
1
Annex 1 : Manufacture of Sterile Products 1
2
Document map 3
Section Number General overview
1. Scope Includes additional areas (other than sterile products) where the
general principles of the annex can be applied.
2. Principle General principles as applied to the manufacture of sterile
products.
3. Pharmaceutical Quality
System (PQS)
Highlights the specific requirements of the PQS when applied
to sterile products.
4. Premises General guidance regarding the specific needs for premises
design and also guidance on the qualification of premises
including the use of Barrier Technology.
5. Equipment General guidance on the design and operation of equipment.
6. Utilities Guidance with regards to the special requirements of utilities
such as water, gas and vacuum.
7. Personnel Guidance on the requirements for specific training, knowledge
and skills. Also gives guidance to the qualification of
personnel.
8. Production and specific
technologies
Discusses the approaches to be taken with regards to aseptic
and terminal sterilization processes. Discusses approaches to
sterilization of products, equipment and packaging
components. Also discusses different technologies such as
lyophilization and Form-Fill-Seal where specific requirements
apply.
9. Viable and non-viable
environmental and process
monitoring
This section differs from guidance given in section 4 in that the
guidance here applies to ongoing routine monitoring with
regards to the design of systems and setting of action limits
alert levels and reviewing trend data.
The section also gives guidance on the requirements of Aseptic
Process Simulation (APS).
10. Quality control (QC) Gives guidance on some of the specific Quality Control
requirements relating to sterile products.
11. Glossary Explanation of specific terminology.
4
5

2
1 Scope 6
7
The manufacture of sterile products covers a wide range of sterile product types (active substance, 8
sterile excipient, primary packaging material and finished dosage form), packed sizes (single unit to 9
multiple units), processes (from highly automated systems to manual processes) and technologies (e.g. 10
biotechnology, classical small molecule manufacturing and closed systems). This Annex provides 11
general guidance that should be used for the manufacture of all sterile products using the principles of 12
Quality Risk Management (QRM), to ensure that microbial, particulate and pyrogen contamination is 13
prevented in the final product. 14
15
QRM applies to this document in its entirety and will not be referred to in specific paragraphs. Where 16
specific limits or frequencies are written, these should be considered as a minimum requirement. They 17
are stated due to regulatory historical experience of issues that have previously been identified and 18
have impacted the safety of patients. 19
20
The intent of the Annex is to provide guidance for the manufacture of sterile products. However, 21
some of the principles and guidance, such as contamination control strategy, design of premises, 22
cleanroom classification, qualification, monitoring and personnel gowning, may be used to support 23
the manufacture of other products that are not intended to be sterile such as certain liquids, creams, 24
ointments and low bioburden biological intermediates but where the control and reduction of 25
microbial, particulate and pyrogen contamination is considered important. Where a manufacturer 26
elects to apply guidance herein to non-sterile products, the manufacturer should clearly document 27
which principles have been applied and acknowledge that compliance with those principles should be 28
demonstrated. 29
30
2 Principle 31
32
2.1 The manufacture of sterile products is subject to special requirements in order to minimize risks of 33
microbial, particulate and pyrogen contamination. The following key areas should be considered: 34
35
i. Facility, equipment and process design should be optimized, qualified and validated 36
according to the relevant sections of the Good Manufacturing Practices (GMP) guide. 37
The use of appropriate technologies (e.g. Restricted Access Barriers Systems (RABS), 38
isolators, robotic systems, rapid microbial testing and monitoring systems) should be 39
considered to increase the protection of the product from potential extraneous sources of 40
particulate and microbial contamination such as personnel, materials and the surrounding 41
environment, and assist in the rapid detection of potential contaminants in the 42
environment and product. 43
44
ii. Personnel should have adequate qualifications and experience, training and attitude with a 45
specific focus on the principles involved in the protection of sterile product during the 46
manufacturing, packaging and distribution processes. 47
48
iii. Processes and monitoring systems for sterile product manufacture should be designed, 49
commissioned, qualified and monitored by personnel with appropriate process, engineering 50
and microbiological knowledge. 51
52
2.2 Processes, equipment, facilities and manufacturing activities should be managed in accordance 53
with QRM principles to provide a proactive means of identifying, scientifically evaluating and 54
controlling potential risks to quality. Where alternative approaches are used, these should be 55
supported by appropriate rationales and risk assessment and should meet the intent of this Annex. 56
QRM priorities should include good design of the facility, equipment and process in the first instance, 57
then implementation of well-designed procedures, with monitoring systems as the final element that 58
3
demonstrate that the design and procedures have been correctly implemented and continue to perform 59
in line with expectations. Exclusively monitoring or testing does not give assurance of sterility. 60
61
2.3 Quality Assurance is particularly important, and manufacture of sterile products must strictly 62
follow carefully established and validated methods of manufacture and control. A Contamination 63
Control Strategy (CCS) should be implemented across the facility in order to define all critical control 64
points and assess the effectiveness of all the controls (design, procedural, technical and 65
organisational) and monitoring measures employed to manage risks associated with contamination. 66
The CCS should be actively updated and should drive continuous improvement of the manufacturing 67
and control methods. 68
69
2.4 Contamination control and steps taken to minimize the risk of contamination from microbial and 70
particulate sources are a series of successively linked events and measures. These are typically 71
assessed, controlled and monitored individually but their collective effectiveness should be considered 72
altogether. 73
74
2.5 The development of the CCS requires thorough technical and process knowledge. Potential 75
sources of contamination are attributable to microbial and cellular debris (e.g. pyrogen, endotoxins) as 76
well as particulate matter (e.g. glass and other visible and sub-visible particulates). 77
Elements to be considered within a documented CCS should include (but are not limited to): 78
79
i. Design of both the plant and processes. 80
81
ii. Premises and equipment. 82
83
iv. Personnel. 84
85
v. Utilities. 86
87
vi. Raw material controls – including in-process controls. 88
89
vii. Product containers and closures. 90
91
viii. Vendor approval – such as key component suppliers, sterilization of components and single 92
use systems (SUS), and services. 93
94
ix. For outsourced services, such as sterilization, sufficient evidence should be provided to the 95
contract giver to ensure the process is operating correctly. 96
97
x. Process risk assessment. 98
99
xi. Process validation. 100
101
xii. Preventative maintenance – maintaining equipment, utilities and premises (planned and 102
unplanned maintenance) to a standard that will not add significant risk of contamination. 103
104
xiii. Cleaning and disinfection. 105
106
xiv. Monitoring systems - including an assessment of the feasibility of the introduction of 107
scientifically sound, modern methods that optimize the detection of environmental 108
contamination. 109
110
xv. Prevention – trending, investigation, corrective and preventive actions (CAPA), root cause 111
determination and the need for more comprehensive investigational tools. 112
113

4
xvi. Continuous improvement based on information derived from the above. 114
115
2.6 The CCS should consider all aspects of contamination control and its life cycle with ongoing and 116
periodic review resulting in updates within the quality system as appropriate. 117
118
2.7 The manufacturer should take all steps and precautions necessary to assure the sterility of the 119
products manufactured within its facilities. Sole reliance for sterility or other quality aspects should 120
not be placed on any terminal process or finished product test. 121
122
3 Pharmaceutical Quality System (PQS) 123
3.1 The manufacture of sterile products is a complex activity that requires specific controls and 124
measures to ensure the quality of products manufactured. Accordingly, the manufacturer’s PQS 125
should encompass and address the specific requirements of sterile product manufacture and ensure 126
that all activities are effectively controlled so that microbial, particulate and pyrogen contamination is 127
minimized in sterile products. In addition to the PQS requirements detailed in Chapter 1 of the GMPs, 128
the PQS for sterile product manufacture should also ensure that: 129
130
i. An effective risk management system is integrated into all areas of the product life cycle 131
with the aim to minimize microbial contamination and to ensure the quality of sterile 132
products manufactured. 133
134
ii. The manufacturer has sufficient knowledge and expertise in relation to the products 135
manufactured and the equipment, engineering and manufacturing methods employed that 136
have an impact on product quality. 137
138
iii. Root cause analysis of procedural, process or equipment failure is performed in such a 139
way that the risk to product is correctly understood and suitable corrective and 140
preventative actions (CAPA) are implemented. 141
142
iv. Risk management is applied in the development and maintenance of the CCS, to identify, 143
assess, reduce/eliminate (where applicable) and control contamination risks. Risk 144
management should be documented and should include the rationale for decisions taken 145
in relation to risk reduction and acceptance of residual risk. 146
147
v. The risk management outcome should be reviewed regularly as part of on-going quality 148
management, during change control and during the periodic product quality review. 149
150
vi. Processes associated with the finishing and transport of sterile products should not 151
compromise the sterile product. Aspects that should be considered include: container 152
integrity, risks of contamination and avoidance of degradation by ensuring that products 153
are stored and maintained in accordance with the registered storage conditions. 154
155
vii. Persons responsible for the quality release of sterile products have appropriate access to 156
manufacturing and quality information and possess adequate knowledge and experience 157
in the manufacture of sterile products and their critical quality attributes. This is in order 158
to allow such persons to ascertain that the sterile products have been manufactured in 159
accordance with the registered specifications and are of the required quality. 160
161
3.2 All non-conformities, such as sterility test failures, environmental monitoring excursions or 162
deviations from established procedures should be investigated. The investigation should determine the 163
potential impact upon process and product quality and whether any other processes or batches are 164
potentially impacted. The reason for including or excluding a product or batch from the scope of the 165
investigation should be clearly justified and recorded. 166
167

