respiratory variation in volumes and flows, IVC dilatation due to
increased central venous pressure
Treatment
o Initial priority is to stabilize cardiorespiratory status and to restore adequate cardiac
output by removal of pericardial fluid (pericardiocentesis)
o Temporizing measures can be used such as IV fluids or sympathomimetics
o Corticosteroids are the mainstay of acute treatment for life-threatening conditions
o Other immunosuppressive agents may be added if there is insufficient improvement or if
required to treat an underlying rheumatic disease and may include NSAIDs, Colchicine,
and Anti-IL-1 agents
References:
1. Imazio M, et al. Contemporary management of pericardial effusion: practical aspects for
clinical practice. Postgrad Med 2017; 129(2):178-186.
2. Mok GC, Menahem S. Large pericardial effusions of inflammatory origin in childhood.
Cardiol Young 2003; 13(2):131-6.
3. Maharaj SS, Chang SM. Cardiac tamponade as the initial presentation of systemic lupus
erythematosus: a case report and review of the literature. Pediatr Rheumatol Online J
2015; 13:9.
13G. Kawasaki Disease (KD) Shock Syndrome (KDSS)
Uncommon but life-threatening complication of KD
Occurs in <10% of children diagnosed with KD; older age of onset
Children often present with shock before the KD diagnosis is made and more likely to have
incomplete presentation
May have more prominent inflammatory markers in early phase and higher risk of coronary
artery dilatation
Clinical Presentation
o Hemodynamic instability with tachycardia, hypotension and poor peripheral perfusion
o May have muffled heart sounds or gallop rhythm
o Typically associated with more severe manifestations of KD, although not necessarily
longer duration of fever
o KDSS is a multisystem disease with impaired cardiac function, gastrointestinal
symptoms (e.g. vomiting), respiratory failure, encephalopathy, and acute renal injury
o More likely to demonstrate IVIG resistance

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 104
Diagnostic Investigations
o Compared to children with KD who are hemodynamically stable, children with KDSS are
more likely to have:
Higher CRP and ESR
Higher white blood cell and neutrophil counts with bands
Lower hemoglobin and platelet counts
Lower sodium and albumin levels
Consumptive coagulopathy with low platelet counts, increased D-dimers and
prolonged PTT
Emerging data shows higher levels of IL-6, IL-10 and IFN-ᵞ (if testing available at
center) may be useful to distinguish KDSS from KD
o ECG typically shows sinus tachycardia
o Echocardiography:
Impaired left ventricular systolic function with a lower ejection fraction and mitral
regurgitation
More likely to develop coronary artery abnormalities
Treatment
o Initial priority is to stabilize cardiorespiratory status
o Require careful fluid resuscitation – large fluid boluses not recommended as these may
precipitate congestive heart failure
o May require inotropic and/or vasopressor support
o IVIG and ASA remain mainstay of therapy; however, IVIG resistance is more common
and may need to progress to further therapies, such as corticosteroids (see Section 5C)
o If treated early and aggressively, most children survive without sequelae
References:
1. Ma L, et al. Clinical Manifestations of Kawasaki Disease Shock Syndrome. Clin Pediatr
(Phila) 2018; 57(4):428-35.
2. Kanegaye JY, et al. Recognition of a Kawasaki Shock Syndrome. Pediatrics 2009;
123(5):e783-9.
3. Li Y, et al. Kawasaki disease shock syndrome: clinical characteristics and possible use
of IL-6, IL-10 and IFN-γ as biomarkers for early recognition. Pediatr Rheumatol Online J
2019; 17(1):1.
