
FOOD AND DRUG ADMINISTRATION
COMPLIANCE PROGRAM MANUAL
PROGRAM
Date of Issuance: September 29, 2023
Cover Page - PAGE 1
)
7382.845
SUBJECT:
INSPECTION OF MEDICAL DEVICE MANUFACTURERS
IMPLEMENTATION DATE
09/29/2023
DATA REPORTING
PRODUCT CODES PRODUCT/ASSIGNMENT CODES
73-91 82845A; 42845A All Level 1 (Abbreviated) Inspections
82845B; 42845B All Level 2 (Comprehensive) Inspections
82845C; 42845C All Level 3 (Compliance Follow-up)
Inspections
82845G All For- Cause Inspections
82845H Specific Product Risk Assignment
Inspections
82845P Joint FDA/Accredited Person Inspections
82845S Report Time spent on Assessment of
Firm’s Sterilization processes
81010 Report Time spent on MDR
Follow-up
81011 Report Time spent on Assessment of
Firm’s MDR Practices
81845T Report Time spent on Assessment of
Firm’s Tracking Practices
81845R Report Time spent on Assessment of
Firm’s Corrections and Removals
Practices
82016 Unique Device Identifier (UDI)
*Previous editions obsolete.

PROGRAM
7382.845
Page 2 of 59
Contents
Contents .............................................................................................................................................. 2
PART I - BACKGROUND ............................................................................................................... 6
1. FDA REAUTHORIZATION ACT OF 2017 (FDARA) ..................................................... 7
2. THE QUALITY SYSTEM (QS) REGULATION (21 CFR Part 820) ................................ 8
3. THE MDR REGULATION (21 CFR Part 803) .................................................................. 8
4. THE MEDICAL DEVICE TRACKING REGULATION (21 CFR Part 821) .................... 9
5. THE CORRECTIONS AND REMOVAL REGULATION (21 CFR Part 806) ................. 9
6. THE REGISTRATION AND LISTING REGULATION (21 CFR Part 807) .................. 10
7. UNIQUE DEVICE IDENTIFICATION (UDI) SYSTEM (21 CFR PART 801 SUBPART
B AND 21 CFR PART 830) ..................................................................................................... 10
8. MEDICAL DEVICE SINGLE AUDIT PROGRAM (MDSAP) ....................................... 11
9. FOOD AND DRUG OMNIBUS REFORM ACT OF 2022 (FDORA) ............................ 11
10. KEY COMPLIANCE DOCUMENTS .............................................................................. 11
A. Benefit-Risk in Medical Device Product Availability, Compliance, and Enforcement
Decisions ............................................................................................................................... 11
B. Enforcement Policy Related to the Coronavirus Disease 2019 (COVID-19) Public
Health Emergency ................................................................................................................. 12
PART II - IMPLEMENTATION .................................................................................................... 13
1. OBJECTIVES .................................................................................................................... 13
2. PROGRAM MANAGEMENT INSTRUCTIONS ............................................................ 13
A. Priorities for QS Inspections ....................................................................................... 13
B. Planning Instructions .................................................................................................. 14
C. Interactions Between Compliance Programs .............................................................. 14
D. Resource Instructions ................................................................................................. 15
E. Interactions with other Federal Agencies, State and Local Counterparts, and Foreign
Regulatory Authorities .......................................................................................................... 15
3. PRE-ANNOUNCEMENT OF INSPECTIONS ................................................................ 15
4. ANNOTATION OF THE FDA 483 .................................................................................. 16
PART III - INSPECTIONAL .......................................................................................................... 17
1. OPERATIONS ................................................................................................................... 17
A. Inspectional Preparation ............................................................................................. 17
B. Inspectional Strategy .................................................................................................. 17

PROGRAM
7382.845
Page 3 of 59
C. Additional Inspectional Instructions ........................................................................... 24
D. Sampling Records ....................................................................................................... 28
E. Special Instructions Concerning Design Controls ...................................................... 28
F. Inspection of Radiation-Emitting Devices ................................................................. 29
G. Field Exams ................................................................................................................ 30
H. Sample Collection....................................................................................................... 30
2. ADDITIONAL CONSIDERATIONS ............................................................................... 30
A. Registration and Listing.............................................................................................. 30
B. Imports ........................................................................................................................ 31
C. Export-Only Firms ...................................................................................................... 31
D. Electronic Records and Electronic Signatures ............................................................ 32
3. REMARKETED DEVICES .............................................................................................. 32
A. Remanufacturers of Used Devices ............................................................................. 32
B. Third Party Refurbishers/Reconditioners/Servicers of Used Devices ........................ 32
C. Reprocessors of Single Use Devices .......................................................................... 32
D. Hospital Reprocessors ................................................................................................ 32
4. REPORTING ..................................................................................................................... 33
A. Establishment Information Updates ........................................................................... 33
B. Observation and Discussion Item Reporting Requirements ....................................... 33
C. Registration Listing Discrepancies ............................................................................. 33
D. Annotations and Firm Commitments Related to Inspectional Observations and
Discussion Items .................................................................................................................... 33
E. Establishment Inspection Report Writing .................................................................. 34
F. eNSPECT Profiles ...................................................................................................... 34
PART IV - ANALYTICAL ............................................................................................................. 35
1. ANALYZING LABORATORIES ..................................................................................... 35
2. ANALYSES TO BE CONDUCTED ................................................................................. 35
3. METHODOLOGY ............................................................................................................. 35
A. Testing Finished Device Samples for Sterility ........................................................... 35
B. Pre-sterilization Microbial Contamination (Bioburden) ............................................. 36
C. Analysis of Packaging Defects ................................................................................... 36
D. Analysis of Endotoxins ............................................................................................... 37