5
4 Premises 168
169
4.1 The manufacture of sterile products should be carried out in appropriate cleanrooms, entry to 170
which should be through changing rooms that act as airlocks for personnel and airlocks for 171
equipment and materials. Cleanrooms should be maintained to an appropriate cleanliness standard 172
and supplied with air which has passed through filters of an appropriate efficiency. Controls and 173
monitoring should be scientifically justified and capable of evaluating the state of environmental 174
conditions for cleanrooms, airlocks and pass-throughs used for material and equipment transfer. 175
176
4.2 The various operations of component preparation, product preparation and filling should be 177
carried out with appropriate technical and operational separation measures within the cleanroom or 178
facility to prevent mix up and contamination. 179
180
4.3 Restricted Access Barrier Systems (RABS) and isolators are beneficial in assuring the required 181
conditions and minimizing the microbial contamination associated with direct human interventions 182
in the critical zone. Their use should be considered in the CCS. Any alternative approaches to the use 183
of RABS or isolators should be justified. 184
185
4.4 For the manufacture of sterile products there are four grades of cleanroom. 186
187
Grade A zone: The critical zone for high risk operations or for making aseptic connections by 188
ensuring protection by first air (e.g. aseptic processing line, filling zone, stopper bowl, open 189
ampoules and vials). Normally, such conditions are provided by a localised airflow protection, 190
such as unidirectional airflow work stations, RABS or isolators. The maintenance of 191
unidirectional airflow should be demonstrated and qualified across the whole of the Grade A 192
zone. Direct intervention (e.g. without the protection of barrier and glove port technology) into 193
the Grade A zone by operators should be minimized by premises, equipment, process and 194
procedural design. 195
196
Grade B area: For aseptic preparation and filling, this is the background cleanroom for the 197
Grade
A zone (where it is not an isolator). When transfer holes are used to transfer filled, 198
closed products to an adjacent cleanrooms of a lower grade, airflow visualization studies should 199
demonstrate that air does not ingress from the lower grade cleanrooms to the Grade B. Pressure 200
differentials should be continuously monitored. Cleanrooms of lower grade than Grade B can 201
be considered where isolator technology is used (refer to paragraph 4.22). 202
203
Grade C and D area: These are cleanrooms used for carrying out less critical stages in the 204
manufacture of aseptically filled sterile
products but can be used for the preparation /filling 205
of terminally sterilized products. (See section 8 for the specific details on terminal sterilization 206
activities). 207
208
4.5 In cleanrooms, all exposed surfaces should be smooth, impervious and unbroken in order
to 209
minimize the shedding or accumulation of particulates or micro-organisms and to permit the
210
repeated application of cleaning, disinfectant and sporicidal agents where used. 211
212
4.6 To reduce accumulation of dust and to facilitate cleaning there should be no recesses that are 213
difficult to clean effectively therefore projecting ledges, shelves, cupboards and equipment should be 214
kept to a minimum. Doors should be designed to avoid recesses that cannot be cleaned. 215
216
4.7 Materials used in cleanrooms should be selected to minimize generation of particles. 217
218
4.8 Ceilings should be designed and sealed to prevent contamination from the space above them. 219
220
4.9 Sinks and drains are prohibited in Grade A zone and Grade B area. In other cleanrooms, air 221
breaks should be fitted between the machine or sink and the drains. Floor drains in lower grade 222
6
cleanrooms should be fitted with traps or water seals designed to prevent back flow and should be 223
regularly cleaned, disinfected and maintained. 224
225
4.10 The transfer of equipment and materials into and out of the cleanrooms and critical zones is one 226
of the greatest potential sources of contamination. Any activities with the potential to compromise 227
the cleanliness of cleanrooms or the critical zone should be assessed and if they cannot be 228
eliminated, appropriate controls should be implemented. 229
230
4.11 The transfer of materials, equipment, and components into an aseptic processing area should be 231
carried out via a unidirectional process. Where possible, items should be sterilized and passed into 232
the area through double-ended sterilizers (e.g. through a double-door autoclave or depyrogenation 233
oven/tunnel) sealed into the wall. Where sterilization on transfer of the items is not possible, a 234
procedure which achieves the same objective of not introducing contaminant should be validated and 235
implemented, (e.g. using an effective transfer disinfection, rapid transfer systems for isolators or, for 236
gaseous or liquid materials, a bacteria-retentive filter). 237
238
4.12 Airlocks should be designed and used to provide physical separation and to minimize microbial 239
and particulate contamination of the different areas, and should be present for material and personnel 240
moving between different grades. Wherever possible, airlocks used for personnel movement should 241
be separated from those used for material movement. Where this is not practical, time-based 242
separation of movement (personnel /material) by procedure should be considered. Airlocks should be 243
flushed effectively with filtered air to ensure that the grade of the cleanroom is maintained. The final 244
stage of the airlock should, in the “at rest” state, be of the same cleanliness grade (viable and non-245
viable) as the cleanroom into which it leads. The use of separate changing rooms for entering and 246
leaving Grade B cleanrooms is desirable. Where this is not practical, time-based separation of 247
activities (ingress/egress) by procedure should be considered. Where the CCS indicates that the risk 248
of cross-contamination is high, separate changing rooms for entering and leaving production areas 249
should be considered. Airlocks should be designed as follow: 250
251
i. Personnel airlocks: Areas of increasing cleanliness used for entry of personnel (e.g. from 252
Grade D to Grade C to Grade B). In general hand washing facilities should be provided 253
only in the first stage of the changing room and not be present in changing rooms directly 254
accessing Grade B cleanrooms. 255
256
ii. Material airlocks: used for materials and equipment transfer. 257
258
• Only materials and equipment that have been included on an approved list, developed 259
during validation of the transfer process, should be allowed to be transferred into the 260
Grade A zone or Grade B cleanroom via an airlock or pass-through hatch. Equipment 261
and materials (intended for use in the Grade A zone) should be protected when 262
transiting through the Grade B cleanroom. Any unapproved items that require transfer 263
should be pre-approved as an exception. Appropriate risk assessment and mitigation 264
measures should be applied and recorded as per the manufacturer's CCS and should 265
include a specific disinfection and monitoring programme approved by quality 266
assurance. 267
268
• Pass-through hatches should be designed to protect the higher grade environment, for 269
example by effective flushing with an active filtered air supply. 270
271
• The movement of material or equipment from lower grade or unclassified area to 272
higher grade clean areas should be subject to cleaning and disinfection commensurate 273
with the risk and in line with the CCS. 274
275
4.13 Both sets of doors for pass-throughs and airlocks (for material and personnel) should not be 276
7
opened simultaneously. For airlocks leading to a Grade A zone and Grade B areas, an interlocking 277
system should be used. For airlocks leading to Grade C and D cleanrooms, a
visual and/or audible 278
warning system should be operated as a minimum. Where required to maintain zone segregation, a 279
time delay between the closing and opening of interlocked doors should be established. 280
281
4.14 Cleanrooms should be supplied with a filtered air supply that maintains a positive pressure 282
and/or an airflow relative to the background environment of a lower grade under all operational 283
conditions and should flush the area effectively. Adjacent rooms of different grades should have 284
pressure differentials of a minimum of 10 pascals (guidance value). Particular attention should be 285
paid to the protection of the critical zone. The recommendations regarding air supplies and pressures 286
may need to be modified where it is necessary to contain certain materials (e.g. pathogenic, highly 287
toxic or radioactive products or live viral or bacterial materials). The modification may include 288
positively or negatively pressurized airlocks that prevent the hazardous material from contaminating 289
surrounding areas. Decontamination of facilities (e.g. the cleanrooms and the heating, ventilation, 290
and air conditioning (HVAC) systems) and the treatment of air leaving a clean area, may be 291
necessary for some operations. Where containment requires air to flow into a critical zone, the 292
source of the air should be from an area of the same grade. 293
294
4.15 Airflow patterns within cleanrooms and zones should be visualised to demonstrate that there is 295
no ingress from lower grade to higher grade areas and that air does not travel from less clean areas 296
(such as the floor) or over operators or equipment that may transfer contaminant to the higher grade 297
areas. Where air movement is shown to be a risk to the clean area or critical zone, corrective actions, 298
such as design improvement, should be implemented. Airflow pattern studies should be performed 299
both at rest and in operation (e.g. simulating operator interventions). Video recordings of the airflow 300
patterns should be retained. The outcome of the air visualisation studies should be considered when 301
establishing the facility's environmental monitoring program. 302
303
4.16 Indicators of pressure differences should be fitted between cleanrooms and/or isolators. Set-304
points and the criticality of pressure differentials should be documented within the CCS. Pressure 305
differentials identified as critical should be continuously monitored and recorded. A warning system 306
should be in place to instantly indicate and warn operators of any failure in the air supply or 307
reduction of pressure differentials (below set limits for those identified as critical). The warning 308
signal should not be overridden without assessment and a procedure should be available to outline 309
the steps to be taken when a warning signal is given. Where alarm delays are set, these should be 310
assessed and justified within the CCS. Other pressure differentials should be monitored and recorded 311
at regular intervals. 312
313
4.17 Facilities should be designed to permit observation of production activities from outside the 314
Grade A zone and Grade B area (e.g. through the provision of windows or remote cameras with a 315
full view of the area and processes to allow observation and supervision without entry). This 316
requirement should be considered when designing new facilities or during refurbishment of existing 317
facilities. 318
319
Barrier Technologies 320
321
4.18 Isolator or RABS technologies, and the associated processes, should be designed to provide 322
protection of the Grade A environment. The entry of materials during processing (and after 323
decontamination) should be minimized and preferably supported by rapid transfer technologies or 324
transfer isolators. 325
326
4.19 The design of the RABS or isolator should take into account all critical factors associated with 327
these technologies including the quality of the air inside and the background environment, the 328
materials and component transfer, the decontamination and/or sterilization processes, the risk factors 329
associated with the manufacturing operations and the operations conducted within the critical zone. 330
331
8
4.20 The critical zone of the RABS or open isolator used for aseptic processes should meet Grade A 332
requirements with unidirectional airflow. In closed isolator systems where airflow may not be 333
unidirectional, it should provide Grade A conditions and be demonstrated to provide adequate 334
protection for exposed products during processing. The design of the RABS and open isolators should 335
ensure a positive airflow from the critical zones to the supporting background environment; (unless 336
containment is required in which case localized air extraction is required to prevent contamination 337
transfer to the surrounding room). Negative pressure isolators should only be used when containment 338
of the product is considered essential and risk control measures are applied to ensure the critical zone 339
is not compromised. 340
341
4.21 For RABS used for aseptic processing, the background environment should meet at least Grade 342
B. The background environment for open isolators should meet Grade C or D, based on a risk 343
assessment. Airflow studies should be performed to demonstrate the absence of air ingress during 344
interventions, such as door openings. 345
346
4.22 The background environment of a closed isolator should correspond to a minimum of Grade D. 347
The disinfection/decontamination programme should be included as a key consideration when 348
performing the risk assessment for the CCS of an isolator. Where additional process risks are 349
identified, a higher grade of background should be considered. The decision as to the supporting 350
background environment should be documented in the CCS. 351
352
4.23 The materials used for glove systems (for both RABS and isolators), as well as other parts of an 353
isolator, should be demonstrated to have good mechanical and chemical resistance. Integrity testing of 354
the barrier systems, and leak testing of the glove system and the isolator should be performed using a 355
methodology demonstrated to be suitable for the task and criticality. The testing should be performed 356
at defined periods, at a minimum at the beginning and end of each batch, and should include a visual 357
inspection following any intervention that may affect the integrity of the system. For single unit batch 358
sizes, integrity may be verified based on other criteria, such as the beginning and end of each 359
manufacturing session. RABS gloves used in Grade A zone should be sterilized before installation 360
and sterilized (or effectively decontaminated by a validated method which achieves the same 361
objective) prior to each manufacturing campaign. The frequency of glove replacement should be 362
defined within the CCS. 363
364
4.24 For RABS and isolator systems, decontamination methods should be validated and controlled 365
within defined cycle parameters. The cleaning process prior to the disinfection step is essential; any 366
residues that remain may inhibit the effectiveness of the decontamination process: 367
368
i. For isolators, the decontamination process should be automated and should include a 369
sporicidal agent in a suitable form (e.g. gaseous, aerosolized or vaporized form) to ensure 370
thorough microbial decontamination of its interior. Decontamination methods (cleaning and 371
sporicidal disinfection) should render the interior surfaces and critical zone of the isolator free 372
of viable microorganisms. 373
374
ii. For RABS systems, the disinfection should include the routine application of a sporicidal 375
agent using a method that has been validated and demonstrated to robustly disinfect the 376
interior and ensure a suitable environment for aseptic processing. 377
378
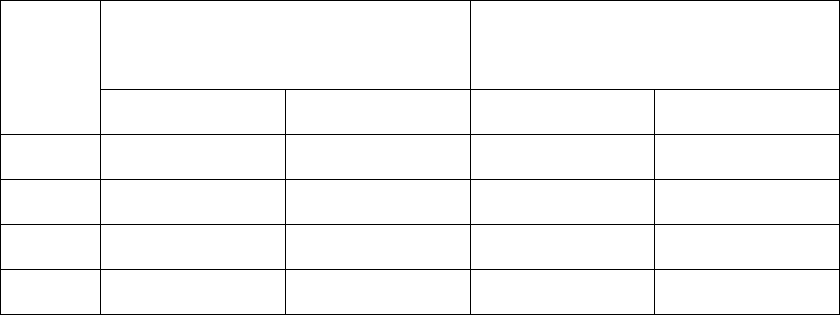
Evidence should also be available to demonstrate that the agent used does not have adverse impact on 379
the product produced within the RABS or isolator. The holding time before use of these systems 380
should be validated. 381
382
Cleanroom and clean air equipment qualification 383
384
4.25 Cleanrooms and clean air equipment such as unidirectional airflow units (UDAFs), 385
RABS and isolators, used for the manufacture of sterile products, should be qualified and 386
9
classified according to the required characteristics of the environment. Each manufacturing 387
operation requires an appropriate environmental cleanliness level in the operational state in 388
order to minimize the risk of particulate or microbial contamination of the product or materials 389
being handled. 390
391
4.26 Cleanrooms and clean air equipment should be qualified using methodology in accordance with 392
the requirements of Annex 15. Cleanroom qualification (including classification) should be clearly 393
differentiated from operational environmental monitoring. 394
395
4.27 Cleanroom Qualification is the overall process of assessing the level of compliance of a 396
classified cleanroom or clean air equipment with its intended use. As part of the qualification 397
requirements of Annex 15, the qualification of cleanrooms and clean air equipment should include 398
(where relevant to the design/operation of the installation): 399
400
i. Installed filter leakage and integrity testing. 401
402
ii. Airflow measurement - Volume and velocity. 403
404
iii. Air pressure difference measurement. 405
406
iv. Airflow direction and visualisation. 407
408
v. Microbial airborne and surface contamination. 409
410
vi. Temperature measurement. 411
412
vii. Relative humidity measurement. 413
414
viii. Recovery testing. 415
416
ix. Containment leak testing. 417
418
4.28 Cleanroom classification is part of a cleanroom qualification and is a method of assessing the 419
level of air cleanliness against a specification for a cleanroom or clean air equipment by measuring 420
the non-viable airborne particulate concentration. Reference for the classification of the cleanrooms 421
and clean air equipment can be found in the ISO 14644 series of standards. 422
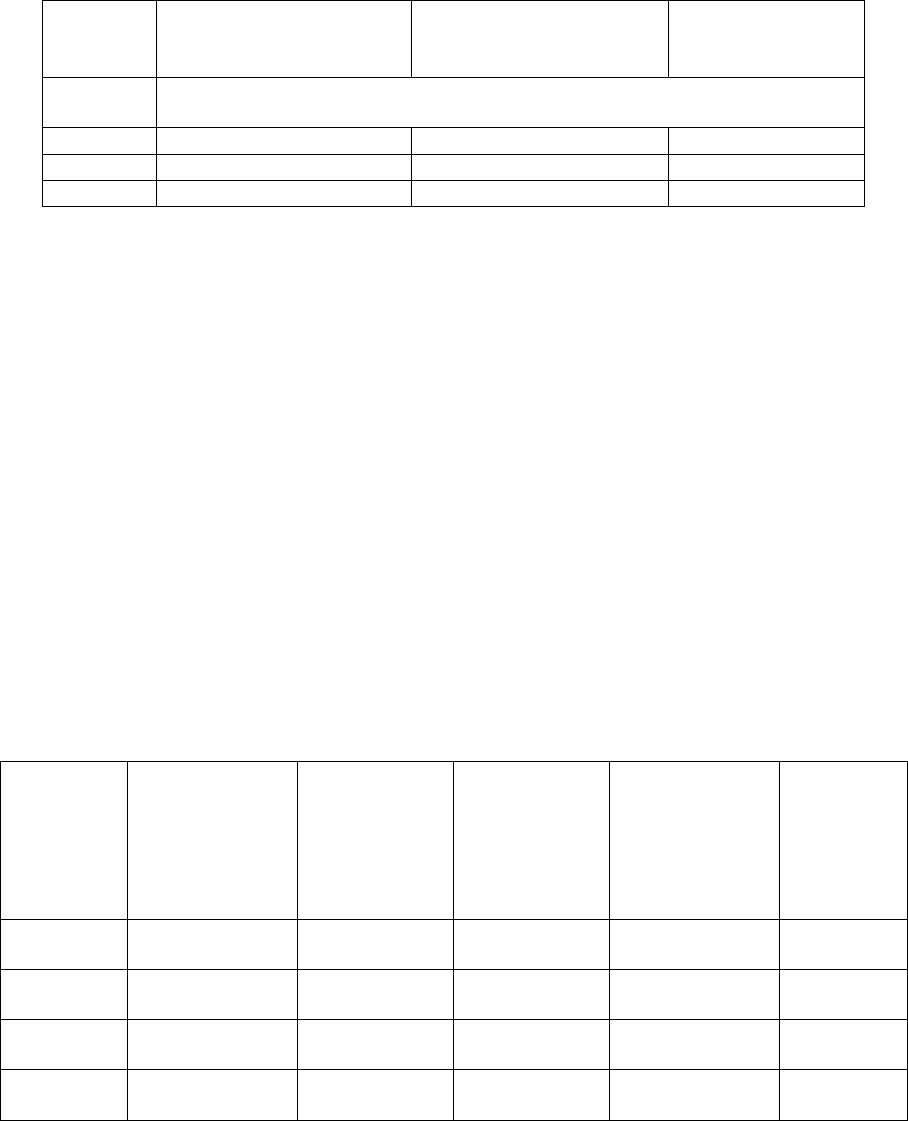
423
4.29 For cleanroom classification, the airborne particulates equal to or greater than 0.5 and 5 µm 424
should be measured. For Grade A zone and Grade B at rest, classification should include 425
measurement of particles equal to or greater than 0.5 µm; however, measurement using a second, 426
larger particle size, e.g. 1 µm in accordance with ISO 14644 may be considered. This measurement 427
should be performed both at rest and in operation. The maximum permitted airborne particulate 428
concentration for each grade is given in Table 1. 429
430
431
10
Table 1: Maximum permitted airborne particulate concentration during classification 432
433
Grade
Maximum limits for particulates
≥ 0.5 µm/m
3
Maximum limits for particulates
≥ 5 µm/m
3
at rest
in operation
at rest
in operation
A
3 520
3 520
Not applicable
Not applicable
B
3 520
352 000
Not applicable
2 900
C
352 000
3 520 000
2 900
29 000
D
3 520
000
Not defined
(
a
)
29 000
Not de
fined
(a)
434
435
(a)
For Grade D, in operation limits are not defined. The company should establish in operation 436
limits based on a risk assessment and historical data where applicable. 437
438
4.30 For classification of the cleanroom, the minimum number of sampling locations and their 439
positioning can be found in ISO 14644 Part 1. In addition, for the aseptic processing room and the 440
background environment (Grade A zone and Grade B area, respectively), sample locations should also 441
consider all critical processing zones such as the point of fill and stopper bowls. Critical processing 442
locations should be based on a documented risk assessment and knowledge of the process and 443
operations to be performed in the area. 444
445
4.31 Clean room classification should be carried out in the “at rest” and “in operation” states. 446
447
i. The definition of “at rest” state is the condition whereby the installation of all the utilities is 448
complete including any functioning HVAC, with the main manufacturing equipment installed 449
as specified and standing by for operation, without personnel in the room. 450
451
ii. The definition of “in operation” state is the condition where the installation of the cleanroom 452
is complete, the HVAC system fully operational, equipment installed and functioning in the 453
manufacturer’s defined operating mode with the maximum number of personnel present 454
performing or simulating routine operational work. In operation classification may be 455
performed during simulated operations or during aseptic process simulations (where worst 456
case simulation is required). 457
458
iii. The particulate limits given in Table 1 above for the “at rest” state should be achieved after 459
a “clean up” period on completion of operations. The "clean up" period should be 460
determined during the classification of the rooms (guidance value of 15 to 20 minutes). 461
462
4.32 The speed of air supplied by unidirectional airflow systems should be clearly justified in the 463
qualification protocol including the location for air speed measurement. Air speed should be designed, 464
measured and maintained to ensure that appropriate unidirectional air movement provides protection 465
of the product and open components at the working height (e.g. where high risk operations and 466
product and/or components are exposed). Unidirectional airflow systems should provide a 467
homogeneous air speed in a range of 0.36 – 0.54 m/s (guidance value) at the working position, unless 468
otherwise scientifically justified in the CCS. Airflow visualization studies should correlate with the air 469
speed measurement. 470
11
4.33 The microbial concentration of the cleanrooms should be determined as part of the cleanroom 471
qualification. The number of sampling locations should be based on a documented risk assessment, 472
including the results of the classification, air visualization studies and knowledge of the process and 473
operations to be performed in the area. The maximum limits for microbial contamination during 474
qualification for each grade are given in Table 2. Qualification should include both at rest and in 475
operation states. 476
477
Table 2: Limits for microbial contamination during qualification 478
Grade
Air sample
cfu/m
3
Settle plates
(diameter 90 mm)
cfu/4 hours
(a)
Contact plates
(diameter 55
mm) cfu/plate
A
(b)
No growth
(b)
B 10 5 5
C 100 50 25
D 200 100 50
(a) Settle plates should be exposed for the duration of operations and changed as required after 4 479
hours. Exposure time should be based on recovery studies and should not allow desiccation of the 480
media used. 481
482
(b) It should be noted that for Grade A, the expected result should be no growth. 483
Note 1: All methods indicated for a specific Grade in the table should be used for qualifying the 484
area of that specific Grade. If one of the methods is not used, or alternative methods are used, the 485
approach taken should be appropriately justified. 486
Note 2: Limits are applied using cfu throughout the document. If different or new technologies 487
are used that present results in a manner different from cfu, the manufacturer should scientifically 488
justify the limits applied and where possible correlate them to cfu. 489
Note 3: For qualification of personnel gowning, the limits given for contact plates and glove prints in 490
Table 7 should apply. 491
Note 4: Sampling methods should not pose a risk of contamination to the manufacturing operations. 492
493
4.34 The requalification of cleanrooms and clean air equipment should be carried out periodically 494
following defined procedures. The requirement for requalification of cleanroom areas is as follows: 495
496
Table 3: Minimum test requirements for the requalification of cleanrooms 497
Grade
Determination
of the
concentration
of airborne
viable and non-
viable particles
Integrity Test
of Terminal
Filters
Airflow
volume
measurement
Verification of
air pressure
difference
between rooms
Air
Velocity
test
A Yes Yes Yes Yes Yes
B Yes Yes Yes Yes *
C Yes Yes Yes Yes *
D Yes Yes Yes Yes *

12
* performed according to a risk assessment documented as part of the CCS. However, required 498
for filling zones (e.g. when filling terminally sterilised products) and background to Grade A 499
RABS. 500
For Grade A & B areas, the maximum time interval for requalification is 6 months. 501
For Grade C & D areas, the maximum time interval for requalification is 12 months. 502
Appropriate requalification consisting of at least the above tests should also be carried out following 503
completion of remedial action implemented to rectify an out-of-compliance equipment or facility 504
condition or after changes to equipment, facility or processes. The significance of a change should be 505
determined through the change management process. Examples of changes to be considered include 506
but are not limited to the following: 507
508
i. Change in the operational use of the cleanroom, or of the operational setting parameters of 509
the HVAC system. 510
ii. Interruption of air movement which affects the operation of the installation. 511
iii. Special maintenance which affects the operation of the installation (e.g. change of final 512
filters). 513
4.35 Other characteristics, such as temperature and relative humidity, should be controlled within 514
ranges that align with product/processing requirements and support maintenance of defined 515
cleanliness standards (e.g. Grade A or B). 516
517
Disinfection
518
519
4.36 The disinfection of cleanrooms is particularly important. They should be cleaned and disinfected 520
thoroughly in accordance with a written programme. For disinfection to be effective, prior cleaning to 521
remove surface contamination should be performed. More than one type of disinfecting agent should 522
be employed to ensure that where they have different modes of action and their combined usage is 523
effective against all bacteria and fungi. Disinfection should include the periodic use of a sporicidal 524
agent. Monitoring should be undertaken regularly in order to assess the effectiveness of the 525
disinfection program and to detect changes in types of microbial flora (e.g. organisms resistant to the 526
disinfection regime currently in use). Cleaning programs should effectively remove disinfectant 527
residues. 528
529
4.37 The disinfection process should be validated. Validation studies should demonstrate the 530
suitability and effectiveness of disinfectants in the specific manner in which they are used and should 531
support the in-use expiry periods of prepared solutions. 532
533
4.38 Disinfectants and detergents used in Grade A zone and Grade B areas should be sterile
prior to 534
use (disinfectants used in Grade C and D may also be required to be sterile). Where the disinfectants 535
and detergents are made up by the sterile product manufacturer, they should be monitored for 536
microbial contamination. Dilutions
should be kept in previously cleaned containers and should only 537
be stored for defined periods. If the disinfectants and detergents are supplied “ready-made” then results 538
from certificates of analysis or conformance can be accepted subject to successful completion of the 539
appropriate vendor qualification. 540
541
4.39 Fumigation or vapour disinfection (e.g. Vapour-phased Hydrogen Peroxide) of cleanrooms and 542
associated surfaces may be useful for reducing microbial contamination in
inaccessible places. 543
544
5 Equipment 545
546
5.1 A written, detailed description of the equipment design should be available (including process and 547
instrumentation diagrams as appropriate). This should form part of the initial qualification package 548
and be kept up to date as part of the ongoing review of the CCS. 549