13H. Renal Crisis in Systemic Sclerosis (SSc)
Rare and potentially life-threatening complication of SSc
High rate of mortality and progression into end-stage renal disease (ESRD)
Incidence of 4-6% in SSc patients, primarily in diffuse SSc
Usually develops within the first 4 years of onset of the disease
Risk factors: presence of anti-RNA polymerase antibodies, rapid progression of skin
thickening, congestive heart failure, high dose glucocorticoids,
Clinical Presentation
o Reflects thrombotic microangiopathy of kidney similar to thrombotic thrombocytopenic
purpura/hemolytic uremic syndrome
o Acute renal failure without warning signs

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 105
o Sudden onset of moderate to severe hypertension (normal blood pressure in 10% of
renal crisis cases)
o May be accompanied by hypertensive encephalopathy, congestive heart failure,
arrhythmia, or acute cerebrovascular event
Diagnostic Investigations
o Elevated creatinine, proteinuria and hematuria
o Thrombotic microangiopathy: hemolytic anemia and thrombocytopenia
o Renal biopsy findings include proliferation and thickening of arcuate and interlobar
arteriole intima, leading to narrowing or full obliteration of vessels
o CXR may demonstrate pulmonary edema
o Eye exam may identify retinal hemorrhages or exudates
o MRI/CT head may show signs of stroke
Treatment
o Rapid (within 72 hr) control of blood pressure
Provides stabilization of renal function in 70% of patients
o ACE inhibitors (Captopril most widely studied)
o Plasma exchange to be considered if intolerant to ACE inhibitor or concomitant
hemolytic microangiopathy
o Adjunctive treatment with endothelin receptor antagonist (e.g. Bosentan) or Eculizumab
(anti-C5a) if refractory
o If ESRD: dialysis and possibly transplantation
References:
1. Gordon SM, et al. Risk Factors for Future Scleroderma Renal Crisis at Systemic
Sclerosis Diagnosis. J Rheumatol 2019;46(1):85-92.
2. Zanatta E, et al. Therapy of Scleroderma Renal Crisis: State of the Art. Autoimmun Rev
2018;17(9):882-9.
3. Guillevin L and Mouthon L. Scleroderma Renal Crisis. Rheum Dis Clin N Am 2015;
41:475-88.
13I. Acute Adrenal Crisis
Many children with rheumatic diseases are treated with systermic glucocorticosteroids in
high doses to achieve disease control or lower doses for prolonged periods of time to
maintain remission
Adrenal crisis may occur during withdrawal of therapy or at times of stress (e.g. illness,
disease flare) requiring additional steroids
Patients at risk of adrenal suppression include those who have used corticosteroids for more
than a 2 week period at >2mg/kg or multiple courses totalling >3 weeks in the previous 6
months
Associated with higher mortality in the pediatric population
Clinical Presentation
o May be variable
o Many signs and symptoms are non-specific and can be mistaken for symptoms of an
intercurrent illness or the underlying condition being treated
o Signs and symptoms include:
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 106
Arthralgias, myalgias, generalized weakness
Headache
Abdominal pain, nausea, vomiting, diarrhea
Fever
Hypotension
Decreased level of consciousness, lethargy
Unexplained hypoglycemia
Hyponatremia
Seizures, coma
Treatment
o Hydrocortisone injection 100 mg/m2 (maximum 100 mg) IV/IM stat with IV normal
saline volume expansion, followed by hydrocortisone 25 mg/m2 every 6 hours
(maximum 25 mg every 6 hours)
o Consult endocrinologist on call for further advice
Prevention
o Stress dosing with hydrocortisone during illness, fever or surgery
o Education of patient and family
References:
1. Levy-Shraga Y, Pinhas-Hamiel O. Novel insights into adrenal insufficiency in childhood.
Minerva Pediatr 2014; 66(6):517-32.
2. Shulman DI, et al. Adrenal insufficiency: still a cause of morbidity and death in childhood.
Pediatr 2007; 119(2):e484-94.
3. Huber BM, et al. Adrenal insufficiency after glucocorticoid withdrawal in children with
rheumatic diseases. Acta Paediatr 2010; 99(12):1889-93.
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 107
SECTION 14 – MEDICATIONS
Medications are listed in alphabetical order by their generic names with the exception of
Corticosteroids and NSAIDs, which are listed by their categories. A table summarizes the
mechanisms of action of the monoclonal antibody (mAb) and fusion protein biologic agents.