PROGRAM
7382.845
Page 4 of 59
E. Antimicrobial Effectiveness Testing .......................................................................... 37
F. Antimicrobial Effectiveness Testing ........................... Error! Bookmark not defined.
PART V - REGULATORY/ADMINISTRATIVE STRATEGY ................................................... 38
1. REGULATORY SIGNIFICANCE FOR COMPLIANCE DECISIONS .......................... 38
A. Inspectional ................................................................................................................. 38
B. Factors Considered by Compliance ............................................................................ 40
2. CLASSIFICATION ........................................................................................................... 41
3. REGULATORY/ADMINISTRATIVE FOLLOW-UP ..................................................... 42
A. Considerations in Determining Regulatory Action .................................................... 42
B. Regulatory Actions ..................................................................................................... 43
C. Violative Compliance Follow-Up Inspections ........................................................... 45
D. Facilitating Review of Regulatory Recommendations for Judicial Actions .............. 47
E. Deciding Responsibility When Taking Regulatory Action - Contract Sterilizers,
Contract Device Manufacturers, and Finished Device Manufacturers .................................. 48
F. Violative Devices Sold to Government Agencies ...................................................... 50
4. MDR REGULATORY/ADMINISTRATIVE FOLLOW UP ........................................... 50
5. TRACKING REGULATORY/ADMINISTRATIVE FOLLOW-UP ............................... 51
6. CORRECTIONS AND REMOVALS REGULATORY/ADMINISTRATIVE FOLLOW-
UP 51
7. REGISTRATION AND LISTING REGULATORY/ADMINISTRATIVE FOLLOW-UP
52
8. UNIQUE DEVICE IDENTIFIER (UDI) REGULATORY/ADMINISTRATIVE
FOLLOW-UP ............................................................................................................................ 52
9. RADIATION EMITTING DEVICE REGULATORY/ADMINISTRATIVE FOLLOW-
UP 53
10. IMPORTS REGULATORY/ADMINISTRATIVE FOLLOW-UP ................................... 53
11. EXPORTS REGULATORY/ADMINISTRATIVE FOLLOW-UP .................................. 54
PART VI - REFERENCES AND PROGRAM CONTACTS ....................................................... 55
1. APPLICABLE REFERENCES ......................................................................................... 55
A. APPLICABLE REFERENCES – SPECIFIC TO STERILIZATION ........................ 55
2. PROGRAM CONTACTS .................................................................................................. 55
A. ORA Contacts ............................................................................................................. 55
B. CDRH Contacts .......................................................................................................... 56
C. CBER Contacts ........................................................................................................... 58

PROGRAM
7382.845
Page 5 of 59
3. FDA WEB SITES .............................................................................................................. 58
A. FDA ............................................................................................................................ 58
B. ORA ............................................................................................................................ 58
C. ORA Office of Medical Device and Radiological Health (OMDRHO) .................... 58
D. CDRH ......................................................................................................................... 58
E. Registration and Listing.............................................................................................. 58
F. Medical Device Reporting (MDR) ............................................................................. 58
G. MedWatch .................................................................................................................. 58
H. Medical Device Tracking ........................................................................................... 59
I. Recalls, Corrections and Removals ............................................................................ 59
J. QSIT Guide................................................................................................................. 59
K. FDA Recognized Standards........................................................................................ 59

PROGRAM
7382.845
Page 6 of 59
PART I - BACKGROUND
This Inspection of Medical Device Manufacturers Compliance Program (CP 7382.845) provides
instruction to FDA field and Center staffs for inspection and administrative/enforcement
activities related to the:
• Statutory requirements in the Federal Food, Drug, and Cosmetic Act (FD&C Act)
including updates to the FD&C Act as required under the FDA Reauthorization Act of
2017 (FDARA) and the Food and Drug Omnibus Reform Act of 2022 (FDORA)
• Quality System (QS) regulation (21 CFR Part 820)
• Medical Device Reporting (MDR) regulation (21 CFR Part 803)
• Medical Device Tracking regulation (21 CFR Part 821)
• Corrections and Removals regulation (21 CFR Part 806)
• Registration and Listing regulation (21 CFR Part 807)
• Unique Device Identifier (UDI) regulation (21 CFR Part 801 Subpart B and 21 CFR
Part 830)
• Medical Device Single Audit Program (MDSAP)
• Key compliance documents, including Factors to Consider Regarding Benefit-Risk in
Medical Device Product Availability, Compliance, and Enforcement Decisions:
Guidance for Industry and Food and Drug Administration Staff, and other
enforcement guidance documents.
This Compliance Program: Inspection of Medical Device Manufacturers CP 7382.845,
supersedes the program of the same name issued on February 2, 2011.
This compliance program encompasses a total product life cycle (TPLC) assessment of medical
devices
1
while making compliance and enforcement decisions informed by benefit-risk,
including reliable information relating to patient perspectives on acceptable benefit-risk when
available.
Under the Quality System (QS) regulation, manufacturers are expected to control their devices
from design stage through postmarket surveillance. Additionally, labelers must comply with the
UDI regulations to ensure adequate identification of medical devices from manufacturing
through distribution. Manufacturing processes, such as sterilization, are required to be
implemented under appropriate controls. This compliance program also provides specific
instruction for MDR, Tracking, Corrections and Removals, and Registration and Listing
1
The Safeguarding Therapeutics Act amended the FD&C Act and redesignated the definition of device to section
201(h)(1) and added the term counterfeit device at section 201(h)(2). “The term ‘‘counterfeit device’’ means a
device which, or the container, packaging, or labeling of which, without authorization, bears a trademark, trade
name, or other identifying mark or imprint, or any likeness thereof, or is manufactured using a design, of a device
manufacturer, processor, packer, or distributor other than the person or persons who in fact manufactured, processed
packed, or distributed such device and which thereby falsely purports or is represented to be the product of, or to
have been packed or distributed by, such other device manufacturer, processor, packer, or distributor.”

PROGRAM
7382.845
Page 7 of 59
regulations and associated activities required of manufacturers and importers after their devices
have been distributed.
While most medical devices subject to Food and Drug Administration (FDA) oversight are
regulated by the Center for Devices and Radiological Health (CDRH), the Center for Biologics
Evaluation and Research (CBER) is also responsible for the regulation of certain medical
devices.
CBER regulates devices which are cleared or approved under the Federal Food, Drug, and
Cosmetic (FD&C) Act’s 510(k) or PMA provisions. Inspections of these devices should be
performed in accordance with Compliance Program 7382.845, Inspection of Medical Device
Manufacturers and under PACs 42845A-C. Examples of devices regulated by CBER include:
• Plasmapheresis machines used to collect, process and/or administer a biological product
• Quality assurance reagents and 510(k) cleared instruments intended for use in conjunction
with licensed IVDs
• Peripheral blood and umbilical cord blood stem cell collection kits
• Leukocyte typing sera
• Computer software with blood bank claims
• HIV test kits with only diagnostic claims
• Automated immunohematology analyzers
In addition, CBER is designated the lead Center in FDA for regulating in vitro diagnostic (IVD)
medical devices intended for screening or confirmatory clinical laboratory testing associated
with blood banking practices and other process testing procedures. These IVD products include
those required for screening of blood, blood products, human cells, tissues, and cellular and
tissue-based products (HCT/Ps), supplemental testing, and related blood banking practices (such
as blood typing and compatibility testing) and are licensed under Section 351 of the Public
Health Service (PHS) Act. Inspections of IVDs licensed by CBER should be performed in
accordance with (PHS) Act. Inspections of IVDs licensed by CBER should be performed in
accordance with Compliance Program 7342.008: Inspection of Licensed In Vitro Diagnostic
Devices .
1. FDA REAUTHORIZATION ACT OF 2017 (FDARA)
FDARA was signed into law on August 18, 2017, amending several sections of the FD&C
Act related to device inspections. However, FDARA does not affect the overarching
authority of the FDA to conduct inspections otherwise permitted in order to ensure