13
550
5.2 Equipment monitoring requirements should be defined in “user requirements specifications” and 551
during early stages of development, and confirmed during qualification. Process and equipment alarm 552
events should be reviewed and approved and evaluated for trends. The frequency at which alarms are 553
assessed should be based on their criticality (with critical alarms reviewed immediately). 554
555
5.3 As far as practicable, equipment, fittings and services should be designed and installed so that 556
operations, maintenance, and repairs can be performed outside the cleanroom. If maintenance has to 557
be performed in the cleanroom, and the required standards of cleanliness and/or asepsis cannot be 558
maintained, then precautions such as restricting access to the work area to specified personnel, 559
generation of clearly defined work protocols and maintenance procedures should be considered. 560
Cleaning, additional disinfection and additional environmental monitoring should also be considered. 561
If sterilization of equipment is required, it should be carried out, wherever possible, after complete 562
reassembly. 563
564
5.4 The cleaning process should be validated to: 565
566
i. Remove any residue or debris that would detrimentally impact the effectiveness of the 567
disinfecting agent used. 568
ii. Minimize chemical, microbial and particulate contamination of the product during the process 569
and prior to disinfection. 570
571
5.5 Direct and indirect contact parts should be sterilized. Direct contact parts are those that the 572
product passes through, such as filling needles or pumps. Indirect product contact parts are 573
equipment parts that come into contact with sterilized critical items and components. 574
575
5.6 All equipment such as sterilizers, air handling systems (including air filtration) and water 576
systems should be subject to qualification, monitoring and planned maintenance. Upon completion 577
of maintenance, their return to use should be approved. 578
579
5.7 Where unplanned maintenance of equipment critical to the sterility of the product is to be carried 580
out, an assessment of the potential impact to the sterility of the product should be performed and 581
recorded. 582
583
5.8 A conveyor belt should not pass through a partition between a Grade A or B area and a
584
processing area of lower air cleanliness, unless the belt itself is continually sterilized (e.g. in a
585
sterilizing tunnel). 586
587
5.9 Particle counters, including sampling tubing, should be qualified. The tubing length should be no 588
greater than 1 meter with a minimum number of bends and bend radius should be greater than 15 cm. 589
Portable particle counters with a short length of sample tubing should be used for classification 590
purpose. Isokinetic sampling heads should be used in unidirectional airflow systems and should be 591
positioned as close as possible to sample air representative of the critical location. 592
593
6 Utilities 594
595
6.1 The nature and extent of controls applied to utility systems should be commensurate with the risk 596
to product quality associated with the utility. The impact should be determined via a risk assessment 597
documented as part of the CCS. 598
599
6.2 In general higher risk utilities are those that: 600
601
i. Directly contact product e.g. water for washing and rinsing, gases and steam for 602
sterilization. 603
604
14
ii. Contact materials that will ultimately become part of the product. 605
606
iii. Contact surfaces that come into contact with the product. 607
608
iv. Otherwise directly impact the product. 609
610
6.3 Utilities should be designed, installed, operated, maintained and monitored in a manner to ensure 611
that the utility functions as expected. 612
613
6.4 Results for critical parameters and critical quality attributes of high risk utilities should be subject 614
to regular trend analysis to ensure that system capabilities remain appropriate. 615
616
6.5 Records of utility installation should be maintained throughout the system’s life-cycle. Such 617
records should include current drawings and schematic diagrams, construction material lists and 618
specifications. Typically, important information includes attributes such as: 619
620
i. Pipeline flow direction, slopes, diameter and length. 621
622
ii. Tank and vessel details. 623
624
iii. Valves, filters, drains, sampling and user points. 625
626
6.6 Pipes, ducts and other utilities should not be present in cleanrooms. If unavoidable, then they 627
should be installed so that they do not create recesses, unsealed openings and surfaces which are 628
difficult to clean. Installation should allow cleaning and disinfection of outer surface of the pipes. 629
630
Water systems 631
632
6.7 Water treatment plant and distribution systems should be designed, constructed and maintained to 633
minimize the risk of particulates, microbial contamination/proliferation and pyrogens (e.g. sloping of 634
piping to provide complete drainage and the avoidance of dead legs), and prevent the formation of 635
biofilms to ensure a reliable source of water of an appropriate quality. Where filters are included in 636
the system, special attention should be given to the monitoring and maintenance of these filters. Water 637
produced should comply with the current monograph of the relevant Pharmacopeia. 638
639
6.8 Water systems should be qualified to maintain the appropriate levels of physical, chemical and 640
microbial control, taking seasonal variation into account. 641
642
6.9 Water flow should remain turbulent through the pipes to minimize the risk of microbial adhesion, 643
and subsequent biofilm formation. 644
645
6.10 Water for injections (WFI) should be produced from water meeting specifications that have been 646
defined during the qualification process, stored and distributed in a manner which minimizes the risk 647
of microbial growth (for example by constant circulation at a temperature above 70°C). Where the 648
WFI is produced by methods other than distillation, further techniques such as nanofiltration and 649
ultra-filtration as well as electrodeionization (EDI) should be considered in conjunction with reverse 650
osmosis (RO) membranes. 651
652
6.11 Where WFI storage tanks are equipped with hydrophobic bacteria retentive vent filters, the 653
filters should be sterilized and the integrity of the filter tested before installation and after removal 654
following use. 655
656
6.12 To minimize the risk of biofilm formation, sterilization or disinfection or regeneration of water 657
systems should be carried out according to a predetermined schedule and when microbial counts 658
exceed action limits. Disinfection of a water system with chemicals should be followed by a 659
15
validated rinsing/flushing procedure. Water should be tested after disinfection/regeneration. The 660
results should be approved before the water system is returned to use. 661
662
6.13 Regular ongoing chemical and microbial monitoring of water systems should be performed. 663
Alert levels should be based on the qualification or a review of ongoing monitoring data that will 664
identify an adverse trend in system performance. Sampling programs should reflect the requirements 665
of the CCS and include: 666
667
i. All points of use, at a specified interval, to ensure that representative water samples are 668
obtained for analysis on a regular basis. 669
670
ii. Potential worst case sampling locations. 671
672
iii. A sample from the point at the end of the distribution loop each day that the water is used. 673
674
6.14 Breaches of alert levels should be documented and reviewed, and include investigation of 675
system trends to determine whether the breach is a single (isolated) event or if results are indicative 676
of loss of control or system deterioration. Each breach of action limits should be investigated to 677
determine the root cause of the issue and any impact on the quality of products and manufacturing 678
processes as a result of the potential use of the water. 679
680
6.15 WFI systems should include continuous monitoring systems such as Total Organic Carbon 681
(TOC) and conductivity, (unless justified otherwise) as these may give a better indication of overall 682
system performance than discrete sampling. Sensor locations should be based on risk and the 683
outcome of qualification. 684
685
Steam used as a direct sterilizing agent 686
687
6.16 Feed water to a pure steam (clean steam) generator should be appropriately purified. Pure steam 688
generators should be designed, qualified and operated in a manner to ensure that the quality of steam 689
produced meets defined chemical and endotoxin levels. 690
691
6.17 Steam used as a direct sterilizing agent should be of suitable quality and should not contain 692
additives at a level which could cause contamination of product or equipment. For a pure steam 693
generator supplying pure steam used for the direct sterilization of materials or product-contact 694
surfaces (e.g. porous hard-good autoclave loads), steam condensate should meet the current 695
monograph for WFI of the relevant Pharmacopeia. A suitable sampling schedule should be in place 696
to ensure that representative pure steam samples are obtained for analysis on a regular basis. Other 697
aspects of the quality of pure steam used for sterilization should be assessed periodically against 698
validated parameters. These parameters should include the following: non-condensable gases, 699
dryness value (dryness fraction) and superheat. 700
701
Gases and vacuum systems 702
703
6.18 Gases that come in direct contact with the product/primary container surfaces should be of 704
appropriate chemical, particulate and microbial quality. All relevant parameters, including oil and 705
water content, should be specified, taking into account the use and type of the gas, the design of the 706
gas generation system and, where applicable, comply with the appropriate Pharmacopoeia 707
monographs. 708
709
6.19 Gases used in aseptic processes should be filtered through a sterilizing filter (with a nominal pore 710
size of a maximum of 0.22 µm) at the point of use. Where the filter is used on a batch basis (e.g. for 711
filtration of gas used for overlay of aseptically filled products) or as product vessel vent filter, then the 712
filter should be integrity tested and the results included as part of the batch certification process. Any 713
transfer pipework or tubing that is located after the final sterilizing filter should be sterilized. When 714

16
gases are used in the process, microbial monitoring of the gas should be performed periodically at the 715
point of use. 716
717
6.20 Where backflow from vacuum or pressure systems poses a potential risk to the product, there 718
should be mechanism(s) to prevent backflow when the vacuum or pressure system is shut off. 719
720
Heating and cooling and hydraulic systems 721
722
6.21 Major items of equipment associated with hydraulic, heating and cooling systems, e.g. such as 723
those associated with Blow-Fill-Seal equipment should, where possible, be located outside the filling 724
room. Where they are located inside the filling room there should be appropriate controls to contain 725
any spillage and/or cross contamination associated with the hydraulic system fluids. Where possible, 726
the system should be at a lower pressure than the processed fluid. 727
728
6.22 Any leaks from these systems that would present a risk to the product should be detectable (i.e. 729
an indication system for leakage). 730
731
6.23 For both vacuum and cooling systems there should be periodic cleaning/disinfection as 732
determined in the CCS. 733
734
7 Personnel 735
7.1 The manufacturer should ensure that there are sufficient appropriate personnel, suitably qualified, 736
trained and experienced in the manufacture and testing of sterile products, and any of the specific 737
manufacturing technologies used in the site’s manufacturing operations, to ensure compliance with 738
GMP applicable to the manufacture and handling of sterile products. 739
740
7.2 Only the minimum number of personnel required should be present in cleanrooms. The 741
maximum number of operators in cleanrooms should be determined, documented and validated 742
during activities such as initial qualification and aseptic process simulations, so as not to 743
compromise sterility assurance. This is particularly important during aseptic processing. 744
745
7.3 Non-essential processes such as product inspection and in process testing should be conducted 746
outside the clean areas wherever possible. 747
748
7.4 All personnel including those performing cleaning, maintenance, monitoring and those that 749
access cleanrooms should receive regular training, gowning qualification and assessment in 750
disciplines relevant to the correct manufacture of sterile products. This training should include the 751
basic elements of microbiology, hygiene, with a specific focus on cleanroom practices, 752
contamination control, aseptic techniques and the protection of sterile products (for those operators 753
entering the Grade B cleanrooms and/or intervening into the Grade A zone) and the potential safety 754
implications to the patient if product is not sterile. The level of training should be based on the 755
criticality of the function and area in which the personnel are working. 756
757
7.5 The personnel working in a Grade A zone and Grade B areas should be trained for aseptic 758
gowning and aseptic practices. Compliance with aseptic gowning procedures should be assessed and 759
confirmed, periodically reassessed at least annually and should involve both visual and microbial 760
assessment (using monitoring locations such as hands, arms, chest and forehead. Refer to paragraph 761
9.30 for the expected limits). The unsupervised access to Grade A zone and Grade B areas where 762
aseptic operations are or will be conducted should be restricted to appropriately qualified personnel, 763
who have passed the gowning assessment and have participated in a successful aseptic process 764
simulation (APS). 765
766
7.6 Unqualified personnel (e.g. building and maintenance contractors and regulatory inspectors) 767
17
should not enter Grade B cleanrooms or Grade A zones in operation. If needed in exceptional cases, 768
manufacturers should establish written procedures outlining the process by which unqualified 769
personnel are brought into the Grade B and A areas. Access by these persons should be assessed and 770
recorded in accordance with the PQS. An authorized person from the manufacturer should supervise 771
the unqualified personnel during their activities and should assess the impact of these activities on 772
the cleanliness of the area. 773
774
7.7 There should be systems in place for disqualification of personnel from entry into cleanrooms 775
based on aspects including ongoing assessment and/or identification of an adverse trend from the 776
personnel monitoring program and/or after participation in a failed APS. Once disqualified, 777
retraining and requalification should be completed before permitting the operator to have any further 778
involvement in aseptic practices. For operators entering Grade B cleanrooms or performing 779
intervention into Grade A zone, this requalification should include consideration of participation in a 780
successful APS. 781
782
7.8 High standards of personal hygiene and cleanliness are essential to prevent excessive shedding or 783
increased risk of introduction of microbial contamination. Personnel involved in the manufacture of 784
sterile products should be instructed to report any specific health conditions or ailments which may 785
cause the shedding of abnormal numbers or types of contaminants and therefore preclude cleanroom 786
access. Health conditions and actions to be taken with regard to personnel who could be introducing 787
an undue microbial hazard should be provided by the designated competent person and described in 788
procedures. 789
790
7.9 Staff who have been engaged in the processing of human or animal tissue materials or of cultures 791
of micro-organisms, other than those used in the current manufacturing process, or any activities that 792
may have a negative impact to quality (e.g. microbial contamination), should not enter clean areas 793
unless clearly defined and effective decontamination and entry procedures have been followed. 794
795
7.10 Wristwatches, make-up, jewellery, other personal items such as mobile phones and any other 796
non-essential items should not be allowed in clean areas. Electronic devices used in cleanrooms, e.g. 797
mobile phones and tablets, that are supplied by the company solely for use in the cleanrooms, may 798
be acceptable if suitably designed to permit cleaning and disinfection commensurate with the Grade 799
in which they are used. The use and disinfection of such equipment should be included in the CCS. 800
801
7.11 Cleanroom gowning and hand washing should follow a written procedure designed to minimize 802
contamination of cleanroom clothing and/or the transfer of contaminants to the clean areas. 803
804
7.12 The clothing and its quality should be appropriate for the process and the grade of the
805
working area. It should be worn in such a way as to protect the product from contamination. When the 806
type of clothing chosen needs to provide the operator protection from the product, it should not 807
compromise the protection of the product from contamination. Garments should be visually checked 808
for cleanliness and integrity immediately prior to gowning and prior to entry to the cleanroom. Gown 809
integrity should also be checked upon exit. For sterilized or effectively decontaminated garments and 810
eye coverings, particular attention should be taken to ensure they have been processed, are within 811
their specified hold time and that the packaging is visually inspected to ensure it is integral before use. 812
Reusable garments (including eye coverings) should be replaced if damage is identified or at a set 813
frequency that is determined during qualification studies. . Damage to garments may not be identified 814
by visual inspection alone, so the qualification should consider any necessary garment testing 815
requirements. 816
817
7.13 Clothing should be chosen to prevent shedding due to operators moving excessively (when 818
cold) or sweating (when hot). 819
820
7.14 The description of clothing required for each grade is given below: 821
822
18
i. Grade A / B: Dedicated garments to be worn under a sterilized suit. Sterile headgear should 823
enclose all hair (including facial hair) and where separate from the rest of the gown, it 824
should be tucked into the neck of the sterile suit. A sterile face mask and sterile eye 825
coverings (e.g. goggles) should be worn to cover and enclose all facial skin and prevent the 826
shedding of droplets and particulates. Appropriate sterilized, non-powdered, rubber or 827
plastic gloves and sterilized footwear (such as overboots) should be worn. Trouser-legs 828
should be tucked inside the footwear and garment sleeves into the gloves. The protective 829
clothing should minimize shedding of fibres or particulate matter and retain particulates 830
shed by the body. Garments should be packed and folded in such a way as to allow operators 831
to gown without contacting the outer surface of the garment. 832
833
ii. Grade C: Hair, beards and moustaches should be covered. A single or two-piece trouser suit 834
gathered at the wrists and with high neck and appropriately disinfected shoes or overshoes 835
should be worn. They should minimize the shedding of fibres and particulate
matter. 836
837
iii. Grade D: Hair, beards and moustaches should be covered. A general protective suit
and 838
appropriately disinfected shoes or overshoes should be worn. Appropriate measures should 839
be
taken to avoid any ingress of contaminants from outside the clean area. 840
841
iv. Gloves should be worn in Grade C and D areas when performing activities considered to be a 842
contamination risk as defined by the CCS. 843
844
7.15 Outdoor clothing (other than personal underwear) should not be brought into changing rooms 845
leading directly to Grade B and C
clean
rooms. Facility suits, covering the full length of the arms 846
and the legs, and socks covering the feet, should be worn before entry to change rooms for Grades B 847
and C. Facility suits and socks should not present a risk of contamination to the gowning area or 848
processes. 849
850
7.16 Every operator entering Grade B or A areas should gown into clean, sterilized protective 851
garments (including eye coverings and masks) of an appropriate size at each entry. The maximum 852
duration of each garment use should be defined as part of the garment qualification. 853
854
7.17 Garments and gloves should be changed immediately if they become damaged and present any 855
risk of product contamination. Gloves should be regularly disinfected during operations. 856
857
7.18 Clean area clothing should be cleaned in a dedicated laundry facility using a qualified process 858
ensuring that the clothing is not damaged and/or contaminated by fibres and particles during the 859
laundry process. Inappropriate handling and use of clothing will damage fibres and may increase the 860
risk of shedding of particles. After washing and before packing, garments should be visually 861
inspected for damage. The garment management processes should be evaluated and determined as 862
part of the garment qualification program. 863
864
7.19 Activities in clean areas that are not critical to the production processes should be kept to a 865
minimum, especially when aseptic operations are in progress. Movement of personnel should be 866
slow, controlled and methodical to avoid
excessive shedding of particulates and organisms due to 867
over-vigorous activity. Operators performing aseptic operations should adhere to aseptic technique 868
at all times to prevent changes in air currents that introduce air of lower quality into the critical zone. 869
Movement adjacent to the critical zone should be restricted and the obstruction of the path of the 870
unidirectional (first air) airflow should be avoided. Airflow visualisation studies should be 871
considered as part of the operator’s training programme. 872
873
874

19
8 Production and Specific Technologies 875
876
Terminally sterilized products 877
878
8.1 Preparation of components and materials should be performed in at least a Grade D 879
cleanroom in order to limit the risk of microbial, pyrogen and particulate contamination, so that the 880
product is suitable for sterilization. Where the product is at a high or unusual risk of microbial
881
contamination (e.g. the product actively supports microbial growth, the product must
be held for 882
long periods before filling or the product is not processed mostly in closed
vessels), then 883
preparation should be carried out in a Grade C environment. Preparation of ointments, creams, 884
suspensions and emulsions should be carried out in a Grade C environment before
terminal 885
sterilization. 886
887
8.2 Primary packaging containers and components should be cleaned using validated processes to 888
ensure that particulate, pyrogen and bioburden contamination is appropriately controlled. 889
890
8.3 Filling of products for terminal sterilization should be carried out in at least a Grade C
891
environment. 892
893
8.4 Where the product is at an unusual risk of contamination from the environment because, for 894
example, the filling operation is slow, the containers are wide necked or are necessarily exposed for 895
more than a few seconds before closing, then the product should be filled in a Grade A zone with at least 896
a Grade C background. 897
898
8.5 Processing of the bulk solution should include a filtration step with a microorganism retaining 899
filter, where possible, to reduce bioburden levels and particulates prior to filling into the final 900
product containers and there should be a maximum permissible time between preparation and filling. 901
902
8.6 Examples of operations to be carried out in the various grades are given in Table 4.
903
904
Table 4: Examples of operations and grades for terminally sterilized preparation and 905
processing operations 906
A Filling of products, when unusually at risk.
C Preparation of solutions, when unusually at risk. Filling of products.
D Preparation of solutions and components for subsequent filling.
907
Aseptic preparation and processing 908
909
8.7 Aseptic preparation and processing is the handling of sterile product, containers and/or devices in 910
a controlled environment in which the air supply, materials and personnel are regulated to prevent 911
microbial, pyrogenic and particulate contamination. 912
913
8.8 The aseptic process should be clearly defined. The risks associated with the aseptic process, and 914
any associated requirements, should be identified, assessed and appropriately controlled. The site’s 915
CCS should clearly define the acceptance criteria for these controls, requirements for monitoring and 916
the review of their effectiveness. Methods and procedures to control these risks should be described 917
and implemented. Accepted residual risks should be formally documented. 918
919
8.9 Precautions to minimize microbial, pyrogenic and particulate contamination should be taken, 920
as per the site’s CCS, during the preparation of the aseptic environment, during all processing stages
921
(
including the stages before and after bulk product sterilization), and until the product is sealed in its 922
final container. The presence of materials liable to generate particulates and fibres should be minimized 923
in cleanrooms. 924