Abatacept
o Class: biologic agent (see Biologic agents for summary table)
o Mechanism of action: Selectively inhibits co-stimulatory signal for T-cell activation
o Dose: 10 mg/kg/dose if <75 kg; 750 mg if 75-100 kg; or 1000 mg if >100 kg via IV every
2 weeks for 3 doses then every 4 weeks thereafter
o Side effects: infusion reactions, anaphylaxis, GI upset, bronchospasm, infections,
potential risk of future malignancy (very rarely)
Adalimumab
o Class: biologic agent (see Biologic agents for summary table)
o Mechanism of action: recombinant mAb that binds to circulating and cell surface TNFα
o Dose: 24 mg/m
2
/dose if <15 kg; 20 mg if 15-30 kg; or 40 mg if >30 kg via SC injection
every 2 weeks (can be given weekly when clinically indicated)
o Side effects: injection site reactions, headaches, infections, cytopenias, potential risk of
future malignancy, demyelinating disease, new or worsening heart failure (adult patients)
o Monitoring: CBC, differential, AST, ALT, creatinine every 3-6 months
Anakinra
o Class: biologic agent (see Biologic agents for summary table)
o Mechanism of action: human recombinant form of IL-1 receptor antagonist (IL-1Ra)
o Dose: 1-2 mg/kg/dose (max 100 mg) SC daily; in sJIA, may titrate up to 4 mg/kg/dose
(max 200 mg) SC daily
o Side effects: injection site reactions, flu-like symptoms, infections
o Monitoring: Neutrophil count prior to initiating; monthly for 3 months; then quarterly
Azathioprine
o Class: antimetabolic agent; purine analogue
o Mechanism of action: interferes with DNA synthesis; inhibits T cells and monocytes
o Dose: 2-3 mg/kg/day (max 150 mg) PO daily
o Side effects: nausea, diarrhea, oral ulcers, rash, cytopenias, pancreatitis, hepatotoxicity
o Monitoring: CBC, differential and liver enzymes every 2 weeks until achieve stable dose
then every 2 months; consider thiopurine methyltransferase (TPMT) genetic testing if
abnormally low CBC (e.g., neutropenia) unresponsive to dose reduction
Belimumab
o Class: biologic agent (see Biologic agents for summary table)
o Mechanism of action: human IgG1 neutralizing monoclonal antibody against B-
lymphocyte stimulating factor (also known as B-lymphocyte simulator [BLyS])
o Dose: 10 mg/kg via IV every 2 weeks for 3 doses then every 4 weeks
o Side effects: infusion reactions, nausea, diarrhea, headaches, infections, potential risk of
future malignancy (very rarely)
o Monitoring: CBC (e.g., leukopenia) and liver enzymes with each infusion
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 108
Biologic agents
Biologic Class
Medication
Mechanism of Action
Approved
in
pediatrics
(Health
Canada)
B cell depletion
Belimumab
Human monoclonal antibody directed against BLyS
Inhibits BLyS-induced proliferation of B cells and decreases
survival of autoreactive B cells
Yes
Rituximab
Chimeric mouse-human monoclonal antibody directed
against CD20 on pre-B and mature B cells
Selectively depletes B cells
No
IL-1 inhibitors
Anakinra
IL-1 receptor antagonist
Blocks IL-1 receptor to prevent pro-inflammatory signaling
(both IL-1α and IL-1β)
Yes
Canakinumab
Human monoclonal antibody directed against IL-1β
Binds to IL-1β to prevent pro-inflammatory signaling
Yes
Rilonacept
Fully human dimeric fusion protein consisting of extracellular
portion of IL-1 receptor and constant region of human
immunoglobulin
Binds to IL-1 to prevent pro-inflammatory signaling
No
IL-6 inhibitor
Tocilizumab
Humanized monoclonal antibody against IL-6 receptor
Blocks IL-6 mediated pro-inflammatory signaling
Yes
IL-12/IL-23
inhibitor
Ustekinumab
Humanized monocloncal antibody against IL-12 and IL-23
Blocks IL-12/23 to prevent pro-inflammatory signaling
No
IL-17A inhibitor
Secukinumab
Humanized monocloncal antibody against IL-17A
Blocks IL-17A mediated pro-inflammatory signaling, which is
mainly produced by T helper 17 cells
No
T cell
co-stimulatory
modulator
Abatacept
Fusion protein consisting of extracellular portion of CTLA-4
and constant region of human immunoglobulin
Blocks co-stimulation and activation of T cells
Yes
TNF inhibitors
Adalimumab
Human monoclonal antibody directed against circulating and
membrane-bound TNFα
Binds to TNFα to block pro-inflammatory signaling