PROGRAM
7382.845
Page 8 of 59
compliance with the FD&C Act. FDARA requires the inspection of device manufacturers in
accordance with a risk-based schedule and requires the adoption of uniform processes and
standards for domestic and foreign inspections, other than for-cause inspections.
Additionally, FDARA amends the FD&C Act to specify a process for persons denied a
Certificate to Foreign Government (CFG) for a device that is exported from the United
States. Other amendments to the FD&C Act include an agency requirement to provide
nonbinding feedback in certain circumstances after an FDA inspection of a device
establishment, as well as FDA’s recognition of auditing organizations established by
governments to facilitate international harmonization for the purposes of conducting
inspections.
2. THE QUALITY SYSTEM (QS) REGULATION (21 CFR Part 820)
Manufacturers establish and follow quality systems to help ensure that their products
consistently meet applicable requirements and specifications. The quality systems for FDA-
regulated products (food, drugs, biologics, and devices) are known as Current Good
Manufacturing Practices (CGMPs). CGMP requirements for devices in Part 820 (21 CFR
Part 820) were first authorized by section 520(f) of the FD&C Act (21 U.S.C. 360j(f)), which
was among the authorities added by the Medical Device Amendments of 1976. Under
section 520(f) of the FD&C Act, the FDA issued a final rule in the Federal Register of July
21, 1978 (43 FR 31 508), prescribing CGMP requirements for the methods used in, and the
facilities and controls used for, the manufacture, packing, storage, and installation of medical
devices. This regulation became effective on December 18, 1978.
The Safe Medical Devices Act of 1990 (the SMDA), enacted on November 28, 1990,
amended section 520(f) of the FD&C Act, providing the FDA with the authority to add
preproduction design controls to the CGMP regulation. This change in law was based on
findings that a significant proportion of device recalls were attributed to faulty design of
product. The SMDA also added section 803 to the act (21 U.S.C. 383), which, among other
things, encourages the FDA to work with foreign countries toward mutual recognition of
CGMP requirements. As a result, the FDA undertook adding design controls, as authorized
by the SMDA, to the CGMP regulation to benefit the public and industry and to achieve
greater consistency with other international standards and quality system requirements. The
FDA published the revised CGMP requirements in the final rule entitled “Quality System
Regulation” in the Federal Register of October 7, 1996 (61 FR 52602). This regulation
became effective on June 1,1997 and remains in effect.
3. THE MDR REGULATION (21 CFR Part 803)
The first Medical Device Reporting (MDR) regulation was published as final on December
13, 1984. As a result of changes mandated by the SMDA, and the Medical Device
Amendments of 1992, the 1984 MDR regulations (21 CFR Parts 803 & 807) were revised
and published again on December 11, 1995 (60 FR 63578). The FDA Modernization Act of
1997 made additional changes and a revised MDR regulation was proposed in May 1998 (63
FR 26129). The final revised MDR regulation was published in the Federal Register on

PROGRAM
7382.845
Page 9 of 59
January 26, 2000 (65 FR 4112). This latest version of MDR regulation includes reporting
requirements for manufacturers, user facilities, and importers. MDR reporting for medical
device distributors (except importers) was revoked by the FDA Modernization Act of 1997.
Distributors are, however, still required to maintain complaint records, per 21 CFR part
803.18(d)(1-3). 21 CFR Part 803 also requires manufacturers of medical devices, including
in vitro diagnostic devices, to report to the FDA whenever the manufacturer or importer
receives, or otherwise becomes aware of, information that reasonably suggests that one of its
marketed devices: (1) may have caused or contributed to a death or serious injury or, (2) has
malfunctioned and the device, or any other device marketed by the manufacturer or importer,
would be likely to cause or contribute to a death or serious injury if the malfunction were to
recur. NOTE: Importers* (initial distributors) of medical devices are subject to 21 CFR Part
803 (65 FR 4112). The Voluntary Malfunction Summary Reporting (VMSR) program was
established in 2018 and permits manufacturers to report certain device malfunction medical
device reports (MDRs) in summary form on a quarterly basis. It reflects changes made by
Section 227 of the Food and Drug Administration Amendments Act of 2007 and the goals for
streamlining malfunction reporting outlined in the commitment letter agreed to by the FDA
and industry, and submitted to Congress, as referenced in the Medical Device User Fee
Amendments of 2017 (MDUFA IV Commitment Letter). The overarching principles for the
VMSR program are described in an August 17, 2018, notification (83 FR 40973).
* Per 21 CFR part 803.3(j) Importer means any person who imports a device into the US and
who furthers the marketing of a device from the original place of manufacture to the person
who makes final delivery or sale to the ultimate user, but who does not repackage or
otherwise change the container, wrapper, or labeling of the device or device package.
4. THE MEDICAL DEVICE TRACKING REGULATION (21 CFR Part 821)
Under the authority of Section 519(e)(1) of the FD&C Act, the agency may issue a written
tracking “order” that directs a manufacturer to implement a tracking program that meets the
requirements of 21 CFR Part 821. Devices subject to tracking may include those that are
permanently implanted or are life-sustaining/life-supporting devices used outside a device
user facility as these devices are considered reasonably likely to cause serious adverse health
consequences if they fail. The regulation is intended to ensure that, in the event of a recall or
safety alert, a tracked device can be traced by the manufacturer, from the device
manufacturing facility to the end user or patient. Note that 21 CFR Part 821 does not include
a current list of devices to be tracked. Questions regarding the tracking status of a device
should be directed to the “PMA, HDE, Presubmission & Device Tracking Lifecycle Team”
in CDRH at [email protected].
5. THE CORRECTIONS AND REMOVAL REGULATION (21 CFR Part 806)
Provisions in the (SMDA) of 1990 required reports and records of corrections and removals
under section 519(g) of the Act (21 U.S.C. 360i(g)). Section 519(g) of the Act was enacted
because Congress was concerned that device manufacturers, distributors, and importers were
carrying out product corrections or removals without notifying the FDA, or not notifying the