20
925
8.10 Where possible, the use of equipment such as RABS, isolators or other systems, should be 926
considered in order to reduce the need for critical interventions into the Grade A zone and to minimize 927
the risk of contamination. Robotics and automation of processes can also be considered to eliminate 928
direct human critical interventions (e.g. dry heat tunnel, automated lyophilizer loading, sterilization in 929
place). 930
931
8.11 Examples of operations to be carried out in the various environmental grades are given in the 932
Table 5. 933
934
Table 5: Examples of operations and grades for aseptic preparation and processing operations 935
936
Grade A
Critical zone for
- Aseptic assembly of filling equipment.
- Connections made under aseptic conditions (where sterilize
d product contact
surfaces are exposed) that are post the final sterilizing filter. T
hese
connections should be sterilized by steam-in-place whenever feasible.
- Aseptic compounding and mixing.
- Replenishment of sterile bulk product, containers and closures.
- Removal and cooling of unprotected (e.g. with no packaging) items
from
sterilizers.
- Staging and conveying of sterile primary packaging components.
- Aseptic filling, sealing of containers such as ampoules, vial closure,
transfer
of open or partially stoppered vials.
- Loading of a lyophilizer.
Grade B
Background support for the Grade A zone (when not in an isolator).
- Transport, while protected from the surrounding environment,
of equipment,
components and ancillary items for introduction into the Grade A zone.
Grade C
- Preparation of solutions to be filtered including weighing.
Grade D
- Cleaning of equipment.
- Handling of components, equipment and accessories after washing.
- Assembly
of cleaned components, equipment and accessories prior to
sterilization.
- Assembly of closed and sterilized SUS using intrinsic aseptic connectors.
937
8.12 For sterile products that cannot be filtered, the following should be considered: 938
939
i. All product and component contact equipment should be sterilized prior to use. 940
941
ii. All raw materials should be sterilized and aseptically added or subsequently sterilized by 942
filtration. 943
944
iii. Bulk solutions should be sterilized by a validated process, e.g. by heat, chemical sterilization 945
or via sterile filtration. 946
947
iv. All materials added to the sterile bulk product should be sterilized prior to addition. 948
949
8.13 The unwrapping, assembly and preparation of sterilized equipment, components and ancillary 950
items and the preparation and filling of the sterile product should be treated as an aseptic process and 951
performed in a Grade A zone with a Grade B background. Where an isolator or RABS is used, the 952
21
background should be in accordance with paragraphs 4.21 & 4.22. 953
954
8.14 Preparation and filling of sterile products such as ointments, creams, suspensions and 955
emulsions should be
performed in a Grade A zone with a Grade B background when the product and 956
components are exposed to the environment and
the product
is not subsequently filtered (via a 957
sterilizing filter) or terminally sterilized. Where an isolator or RABS is used, the background should 958
be in accordance with paragraphs 4.21 & 4.22. 959
960
8.15 Aseptic connections should be performed in a Grade A zone with a Grade B background unless 961
subsequently sterilized in place or conducted with validated intrinsic sterile connection devices that 962
minimize any potential contamination from the immediate environment. Where an isolator or RABS 963
is used, the background should be in accordance with paragraphs 4.21 & 4.22. Aseptic connections 964
should be appropriately assessed and their effectiveness verified. For requirements regarding intrinsic 965
sterile connection devices refer to paragraph 8.120. 966
967
8.16 Aseptic manipulations (including non-intrinsic aseptic connections) should be minimized 968
through the use of engineering design solutions such as preassembled and sterilized equipment. 969
Whenever feasible, product contact piping and equipment should be pre-assembled, and sterilized in 970
place. 971
972
8.17 There should be an authorized list of allowed interventions, both inherent and corrective, that 973
may occur during production. The procedures listing the types of inherent and corrective 974
interventions, and how to perform them, should be updated, as necessary to ensure consistency with 975
the actual manufacturing activities. In the event that an unauthorized intervention is required, details 976
of the intervention conducted should be recorded and fully assessed under the manufacturer's PQS. 977
978
8.18 The duration of each aspect of aseptic preparation and processing should be limited to a defined 979
and validated maximum time, including: 980
981
i. The holding time between equipment, component, and container cleaning, drying and 982
sterilization. 983
984
ii. The holding time for sterilized equipment, components, and containers before use and 985
during filling/assembly. 986
987
iii. The holding time for a decontaminated environment, such as the RABS and isolator before 988
and during filling /assembly. 989
990
iv. The time between the start of the preparation of a product and its sterilization or filtration 991
through a microorganism-retaining filter (if applicable), through to the end of the aseptic 992
filling process. There should be a maximum permissible time for each product that takes 993
into account its composition and the prescribed method of storage. 994
995
v. The holding time for sterilized product prior to filling. 996
997
vi. The aseptic processing time. 998
999
vii. The filling time. 1000
1001
viii. The maximum exposure time of sterilized containers and closures in the critical processing 1002
zone (including filling) prior to closure. 1003
1004
8.19 Aseptic operations (including APS) should be observed at a regular basis by personnel with 1005
specific expertise in aseptic processing to verify the correct performance of operations and address 1006
inappropriate practices if detected. 1007
22
1008
Finishing of sterile products 1009
1010
8.20 Open primary packaging containers (including partially stoppered vials or prefilled syringes) 1011
should be maintained under Grade A conditions with Grade B background (e.g. Barrier Technology), 1012
or under Grade A conditions with physical segregation from operators (e.g. UDAF carts) until the 1013
stopper is fully inserted. 1014
1015
8.21 Containers should be closed by appropriately validated methods. Containers closed by fusion, 1016
e.g. Blow-fill-seal (BFS), Form-Fill-Seal (FFS), Small and Large Volume Parenteral 1017
(SVP & LVP) bags, glass or plastic ampoules, should be subject to 100% integrity testing. 1018
Samples of containers closed by other methods should be taken and checked for integrity using 1019
validated methods. The frequency of testing should be based on the knowledge and experience of the 1020
container and closure systems being used. A scientifically valid sampling plan should be utilized. 1021
The sample size should be based on information such as supplier approval, packaging component 1022
specifications and process knowledge. It should be noted that visual inspection alone is not 1023
considered as an acceptable integrity test method. 1024
1025
8.22 Containers sealed under vacuum (where the vacuum is necessary for the product stability) 1026
should be tested for maintenance of vacuum after
an appropriate pre-determined period and during 1027
shelf life. 1028
1029
8.23 The container closure integrity validation should take into consideration any transportation or 1030
shipping requirements that may negatively impact the integrity of the container (e.g. by 1031
decompression or temperature extremes). 1032
1033
8.24 Where the equipment used to crimp vial caps can generate large quantities of non-viable 1034
particulates, measures to prevent particulate contamination such as locating the equipment at a 1035
physically separate station equipped with adequate air extraction should be taken. 1036
1037
8.25 Vial capping can be undertaken as an aseptic process using sterilized caps or as a clean
1038
process outside the aseptic core. Where the latter approach is adopted, vials should be
1039
protected by Grade A conditions up to the point of leaving the aseptic processing area, and 1040
thereafter stoppered vials should be protected with a Grade A air supply until the cap has been 1041
crimped. Where capping is a manual process it should be performed under Grade A conditions either 1042
in an appropriately designed isolator or into Grade A zone with a Grade B background. 1043
1044
8.26 Where capping of aseptically filled sterile product is conducted as a clean process with Grade A 1045
air supply protection, vials with missing or displaced stoppers should be rejected prior to capping. 1046
Appropriately qualified, automated methods for stopper height detection should be in place. 1047
1048
8.27 Where
human intervention is required at the capping station, appropriate technological and 1049
organizational measures should be used to prevent direct contact with the vials and to minimize 1050
microbial contamination. 1051
1052
8.28 RABS and isolators may be beneficial in assuring the required
conditions and minimizing the 1053
microbial contamination associated with direct human interventions into the capping operation. 1054
1055
8.29 All filled containers of parenteral products should be inspected individually for extraneous
1056
contamination or other defects. Defect classification and criticality should be determined during 1057
qualification and based on risk and historical knowledge. Factors to consider include, but are not 1058
limited to, the potential impact of the defect to the patient and the route of administration. Different 1059
defect types should be categorized and batch performance analysed. Batches with unusual levels of 1060
defects, when compared with routine defect numbers for the process (based on historical and trend 1061
data), should lead to an investigation. A defect library should be generated and maintained which 1062
23
captures all known classes of defects. The defect library should be used for the training of production 1063
and quality assurance personnel. Critical defects should not be identified during any subsequent 1064
sampling and inspection of acceptable containers. Any critical defect identified should trigger an 1065
investigation as it indicates a possible failure of the original inspection process. 1066
1067
8.30 When inspection is done manually, it should be performed under
suitable and controlled 1068
conditions of illumination and background. Inspection rates should be appropriately controlled and 1069
qualified. Operators performing the
inspection should undergo visual inspection qualification (whilst 1070
wearing corrective lenses, if these are normally worn) at least annually. The qualification should be 1071
undertaken using appropriate samples from the manufacturer's defect library sets and taking into 1072
consideration worst case scenarios (e.g. inspection time, line speed where the product is transferred to 1073
the operator by a conveyor system, container size or fatigue at the end of shift) and should include 1074
consideration of eyesight checks. Operator distractions should be minimized and frequent breaks, of 1075
an appropriate duration, from inspection should be taken. 1076
1077
8.31 Where automated methods of inspection are used, the process should be validated to detect 1078
known defects (which may impact the product quality, safety or efficacy) and be equal to, or better 1079
than, manual inspection methods. The performance of the equipment should be challenged using 1080
representative defects prior to start up and at regular intervals. 1081
1082
8.32 Results of the inspection should be recorded and defect types and numbers trended. Reject levels 1083
for the various defect types should also be trended based on statistical principles. Impact to product on 1084
the market should be assessed as part of the investigation when adverse trends are observed. 1085
1086
Sterilization 1087
1088
8.33 Where possible, finished product should be terminally sterilized, using a validated and controlled 1089
sterilization process, as this provides a greater assurance of sterility than a validated and controlled 1090
sterile filtration process and/or aseptic processing. Where it is not possible for a product to undergo 1091
terminal sterilization, consideration should be given to using terminal bioburden reduction steps, such 1092
as heat treatments (e.g. pasteurization), combined with aseptic process to give improved sterility 1093
assurance. 1094
1095
8.34 The selection, design and location of the equipment and cycle/programme used for sterilization 1096
should be based on scientific principles and data which demonstrate repeatability and reliability of the 1097
sterilization process. Critical parameters should be defined, controlled, monitored and recorded. 1098
1099
8.35 All sterilization processes should be validated. Validation studies should take into account the 1100
product composition, storage conditions and maximum time between the start of the preparation of a 1101
product or material to be sterilized and its sterilization. Before any sterilization process is adopted, its 1102
suitability for the product and equipment, and its efficacy in consistently achieving the desired 1103
sterilizing conditions in all parts of each type of load to be processed should be validated notably by 1104
physical measurements and where appropriate by biological indicators (BI). For effective sterilization, 1105
the whole of the product, and surfaces of equipment and components should be subject to the required 1106
treatment and the process should be designed to ensure that this is achieved. 1107
1108
8.36 Particular attention should be given when the adopted sterilization method is not described in the 1109
current edition of the Pharmacopoeia, or when it is used for a product which is not a simple aqueous 1110
solution. Where possible, heat sterilization is the method of choice. 1111
1112
8.37 Validated loading patterns should be established for all sterilization processes and should be 1113
subject to periodic revalidation. Maximum and minimum loads should also be considered as part of 1114
the overall load validation strategy. 1115
1116
24
8.38 The validity of the sterilizing process should be reviewed and verified at scheduled intervals 1117
based on risk. Heat sterilization cycles should be revalidated with a minimum frequency of at least 1118
annually. 1119
1120
8.39 Routine operating parameters should be established and adhered to for all sterilization 1121
processes, e.g. physical parameters and loading patterns. 1122
1123
8.40 There should be mechanisms in place to detect a sterilization cycle that does not conform to the 1124
validated parameters. Any failed sterilization or sterilization that deviated from the validated process 1125
(e.g. have longer or shorter phases such as heating cycles) should be investigated. 1126
1127
8.41 Suitable BIs placed at appropriate locations may be considered as an additional method to 1128
support the validation of the sterilization process. BIs should be stored and used according to the 1129
manufacturer’s instructions. Where BIs are used to support validation and/or to monitor a 1130
sterilization process (e.g. for ethylene oxide), positive controls should be tested for each sterilization 1131
cycle. If BIs are used, strict precautions should be taken to avoid transferring microbial contamination to 1132
the manufacturing or other testing processes. BI results in isolation do not give assurance of 1133
sterilization and should not be used to override other critical parameters and process design 1134
elements. 1135
1136
8.42 The reliability of BIs is important. Suppliers should be qualified and transportation and storage 1137
conditions should be controlled in order that BI quality is not compromised. Prior to use of a new 1138
batch/lot of BIs, the population and identity of the indicator organism of the batch/lot should be 1139
verified. For other critical parameters, e.g. D-value, Z- value, the batch certificate provided by the 1140
qualified supplier can normally be used. 1141
1142
8.43 There should be a clear means of differentiating products, equipment and components, which 1143
have not been subjected to the sterilization process from those which have. Containers used to carry 1144
products such as baskets or trays, items of equipment and/or components should be clearly labelled 1145
(or electronically tracked) with the material name, product batch number and an indication of 1146
whether or not it has been sterilized. Indicators such as autoclave tape, or irradiation indicators may 1147
be used, where appropriate, to
indicate whether or not a batch (or sub-batch) has passed through a 1148
sterilization process. However, these indicators show only that the sterilization process has occurred, 1149
they do not indicate product sterility or achievement of the required sterility assurance level. 1150
1151
8.44 Sterilization records should be available for each sterilization run. Each cycle should have a 1152
unique identifier. They should be reviewed and approved as part of the batch certification procedure. 1153
1154
8.45 Where possible, materials, equipment and components should be sterilized by validated methods 1155
appropriate to the specific material. Suitable protection after sterilization should be provided to 1156
prevent recontamination. If sterilized items are not used immediately after sterilization, these should 1157
be stored using appropriately sealed packaging. A maximum hold time should also be established. 1158
Where justified, components that have been packaged with multiple sterile packaging layers need not 1159
be stored in a cleanroom if the integrity and configuration of the sterile pack allows the items to be 1160
readily disinfected during transfer by operators into the Grade A zone, (e.g. by the use of multiple 1161
sterile coverings that can be removed at each transfer from lower to higher grade). Where protection is 1162
achieved by containment in sealed packaging, this packaging process should be undertaken prior to 1163
sterilization. 1164
1165
8.46 Where materials, equipment, components and ancillary items are sterilized in sealed packaging 1166
and then transferred into the Grade A zone, this should be done using appropriate, validated methods 1167
(for example, airlocks or pass-through hatches) with accompanying disinfection of the exterior of the 1168
sealed packaging. The use of rapid transfer port technology should also be considered. These methods 1169
should be demonstrated to effectively control the potential risk of contamination of the Grade A zone 1170
and Grade B area and, likewise, the disinfection procedure should be demonstrated to be effective in 1171
25
reducing any contamination on the packaging to acceptable levels for entry of the item into the Grade 1172
B and Grade A areas. 1173
1174
8.47 Where materials, equipment, components and ancillary items are sterilized in sealed packaging 1175
or containers, the packaging sealing process should be validated. The validation should consider the 1176
integrity of the sterile protective barrier system and the maximum hold time before sterilization and 1177
maximum shelf life assigned to the sterilized items. The integrity of the sterile protective barrier 1178
system for each of the sterilized items should be confirmed prior to use. 1179
1180
8.48 For materials, equipment, components and ancillary items that are necessary for aseptic 1181
processing but cannot be sterilized, an effective and validated disinfection and transfer process should 1182
be in place. These items, once disinfected, should be protected to prevent recontamination. These 1183
items, and others representing potential routes of contamination, should be included in the 1184
environmental monitoring program. 1185
1186
1187
Sterilization by heat 1188
1189
8.49 Each heat sterilization cycle should be recorded either electronically or by hardcopy, on 1190
equipment with suitable accuracy and
precision. Monitoring and recording systems should be 1191
independent of the controlling system (e.g. by the use of duplex/double probes). 1192
1193
8.50 The position of the temperature probes used for controlling and/or recording should
be 1194
determined during the validation which should include heat distribution and penetration studies 1195
and, where applicable, also checked against a
second independent temperature probe located at the 1196
same position. 1197
1198
8.51 Sufficient time should be allowed for the whole of the load to reach the required
temperature 1199
before measurement of the sterilizing time-period starts. For sterilization cycles controlled by 1200
using a reference probe within the load, specific consideration should be given to ensuring the 1201
load probe temperature is controlled within defined temperature range prior to cycle 1202
commencement. 1203
1204
8.52 After completion of the high temperature phase of a heat sterilization cycle, precautions should 1205
be taken
against contamination of a sterilized load during cooling. Any cooling liquid or gas that 1206
comes in contact with the product or sterilized material should be sterilized. 1207
1208
8.53 In those cases where parametric release has been authorized, a robust system should be applied 1209
to the product lifecycle validation and the routine monitoring of the manufacturing process. This 1210
system should be periodically reviewed. Further guidance regarding parametric release is provided in 1211
Annex 17. 1212
1213
Moist heat sterilization 1214
1215
8.54 Moist heat sterilization utilises steam or superheated water, typically at lower temperatures and 1216
shorter duration than dry heat processes, in order to sterilize a product or article. Moist heat 1217
sterilization of hard goods or porous loads is primarily effected by latent heat of condensation of 1218
clean steam and the quality of steam is therefore important to provide consistent results. For aqueous 1219
liquid-filled containers, energy from moist heat is transferred through conduction and/or convection 1220
to the content of the container without direct contact with the autoclave steam. In these cases, time 1221
and temperature are the key parameters and steam quality does not have the same impact to the 1222
process. Moist heat sterilization processes may be utilized to sterilize or control bioburden (for non-1223
sterile applications) of thermally stable materials, articles or products and is the preferred method of 1224
sterilization, where possible. Moist heat sterilization can be achieved using steam, (direct or indirect 1225
contact), but also includes other systems such as superheated water systems. Superheated systems 1226
26
are typically used for the terminal sterilization of product in flexible containers where the pressure 1227
differentials associated with the steam would cause damage to the primary container. 1228
1229
8.55 For porous cycles (hard goods) time, temperature and pressure should be used to monitor the 1230
process. Each item sterilized should be inspected for damage, packaging material integrity and 1231
moisture on removal from the autoclave. Any item found not to be fit for purpose should be 1232
removed from the manufacturing area and an investigation performed. 1233
1234
8.56 For autoclaves fitted with a drain at the bottom of the chamber, the temperature should be 1235
recorded at this position throughout the sterilization period. For steam in place systems, the 1236
temperature should be recorded at condensate drain locations throughout the sterilization period. 1237
1238
8.57 Validation of porous cycles should include a calculation of equilibration time, exposure time, 1239
correlation of pressure and temperature and maximum temperature range during exposure. 1240
Validation of fluid cycles should include temperature, time and/or F
o
. These critical processing 1241
parameters should be subject to defined limits (including appropriate tolerances) and be confirmed as 1242
part of the sterilization validation and routine cycle acceptance criteria. 1243
1244
8.58 Leak tests on the sterilizing system should be carried out periodically (normally weekly) when a 1245
vacuum phase is part of the
cycle or the system is returned, post-sterilization, to a pressure lower 1246
than the environment surrounding the sterilized system. 1247
1248
8.59 There should be adequate assurance of air removal prior to and during sterilization when the 1249
sterilization process includes air purging (e.g. porous autoclave loads, lyophilizer chambers). For 1250
autoclaves, this should include an air removal test cycle (normally performed on a daily basis) or an 1251
air detector system. Loads to be sterilized should be designed to support effective air removal and be 1252
free draining to prevent the build-up of condensate. 1253
1254
8.60 The items to be sterilized, other than products in sealed containers, should be dry, wrapped in a
1255
material which allows removal of air and penetration of steam and prevents
recontamination after 1256
sterilization. All loaded items should be dry upon removal from the sterilizer. Load dryness should 1257
be confirmed by visual inspection as a part of the sterilization process acceptance. 1258
1259
8.61 If it is necessary to wet equipment using WFI (e.g. ultrafiltration membrane) prior to the 1260
sterilization process, then a risk-based assessment should be carried out to demonstrate the 1261
acceptable dryness level that will not impact the sterility of the equipment sterilized and the product 1262
sterility assurance level. The hold time between the wetting phase and sterilization should be 1263
justified and validated. 1264
1265
8.62 Distortion and damage of non-rigid containers that are terminally sterilized, such as containers 1266
produced by Blow-Fill-Seal or Form-Fill-Seal technologies, should be prevented by appropriate cycle 1267
design and control (for instance setting correct pressure, heating and cooling rates and loading 1268
patterns). 1269
1270
8.63 Where steam in place systems are used (e.g. for fixed pipework, vessels and lyophilizer 1271
chambers), the system should be appropriately designed and validated to assure all parts of the 1272
system are subjected to the required treatment. The system should be monitored for temperature, 1273
pressure and time at appropriate locations during routine use to ensure all areas are effectively and 1274
reproducibly sterilized. These locations should be demonstrated as being representative of, and 1275
correlated with, the slowest to heat locations during initial and routine validation. Once a system has 1276
been sterilized by steam in place it should remain integral and held under positive pressure prior to 1277
use. 1278
1279
8.64 For systems using superheated water rather than steam, as the sterilizing agent, the heated water 1280
should consistently reach all of the required contact points. Initial qualification studies should 1281
27
include temperature mapping of the entire load. There should be routine checks on the equipment to 1282
ensure that nozzles (where the water is introduced) are not blocked and drains remain free from 1283
debris. 1284
1285
8.65 For the qualification of superheated systems it should be demonstrated that all parts of the load 1286
meet the minimum required temperature and that routine monitoring probes are located in the worst 1287
case positions identified during the qualification process. 1288
1289
Dry heat sterilization 1290
1291
8.66 Dry heat sterilization is of particular use in the removal of thermally robust contaminants such as 1292
pyrogens and is often used in the preparation of components for aseptic filling. The combination of 1293
time and temperature to which product, components and equipment are exposed should produce an 1294
adequate and reproducible level of lethality and/or pyrogen (endotoxin) inactivation/removal when 1295
operated routinely within the established limits. 1296
1297
8.67 Dry heat sterilization/depyrogenation tunnels should be configured to ensure that airflow protects 1298
the integrity and performance of the Grade A sterilizing zone by maintaining pressure differentials 1299
and airflow through the tunnel from the higher grade area to the lower grade area. Airflow patterns 1300
should be visualised and correlated with temperature studies. The impact of any airflow change 1301
should be assessed to ensure the heating profile is maintained. All air supplied to the tunnel should 1302
pass through at least a HEPA filter and periodic tests should be performed to demonstrate air filter 1303
integrity (at least biannually). Any tunnel parts that come into contact with sterilized components 1304
should be appropriately sterilized or disinfected. Critical process parameters that should be considered 1305
during validation and/or routine processing should include, but may not be limited to: 1306
1307
i. Belt speed or dwell time within the sterilizing zone. 1308
1309
ii. Temperature – minimum and maximum temperatures. 1310
1311
iii. Heat penetration of the material/article. 1312
1313
iv. Heat distribution/uniformity. 1314
1315
v. Airflows – correlated with the heat distribution and penetration studies. 1316
1317
8.68 When a thermal depyrogenation process is used for any component or product contact 1318
equipment, validation studies should be performed to demonstrate that the process provides a suitable 1319
F
h
value and results in a minimum 3 log reduction in endotoxins concentration. 1320
1321
8.69 Containers inoculated with endotoxin should be used during validation and should be carefully 1322
managed with a full reconciliation performed. Containers should be representative of the materials 1323
normally processed. Endotoxin quantification and recovery efficiency should also be demonstrated 1324
through biological measurement. 1325
1326
8.70 Dry heat ovens are typically employed to sterilize or depyrogenate primary packaging 1327
components, finished materials or active substances but may be used for other processes. They should 1328
be maintained at a positive pressure relative to lower grade areas throughout the sterilization and post 1329
sterilization hold process. All air entering the oven should pass through a sterilizing filter. Critical 1330
process parameters that should be considered in qualification and/or routine processing should 1331
include, but may not be limited to: 1332
1333
i. Temperature. 1334
1335
ii. Exposure period/time. 1336
28
1337
iii. Chamber pressure (for maintenance of over pressure). 1338
1339
iv. Air speed. 1340
1341
v. Air quality within the oven. 1342
1343
vi. Heat penetration of material/article (slow to heat spots). 1344
1345
vii. Heat distribution/uniformity. 1346
1347
8.71 For dry heat sterilization of starting materials and intermediates, the same principles should be 1348
applied. Consideration should also be given to factors affecting heat penetration such as the container 1349
type, size and packing matrix. 1350
1351
Sterilization by radiation 1352
1353
8.72 Guidance regarding ionising radiation sterilization can be found within Annex 12. 1354
1355
8.73 Sterilization by radiation is used mainly for the sterilization of heat sensitive materials and
1356
products. Ultraviolet irradiation is not an acceptable method of sterilization. 1357
1358
8.74 Validation procedures should ensure that the effects of variations in density of the product and 1359
packages are considered. 1360
1361
Sterilization with ethylene oxide 1362
1363
8.75 This method should only be used when no other method is practicable. During process 1364
validation, it should be shown that there is no damaging effect on the product and that the
1365
conditions and time allowed for degassing result in the reduction of any residual ethylene oxide 1366
(EO) gas and reaction
products to defined acceptable limits for the given product or material. 1367
1368
8.76 Direct contact between gas and microbial cells is essential, precautions should be taken to avoid 1369
the presence of organisms likely to be enclosed in material such as crystals or dried protein. The 1370
nature, porosity and quantity of packaging materials can significantly affect the process. 1371
1372
8.77 Before exposure to the gas, materials should be brought into equilibrium with the
1373
humidity and temperature required by the process. The time required for this should be
1374
balanced against the opposing need to minimize the time before sterilization. 1375
1376
8.78 Each sterilization cycle should be monitored with suitable BIs, using the
appropriate number of 1377
test units distributed throughout the load at defined locations that have been shown to be worst case 1378
during validation. 1379
1380
8.79 Critical process variables that could be considered as part of the sterilization process validation 1381
and routine monitoring include, but are not limited to: 1382
1383
i. EO gas concentration. 1384
1385
ii. EO gas pressure. 1386
1387
iii. Amount of EO gas used. 1388
1389
iv. Relative humidity. 1390
1391
29
v. Temperature. 1392
1393
vi. Exposure time. 1394
1395
8.80 After sterilization, the load should be aerated to allow EO gas and/or its reaction products to 1396
desorb from the packaged product to predetermined levels. Aeration can occur within a sterilizer 1397
chamber and/or in a separate aeration chamber or aeration room. The aeration phase should be 1398
validated as part of the overall EO sterilization process validation. 1399
1400
Filter sterilization of products which cannot be sterilized in their final container 1401
1402
8.81 If the product cannot be sterilized in the final container, solutions or liquids should be sterilized 1403
by filtration through a sterile sterilizing grade filter (with a nominal pore size of 0.22 µm (or less) 1404
that has been appropriately validated to obtain a sterile filtrate) and subsequently aseptically filled 1405
into a previously sterilized container. The selection of the filter used should ensure that it is 1406
compatible with the product and as described in the marketing authorization (refer to paragraph 1407
8.125). 1408
1409
8.82 Suitable bioburden reduction prefilters and/or sterilizing grade filters may be used at multiple 1410
points during the manufacturing process to ensure a low and controlled bioburden of the liquid prior 1411
to the primary sterilizing grade filter. Due to the potential additional risks of a sterile filtration 1412
process, as compared with other sterilization processes, a second filtration through a sterile sterilizing 1413
grade filter, immediately prior to filling, should be considered as part of an overall CCS. 1414
1415
8.83 The selection of components for the filtration system and their interconnection and arrangement 1416
within the filtration system, including pre-filters, should be based on the critical quality attributes of 1417
the product, justified and documented. The filtration system should minimize the generation of fibres 1418
and particulates, not cause or contribute to unacceptable levels of impurities, or possess characteristics 1419
that otherwise alter the quality and efficacy of the product. Similarly, the filter characteristics should 1420
be compatible with the fluid and not be adversely affected by the product to be filtered. Adsorption of 1421
product components and extraction/leaching of filter components should be evaluated (refer to 1422
paragraph 8.125). 1423
1424
8.84 The filtration system should be designed to: 1425
1426
i. Allow operation within validated process parameters. 1427
1428
ii. Maintain the sterility of the filtrate. 1429
1430
iii. Minimize the number of aseptic connections required between the sterilizing filter and the 1431
final filling of the product. 1432
1433
iv. Allow cleaning procedures to be conducted as necessary. 1434
1435
v. Allow sterilization procedures, including sterilization in place, to be conducted as necessary. 1436
1437
vi. Permit in-place integrity testing, of the 0.22 µm sterilizing filter, preferably as a closed 1438
system, prior to filtration as necessary. In-place integrity testing methods should be selected 1439
to avoid any adverse impact on the quality of the product. 1440
1441
8.85 Sterile filtration of liquids should be validated in accordance with European (or other relevant) 1442
Pharmacopeia requirements. Validation can be grouped by different strengths or variations of a 1443
product but should be done under worst case conditions. The rationale for grouping should be 1444
justified and documented. 1445
30
1446
8.86 During filter validation, wherever possible, the product to be filtered should be used for 1447
bacterial retention testing of the sterilizing filter. Where the product to be filtered is not suitable for 1448
use in bacterial retention testing, a suitable surrogate product should be justified for use in the test. 1449
The challenge organism used in the bacterial retention test should be justified. 1450
1451
8.87 Filtration parameters that should be considered and established in validation and monitored in 1452
routine processing should include, but are not limited to: 1453
1454
1455
i. The wetting fluid used for filter integrity testing should be based on the filter manufacturer’s 1456
recommendation or the fluid to be filtered. The appropriate integrity test value specification 1457
should be established. 1458
ii. 1459
If the system is flushed or integrity tested in-situ with a fluid other than the product, 1460
appropriate actions are taken to avoid any deleterious effect on product quality. 1461
1462
iii. Filtration process conditions including: 1463
1464
• Fluid pre-filtration holding time and effect on bioburden. 1465
1466
• Filter conditioning, with fluid if necessary. 1467
1468
• Maximum filtration time/total time filter is in contact with fluid. 1469
1470
• Maximum operating pressure. 1471
1472
• Flow rate. 1473
1474
• Maximum filtration volume. 1475
1476
• Temperature. 1477
1478
• The time taken to filter a known volume of bulk solution and the
pressure difference 1479
to be used across the filter. 1480
1481
Note: Results of these checks should be included in the batch record. Any
significant difference in 1482
parameters from those validated to those observed during routine manufacturing should be noted 1483
and
investigated. 1484
1485
8.88 The integrity of the sterilized filter assembly should be verified by integrity testing before use, to 1486
check for damage and loss of integrity caused by the filter preparation prior to use. A sterilizing grade 1487
filter that is used to sterilize a fluid should be subject to a non-destructive integrity test post-use prior 1488
to removal of the filter from its housing. Test results should correlate to the microbial retention 1489
capability of the filter established during validation. Examples of tests that are used include bubble 1490
point, diffusive flow, water intrusion or pressure hold test. It is recognized that pre-use post 1491
sterilization integrity testing (PUPSIT) may not always be possible after sterilization due to process 1492
constraints (e.g. the filtration of very small volumes of solution). In these cases, an alternative 1493
approach may be taken providing that a thorough risk assessment has been performed and compliance 1494
is achieved by the implementation of appropriate controls to mitigate any risk of non-sterility. Points 1495
to consider in such a risk assessment should include but are not be limited to: 1496
1497
i. In depth knowledge and control of the sterilization process to ensure that the potential for 1498
damage to the filter is minimized. 1499
31
1500
ii. In depth knowledge and control of the supply chain to include: 1501
• Contract sterilization facilities. 1502
• Defined transport mechanisms. 1503
• Packaging of the sterilized filter, to prevent damage to the filter during transportation 1504
and storage. 1505
iii. In depth process knowledge such as: 1506
• The specific product type, including particulate burden and whether there exists any 1507
risk of impact on filter integrity values, such as the potential to alter integrity testing 1508
values and therefore prevent the detection of a non-integral filter during a post-use 1509
filter integrity test. 1510
• Pre-filtration and processing steps, prior to the sterilizing filter, which would remove 1511
particulate burden and clarify the product prior to the sterile filtration. 1512
1513
8.89 The integrity of critical sterile gas and air vent filters (that are directly linked to the sterility of 1514
the product) should be verified by testing after use, with the filter remaining in the filter assembly. 1515
1516
8.90 The integrity of non-critical air or gas vent filters should be confirmed and recorded at 1517
appropriate intervals. Where gas filters are in place for extended periods such as vent filters, integrity 1518
testing should be carried out pre and post-use. The maximum duration of use should be specified and 1519
monitored based on risk (e.g. considering the maximum number of uses and sterilization cycles 1520
permitted). 1521
1522
8.91 For gas filtration, attention should be paid to avoiding unintended moistening or wetting of the 1523
filter or filter equipment. This can be achieved by the use of hydrophobic filters. 1524
1525
8.92 If the sterilizing filtration process has been validated as a system consisting of multiple filters to 1526
achieve the sterility for a given fluid, the filtration system is considered to be a single sterilizing unit 1527
and all filters within the system should satisfactorily pass integrity testing after use. 1528
1529
8.93 In a redundant filtration system (where a second filter is present as a backup but the sterilizing 1530
process is validated as only requiring one filter), post-use integrity test of the primary sterilizing filter 1531
should be performed and if demonstrated to be integral, then a post-use integrity test of the secondary 1532
filter is not necessary. However, in the event of a failure of the post-use integrity test on the primary 1533
filter, a risk assessment should be carried out to determine the acceptability of performing a post-use 1534
integrity test on the secondary (redundant) filter. 1535
1536
8.94 Bioburden samples should be taken from the bulk product and immediately prior to the final 1537
sterile filtration. Systems for taking samples should be designed so as not to introduce contamination. 1538
1539
8.95 Liquid sterilizing filters should be discarded after the processing of a single lot and the same 1540
filter should not be used for more than one working day unless such use has been validated. 1541
8.96 Where campaign manufacture of a product has been appropriately justified in the CCS and 1542
validated, the filter user should: 1543
i. Assess and document the risks associated with the duration of filter use for the sterile 1544
filtration process for a given fluid. 1545
ii. Conduct and document effective validation and qualification studies to demonstrate that the 1546
duration of filter use for a given sterile filtration process and for a given fluid does not 1547
compromise performance of the sterilizing filter or filtrate quality. 1548
32
iii. Document the maximum validated duration of use for the filter and implement controls to 1549
ensure that filters are not used beyond the validated maximum duration. Records of these 1550
controls should be maintained. 1551
iv. Implement controls to ensure that filters contaminated with fluid or cleaning agent residues, 1552
or considered defective in any other way, are removed from use. 1553
Form-Fill-Seal 1554
1555
8.97 Form-Fill-Seal (FFS) units include blow moulding from thermoplastic granulate and 1556
thermoforming from thermoplastic film, typically known as Blow-Fill-Seal (BFS) and Vertical-Form-1557
Fill-Seal (VFFS), respectively. VFFS process is an automated filling process, typically for terminally 1558
sterilized products, that may utilize a single or dual web system which constructs the primary 1559
container out of a flat roll of thermoplastic film while simultaneously filling the formed bags with 1560
product and sealing the filled bags in a continuous process. All such containers are considered to be 1561
closed through sealing by fusion and, as such, fall under the requirement to perform 100% integrity 1562
testing (refer to paragraph 8.21). 1563
1564
8.98 Process parameters relating to seal integrity should be qualified and appropriately controlled. 1565
1566
8.99 Critical parameters include, but are not limited to: 1567
1568
i. Seal strength. 1569
1570
ii. Seal uniformity. 1571
1572
iii. Sealing temperatures. 1573
1574
iv. Sealing pressures. 1575
1576
v. Sealing times. 1577
1578
vi. Dwell time for filling. 1579
1580
8.100 Seal strength and uniformity should be monitored routinely. 1581
1582
8.101 The controls identified during qualification should be in alignment with the site’s CCS. Aspects 1583
to be considered include but are not limited to: 1584
1585
i. Determination of the boundaries of the critical zone. 1586
1587
ii. Environmental control and monitoring, both of the machine and the background in which it 1588
is placed. 1589
1590
iii. Integrity testing of the product filling lines. 1591
1592
iv. Integrity testing of the cooling system. 1593
1594
v. Duration of the batch or filling campaign. 1595
1596
vi. Control of polymer starting material (including resin pellets). 1597
1598
vii. Cleaning-in-place and sterilization-in-place of equipment in direct contact to the formulation 1599
(product filling lines); sterilization-in-place of sterile air pathways. 1600
1601
33
Blow-Fill-Seal 1602
1603
8.102 Blow-Fill-Seal (BFS) units are purpose built machines in which, in one continuous operation, 1604
containers are formed from a thermoplastic granulate, filled and then sealed by one automatic 1605
machine. Air that makes contact with critical surfaces of the container during extrusion, formation or 1606
sealing of the moulded container should undergo appropriate filtration. 1607
1608
8.103 For shuttle type equipment used for aseptic filling, the area between parison cutting and mould 1609
sealing should be covered by a flow of filtered air to provide Grade A conditions at the critical zone. 1610
The equipment should be installed in at least a Grade C environment, provided that Grade A/B 1611
clothing is used. The filling environment should meet Grade A for viable and non-viable limits at rest 1612
and the viable limit only when in operation. 1613
1614
8.104 For rotary-type equipment, used for aseptic filling, the filling environment should be designed 1615
to meet Grade A conditions. Other sterility assurance controls such as monitoring of critical 1616
parameters and alarms during each batch and parison support filter integrity testing should be 1617
considered. 1618
1619
8.105 The environmental control and monitoring program should take into consideration the moving 1620
parts and complex airflow paths generated by the BFS process and the effect of the high heat outputs 1621
of the process, e.g. by performing smoke studies and/or other equivalent studies. Environmental 1622
monitoring should be applied taking into consideration elements such as air-filter configuration, air-1623
filter integrity, cooling systems integrity, equipment design and installation. 1624
1625
8.106 Blow-Fill-Seal equipment used for the manufacture of products which are terminally sterilized 1626
should be installed in at least a Grade D environment. The conditions at the point of fill should 1627
comply with the environmental requirements of paragraphs 8.3 and 8.4. 1628
1629
8.107 External particulate and microbial contamination of the polymer should be prevented by 1630
appropriate design, control, and maintenance of the polymer storage, sampling and distribution 1631
systems. The capability of the extrusion system to provide appropriate sterility assurance for the 1632
moulded container should be fully understood and validated. The sampling frequency, the bioburden 1633
and, where applicable, endotoxins levels of the raw polymer should be defined and controlled within 1634
the CCS. 1635
1636
8.108 Interventions requiring cessation of filling and/or extrusion, moulding and sealing and, where 1637
required, re-sterilization of the filling machine should be clearly defined and well described in the 1638
aseptic filling procedure, and included in the APS (refer to paragraphs 9.36, 9.37 and 9.38). 1639
1640
8.109 The moulds used to form containers are considered critical equipment and any changes or 1641
modification to moulds should result in an assessment of finished product container integrity, and 1642
should be supported by validation. 1643
1644
Lyophilization 1645
1646
8.110 Lyophilization is a critical process step and all activities that can affect the sterility of the 1647
product or material need to be regarded as extensions of the aseptic processing of the sterilized 1648
product. The lyophilization equipment and its processes should be designed to ensure that product or 1649
material sterility is maintained during lyophilization by preventing microbial and particulate 1650
contamination between the filling of products for lyophilization, and completion of lyophilization 1651
process. All control measures in place should be determined by the site’s CCS. 1652
1653
8.111 The sterilization of lyophilizers and associated equipment, (e.g. trays, vial support rings) should 1654
be validated and holding times between sterilization cycles appropriately challenged during aseptic 1655
process simulations. The lyophilizer should be sterilized regularly, based on system design. Re-1656
34
sterilization should be performed following maintenance or cleaning. Sterilized lyophilizers and 1657
associated equipment should be protected from contamination after sterilization. 1658
1659
8.112 Lyophilizers that are manually loaded or unloaded should normally be sterilized before each 1660
load. For lyophilizers loaded by automated closed systems or located within systems that exclude 1661
operator intervention, the frequency of sterilization should be justified and documented as part of the 1662
CCS. 1663
1664
8.113 The integrity of the lyophilizer system should be maintained following sterilization and during 1665
use. The filter used to maintain lyophilizer integrity should be sterilized before each use of the system 1666
and its integrity testing results should be part of the batch certification. The frequency of vacuum/leak 1667
integrity testing of the chamber should be documented and the maximum permitted leakage of air into 1668
the lyophilizer should be specified and checked at the start of every cycle. 1669
1670
8.114 Lyophilization trays should be checked regularly to ensure that they are not misshapen or 1671
damaged. 1672
1673
8.115 Points to consider for the design of loading (and unloading, where the lyophilised material is 1674
not in a sealed container (e.g. open tray dried materials), include but are not limited to: 1675
1676
i. The loading pattern within the lyophilizer should be specified and documented. 1677
1678
ii. The transfer of partially closed containers to a lyophilizer should be undertaken under Grade 1679
A conditions at all times and handled in a manner designed to minimize direct operator 1680
intervention. Technologies such as conveyor systems, portable transfer systems (e.g. clean air 1681
transfer carts, portable unidirectional airflow workstations) should be used to ensure that the 1682
cleanliness of the system used to transfer the partially closed containers is maintained). 1683
Alternatively, where supported by validation, containers closed in the Grade A zone and not 1684
reopened whilst in the Grade B may be used to protect partially stoppered vials (e.g. sealed 1685
sterilized trays). 1686
1687
iii. Airflow patterns should not be adversely affected by transport devices and venting of the 1688
loading zone. 1689
1690
iv. Unsealed containers (such as partially stoppered vials) should be maintained under Grade A 1691
conditions and should normally be separated from operators by physical barrier technology or 1692
any other appropriate measures. 1693
1694
v. Where seating of the stoppers is not completed prior to opening the lyophilizer chamber, 1695
product removed from the lyophilizer should remain under Grade A conditions during 1696
subsequent handling. 1697
1698
vi. Utensils used during transfer to and loading and unloading of the lyophilizer (such as trays, 1699
bags, placing devices, tweezers, etc.) should be subject to a validated sterilization process. 1700
1701
Closed systems 1702
1703
8.116 Closed systems can be single use systems (i.e. disposable systems) and fixed systems (such as 1704
vessels with fixed pipework). Guidance in this section is equally applicable to both systems. 1705
1706
8.117 The use of closed systems can reduce the risk of extraneous contamination such as microbial, 1707
particulate and chemical from the adjacent environment. Closed systems should always be designed to 1708
reduce the need for, and complexity of manual interventions. 1709
1710
8.118 It is critical to ensure the sterility of all product contact surfaces of closed systems used for 1711
35
aseptic processing. The design and selection of any closed system used for aseptic processing should 1712
ensure maintenance of sterility. Connection of sterile equipment (e.g. tubing / pipework) to the 1713
sterilized product pathway after the final sterilizing filter should be designed to be connected 1714
aseptically (e.g. by intrinsic aseptic connectors or fusion systems). 1715
1716
8.119 Appropriate measures should be in place to ensure the integrity of components used in aseptic 1717
connections. The means by which this is achieved should be determined and captured in the CCS. 1718
Appropriate system integrity tests should be considered when there is a risk of compromising product 1719
sterility. Supplier assessment should include the collation of data in relation to potential failure modes 1720
that may lead to a loss of system sterility. 1721
1722
8.120 The background in which closed systems are located should be based on their design and the 1723
processes undertaken. For aseptic processing and where there are any risks that system integrity may 1724
be compromised, the system should be located in a Grade A zone. If the system can be shown to 1725
remain integral at every usage (e.g. via pressure testing and/or monitoring) then a lower classified area 1726
may be used. If the closed system is opened (e.g. for maintenance of a bulk manufacturing line) then 1727
this should be performed in a classified area appropriate to the materials (e.g. Grade C for terminally 1728
sterilization processes, or Grade A for aseptic processing) or be subject to further cleaning and 1729
disinfection (and sterilization in case of aseptic processes). 1730
1731
Single use systems (SUS) 1732
1733
8.121 SUS are those technologies used in manufacture of sterile products which are used as an 1734
alternative to reusable equipment. SUS can be individual components or made up of multiple 1735
components such as bags, filters, tubing, connectors, valves, storage bottles and sensors. 1736
1737
8.122 There are some specific risks associated with SUS which should be assessed as part of the CCS. 1738
These risks include but are not limited to: 1739
1740
i. The interaction between the product and product contact surface (such as adsorption, or the 1741
formation of leachables and extractables). 1742
1743
ii. The fragile nature of the system compared to fixed reusable systems. 1744
1745
iii. The increase in the number and complexity of manual operations (including inspection and 1746
handling of the system) and connections made. 1747
1748
iv. The complexity of the assembly. 1749
1750
v. The performance of the pre-use integrity test for sterilizing grade filters (refer to paragraph 1751
8.88). 1752
1753
vi. The risk of holes and leakage. 1754
1755
vii. The potential for compromising the system at the point of opening the outer packaging. 1756
1757
viii. The risk of particulate contamination. 1758
1759
8.123 Sterilization processes for SUS should be validated and shown to have no adverse impact on 1760
system performance. 1761
1762
8.124 Assessment of suppliers of disposable systems including sterilization is critical to the selection 1763
and use of these systems. For sterile SUS, verification of sterility should be performed as part of the 1764
supplier qualification and on receipt and use of each unit. 1765
1766