May result in cell lysis in presence of complement
Yes
Certolizumab
PEGylated Fab fragment of humanized monoclonal antibody
directed against TNFα
Binds to TNFα to block pro-inflammatory signaling
No
Etanercept
Soluble fusion protein consisting of extracellular portion of
TNFα receptor and the constant region of human
immunoglobulin
Binds to circulating (but not membrane-bound) TNFα to block
pro-inflammatory signaling
Yes
Golimumab
Human monoclonal antibody directed against TNFα
Binds to TNFα to block pro-inflammatory signaling
No
Infliximab
Monoclonal human-mouse antibody directed against
circulating and membrane-bound TNFα
Binds to TNFα to block pro-inflammatory signaling
Enables antibody-dependent and complement-dependent
cytotoxicity
Yes
TNF: tumor necrosis factor; IL: interleukin; BLyS: B-lymphocyte stimulator; CTLA-4: cytotoxic T lymphocyte-
associated antigen-4; Note: suffix of monoclonal antibody (mAb) = -mab

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 109
Biosimilars
o A biosimilar is a newer type of biologic medication that is designed to be identical to an
existing biologic medication, but is created using a different process
o Uncertain whether biosimilars will have identical effects to reference biologic product
since even minor modifications may alter pharmacokinetic, immunogenetic,
glycosylation, sialylation, stability, safety, and efficacy
o Biosimilars often more cost-effective than the biologic agents that they replicated
o Rare events and long-term safety will be assessed in postmarketing surveillance studies
o Currently approved Anti-TNF biosimilars
Infliximab biosimilars: Remsina, Inflectra and Renflexis are approved for adults in
Canada, but not yet mandated for use in children
Adalimumab biosimilar: Hadlima approved for adults in Canada
Etanercept biosimilars: Brezys and Erelzi are approved for adults in Canada, while
Erelzi is mandated for use by certain Canadian provinces for children over 62 kg
(dose 50 mg SC weekly)
Canakinumab
o Class: biologic agent (see Biologic agents for summary table)
o Mechanism of action: fully human mAb targeting IL-1β
o Dose: sJIA 4 mg/kg/dose SC every 4 weeks; CAPS 2-4 mg/kg if 15-40 kg; or 150
mg (may consider 300 mg) if >40 kg via SC injection every 8 weeks
o Side effects: injection site reactions, headache, flu-like symptoms, GI upset, infections
Colchicine
o Class: alkaloid
o Mechanism of action: binds to microtubules to prevent activation, proliferation and
functioning of inflammatory cells
o Dose: 0.6-1.8 mg/day; may divide into twice daily doses if side effects
o Side effects: nausea, vomiting, diarrhea, cytopenias, rhabdomyolysis, renal failure
o Monitoring: CBC, differential, renal function
Corticosteroids
o Potent anti-inflammatory agents
o Mechanism of action: multiple anti-inflammatory actions including binding to transcription
factors (such as NF-κB) to block production of pro-inflammatory proteins; binding to
enzymes to block function of inflammatory cells; and direct inhibition of cytokines
o Commonly used corticosteroids
Prednisone (PO tablets), prednisolone (PO liquid) – equivalent
Methylprednisolone (IV) – very similar to prednisone/prednisolone
Dexamethasone (PO or IV) – superior blood-brain barrier penetration, more potent
Triamcinolone hexacetonide (intra-articular)
o Dose: depends on indication and severity of inflammation
o Side effects:
Early: increased appetite, GI upset, gastritis, mood and behaviour changes
Late: infections, Cushing syndrome (truncal obesity, moon facies, cutaneous striae),
acne, growth suppression, osteoporosis, AVN, psychosis, hypertension,
dyslipidemia, hyperglycemia, myopathy, cataracts, glaucoma
o Monitoring: clinical (including blood pressure); consider monitoring bone health carefully
if long-term corticosteroids are used
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 110
Cyclophosphamide
o Class: cytotoxic alkalating agent
o Mechanism of action: alkylating metabolites prevent cell division by crosslinking DNA
and RNA strands, particularly affecting lymphocytes (B and T cells)
o Dose: 500-1000 mg/m
2
/dose IV every 2 to 4 weeks up to 6 months