PROGRAM
7382.845
Page 10 of 59
agency in a timely fashion. The Corrections and Removal regulation, 21 CFR Part 806, was
promulgated to meet these provisions and took effect on May 18, 1998. The regulation
initially required manufacturers, distributors, and importers to report promptly to the FDA
any corrections or removals of devices being undertaken to reduce risk to health. The Food
and Drug Administration Modernization Act (FDAMA) of 1997 amended section 519(g) of
the Act (21 U.S.C. 360i(g)) to eliminate the requirement for distributors to report corrections
and removals. The revised 21 CFR Part 806 was published in the Federal Register and
became effective December 21, 1998 (63 FR 42229). The regulation, 21 CFR Part 806,
requires that device manufacturers and importers report promptly to the FDA any correction
or removal of a device undertaken: (1) to reduce a risk to health posed by the device; or (2) to
remedy a violation of the act caused by a device which may present a risk to health. Device
manufacturers and importers are also required to keep records of all corrections and
removals, including those not required to be reported to the FDA under Section 519(g)(1)(B)
of the Act.
6. THE REGISTRATION AND LISTING REGULATION (21 CFR Part 807)
The Registration and Listing regulation, 21 CFR Part 807, was promulgated to meet
requirements of the Medical Device Amendments of 1976 (42 FR 42526). Owners or
operators of establishments that are involved in the production and distribution of medical
devices intended for use in the United States are required to register annually with the FDA,
and as general rule, establishments required to register with the FDA are also required to list
the devices they make and the activities that are performed on those devices. Registration and
listing provide the FDA with the location of medical device establishments and the devices
manufactured at those establishments.
Who Must Register, List and Pay the Fee | FDA describes the requirements for registration
and listing based on the type of activity performed at that establishment, including which
types of activities require payment of the establishment registration fee.
7. UNIQUE DEVICE IDENTIFICATION (UDI) SYSTEM (21 CFR PART 801
SUBPART B AND 21 CFR PART 830)
The FDA established the UDI system to adequately identify medical devices sold in the
United States, from manufacturing through distribution, under section 519(f) of the F D&C
Act. Benefits of the UDI system include, but are not limited to, simplifying the integration of
device use; providing for more rapid identification of medical devices with adverse events;
providing for more rapid development of solutions to reported problems and efficient
resolution of device recalls; and providing better focused and more effective FDA safety
communications. In the UDI final rule (78 FR 58785), device labelers (typically
manufacturers) are required to: 1) include a UDI, issued under an FDA-accredited issuing
agency’s UDI system, on device labels, device packages, and in some cases, directly marked
on the device; and 2) submit device information to the Global Unique Device Identification
Database (GUDID). AccessGUDID is a searchable database of device information
(including, for instance, device identifier on the label, device name, premarket submission
numbers) available to the public. The Unique Device Identifier System: Frequently Asked

PROGRAM
7382.845
Page 11 of 59
Questions, Vol. 1, provides guidance to industry and FDA staff, and provides clarification on
key provisions of the UDI Rule.
8. MEDICAL DEVICE SINGLE AUDIT PROGRAM (MDSAP)
In 2012, the FDA started working on the Medical Device Single Audit Program (MDSAP)
with other global regulators within the International Medical Device Regulators Forum
(IMDRF). From January 2, 2014, to December 31, 2016, the FDA announced participation
within the MDSAP consortium’s pilot program (78 FR 68853). On January 1, 2017, the FDA
announced participation in the operational phase of MDSAP, which included opening
applications for additional auditing organizations beyond the pilot phase auditing
organizations (80 FR 78741). Additionally, each regulator within the consortium committed
to utilizing the MDSAP audit reports submitted under the program.
The program allows an MDSAP-recognized Auditing Organization to conduct a single
regulatory audit of a medical device manufacturer that satisfies the relative requirements of
the regulatory authorities participating in the program. An up-to-date list of international
partners participating in MDSAP are listed at: https://www.fda.gov/medical-devices/cdrh-
international-programs/medical-device-single-audit-program-mdsap.
The FDA utilizes MDSAP audit reports as a substitute for routine agency inspections for
firms that volunteer to participate in the MDSAP. MDSAP audit reports submitted by
MDSAP Auditing Organizations that include the United States as a jurisdiction are reviewed
and classified by the FDA. Frequently Asked Questions can be found at:
https://www.fda.gov/media/161094/download. Firms with activities related to the Electronic
Product Radiation Control (EPRC) provisions of the Act continue to be subject to FDA
inspections for the EPRC activities. All firms continue to be subject to FDA for-cause
inspections.
9. FOOD AND DRUG OMNIBUS REFORM ACT OF 2022 (FDORA)
704(a)(4) of the FD&C Act, as revised by the Food and Drug Omnibus Reform Act of 2022
(FDORA)
2
, gives FDA authority to request (and requires establishments to provide) any
records or other information that FDA may inspect under section 704 of the FD&C Act, in
advance of or in lieu of inspections of specified establishments.
10. KEY COMPLIANCE DOCUMENTS
A. Benefit-Risk in Medical Device Product Availability, Compliance, and Enforcement
Decisions
In 2012, the FDA issued the first guidance document describing factors considered in
making benefit-risk determinations in certain premarket submissions
3
. The FDA may
2
FDORA was enacted as part of the Consolidated Appropriations Act, 2023, Pub. L. No. 117-328 (2022). FDORA
sections 3611(b)(1)(A), and 3612(a) included device establishments (in addition to those for drugs) as
establishments that are subject to mandatory requests for records or other information under 704(a)(4) (21 U.S.C.
374(a)(4)).

PROGRAM
7382.845
Page 12 of 59
also consider benefit and risk factors in prioritizing resources for compliance and
enforcement efforts to maximize medical device quality and patient safety. The FDA
describes the general framework for medical device decision making related to product
availability, compliance, and enforcement in its December 2016 guidance document,
Factors to Consider Regarding Benefit-Risk in Medical Device Product Availability,
Compliance and Enforcement Decisions: Guidance for Industry and Food and Drug
Administration Staff.
The FDA may consider benefit-risk factors during the following situations:
• Evaluation of device shortage situations.
• Selection of the appropriate regulatory engagement mechanism following an
inspection during which regulatory non-compliance was observed.
• Evaluation of recalls.
• Consideration of petitions for variance from those sections of the Quality System
(QS) regulation (21 CFR Part 820) for which there were inspectional observations
during a PMA pre-approval inspection.
When making medical device product availability, compliance, and enforcement
decisions informed by benefit-risk, the FDA may consider relevant, reliable information
relating to patient perspectives, including what constitutes meaningful benefit, what
constitutes risk, and what options patients are willing to accept, as well as what
alternatives are available.
B. Enforcement Policy Related to the Coronavirus Disease 2019 (COVID-19) Public
Health Emergency
To address critical public health needs during the COVID-19 public health emergency
(PHE), the FDA issued numerous enforcement policy guidance documents that were
made effective immediately. In addition, as it relates these guidance documents, on
2/9/2023, the HHS Secretary renewed the COVID-19 public health emergency
declaration issued under section 319 of the PHS Act, effective 2/11/2023. The declaration
expired at the end of the day on 5/11/2023. On 3/27/2023, FDA finalized two guidances:
Transition Plan for Medical Devices That Fall Within Enforcement Policies Issued
During the Coronavirus Disease 2019 (COVID-19) Public Health Emergency and
Transition Plan for Medical Devices Issued Emergency Use Authorizations (EUAs)
Related to Coronavirus Disease 2019 (COVID-19). The guidances outline the FDA's
general recommendations to transition from certain policies adopted and operations
implemented during the COVID-19 pandemic to normal operations. It is important for
ORA and CDRH staff to be aware of the scope and length of applicability of these
policies when implementing this compliance program, CP 7382.845.