36
8.125 The adsorption and reactivity of the product with product contact surfaces should be evaluated 1767
under process conditions. 1768
1769
8.126 The extractable and leachable profile of the SUS and any impact on the quality of the product 1770
especially where the system is made from polymer-based materials should be evaluated. An 1771
assessment should be carried out for each component to evaluate the applicability of the extractable 1772
profile data. For components considered to be at high risk from leachables, including those that may 1773
absorb processed materials or those with extended material contact times, an assessment of leachable 1774
profile studies, including safety concerns, should be taken into consideration. If applying simulated 1775
processing conditions, these should accurately reflect the actual processing conditions and be based 1776
on a scientific rationale. 1777
1778
8.127 SUS should be designed to maintain integrity throughout processing under the intended 1779
operational conditions. Attention to the structural integrity of the single use components is necessary 1780
where these may be exposed to more extreme conditions (e.g. freezing and thawing processes) either 1781
during routine processing or transportation. This should include verification that intrinsic aseptic 1782
connections (both heat sealed and mechanically sealed) remain integral under these conditions. 1783
1784
8.128 Acceptance criteria should be established and implemented for SUS corresponding to the risks 1785
or criticality of the products and its processes. On receipt, each piece of SUS should be checked to 1786
ensure that they have been manufactured, supplied and delivered in accordance with the approved 1787
specification. A visual inspection of the outer packaging (e.g. appearance of exterior carton, product 1788
pouches), label printing, and review of attached documents (e.g. certificate of conformance and proof 1789
of sterilization) should be carried out and documented prior to use. 1790
1791
8.129 Critical manual handling operations of SUS such as assembly and connections should be 1792
subject to appropriate controls and verified during the APS. 1793
1794
9 Viable and non-viable environmental & process monitoring 1795
1796
General 1797
1798
9.1 The site’s environmental and process monitoring program forms part of the overall CCS and is 1799
used to monitor the controls designed to minimize the risk of microbial and particulate contamination. 1800
It should be noted that the reliability of each of the elements of the monitoring system (viable, non-1801
viable and APS) when taken in isolation is limited and should not be considered individually to be an 1802
indicator of asepsis. When considered together, their reliability is dependent on the design, validation 1803
and operation of the system that they are monitoring. 1804
1805
9.2 This program is typically comprised of the following elements: 1806
i. Environnemental monitoring – non-viable particles. 1807
1808
ii. Environmental and personnel monitoring – viable particles. 1809
1810
iii. Aseptic process simulation (aseptically manufactured product only). 1811
1812
9.3 The information from these systems should be used for routine batch certification and for periodic 1813
assessment during process review or investigation. This applies for both terminal sterilization and 1814
aseptic processes, however, the criticality of the impact may differ depending upon the product and 1815
process type. 1816
1817
Environmental monitoring 1818
1819
9.4 Risk assessments should be performed in order to establish a comprehensive environmental 1820
37
monitoring program, i.e. sampling locations, frequency of monitoring, monitoring method used and 1821
incubation conditions (e.g. time, temperature(s), aerobic and/or anaerobic conditions). These risk 1822
assessments should be conducted based on detailed knowledge of; the process inputs and final 1823
product, the facility, equipment, specific processes, the operations involved, historical monitoring 1824
data, monitoring data obtained during qualification and knowledge of typical microbial flora isolated 1825
from the environment. Consideration of other information such as air visualization studies should 1826
also be included. These risk assessments should be reviewed regularly in order to confirm the 1827
effectiveness of the site’s environmental monitoring program. The monitoring program should be 1828
considered in the overall context of the trend analysis and the CCS for the site. 1829
1830
9.5 Routine monitoring of cleanrooms, clean air equipment and personnel should be performed in 1831
operation throughout all critical stages, including equipment set-up. 1832
1833
9.6 The monitoring of Grade A zones should demonstrate the maintenance of aseptic processing 1834
conditions during critical operations. Monitoring should be performed at locations posing the highest 1835
risk of contamination to the sterile equipment surfaces, container, closures and product. The selection 1836
of monitoring locations and the orientation and positioning of sampling devices should be justified 1837
and appropriate to obtain reliable data from the critical zones. 1838
1839
9.7 Sampling methods should not pose a risk of contamination to the manufacturing operations. 1840
1841
9.8 Appropriate alert levels and action limits should be set for the results of viable and non-viable 1842
particle monitoring. Alert levels should be established based on results of cleanroom qualification 1843
tests or trend data and should be subject to periodic review. 1844
1845
9.9 Alert levels for Grade A (non-viable particles only) Grade B, Grade C and Grade D should be set 1846
such that adverse trends (e.g. a numbers of events or individual events that indicate a deterioration of 1847
cleanliness) are detected and addressed. 1848
1849
9.10 Monitoring procedures should define the approach to trending. Trends can include, but are not 1850
limited to: 1851
1852
i. Increasing numbers of action limit or alert level breaches. 1853
1854
ii. Consecutive breaches of alert levels. 1855
1856
iii. Regular but isolated breaches of action limits that may have a common cause, for example 1857
single excursions that always follow planned preventative maintenance. 1858
1859
iv. Changes in microbial flora type and numbers and predominance of specific organisms. 1860
Particular attention should be given to objectionable organisms or those that can be difficult 1861
to control such as spore-forming microorganisms. 1862
1863
9.11 The monitoring of Grade C and D cleanrooms in operation should be performed based on data 1864
collected during qualification and historical data to allow effective trend analysis. The requirements 1865
of alert levels and action limits will depend on the nature of the operations carried out. Action limits 1866
may be more stringent than those listed in Table 6 and Table 7. 1867
1868
9.12 If action limits are exceeded, operating procedures should prescribe a root cause investigation, 1869
an assessment of the potential impact to product and requirements for corrective and preventive 1870
actions. If alert levels are exceeded, operating procedures should prescribe assessment and follow-1871
up, which should include consideration of an investigation and/or corrective actions to avoid any 1872
further deterioration of the environment. 1873
1874
9.13 Results from environmental monitoring should be considered when reviewing batch 1875