o Side effects:
Short-term: nausea, vomiting, anorexia, alopecia, oral ulcers, cytopenias,
opportunistic infections, hemorrhagic cystitis, SIADH, teratogenicity, gonadal
dysfunction
Long-term: bladder fibrosis, bladder carcinoma, fertility issues, malignancy
o Monitoring: CBC, differential on day of infusion and then days 7, 10 and 14 after infusion
to monitor cytopenias
o Special consideration: prophylaxis
Mesna administered with infusion to prevent hemorrhagic cystitis
Cotrimazole (trimethoprim-sulfamethoxazole) given 3 times weekly to prevent
opportunistic infection by Pneumocystis jirovecii
Cyclosporine
o Class: immunomodulatory agent
o Mechanism of action: inhibits calcineurin leading to inhibition of nuclear factor of
activated T cells (NF-AT) resulting in profound inhibition of T cell proliferation and
cytokine production
o Dose: 3-5 mg/kg/day PO divided twice daily; may be given by IV in MAS
o Side effects: hypertension, headache, nausea, vomiting, myalgias, renal toxicity,
hepatotoxicity, GI upset, tremor, paresthesias, gingival hyperplasia, hirsutism
o Monitoring: BP, renal function, urinalysis, CBC, differential, and liver enzymes monthly
Etanercept
o Class: biologic agent (see Biologic agents for summary table)
o Mechanism of action: fully human dimeric fusion protein that binds to circulating TNFα
o Dose: 0.4 mg/kg/dose (max 25 mg) twice weekly or 0.8 mg/kg/dose (max 50 mg) weekly
via SC injection
o Side effects: injection site reactions, headaches, flu-like symptoms, infections,
cytopenias, potential risk of future malignancy, demyelinating disease, new or worsening
heart failure (adult patients)
o Monitoring: CBC, differential, AST, ALT, creatinine every 3-6 months
Hydroxychloroquine
o Class: disease-modifying antirheumatic drug (DMARD); antimalarial agent
o Mechanism of action: interferes with antigen processing and antigen-antibody
interactions, inhibits nucleic acid and protein synthesis
o Dose: up to 6.5 mg/kg/day (max 400 mg) PO daily
o Side effects: nausea, anorexia, skin rash, headache, dizziness, photosensitivity, retinal
toxicity
o Monitoring: eye examinations every 12 months to assess for retinal deposits
Infliximab
o Class: biologic agent (see Biologic agents for summary table)
o Mechanism of action: monoclonal chimeric human-mouse antibody that binds to
circulating and cell surface anti-TNFα
A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 111
o Dose: 6-10 mg/kg/dose on week 0, 2, 6 then every 4 to 8 weeks (may occasionally
require higher doses)
o Side effects: injection site reactions, headaches, infections, cytopenias, potential risk of
future malignancy, demyelinating disease, new or worsening heart failure
o Monitoring: CBC, differential, AST, ALT, creatinine every 3-6 months
o Special consideration: human anti-chimeric antibodies (HACAs) can develop and
decrease efficacy and increase risk of infusion reactions; incidence is lower in patients
receiving continuous (rather than intermittent) therapy and concomitant
immunosuppressive therapy (e.g., methotrexate)
IVIG
o Class: biologic agent; plasma-derived protein
o Mechanism of action: multiple anti-inflammatory mechanisms including inhibition of
antibody-mediated cytotoxicity; attenuation of complement-mediated damage;
modulation of cytokine production; and neutralization of superantigens
o Dose: 2 g/kg/dose IV
o Side effects: infusion reactions, haemolysis, aseptic meningitis 18-36 hours post infusion
(headache and vomiting), cough, acute renal failure
o Special consideration: need to delay future immunizations with live-virus vaccines by 11
months due to possible inefficacy of subsequent vaccines for this time period
Leflunomide
o Class: disease-modifying antirheumatic drug (DMARD)
o Mechanism of action: inhibits enzyme involved in DNA synthesis and interferes with
lymphocyte proliferation
o Dose: 10 mg PO every other day for patients <20 kg, 10 mg PO daily for patients 20-40
kg, 20 mg PO daily for patients >40 kg
o Side effects: oral ulcers, nausea, vomiting, allergic rash, alopecia, leukopenia,
hepatotoxicity, teratogenicity
o Monitoring: CBC, differential, liver enzymes, creatinine at baseline, in 2-4 weeks after