PROGRAM
7382.845
Page 13 of 59
PART II - IMPLEMENTATION
1. OBJECTIVES
The goal of this program is to advance and continually improve the quality, safety, and
effectiveness of medical devices to meet patient needs. The objectives of this program are
meant to guide FDA staff: 1) to conduct inspections of manufacturers and identify
manufacturers who are not in compliance with the regulations noted below, and 2) to bring
these firms into compliance through voluntary, administrative, and/or regulatory means, as
appropriate.
Those pertinent regulations guiding our work include the following:
• Quality System (QS) regulation
• Medical Device Reporting (MDR) regulation
• Medical Device Tracking regulation
• Corrections and Removals (C&R) regulation
• Registration and Listing regulation
• Unique Device Identifier (UDI) regulation
2. PROGRAM MANAGEMENT INSTRUCTIONS
A. Priorities for QS Inspections
ORA divisions should target coverage of manufacturers of Class II and Class III devices
utilizing a risk-based methodology with consideration of the following:
(1) Pre-Market inspections under Medical Device User Fee Amendments (MDUFA.)
Inspections of manufacturers of devices with a pending PMA approval will be
assigned under the PMA Compliance Program 7383.001.
(2) Manufacturers of Class III devices that have never been inspected
(3) Manufacturers of high-risk devices which can be identified by:
(a) Product Codes for implantable, life-sustaining devices
(b) Devices with a higher frequency of recalls and MDRs
(c) Newly marketed devices such as recent 510(k) clearances or De Novo
classification
(4) Compliance Follow-up and For-cause Inspections

PROGRAM
7382.845
Page 14 of 59
B. Planning Instructions
(1) The following guidelines are suggested for implementing this compliance program:
(a) This compliance program should be used for QS inspections of devices.
(b) The profile information should be updated in eNSpect for QS inspections.
(2) Decisions regarding the size and composition of the inspection team should consider
factors such as the complexity of products manufactured, the type and extent of
deficiencies identified on previous inspections, new indicators of risk to patient safety
(recalls, consumer complaints, MDR increase), and the firm’s use of novel or new
manufacturing processes. ORA may contact CDRH (or CBER if CBER regulated
medical device) prior to, or during inspections, to discuss specific technical issues.
Additionally, ORA may include ORA Subject Matter Experts (SMEs) as part of team
inspections and may also request CDRH, CDER or CBERon-site SME participation.
The inspection team may also include ORA trainees to achieve FDA training
objectives. For combination products, ORA will work with the appropriate medical
product programs and Centers to determine the need for a team inspection.
(3) Many large firms have several manufacturing facilities located within a division or in
more than one division. Information should be reviewed to determine if:
(a) The firm’s quality system overlaps across multiple firm locations (e.g.,
procedures for a particular QS element such as complaint handling or design
control are the same at different locations), or
(b) The firm has segregated quality system functions across their organization (e.g.,
complaint handling at one site and design control at another site), or
(c) The firm has critical manufacturing responsibilities across multiple firm locations
(e.g., manufacturing steps for the same product family at different locations across
the same firm).
If any one of the above criteria apply, Investigations Branch (IB) should
contact the appropriate management or division to determine if additional
domestic or foreign sites should be inspected.
(4) Class I Device Manufacturers
Class I manufacturers should not be routinely scheduled for inspection and should
receive lowest inspectional priority unless addressed by a special, “for-cause”
assignment or when a health hazard is apparent. Use the following link to determine if
a device is Class I exempt from QS requirements:
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPCD/classification.cfm
C. Interactions Between Compliance Programs
CBER is designated the lead Center in FDA for regulating in vitro diagnostic (IVD)

PROGRAM
7382.845
Page 15 of 59
medical devices intended for screening or confirmatory clinical laboratory testing
associated with blood banking practices and other process testing procedures. These
IVD products include those required for screening of blood, blood products, human
cells, tissues, and cellular and tissue-based products (HCT/Ps), supplemental testing,
and related blood banking practices (such as blood typing and compatibility testing) and
are licensed under Section 351 of the Public Health Service (PHS) Act. Inspections of
IVDs licensed by CBER should be performed in accordance with Compliance Program
7342.008: Inspection of Licensed In Vitro Diagnostic Devices.
For guidance, see the Intercenter Agreement between the Center for Biologics
Evaluation and Research, and the Center for Devices and Radiological Health, dated
October 31, 1991.
Combination Products:
Drug Device combination products will be inspected under, Inspections of CDER-led
or CDRH-led Combination Products, CP 7356.000.
D. Resource Instructions
When possible, inspection of laser devices should be conducted by Electro-Optical
Specialists (EOS), whose time is reported under PAC 86001. EOSs should conduct the
QS portion of this program if they are trained on Quality System Inspection Technique
and QS Regulation.
E. Interactions with other Federal Agencies, State and Local Counterparts, and
Foreign Regulatory Authorities
Under the Medical Device Single Audit Program (MDSAP), a recognized Auditing
Organization is to report a public health threat, fraudulent activity, or counterfeit
product to MDSAP regulatory authorities within five working days following the
conclusion of an MDSAP audit. Additionally, if the MDSAP audit should reveal any of
the above-mentioned conditions, the Auditing Organization must submit the audit
report documentation to regulatory authorities for evaluation within 45 calendar days
following the audit end date.
CDRH can exchange regulatory information that is publicly available with foreign
regulatory counterparts as part of its International Program. The FDA may also share
certain kinds of non-public information with FDA counterparts in foreign countries and
international organizations, as part of cooperative law enforcement or regulatory
activities. To facilitate this type of information sharing, a Confidentiality Commitment
must be in place between the FDA and the external party.
3. PRE-ANNOUNCEMENT OF INSPECTIONS

PROGRAM
7382.845
Page 16 of 59
Refer to Guide to Inspections of Quality Systems, August 1999, and IOM Chapter 5 Pre-
Inspectional Activities. Review Update of Device Establishment Inspection Processes and
Standards, GFI of June 2020.
4. ANNOTATION OF THE FDA 483
Annotation of the FDA 483 should occur for all medical device inspections unless the
manufacturer declines. Refer to IOM Chapter 5.