38
documentation for finished product batch certification. 1876
1877
Environmental monitoring- non-viable particles 1878
1879
9.14 Non-viable particulate monitoring systems should be established to obtain data for assessing 1880
potential contamination risks and to ensure the maintenance of the environment for sterile operations 1881
in a qualified state. 1882
1883
9.15 The limits for environmental monitoring of airborne particulate concentrations for each graded 1884
area are given in Table 6. 1885
1886
Table 6: Limits for airborne particulate concentration for the monitoring of non-viable 1887
contamination. 1888
1889
Grade
M
aximum limits for
pa
rticulate
s
≥ 0.5 μm/m
3
M
aximum limits for
particulate
s
≥ 5 μm/m
3
at rest
in operation
at rest
in operation
A
3 520
3 520
29
29
B
3 520
352 000
29
2 900
C
352 000
3 520 000
2 900
29 000
D
3 520
000
Not defined
(
a
)
29 000
Not defined
(
a
)
1890
1891
1892
(a)
For Grade D, in operation limits are not defined. The company should establish in operation 1893
limits based on a risk assessment and on historical data, where applicable. 1894
1895
Note 1: The particulate limits given in the table for the “at rest” state should be achieved after 1896
a short “clean up” period (defined during qualification with a guidance value of 15 to 20 1897
minutes) in an unmanned state, after the completion of operations (refer to paragraph 4.30 and 1898
4.31). 1899
1900
Note 2: With regards to the monitoring of airborne particulates ≥5 μm particulate 1901
concentration, the limit of 29 (Grade A) is selected due to the limitations of monitoring 1902
equipment. Alert levels should be set based on historical data, such that frequent sustained 1903
counts below the action limit which may be indicative of system contamination or 1904
deterioration should trigger an investigation. For the Grade A zone and Grade B area the 1905
importance of monitoring the ≥5 μm particulates is to identify negative trends as defined in the 1906
manufacturer's CCS. 1907
1908
9.16 For the Grade A zone, particulate monitoring should be undertaken for the full duration of 1909
critical
processing, including equipment assembly. 1910
1911
9.17 The Grade A zone should be monitored continuously (for particulates ≥0.5 and ≥5 µm) and 1912
with a suitable sample flow rate (at least 28 litres (1ft
3
) per minute) so that all interventions, transient 1913
events and any system deterioration is captured. The system should frequently correlate each 1914
individual sample result with the limits in Table 6 at such a frequency that any potential excursion 1915
can be identified and responded to in a timely manner. Alarms should be triggered if alert levels are 1916
exceeded. Procedures should define the actions to be taken in response to alarms including the 1917
consideration of additional microbial monitoring. 1918
39
1919
9.18 It is recommended that a similar system be used for Grade B area although the sample frequency 1920
may be decreased. The Grade B zone should be monitored at such a frequency and with suitable 1921
sample size that the programme captures any increase in levels of contamination and system 1922
deterioration. If alert or action levels are exceeded, alarms should be triggered. 1923
1924
9.19 The selection of the monitoring system should take i n t o account any risk presented by 1925
the
materials used in the manufacturing operation (for example, those involving live organisms, 1926
powdery products or radiopharmaceuticals) that may give rise to biological or chemical hazards. 1927
1928
9.20 In the case where contaminants are present due to the processes involved and would potentially 1929
damage the particle counter or present a hazard (e.g. live organisms, powdery products and radiation 1930
hazards), the frequency and strategy employed should be such as to assure the environmental 1931
classification both prior to and post exposure to the risk. An increase in viable particle monitoring 1932
should be considered to ensure comprehensive monitoring of the process. Additionally, monitoring 1933
should be performed during simulated operations. Such operations should be performed at appropriate 1934
intervals. The approach should be defined in the CCS. 1935
1936
9.21 The size of monitoring samples taken using automated systems will usually be a
function of the 1937
sampling rate of the system used. It is not necessary for the sample volume to
be the same as that 1938
used for formal classification of cleanrooms and clean air equipment. Monitoring sample volumes 1939
should be justified. 1940
1941
9.22 The occasional indication of macro particulate counts, especially ≥ 5 µm, may be considered to 1942
be false counts due to electronic noise, stray light, coincidence, etc. However, consecutive or regular 1943
counting of low levels may be indicative of a possible contamination event and should be 1944
investigated. Such events may indicate early failure of the room air supply filtration system, filling 1945
equipment failure, or may also be diagnostic of poor practices during machine set-up and routine 1946
operation. 1947
1948
9.23 Monitoring conditions such as frequency, sampling volume or duration, alert levels and action 1949
limits and corrective actions (including an investigation) should be established in each 1950
manufacturing area based on data generated during the initial qualification process, ongoing routine 1951
monitoring and periodic review of data. 1952
1953
Environmental and personnel monitoring-viable particles 1954
1955
9.24 Where aseptic operations are performed, microbial monitoring should be frequent using a 1956
combination of methods such as settle plates, volumetric air sampling, glove, gown and surface 1957
sampling (e.g. swabs and contact plates). The method of sampling used should be justified within 1958
the CCS and should be demonstrated not to have a detrimental impact on Grade A and B airflow 1959
patterns. 1960
1961
9.25 Monitoring should include sampling of personnel at periodic intervals during the process. 1962
Sampling of personnel should be performed in such a way that it will not compromise the process. 1963
Particular consideration should be given to monitoring personnel following involvement in critical 1964
interventions and on each exit from the Grade B cleanroom. 1965
1966
9.26 Viable particle monitoring should also be performed within the cleanrooms when normal 1967
manufacturing operations are not occurring (e.g. post disinfection, prior to start of manufacturing, 1968
on completion of the batch and after a shutdown period), and in associated rooms that have not 1969
been used, in order to detect potential incidents of contamination which may affect the controls 1970
within the cleanrooms. In case of an incident, additional sample locations may be used as a 1971
verification of the effectiveness of a corrective action (i.e. cleaning and disinfection). 1972
1973