starting and then every 8-12 weeks
o Special consideration: need to discuss alcohol avoidance and birth control
Methotrexate
o Class: disease-modifying antirheumatic drug (DMARD)
o Mechanism of action: inhibitor of dihydrofolate reductase enzyme (folate pathway) and
DNA synthesis
o Dose: 10-15 mg/m
2
/dose (max 25 mg) PO or SC weekly (note: often better response
and fewer side effects with SC route)
o Side effects: GI upset, oral ulcers, hepatotoxicity, bone marrow suppression,
teratogenicity
o Monitoring: CBC, differential, liver enzymes, creatinine at baseline, in 2-4 weeks and
then every 8-12 weeks
o Special considerations: need to discuss alcohol avoidance and birth control; administer
with folic or folinic acid to minimize side effects
Mycophenolate mofetil
o Class: antimetabolic agent
o Mechanism of action: inhibits enzyme in DNA synthesis leading to inhibition of B and T
cell proliferation

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 112
o Dose: 800-1200 mg/m
2
/day (max 3000 mg/day) PO divided twice daily
Typical starting dose is 250 mg daily
May use drug levels (MMF kinetics) to optimize dose where available
o Side effects: GI upset, headaches, cytopenias, infections, teratogenicity
o Monitoring: CBC, differential, liver enzymes, creatinine at baseline, in 2-4 weeks and
then every 8-12 weeks
Non-steroidal anti-inflammatory drugs (NSAIDs)
o First-line anti-inflammatory agents for arthritis
o Mechanism of action: inhibit cyclooxygenase (COX) to block production of pro-
inflammatory prostaglandins
o Dose: see table below for doses of commonly used NSAIDs
o Side effects: abdominal pain, nausea, vomiting, diarrhea, constipation, pseudoporphyria
(Naprosyn), gastritis, GI bleeding, rare renal toxicity, rare hepatotoxicity, rare ototoxicity
o Monitoring: no routine monitoring necessary if NSAIDs used as monotherapy, but
hemoglobin, renal function and liver enzymes with clinic visits
NSAID
Dose
Comments
Aspirin/ASA
High dose (anti-inflammatory): 50-100 mg/kg/day
PO div QID
Low dose (anti-platelet): 3-5 mg/kg/day PO OD
Used mostly in the setting of
Kawasaki disease and acute
rheumatic fever
Celecoxib
50 mg PO BID if 10-25 kg
100 mg PO BID if >25 kg
Selective COX-2 inhibitor;
expensive
Ibuprofen
20-40 mg/kg/day PO div TID or QID
Commonly used in childhood JIA
Indomethacin
2-3 mg/kg/day (max 150 mg/day) PO div TID
Commonly used in ERA and sJIA,
can be compounded into liquid
Meloxicam
0.125 mg/kg (max 15 mg/day) PO daily
Used in JIA, can be compounded
into liquid
Naproxen
20 mg/kg/day (max 500 mg/dose) PO div BID
Frequently used in childhood JIA
Piroxicam
0.4 mg/kg (max 20 g/day) PO daily
Used in JIA, capsule may be
opened and sprinkled on food
ERA: enthesitis related arthritis; sJIA: systemic juvenile idiopathic arthritis; COX = cyclooxygenase
Pamidronate
o Class: bisphosphonate
o Mechanism of action: inhibits bone resorption, decreases mineralization by inhibiting
osteoclast activity
o Dose: 1 mg/kg/day if >3 yrs (max 60 mg/day) monthly for 3 months (note: first dose to be
given over 2 days)
o Side effects: bone pain, fever, headaches, lethargy, fetal toxicity, unclear risk of
osteonecrosis of the jaw
o Monitoring: calcium, phosphate, creatinine, ALP and PTH prior to each infusion

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 113
Rilonacept
o Class: biologic agent (see Biologic agents for summary table)
o Mechanism of action: fully human dimeric fusion protein that blocks IL-1 by acting as a
soluble decoy receptor; also known as “IL-1 Trap”
o Dose: loading dose 4.4 mg/kg/dose (max 320 mg) then 2.2 mg/kg/dose SC weekly (max
160mg)
o Side effects: injection reactions, infections, dyslipidemia, potential risk of future
malignancy
o Monitoring: CBC and liver transaminases after 4 weeks and then every 3 months; serum
lipid monitoring added 8-12 weeks after initiation
Rituximab
o Class: biologic agent (see Biologic agents for summary table)
o Mechanism of action: chimeric mouse-human monoclonal antibody that binds to the B
cell CD20 receptor (on pre-B and mature B cells but not on stem cells or plasma cells)
o Dose: dosing may depend on indication; 375 mg/m