PROGRAM
7382.845
Page 17 of 59
PART III - INSPECTIONAL
1. OPERATIONS
A. Inspectional Preparation
Prior to initiating an inspection, review any assignment instructions, follow information
listed in the IOM Chapter 5, Pre-Inspectional Activities and Quality System Inspection
Technique (QSIT) Guide, and follow current instructions for verification and
communication for MDSAP firms. Available post-market information and quality data
should be reviewed as a part of the preparation for the inspection to facilitate time spent
at the facility. Such information and data available for your review includes, but is not
limited to:
• FDA-received complaints and MDRs.
• Newly marketed products and new clearances.
• Recalls.
• Inspection, investigation, remote regulatory assessment (RRA) (as
applicable)
3
, and compliance history, including previously issued FDA–483
observation(s), along with firm’s response and applicable compliance
actions.
• Safety communications.
• Post approval studies.
• Related firms or contracted facilities.
• Registration and listing data
Any concerns you identify as a result of the review of post-market information and
quality data should be followed up during the inspection.
If during preparation you find quality data or other information of concern,
communicate with your supervisor about the potential need for an expansion of
assignment type. Adequate preparation will assist in identifying the type of inspection
to perform and in determining whether pre-announcement is required. (For example, an
assignment for an abbreviated inspection may need to be changed to an assignment for
a comprehensive inspection or be reassigned as a for-cause inspection.)
B. Inspectional Strategy
The inspection will assess the firm’s systems, processes, and procedures to ensure
3
An RRA is an examination of an FDA-regulated establishment and/or its records, conducted entirely remotely, to
evaluate compliance with applicable FDA requirements. RRAs assist in protecting human and animal health,
informing regulatory decisions, and verifying certain information submitted to the Agency

PROGRAM
7382.845
Page 18 of 59
that the firm’s quality system is effectively established (which means defined,
documented, and implemented) and effectively maintained.
The QS inspection should generally start with a walk through of the facility to become
familiar with the firm’s operations and general state of control.
QS inspections should include the assessment of post market information and
quality data for distributed devices. This includes but is not limited to the review
of:
• Corrections and removals.
• MDRs and Complaints
• Significant changes in device specifications or in the manufacturing
specifications.
• Corrections following previous FDA-483 observation(s), to include the
corrections, corrective actions, or preventive actions for the observation(s)
and the related system(s).
• Newly marketed products, applicable approvals, and/or clearances.
• Significant changes to marketed products, including labeling and UDI, or
significant changes to manufacturing processes.
(1) Quality System (QS) Inspections
QS inspections should generally be conducted using the Quality System Inspection
Technique (QSIT), as described in the Guide to Inspections of Quality Systems,
August 1999.
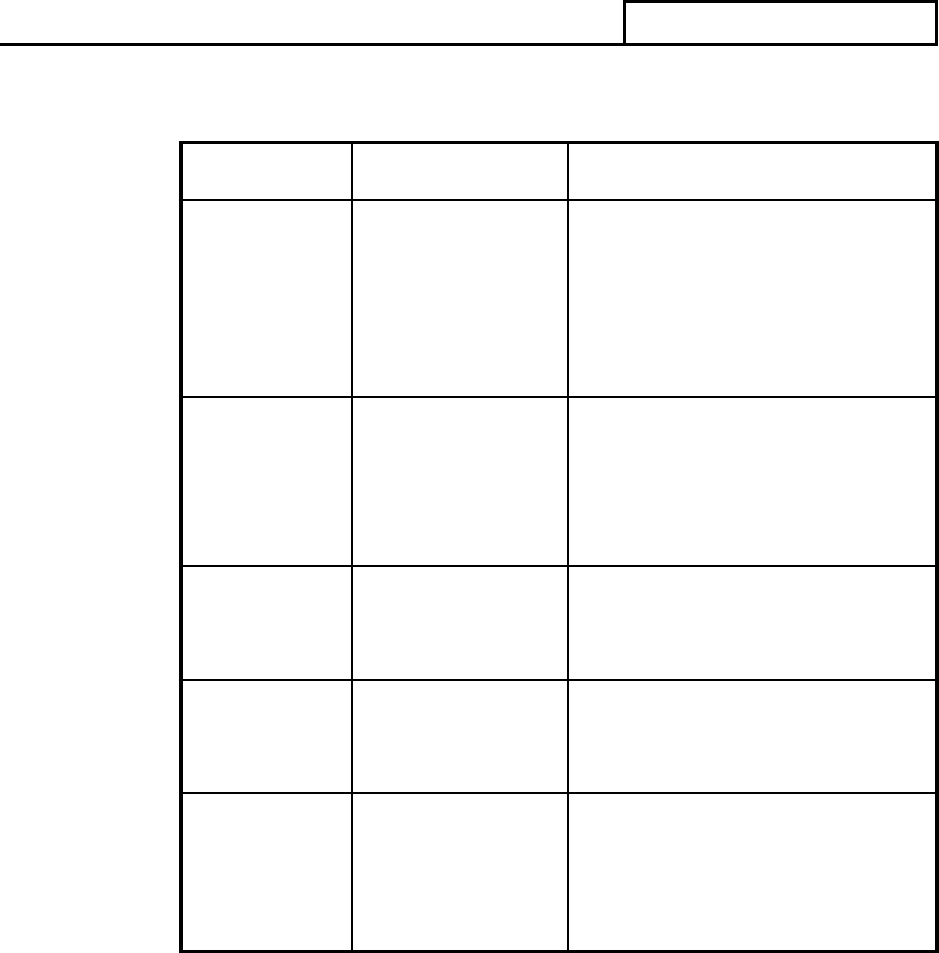
QSIT can be scaled to meet the needs of each particular inspection. The table below
correlates to the level and type of inspection.
PROGRAM
7382.845
Page 19 of 59
TABLE III.A.1 Inspection Guidance
Inspection
Level
Type of
Inspection
Guide to Inspections
1
Abbreviated
QSIT – Two subsystems;
Corrective and Preventive
Actions (CAPA) plus
Production and Process
Controls (P&PC) or Design
Controls
(PAC 82845A)
2
Comprehensive
QSIT - The four major
subsystems; Management
Controls, Design Controls,
CAPA and P&PC
(PAC 82845B)
3
Compliance
Follow-up
As directed by inspectional
guidance and elements of
QSIT
(PAC 82845C)
Special
For-Cause
As directed by inspectional
guidance and elements of
QSIT
(PAC 82845G)
Special
Specific Product
Risk Assignment
As directed by CDRH
inspection assignment and
elements of QSIT
(PAC 82845H)
The QS regulation can be grouped into seven subsystems; however, the following four
subsystems are considered major subsystems and comprise the foundation of a firm’s
quality management system:
• Management Controls.
• Design Controls.
• Corrective and Preventive Actions (CAPA).
• Production and Process Controls (P&PC).
MDR, Corrections and Removals, and Tracking requirements (where applicable)
should be reviewed when covering the CAPA subsystem. The three remaining