40
9.27 Continuous viable air monitoring in the Grade A zone (e.g. air sampling or settle plates) should 1974
be undertaken for the full duration of critical processing, including equipment (aseptic set-up) 1975
assembly and filling operations. A similar approach should be considered for Grade B cleanrooms 1976
based on the risk of impact on the aseptic processing. The monitoring should be performed in such a 1977
way that all interventions, transient events and any system deterioration would be captured and any 1978
risk caused by interventions of the monitoring operations is avoided. 1979
1980
9.28 The adoption of suitable rapid or automated monitoring systems should be considered by 1981
manufacturers in order to expedite the detection of microbiological contamination issues and to 1982
reduce the risk to product. These rapid and automated microbial monitoring methods may be adopted 1983
after validation has demonstrated their equivalency or superiority to the established methodology. 1984
1985
9.29 Sampling methods and equipment used should be fully understood and procedures should be in 1986
place for the correct operation and interpretation of results obtained. The recovery efficiency of the 1987
sampling methods chosen should be qualified. 1988
1989
9.30 Action limits for viable particle contamination are shown in Table 7 1990
1991
Table 7: Maximum action limits for viable particle contamination 1992
1993
Grade
Air sample
cfu/m
3
Settle plates
(diam. 90 mm)
cfu/4 hours
(a)
Contact plates
(diam. 55mm),
cfu/ plate
(c)
Glove print,
Including 5 fingers on
both hands
cfu/ glove
A
No growth
(b)
B 10 5 5 5
C 100 50 25 -
D 200 100 50 -
1994
(a)
Settle plates should be exposed for the duration of operations and changed as required after 1995
4 hours (exposure time should be based on validation including recovery studies and it should 1996
not have any negative effect on the suitability of the media used). Individual settle plates may 1997
be exposed for less than 4 hours. 1998
(b)
It should be noted that for Grade A, any growth should result in an investigation. 1999
2000
(c)
Contact plate limits apply to equipment room and gown surfaces within the Grade A zone 2001
and Grade B area. Routine gown monitoring is not normally required for Grade C and D areas, 2002
depending on their function. 2003
2004
Note 1: It should be noted that the types of monitoring methods listed in the table above are 2005
examples and other methods can be used provided they meet the intent of providing 2006
information across the whole of the critical process where product may be contaminated (e.g. 2007
aseptic line set-up, filling and lyophilizer loading). 2008
2009
Note 2: Limits are applied using cfu throughout the document. If different or new technologies 2010
are used that present results in a manner different from cfu, the manufacturer should 2011
scientifically justify the limits applied and where possible correlate them to cfu. 2012
2013
9.31 Microorganisms detected in Grade A zone and Grade B area should be identified to species level 2014
and the potential impact of such microorganisms on product quality (for each batch implicated) and 2015
overall state of control should be evaluated. Consideration should also be given to the identification of 2016
microorganisms detected in Grade C and D areas (for example where action limits or alert levels are 2017
41
exceeded or where atypical or potentially objectionable microorganisms are recovered). The approach 2018
to organism identification and investigation should be documented. 2019
2020
9.32 Personnel gloves (and any part of the gown that may potentially have direct impact on the 2021
product sterility (e.g. the sleeves if these enter a critical zone) should be monitored for viable 2022
contamination after critical operations and on exit from the cleanroom. Other surfaces should be 2023
monitored at the end of an operation. 2024
2025
9.33 Microbial monitoring of personnel in the Grade A zone and Grade B area should be performed to 2026
assess their aseptic behaviour. Where filling operations are manual in nature e.g. hand filling, the 2027
process in its entirety may be considered as one critical intervention. In these cases, the frequency of 2028
microbial monitoring of gowning should be based on scientific principles and justified as part of the 2029
CCS. Where monitoring is routinely performed by manufacturing personnel, consideration should be 2030
given to periodic monitoring under the supervision of the quality unit. 2031
2032
Aseptic process simulation (APS) (also known as media fill) 2033
2034
9.34 Periodic verification of the effectiveness of the controls in place for aseptic processing should 2035
include a process simulation test using a sterile nutrient media and/or surrogate in place of the 2036
product. Selection of an appropriate nutrient media should be made based on the ability of the media 2037
and/or surrogate to imitate product characteristics at all processing stages. Where processing stages 2038
may indirectly impact the viability of any introduced microbial contamination, (e.g. sterile aseptically 2039
produced semi-solids, powders, solid materials, microspheres, liposomes and other formulations 2040
where product is cooled or heated or lyophilized), alternative procedures that represent the operations 2041
as closely as possible can be developed and justified. Where surrogate materials, such as buffers, are 2042
used in parts of the process simulation, the surrogate material should not inhibit the growth of any 2043
potential contamination. 2044
2045
9.35 The process simulation test should imitate as closely as possible the routine aseptic 2046
manufacturing process and include all the critical manufacturing steps, specifically: 2047
2048
i. Process simulation tests should assess all aseptic operations performed subsequent to the 2049
sterilization and decontamination cycles of materials utilised in the process to the point 2050
where the container is sealed. 2051
2052
ii. For non-filterable formulations, any additional aseptic steps should be assessed. 2053
2054
iii. Where aseptic manufacturing is performed under an inert atmosphere, the inert gas should 2055
be substituted with air in the process simulation unless anaerobic simulation is intended. 2056
2057
iv. Processes requiring the addition of sterile powders should use an acceptable surrogate 2058
material in containers identical to those used in the process under evaluation. 2059
2060
v. Separate simulations of individual unit operations (e.g. processes involving drying, 2061
blending, milling and subdivision of a sterile powder) should generally be avoided. Any use 2062
of individual simulations should be supported by a documented justification and ensure that 2063
the sum total of the individual simulations continues to fully cover the whole process. 2064
2065
vi. The process simulation procedure for lyophilized products should represent the entire 2066
aseptic processing chain including filling, transport, loading, chamber dwell, unloading and 2067
sealing under specified, documented and justified conditions representing worst case 2068
operating parameters. 2069
2070
vii. The lyophilization process simulation should duplicate all aspects of the process, except 2071
those that may affect the viability or recovery of contaminants. For instance, boiling-over or 2072
actual freezing of the solution should be avoided. Factors to consider in determining APS 2073
42
design include, where applicable: 2074
2075
• The use of air to break vacuum instead of nitrogen. 2076
2077
• Replicating the maximum interval between sterilization of the lyophilizer and its 2078
use. 2079
2080
• Replicating the maximum period of time between sterilization and lyophilization. 2081
2082
• Quantitative aspects of worst case situations, e.g. loading the largest number of 2083
trays, replicating the longest duration of loading where the chamber is open to the 2084
environment. 2085
2086
9.36 The process simulation testing should take into account various aseptic manipulations and 2087
interventions known to occur during normal production as well as worst case situations, including: 2088
2089
i. Inherent interventions representative of the routine process at the maximum accepted 2090
frequency per number of filled units (e.g. loading of vials into a lyophilizer). 2091
2092
ii. Corrective interventions, that occur frequently during routine production, in a representative 2093
number and with the highest degree of acceptable intrusion (e.g. correcting jammed 2094
stoppers). 2095
2096
9.37 Interventions should not be designed or selected to justify poor process or facility design or to 2097
assess unacceptable interventions that rarely occur and which should lead to a thorough investigation 2098
and product assessment when they do occur. 2099
2100
9.38 In developing the process simulation test plan, consideration should be given to the following: 2101
2102
i. Identification of worst case conditions covering the relevant variables, such as container size 2103
and line speed, and their impact on the process. The outcome of the assessment should 2104
justify the variables selected. 2105
2106
ii. Determining the representative sizes of container/closure combinations to be used for 2107
validation. Bracketing or matrix approach may be considered for validation of the same 2108
container/closure configuration for different products where process equivalence is 2109
scientifically justified. 2110
2111
iii. The volume filled per container, which should be sufficient to ensure that the media contacts 2112
all equipment and component surfaces that may directly contaminate the sterile product. The 2113
volume used should provide sufficient headspace to support potential microbial growth and 2114
ensure that turbidity can be detected during inspection. 2115
2116
iv. Maximum permitted holding times for sterile product and associated sterile components and 2117
equipment exposed during the aseptic process. 2118
2119
v. The method of detection of microbial contamination should be scientifically justified to 2120
ensure that any contamination is detectable. 2121
2122
vi. The selected nutrient media should be capable of growing a designated group of reference 2123
microorganisms as described by the relevant pharmacopeia and suitably representative local 2124
isolates and supporting recovery of low numbers of these microorganisms. 2125
2126
vii. The requirement for substitution of any inert gas used in the routine aseptic
manufacturing 2127
process by air unless anaerobic simulation is intended. In these situations, inclusion of 2128
43
occasional anaerobic simulations as part of the overall validation strategy should be 2129
considered (refer to paragraph 9.35 point iii). 2130
2131
viii. The process simulation should be of sufficient duration to challenge the process, the 2132
operators that perform interventions, shift changes and the capability of the processing 2133
environment to provide appropriate conditions for the manufacture of a sterile product. 2134
2135
ix. Where the manufacturer operates different shifts then the APS should be designed to capture 2136
specific factors (e.g. for those manufacturing during a night or extended shift, fatigue should 2137
be considered). 2138
2139
x. Simulating normal aseptic manufacturing interruptions where the process is idle (e.g. shift 2140
changeovers, recharging dispensing vessels, introduction of additional equipment, etc.). 2141
2142
xi. Ensuring that environmental monitoring is conducted as required for routine production, and 2143
throughout the entire duration of the process simulation. 2144
2145
xii. Where campaign manufacturing occurs, such as in the use of Barrier Technologies or 2146
manufacture of sterile active substances, consideration should be given to designing and 2147
performing the process simulation so that it simulates the risks associated with both the 2148
beginning and the end of the campaign and demonstrating that the campaign duration does 2149
not pose any risk. The performance of "end of production or campaign APS" may be used as 2150
additional assurance or investigative purposes; however, their use should be justified in the 2151
CCS and should not replace routine APS. If used, it should be demonstrated that any 2152
residual product does not negatively impact the recovery of any potential microbial 2153
contamination. 2154
2155
9.39 For sterile active substances, batch size should be large enough to represent routine operation, 2156
simulate intervention operation at the worst case, and cover potential contact surfaces. In addition, all 2157
the simulated materials (surrogates or growth medium) should be subjected to microbial evaluation. 2158
The simulation materials should be sufficient to satisfy the evaluation of the process being simulated 2159
and should not compromise the recovery of micro-organisms. 2160
2161
9.40 Process simulation tests should be performed as part of the initial validation, with at least three 2162
consecutive satisfactory simulation tests that cover all working shifts that the aseptic process may 2163
occur in, and after any significant modification to operational practices, facilities, services or 2164
equipment (e.g. modification to the HVAC system, equipment, major facility shut down, changes to 2165
process, number of shifts and numbers of personnel etc.). Normally, process simulation tests (periodic 2166
revalidation) should be repeated twice a year (approximately every six months) for each aseptic 2167
process, each filling line and each shift. Each operator should participate in at least one successful 2168
APS annually. Consideration should be given to performing an APS after the last batch prior to shut 2169
down, before long periods of inactivity or before decommissioning or relocation of a line. 2170
2171
9.41 Where manual operation (e.g. aseptic compounding or filling) occurs, each type of container, 2172
container closure and equipment train should be initially validated with each operator participating in 2173
at least 3 consecutive successful APS and revalidated with one APS approximately every 6 months for 2174
each shift. The APS batch size should mimic that used in the routine aseptic manufacturing process. 2175
2176
9.42 The number of units processed (filled) for APS tests should be sufficient to effectively simulate 2177
all activities that are representative of the aseptic manufacturing process. Justification for the number 2178
of units to be filled should be clearly captured in the PQS. Typically, a minimum of 5000 to 10000 2179
units are filled. For small batches (e.g. those under 5000 units), the number of containers for media fill 2180
should at least equal the size of the production batch. 2181
2182
9.43 Filled APS units should be agitated, swirled or inverted before incubation to ensure contact of 2183
the media with all interior surfaces in the container. Units with cosmetic defects or those who have 2184
44
gone through non-destructive in process control checks should be identified and incubated. Units 2185
discarded during the process simulation and not incubated should be comparable with units discarded 2186
during a routine fill. Examples may include those normally discarded after the set-up process or due to 2187
an intervention or where the integrity of the unit is compromised as would be identified by the routine 2188
inspection process for the product. 2189
2190
9.44 Where processes have materials that contact the product contact surfaces but are then discarded, 2191
the discarded material should be simulated with nutrient media and be incubated as part of the APS, 2192
unless it can be clearly demonstrated that this waste process would not impact the sterility of the 2193
product. 2194
2195
9.45 Filled APS units should be incubated in a clear container to ensure visual detection of microbial 2196
growth. Where the product container is not clear (i.e. amber glass, opaque plastic), clear containers of 2197
identical configuration may be substituted to aid in the detection of contamination. When a clear 2198
container of identical configuration cannot be substituted, a suitable method for the detection of 2199
microbial growth should be developed and validated. Microorganisms isolated from contaminated 2200
units should be identified to at least genus, and to the species level when practical, to assist in the 2201
determination of the likely source of the contaminant. The selection of the incubation conditions and 2202
duration should be scientifically justified and validated to provide an appropriate level of sensitivity 2203
of detection of microbial contamination. 2204
2205
9.46 Filled APS units should be incubated without unnecessary delay to achieve the best possible 2206
recovery of potential contamination. 2207
2208
9.47 On completion of incubation: 2209
2210
i. Filled APS units should be inspected by staff, who have been trained and qualified in the 2211
visual inspection procedures, under conditions similar to those for visual inspection, that 2212
facilitate the identification of any microbial contamination. 2213
2214
ii. Samples of these units should undergo positive control by inoculation with a suitable range of 2215
reference organisms and local isolates. 2216
2217
9.48 The target should be zero growth. Any contaminated unit should result in a failed process 2218
simulation and the following actions should occur: 2219
2220
i. An investigation to determine the most probable root causes. 2221
2222
ii. Determination and implementation of appropriate corrective measures. 2223
2224
iii. A sufficient number of successful, consecutive repeat media fills (normally a minimum of 3) 2225
should be conducted in order to demonstrate that the process has been returned to a state of 2226
control. 2227
2228
iv. A prompt review of all appropriate records relating to aseptic production since the last 2229
successful APS. The outcome of the review should include a risk assessment of potential 2230
sterile breaches in batches manufactured since the last successful process simulation. All other 2231
batches not released to the market should be included in the scope of the investigation. Any 2232
decision regarding their release status should consider the investigation outcome. 2233
2234
v. All products that have been manufactured on a line subsequent to a process simulation failure 2235
should be quarantined until a successful resolution of the process simulation failure has 2236
occurred. 2237
2238
vi. Production should resume only after completion of successful revalidation. 2239