2
once weekly for 2-4 doses or 750
mg/m
2
on days 1 and 15; for polyarticular RF-positive JIA patients 500 mg/m
2
(max 1000
mg) every 2 weeks for 2 doses or 375 mg/m
2
once weekly for 4 doses
o Side effects: infusion reactions, allergic reaction, hypogammaglobulinemia, infection,
potential risk of future malignancy, progressive multifocal leukoencephalopathy (PML)
o Monitoring: screen for hepatitis B; check B cell numbers before and 1 month after
infusion; quantitative immunoglobulins every 3 months; follow liver transaminases
o Special considerations:
Prophylaxis with cotrimazole (trimethoprim-sulfamethoxazole) given 3 times weekly
to prevent opportunistic infection by Pneumocystis jirovecii
Human anti-chimeric antibodies (HACAs) can develop and decrease efficacy and
increase risk of infusion reactions
Recommend screening for hepatitis status
Recommend pre-medication with Hydrocortisone
Sulfasalazine
o Class: disease-modifying antirheumatic drug (DMARD); analogue of 5-ASA linked to a
sulfonamide
o Mechanism of action: inhibits enzymes and transcription factors involved in production of
pro-inflammatory cytokines
o Dose: 50 mg/kg/day (max 3 g daily) PO divided twice daily; typically start at 10
mg/kg/day and increase weekly over 4 weeks to target dose
o Side effects: nausea, vomiting, rash, oral ulcers, photosensitivity, cytopenias,
hypogammaglobulinemia, hepatotoxicity, allergy (Stevens-Johnson syndrome)
o Monitoring: CBC, differential and liver enzymes every 2 months, immunoglobulin levels
every 6 months
o Special consideration: contraindicated if history of allergy to sulfonamide antibiotics
Tocilizumab
o Class: biologic agent (see Biologic agents for summary table)
o Mechanism of action: humanized monoclonal antibody that binds both soluble and
membrane-bound IL-6 receptor
o Dose:
Systemic JIA: 12 mg/kg/dose if <30 kg; or 8 mg/kg/dose (max 800 mg) if ≥ 30 kg via
IV every 2 weeks

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 114
Polyarticular JIA: 10 mg/kg/dose if <30 kg; or 8 mg/kg/dose (max 800 mg) if ≥ 30 kg
via IV every 4 weeks
o Side effects: infusion reactions, headaches, GI upset, hepatotoxicity, dyslipidemia,
neutropenia, thrombocytopenia, GI perforation, infection, potential risk of future
malignancy
o Monitoring: AST, ALT, absolute neutrophil count at baseline, at second infusion and then
every 2-4 weeks; lipid panel 4-8 weeks after start of treatment then every 6 months
Tofacitinib
o Class: Janus Kinase inhibitor
o Mechanism of action: interferes with Jak-stat system (Jak 3 and Jak 1) and subsequent
production of selective interleukins and interferons
o Dose: dosing in adult rheumatoid arthritis 5 mg PO twice daily
o Side effects: infections, viral reactivation, anemia, thrombocytopenia, neutropenia,
lymphopenia, hypercholesterolemia, increased liver transaminases, GI perforation,
potential risk of future malignancy
Secukinumab
o Class: biologic agent (see Biologic agents for summary table)
o Mechanism of action: humanized monoclonal antibody that binds soluble and membrane
bound IL-17A
o Dose: for ankylosing spondylitis and psoriatic arthritis 150 mg SC every week for 4
weeks and then every 4 weeks
o Side effects: infections, flu-like symptoms, injection site reactions, potential risk of future
malignancy, may un-mask or lead to flares of inflammatory bowel disease
Ustekinumab
o Class: biologic agents (see Biologic agents for summary table)
o Mechanism of action: humanized monoclonal antibody that binds soluble and membrane
bound IL-12 and IL-23
o Dose: for psoriasis 0.75 mg/kg if <60 kg, 45 mg if 60-100 kg or 90 mg for >100 kg; given
by SC injection at baseline, in 4 weeks and then every 12 weeks
o Side effects: infections, injection site reactions, potential risk of future malignancy, rare
reports of posterior reversible encephalopathy syndrome

A RESIDENT’S GUIDE TO PEDIATRIC RHEUMATOLOGY
© Copyright of The Hospital For Sick Children Page 115
APPENDIX – HELPFUL RESOURCES IN PEDIATRIC RHEUMATOLOGY
Textbooks
Petty RE, Laxer RM, Lindsley CB, Wedderburn L. Textbook of Pediatric Rheumatology, Seventh
Edition. 2015: Saunders Elsevier Publishing.