PROGRAM
7382.845
Page 20 of 59
subsystems (Facilities and Equipment Controls, Materials Controls and
Document/Records/Change Controls) cut across a firm’s quality management system
and are evaluated while covering the four major subsystems.
Inspection resources should be primarily focused on facilities that represent a
potential risk of harm. For instance, if during the inspection of a manufacturer the
investigator determines that the firm only makes Class I QS exempt devices, review
and determine the adequacy of the firm's complaint handling system and MDR
practices, and then terminate the inspection.
Medical device firms may have several manufacturing facilities located across
the country, or globe. It may be necessary to request inspectional assistance or
follow-up from another division in the following instances: if the inspection
finds concerns that may relate to additional locations, the firm’s quality system
overlaps across multiple firm locations, the firm has segregated quality system
functions across their organization, or the firm has critical manufacturing
responsibilities across multiple firm locations.
NOTE: Foreign Inspections: All inspection types listed below can be conducted at
foreign facilities. However, inspections conducted for surveillance purposes are
generally conducted as Level 2 and may include special instructions contained in the
inspection assignment.
(2) Level 1 Inspections - PAC 82845A/42845A
4
(Abbreviated)
Level 1 inspections are Abbreviated Inspections. Generally, inspections conducted at
foreign facilities are not conducted as abbreviated inspections.
This level of inspection (CAPA plus P&PC, or CAPA plus Design Controls) may be
used for surveillance and initial inspections of all firms, other than firms that
manufacture Class III devices. However, it is recommended that initial inspections of
Class II manufacturers utilize a Level 2 Comprehensive inspection whenever
resources permit. Level 1 inspections should cover the CAPA subsystem, then
P&PC or Design Controls, using the QSIT Guide.
The following should be considered, in no particular order, in determining whether to
select P&PC or Design Controls:
• Any significant design changes or new projects since the previous
establishment inspection,
• CAPA findings during the inspection
4
PACs 42845A-C are for inspections of CBER regulated medical devices that are cleared or approved under the
FD&C Act’s 510k and PMA provisions. ORA’s Office of Biological Products Operations (OBPO) conducts the
inspections of CBER regulated devices under these PACs. OMDRHO staff should not use PACs 42845A-C for
inspections of CDRH regulated devices and should not initiate inspections of CBER regulated devices unless
requested by OBPO.

PROGRAM
7382.845
Page 21 of 59
• Any major process changes,
• Post market information (Ex. Complaints) indicating potential design and/or
process problems
When none of the above activities identify a clear selection, then the P&PC or
Design Controls subsystem should be alternated each inspection.
When an inspection starts out as Level 1 and reveals information or objectionable
conditions that cannot be adequately assessed as a Level 1 inspection, then
expanding to a Level 2 inspection should be considered. Elements to consider when
determining if a Level 1 inspection should be expanded to cover additional
subsystems includes:
• Risk presented by the device(s) when considering essential function,
intended use, and risk if device fails.
• Level of risk presented by the quality system violation(s).
• Quality System violations (such as in design) that may present device
marketing (PMA, 510(k), etc.) violations.
(5) Level 2 Inspections - PAC 82845B/42845B
5
(Comprehensive)
Level 2 inspections are Comprehensive Inspections. The Level 2 inspection is
considered a comprehensive review of the compliance status of the firm.
Level 2 inspections cover all four major subsystems (Management Controls,
Design Controls, CAPA, and P&PC) as explained in the QSIT Guide. The
decision to conduct a Level 2 should be based on signals observed during pre-
inspectional planning or other risk indicators.
Level 2 inspections should be performed for the following:
• All initial inspections of Class III device manufacturers and, where possible,
Class II device manufacturers.
• By assignment.
• Foreign inspections.
• Training.
• When preparation of a Level 1 inspection reveals potential risks or quality
data issues.
• During a Level 1 inspection, if significant quality system deficiencies are
revealed.
5
PACs 42845A-C are for inspections of CBER regulated medical devices that are cleared or approved under the
FD&C Act’s 510k and PMA provisions. ORA’s Office of Biological Products Operations (OBPO) conducts the
inspections of CBER regulated devices under these PACs. OMDRHO staff should not use PACs 42845A-C for
inspections of CDRH regulated devices and should not initiate inspections of CBER regulated devices unless
requested by OBPO.

PROGRAM
7382.845
Page 22 of 59
The Level 2 QSIT approach was developed using the following inspectional
sequence: Management Controls, Design Controls, CAPA, and P&PC. This
inspectional sequence allows the investigator to review design control issues and
how the device specifications were established, before initiating review of the
CAPA subsystem. The subsystems may be inspected in any appropriate and
justifiable sequence in order to perform an effective inspection.
Information from Design Controls and CAPA may be used to select the products
and processes for inspecting production and process controls, and appropriate
linkages.
Deciding which manufacturing processes to inspect should include the following
considerations:
• CAPA indicators of process problems.
• Processes used to manufacture high risk products.
• Processes that have a high risk of causing product failure.
• Processes that require process validation.
• Processes that are new to the manufacturer.
• Processes that cover a variety of process technologies and profile classes.
• Common processes used in multiple products.
• Processes not covered during previous inspections.
(4) Level 3 Inspections - PAC 82845C/42845C
6
(Compliance Follow-Up)
Level 3 compliance follow-up inspections are conducted by assignment using QSIT
as a guide. The assignment may outline previous inspectional findings, the firm’s
commitments to corrections, and any additional signal data needing evaluation.
During a Level 3 inspection:
(a) Verify that adequate correction(s) and corrective action(s) have been implemented
to address the quality system deficiencies previously identified. If the
correction(s) and corrective action(s) were not implemented or were not
implemented effectively, verify that the deficiencies continue to exist.
(b) In addition to the assignment guidance, consider the need to cover additional
subsystems and document any quality system deficiencies observed.
(c) Provide evidence to demonstrate the significance of the deficiencies identified to
support a possible regulatory action.
6
PACs 42845A-C are for inspections of CBER regulated medical devices that are cleared or approved under the
FD&C Act’s 510k and PMA provisions. ORA’s Office of Biological Products Operations (OBPO) conducts the
inspections of CBER regulated devices under these PACs. OMDRHO staff should not use PACs 42845A-C for
inspections of CDRH regulated devices and should not initiate inspections of CBER regulated devices unless
requested by OBPO.

PROGRAM
7382.845
Page 23 of 59
Consider the following factors when verifying the adequacy of corrections:
• The depth and details of investigation performed.
• Identification of the potential cause(s).
• Scope of evaluating and implementing corrections needed throughout the
entire system.
• Evaluation of corrections and if they had any adverse impact.
• Corrections addressed deficiencies with product currently in the field
• Implementation of additional or updated procedures.
• CAPA process was followed correctly
• Validation of processes.
• Verification and/or validation of effectiveness of a CAPA.
• Timeliness of the performed activities of the investigation and
implementation of corrections and corrective actions.
(5) For-Cause Inspections - PAC 82845G
For-cause inspections are carried out in response to specific information that raises
questions, concerns, or problems associated with medical devices or medical device
firms. This information could come to the attention of FDA from any source or
quality data signal and includes, but is not limited to, the following:
• Observations made during prior inspections
• Recalls or market withdrawals
• Complaints or Allegations
• Medical Device Reports (MDRs)
• Suspicions of fraud
• Follow-ups to Remote Regulatory Assessments (RRAs)
• MDSAP Regulatory Audit Review
• When the result of an inspection indicates potential quality issues at
another related facility, such as contract manufacturer, contract sterilizer,
or associated site that uses the same quality system (domestic and
foreign)
• Results of a sample analysis
• Notified by another regulatory agency
For-cause inspections are usually created by CDRH, the OMDRHO Immediate
Office, the OMDRHO Division Compliance or Investigation Branches, CBER, or
the OBPO Division Compliance or Investigation Branches. For-cause inspections
may differ from the typical QSIT approach. The inspectional guidance provided by
the assignment will guide the flow of these inspections; however, elements of the
QSIT Guide may also be utilized.
MDSAP-participating firms may be inspected as part of a For-cause assignment.