45
2240
9.49 APS should be carefully observed by personnel with specific expertise in aseptic processing to 2241
assess the correct performance of operations and address inappropriate practices if detected. 2242
9.50 Where results indicate that an operator may have failed qualification, actions to limit the 2243
operator’s activities, until retrained and requalified, should be taken. 2244
2245
9.51 An aseptic process or filling should be subject to a repeat of the initial validation when: 2246
2247
i. The specific aseptic process has not been in operation for an extended period of time. 2248
2249
ii. There is a change to the process, equipment, procedures or environment that has the 2250
potential to affect the aseptic process or an addition of new product containers or container-2251
closure combinations. 2252
2253
9.52 All process simulation runs should be fully documented and include a reconciliation of units 2254
processed (e.g. units filled, incubated, not incubated, and rejected). All interventions performed 2255
during the process simulations should be recorded, including the start and end of each intervention. 2256
All microbial monitoring data as well as other testing data should be recorded in the APS batch 2257
record. 2258
2259
10 Quality Control (QC) 2260
2261
10.1 It is important that there are personnel with appropriate training and experience in microbiology 2262
and knowledge of the process to support the design of the manufacturing process, environmental 2263
monitoring regime and any investigation assessing the impact of microbiologically linked events to 2264
the safety of the sterile product. 2265
2266
10.2 Specifications for raw materials, components and products should include requirements for 2267
microbial quality when the need for this has been indicated by monitoring and/or by the CCS. 2268
2269
10.3 The bioburden assay should be performed on each batch for both aseptically filled product and 2270
terminally sterilized products and the results considered as part of the final batch review. There should 2271
be defined limits for bioburden immediately before the sterilizing filter or the terminal sterilization 2272
process, which are related to the efficiency of the method to be used. Samples should be taken to be 2273
representative of the worst case scenario (e.g. at the end of hold time). Where overkill sterilization 2274
parameters are set for terminally sterilized products, bioburden should be monitored at suitable 2275
scheduled intervals. 2276
2277
10.4 A pre-sterilization bioburden monitoring program for the product and components should be 2278
developed to support parametric release. The bioburden should be performed for each batch. The 2279
sampling locations of filled units before sterilization should be based on a worst case scenario and be 2280
representative of the batch. Any organisms found during bioburden testing should be identified and 2281
their impact on the effectiveness of the sterilizing process determined. Where appropriate, the level of 2282
pyrogen (endotoxins) should be monitored. 2283
2284
10.5 The sterility test applied to the finished product should only be regarded as the last in a series of 2285
control measures by which sterility is assured. It cannot be used to assure sterility of a product that 2286
does not meet its design, procedural or qualification parameters. The test should be validated for the 2287
product concerned. 2288
2289
10.6 The sterility test should be performed under aseptic conditions. Samples taken for sterility 2290
testing should be representative of the whole of the batch but should in particular include samples 2291
46
taken from parts of the batch considered to be most at risk of contamination, for example: 2292
2293
i. For products which have been filled aseptically, samples should include containers filled at
2294
the beginning, middle and end of the batch and after any significant intervention (e.g. 2295
interventions where the integrity of a barrier is breached (open door)) or an operator 2296
intervention into critical zones. 2297
2298
ii. For products which have been heat sterilized in their final containers, samples taken 2299
should
be
representative of the worst case locations (e.g. the potentially coolest or slowest to 2300
heat part of each load). 2301
2302
iii. For products that are lyophilized, samples taken from different lyophilization loads. 2303
2304
Note: Where the manufacturing process results in sub-batches (e.g. for terminally sterilized 2305
products) then sterility samples from each sub-batch should be taken and a sterility test for each sub-2306
batch performed. Consideration should also be given to performing separate testing for other 2307
finished product tests. 2308
2309
10.7 For some products it may not be possible to perform a sterility test prior to release because the 2310
shelf life of the product is too short to allow completion of a sterility test. In these cases, the CCS 2311
should clearly capture the identified risks, the additional considerations of design of the process and 2312
additional monitoring required to mitigate the identified risks. 2313
2314
10.8 Any process (e.g. Vaporized Hydrogen Peroxide or VH202, Ultra Violet) used to decontaminate 2315
the external surfaces of sterility samples prior to testing should not negatively impact the sensitivity of 2316
the test method. 2317
2318
10.9 Media used for environmental monitoring and APS should be tested for its growth promotion 2319
capability, in accordance with a formal written program. 2320
2321
10.10 Environmental monitoring data and trend data generated for classified areas should be reviewed 2322
as part of product batch certification. A written plan should be available that describes the actions to 2323
be taken when data from environmental monitoring are found out of trend or exceeding the 2324
established limits. For products with short shelf life, the environmental data for the time of 2325
manufacture may not be available; in these cases, the certification should include a review of the most 2326
recent available data. Manufacturers of these products should consider the use of rapid monitoring 2327
systems. 2328
2329
10.11 Where rapid and automated microbial methods are used for general manufacturing purposes, 2330
these methods should be validated for the product(s) or processes concerned. 2331
2332

47
Glossary 2333
2334
Airlock – An enclosed space with interlocked doors, constructed to maintain air pressure control 2335
between adjoining rooms (generally with different air cleanliness standards). The intent of an airlock 2336
is to preclude ingress of particulate matter and microorganism contamination from a lesser controlled 2337
area. 2338
2339
Action limit – An established relevant measure (e.g. microbial, or airborne particulate limits) that, 2340
when exceeded, should trigger appropriate investigation and corrective action based on the 2341
investigation. 2342
2343
Alert level – An established relevant measure (e.g. microbial, or airborne particulate levels) giving 2344
early warning of potential drift from normal operating conditions and validated state, which does not 2345
necessarily give grounds for corrective action but triggers appropriate scrutiny and follow-up to 2346
address the potential problem. Alert levels are established based on historical and qualification trend 2347
data and periodically reviewed. The alert level can be based on a number of parameters including 2348
adverse trends, individual excursions above a set limit and repeat events. 2349
2350
Aseptic processing room – A room in which one or more aseptic activities or processes are performed. 2351
2352
Aseptic Process Simulation (APS) –A simulation of the entire aseptic formulation and filling process 2353
in order to determine the capability of the process to assure product sterility. 2354
2355
Asepsis – A state of control attained by using an aseptic work area and performing activities in a 2356
manner that precludes microbial contamination of the exposed sterile product. 2357
2358
Bacterial retention testing – This test is performed to validate that a filter can remove bacteria from a 2359
gas or liquid. The test is usually performed using a standard organism, such as Brevundimonas 2360
diminuta at a minimum concentration of 10
7
Colony Forming Units/cm
2
. 2361
2362
Barrier – A physical partition that affords aseptic processing area (usually Grade A) protection by 2363
separating it from the background environment. Such systems frequently use in part or totally the 2364
Barrier Technologies known as RABS or isolators. 2365
2366
Bioburden – The total number of microorganisms associated with a specific item such as personnel, 2367
manufacturing environments (air and surfaces), equipment, product packaging, raw materials 2368
(including water), in-process materials, or finished products. 2369
2370
Biological Indicator (BI) – A population of microorganisms inoculated onto a suitable medium (e.g. 2371
solution, container or closure) and placed within a sterilizer or load or room locations to determine the 2372
sterilization or disinfection cycle efficacy of a physical or chemical process. The challenge 2373
microorganism is selected and validated based upon its resistance to the given process. Incoming lot 2374
D value, microbiological count and purity define the quality of the BI. 2375
2376
Blow-Fill-Seal (BFS) – A technology in which containers are formed from a thermoplastic granulate, 2377
filled with product, and then sealed in a continuous, integrated, automatic operation. The two most 2378
common types of BFS machines are the Shuttle type (with Parison cut) and the Rotary type (Closed 2379
Parison) types. 2380
2381
Classified area – An area that contains a number of cleanrooms (see cleanroom definition). 2382
2383
Cleaning – A process for removing contamination e.g. product residues and disinfectant residues. 2384
2385
Clean area – An area with defined particulate and microbiological cleanliness standards usually 2386
containing a number of joined cleanrooms. 2387

48
2388
Cleanroom – A room designed, maintained, and controlled to prevent particulate and microbial 2389
contamination of drug products. Such a room is assigned and reproducibly meets an appropriate air 2390
cleanliness level. Grade A will be referred to as Grade A zone. 2391
2392
Cleanroom classification – A method of assessing the level of air cleanliness against a specification 2393
for a cleanroom or clean air equipment by measuring the non-viable airborne particulate 2394
concentration. 2395
2396
Cleanroom qualification – A method of assessing the level of compliance of a classified cleanroom or 2397
clean air equipment with its intended use. 2398
2399
Closed system – A system in which the sterile product is not exposed to the surrounding environment. 2400
For example, this can be achieved by the use of bulk products holders (such as tanks or bags) that are 2401
connected to each other by pipes or tubes as a system, with the system being sterilized after the 2402
connections are made. Examples of these can be (but are not limited to) large scale reusable systems, 2403
such as those seen in active substance manufacturing, or disposable bag and manifold systems, such 2404
as those seen in the manufacture of biological products. Closed systems, when used in this document, 2405
does not refer to systems such as RABS or isolator systems which are referred to as Barrier 2406
Technologies. 2407
2408
Colony Forming Unit (CFU) – A microbiological term that describes a single detectable colony that 2409
originates from one or more microorganisms. Colony forming units are typically expressed as cfu per 2410
ml for liquid samples, and cfu per cm
2
for samples captured on solid medium such as settle or contact 2411
plates. 2412
2413
Contamination – The undesired introduction of impurities of a microbiological nature (quantity and 2414
type of microorganisms, pyrogens), or of foreign particulate matter, into or onto a raw material, 2415
intermediate, active substance or drug product during production, sampling, packaging or 2416
repackaging, storage or transport with the potential to adversely impact product quality. 2417
2418
Contamination Control Strategy (CCS) – A planned set of controls for microorganisms, pyrogens and 2419
particulates, derived from current product and process understanding that assures process performance 2420
and product quality. The controls can include parameters and attributes related to active substance, 2421
excipient and drug product materials and components, facility and equipment operating conditions, in-2422
process controls, finished product specifications, and the associated methods and frequency of 2423
monitoring and control. 2424
2425
Corrective intervention – An intervention that is performed to correct or adjust an aseptic process 2426
during its execution. These may not occur with the same frequency in the routine aseptic process. 2427
Examples include such as clearing component jams, stopping leaks, adjusting sensors, and replacing 2428
equipment components. Corrective measures should be taken to reduce their extent and frequency. 2429
2430
Critical surfaces – Surfaces that may come directly into contact with, or directly affect, a sterile 2431
product or its containers or closures. Critical surfaces are rendered sterile prior to the start of the 2432
manufacturing operation, and sterility is maintained throughout processing. 2433
2434
Critical zone – A location within the aseptic processing area in which product and critical surfaces are 2435
exposed to the environment. 2436
2437
Critical intervention – An intervention (corrective or inherent) into the critical zone. 2438
2439
D value – The value of a parameter of sterilization (duration or absorbed dose) required to reduce the 2440
number of viable organisms to 10 per cent of the original number. 2441
2442

49
Dead leg – Length of non-circulating pipe (where fluid may remain static) that is greater than 3 2443
internal pipe diameters. 2444
2445
Decommission – When a process, equipment or cleanroom are closed where they will not be used 2446
again. 2447
2448
Decontamination – The overall process of removal or reduction of any contaminants (chemical, waste, 2449
residue or microorganisms) from an area, object, or person. The method of decontamination used (e.g. 2450
cleaning, disinfection, sterilization) should be chosen and validated to achieve a level of cleanliness 2451
appropriate to the intended use of the item decontaminated. 2452
2453
Depyrogenation – A process designed to remove or inactivate pyrogenic material (e.g. endotoxins) to 2454
a specified minimum quantity. 2455
2456
Disinfection – The process by which the reduction of the number of microorganisms is achieved by 2457
the irreversible action of a product on their structure or metabolism, to a level judged to be 2458
appropriate for a defined purpose. 2459
2460
Endotoxin – A pyrogenic product (e.g. lipopolysaccharide) present in the bacterial cell wall. 2461
Endotoxin can lead to reactions in patients receiving injections ranging from fever to death. 2462
2463
Extractables - Chemical entities that migrate from the surface of the process equipment, exposed to an 2464
appropriate solvent at extreme conditions, into the product or material being processed. 2465
2466
First Air – Refers to filtered air that has not been interrupted by items (such as operators) with the 2467
potential to add contamination to the air prior to reaching the critical zone. 2468
2469
Form-Fill-Seal (FFS) – Similar to Blow fill Seal, this involves the formation of a large tube formed 2470
from a flexible packaging material, in the filling machine, and generally the tube is filled to form the 2471
bags. 2472
2473
Gowning qualification – A program that establishes, both initially and on a periodic basis, the 2474
capability of an individual to don the complete sterile gown in an aseptic manner. 2475
2476
Grade A air supply – Air which is passed through a filter qualified as capable of producing Grade A 2477
non-viable quality air, but where there is no requirement to perform continuous non-viable monitoring 2478
or meet Grade A viable monitoring limits and the area itself is not classified. Specifically used for the 2479
protection of fully stoppered vials where the cap has not been crimped and the equipment and 2480
engineering systems that have a direct impact on product quality. 2481
2482
HEPA filter - High efficiency particulate air filter with 0.3 µm particulate retaining efficiency of no 2483
less than 99.95 percent according to the relevant norms (e.g. EN 1822).. 2484
2485
Inherent interventions – An intervention that is an integral part of the aseptic process and is required 2486
for either set-up, routine operation and/or monitoring (e.g. aseptic assembly, container replenishment, 2487
environmental sampling, etc.). Inherent interventions are required by procedure or work instruction 2488
for the execution of the aseptic process. 2489
2490
Integrity test - A test to confirm that a filter (product, gas or HVAC filter) retain their retentive 2491
properties and have not been damaged during handling, installation or processing. 2492
2493
Intrinsic Sterile Connection device – A device that reduces the risk of contamination during the 2494
connection process; these can be mechanical or fusion sealing. 2495
2496

50
Isokinetic sampling head – A sampling head designed to disturb the air as little as possible so that the 2497
same particulates go into the nozzle as would have passed the area if the nozzle had it not been there 2498
i.e. the sampling condition in which the mean velocity of the air entering the sample probe inlet is 2499
nearly the same (± 20 percent) as the mean velocity of the airflow at that location. 2500
2501
Isolator – A decontaminated unit, with an internal work zone meeting Grade A conditions that 2502
provides uncompromised, continuous isolation of its interior from the external environment (e.g. 2503
surrounding cleanroom air and personnel). There are two major types of isolators 2504
2505
i. Closed isolator systems exclude external contamination of the isolator’s interior by 2506
accomplishing material transfer via aseptic connection to auxiliary equipment, rather than 2507
use of openings to the surrounding environment. Closed systems remain sealed throughout 2508
operations. 2509
ii. Open isolator systems are designed to allow for the continuous or semi-continuous ingress 2510
and/or egress of materials during operations through one or more openings. Openings are 2511
engineered (e.g. using continuous overpressure) to exclude the entry of external contaminant 2512
into the isolator. 2513
2514
Leachables – Chemical entities that migrate into products from the product contact surface of the 2515
process equipment or containers under normal condition of use and/or storage. 2516
2517
Local Isolates – Suitably representative microorganisms of the site that are frequently recovered 2518
through environmental monitoring within the classified zone/areas especially Grade A zone and 2519
Grade B area, personnel monitoring or positive sterility test results. 2520
2521
Lyophilization – A physical-chemical drying process designed to remove solvents, by way of 2522
sublimation, from both aqueous and non-aqueous systems, primarily to achieve product or material 2523
stability. Lyophilization is synonymous to the term freeze-drying. 2524
2525
Manual Filling – A filling process where operator intervention is required to complete the filling of 2526
each container (e.g. as occurs during aseptic compounding operations). 2527
2528
Operator - Any individual participating in the processing operation, including line set-up, filler, 2529
maintenance, or other personnel associated with manufacturing activities. 2530
2531
Overkill sterilization – A process that is sufficient to provide at least a 12 log reduction of 2532
microorganisms having a minimum D value of 1 minute. 2533
2534
Pass-through hatch – Synonymous with airlock (refer to airlock definition) but typically smaller in 2535
size. 2536
2537
Pyrogen – A substance that induces a febrile reaction in a patient. 2538
2539
Rapid transfer system (RTP) – A System used for the transfer of items into RABS and isolators that 2540
minimize the risk to the critical zone. An example would be a rapid transfer container with an 2541
alpha/beta port. 2542
2543
Raw material – Any ingredient intended for use in the manufacture of a sterile product, including 2544
those that may not appear in the final drug product. 2545
2546
Restricted Access Barrier System (RABS) – System that provides an enclosed, but not sealed, 2547
environment meeting defined cleanroom conditions (for aseptic processing Grade A, (but where used 2548
for non-sterile applications can be lesser grade) and using a rigid-wall enclosure and air overspill to 2549
separate its interior from the surrounding environment. The inner surfaces of the RABS are 2550
disinfected and decontaminated with a sporicidal agent. Operators use gloves, half suits, rapid transfer 2551

51
systems (RTPs) and other integrated transfer ports to perform manipulations or convey materials to 2552
the interior of the RABS. Depending on the design, doors are rarely or never opened: 2553
2554
i. Active RABS: integral HEPA-filtered air supply. 2555
2556
ii. Passive RABS: air supply by ceiling mounted HEPA-filters. 2557
2558
iii. Closed RABS: where the air is vented in return ducts within the cabinet. 2559
2560
iv. Open RABS: Where there are vents in the barrier that allow air to move from the Grade A 2561
zone to the Grade B area. 2562
2563
Single Use Systems (SUS) – Systems in which product contact components are used only once (i.e. 2564
single use components) to replace reusable equipment such as stainless steel transfer lines or bulk 2565
containers. SUS covered in this document are those that are used in manufacturing processes of sterile 2566
products (e.g. sterile active substance, sterile bio bulk, sterile finished dosage), and are typically made 2567
up of disposable components such as bags, filters, tubing, connectors, storage bottles and sensors. 2568
2569
Sporicidal agent – An agent that destroys bacterial and fungal spores when used in sufficient 2570
concentration for specified contact time. It is expected to kill all vegetative microorganisms. 2571
2572
Sterile Product – For purpose of this guidance, sterile product refers to one or more of the sterilized 2573
elements exposed to aseptic conditions and ultimately making up the sterile active substance or 2574
finished sterile product. These elements include the containers, closures, and components of the 2575
finished drug product. Or, a product that is rendered sterile by a terminal sterilization process. 2576
2577
Sterilizing grade filter – A filter that, when appropriately validated, will remove a defined microbial 2578
challenge from a fluid or gas producing a sterile effluent. Usually such filters have a pore size equal or 2579
less than 0.22 µm (for the purposes of this document 0.2 µm and 0.22 µm are used interchangeably 2580
and deemed equivalent). 2581
2582
Terminal Sterilization – The application of a lethal sterilizing agent or conditions to a product within a 2583
sealed container to achieve a predetermined sterility assurance level (SAL) of 10⁻⁶ or better (i.e. the 2584
theoretical probability of there being a single viable microorganism present on or in a sterilized unit is 2585
equal to or less than 1 x 10
-6
(one in a million)). 2586
2587
Turbulent airflow – Air that is not unidirectional. Turbulent air in cleanrooms should flush the 2588
cleanroom via mixed flow dilution and ensure maintenance of acceptable air quality. 2589
2590
Unidirectional airflow – An airflow moving in a single direction, in a robust and uniform manner, and 2591
at sufficient speed, to reproducibly sweep particulates away from the critical processing or testing 2592
area. 2593
2594
Unidirectional Airflow Unit (UDAF) – A cabinet supplied with filtered unidirectional airflow 2595
(previously referred to as a Laminar Airflow Unit or LAF). 2596
2597
Vertical-Form-Fill-Seal (VFFS) – An automated filling process, typically for terminally sterilized 2598
products, that may utilize a single or dual web system which constructs the primary container out of a 2599
flat roll of thermoplastic film while simultaneously filling the formed bags with product and sealing 2600
the filled bags in a continuous process. 2601
2602
Worst case – A set of conditions encompassing processing limits and circumstances, including those 2603
within standard operating procedures, that pose the greatest chance of process or product failure 2604
(when compared with ideal conditions). Such conditions have the highest potential to, but do not 2605
necessarily always induce, product or process failure. 2606

52
2607
Water system – A system for producing, storing and distributing water, usually compliant to a specific 2608
pharmacopeia grade e.g. purified and water for injection (WFI). 2609
2610