Foster HE, Brogan P. Paediatric Rheumatology. 2018: Oxford University Press.
Laxer RM, Sherry DD, Hashkes PN Pediatric Rheumatology in Clinical Practice. 2
nd
edition.
2016: Springer-Verlag.
Hashkes PJ, Laxer RM, Simon A. Textbook of Autoinflammation. 2019: Springer Nature
Switzerland.
Monographs
Li SC, Higgins GC, Eds. Pediatric Rheumatology. Pediatr Clin North Am 2018; 65(4).
Laxer RM, Sherry DD, Eds. Pediatric Rheumatology. Pediatr Clin North Am 2012; 59(2).
Rouster-Stevens KA, et al. Choosing Wisely: The American College of Rheumatology’s Top 5
for Pediatric Rheumatology. Arthritis Care Res 2014; 66(5):649-57.
Internet Resources for Images in Rheumatology
American College of Rheumatology Image Bank: http://images.rheumatology.org/
Rheumatlas website: http://rheumexamatlas.com/atlas/
Internet Resources for Patients
Paediatric Rheumatology InterNational Trials Organisation (PRINTO) – provides information on
all childhood rheumatic diseases in many languages:
https://www.printo.it/pediatric-rheumatology/IE/info/IE
RheumInfo – provides easy-to-read pictopamphlets on medications
https://rheuminfo.com/
About Kids Health – provides information on multiple childhood rheumatic diseases from a
general pediatric perspective
https://www.aboutkidshealth.ca/
Teens Taking Charge (Managing JIA Online) – provides resources for adolescent JIA patients
https://www.aboutkidshealth.ca/Article?contentid=1087&language=English
Arthritis Australia - provides information on JIA and chronic pain in children
https://arthritisaustralia.com.au/managing-arthritis/arthritis-and-children/
We are interested in your feedback on the guide! If you have comments or questions,
please feel free to contact us via email at pedrheumguide@gmail.com.

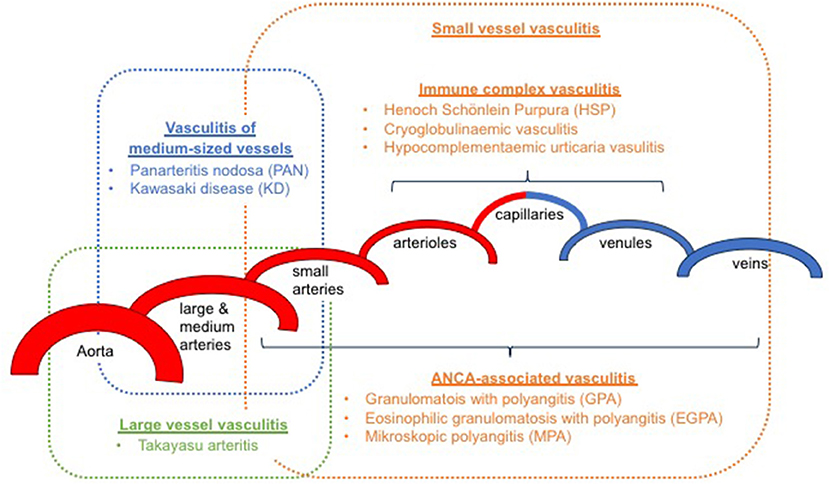{kind=link}