PROGRAM
7382.845
Page 24 of 59
(6) Specific Product Risk Assignments PAC 82845H (SPRA)
Specific product risk assignment requests are initiated by CDRH. The inspectional
guidance provided by the assignment will guide the flow of these inspections;
however, elements of the QSIT Guide may also be utilized.
If a serious public health risk is encountered during a specific product risk assignment
inspection, CDRH and the division compliance branch should be consulted.
MDSAP participating firms may be inspected as part of an SPRA assignment.
(7) Foreign Inspections
All inspection types listed above can be conducted at foreign facilities. However, all
foreign surveillance inspections should be conducted as Level 2 and follow the
special instructions contained in the inspection assignment.
The foreign manufacturer's compliance with registration and listing requirements
should be covered during foreign inspections. The failure of foreign device
manufacturers to list products exported to the United States will subject medical
devices to detention upon entry.
Foreign inspections are subject to time constraints but need to follow the instructions
for a Level 2 inspection as described above. Requests for documents should be made
as early as possible to give foreign firms time to conduct or acquire any necessary
written or oral translations, and to obtain documents that may be located in U.S.
offices. Oral translations need to be documented in the EIR if that information is
utilized in supporting an observation(s).
C. Additional Inspectional Instructions
(1) Satellite Program Areas (QSIT)
Some program areas are considered satellites to the four major quality management
system subsystems.
Refer to the QSIT Guide for details on how to inspect the satellite program areas
and consider additional inspectional objectives, including UDI. Refer to Part V of
this Compliance Program for guidance on regulatory and administrative follow-up
to these programs. Report the time spent on these satellite areas under the
appropriate corresponding PAC.

PROGRAM
7382.845
Page 25 of 59
(2) CAPA Satellites
(a) MDR: 21 CFR Part 803
Cover during all QS inspections. Prior to initiating an inspection, the MDR data
should be reviewed using Manufacturer and User Facility Device Experience
(MAUDE), or by obtaining information from CDRH (or CBER if CBER regulated
medical device) regarding the firm’s current reports.
NOTE: As stated in QSIT, “Confirm any claims for exemption from 806 as a
result of submission under either the MDR regulation or Radiological Health
requirements” Under Correction and removal (21 CFR 806) section of QSIT.
Verify that any medical device reports (MDRs) include unique device identifier
(UDI) that appears on the device label, or on the device package, when known.
(b) Corrections & Removals: 21 CFR 806
Covered during all QS inspections. Determine whether the firm has
initiated any corrections or removals since the previous inspection as
described in the QSIT Guide.
Verify that any correction and removal reports include UDI that appears on the
device label, or on the device package.
(c) Tracking: 21 CFR 821
Cover for all devices that were issued a tracking order for all QS
inspections. To obtain Tracking information, refer to “Medical Device
Tracking Guidance for Industry and FDA Staff” dated March 27, 2014,
by accessing Medical Device Tracking | FDA
Verify the device tracking system documents the unique device identifier (UDI)
that appeared on the device label or on the device package.
(3) Production and Process Control Satellite
When the P&PC subsystem is being inspected, and if the firm
manufactures sterile devices, sterilization should be chosen if not
covered during the previous inspection, unless CAPA indicators of
existing or potential problems are found with any other specific
process.

PROGRAM
7382.845
Page 26 of 59
(a) Sterilization Process Controls coverage is defined in the QSIT
Guide.
The Sterilization Process Controls Decision Flow Chart should be followed at the
following types of facilities:
• Device manufacturers that sterilize their own product
• Device manufacturers that use contract sterilizers
• Contract sterilizers
NOTE: Report time spent covering sterilization processes under PAC 82845S.
(4) Additional Considerations for Purchasing Controls
If the firm has outsourced any activities, the investigator should evaluate the firm’s
purchasing controls program to determine controls they have put in place to manage and
monitor these operations. Examples of quality system operations that may be contracted
out by the manufacturer include:
• Contract manufacturers.
• Design operations.
• Sub-assemblies.
• Contract sterilizers.
• Laboratory testing.
• Labelers/packagers.
• Software developers.
• GMP auditors.
• Call centers.
• Service and repair operations.
Outsourced processes may need to be evaluated to determine how they are integrated into
the firm’s quality system. Some examples include nonconformances and complaints
feeding back into the firm’s CAPA system, specification changes are communicated to
the contracted facility, and receiving and acceptance activities feeding into purchasing
control decisions for continual evaluation of contract or material providers.
Consider if the firm has written agreements and/or written instructions and review these
documents to evaluate the type of controls the firm has put in place. These written
agreements typically include:
• Quality Agreements.
• Contractual Agreements.
• Service Agreements.
• Detailed Instructions

PROGRAM
7382.845
Page 27 of 59
(5) Unique Device Identifier (UDI)
The Unique Device Identification System final rule (78 FR 58825) requires device
labelers (typically, the manufacturer) to include a UDI on device labels and packages,
except where the rule provides for an exception or alternative and requires submitting
device information to the Global Unique Device Identification Database (GUDID.) The
device labeler must also mark the UDI directly on the device if a device is intended to be
used more than once and intended to be reprocessed before each use.
During the inspection review of the Production and Process Controls and/or Design
Controls:
(a) Confirm that the UDI labeling on the device is available in both easily readable,
plain text and Automatic Identification and Data Capture (AIDC) technology. In
addition, request the firm to confirm the AIDC format is of adequate quality and
can be scanned to allow for retrieval of UDI information.
(b) Review the UDI to confirm that the data elements in AccessGUDID - Identify
Your Medical Device (specifically, within the Device Identifier (DI) Information,
Device Characteristics, Alternative and Additional Identifiers, and Customer
Contact sections) match the labeling and UDI information on the device. Ensure
that when an AccessGUDID attribute appears in the medical device labeling, the
values submitted to the GUDID match the values in the labeling.
(c) If a device has been discontinued from distribution, ensure that the DI
Information in AccessGUDID for both the “Commercial Distribution Status” and
“Commercial Distribution End Date” fields is updated.
(d) If a firm is claiming applicability of an exception or alternative to UDI
requirements, please confirm that the Product Codes are listed on the UDI
website, Unique Device Identification System (UDI System).
(e) When inspecting design controls, ensure that design reviews include reviewing
the UDI to determine if a new UDI needs to be assigned, due to the type and
extent of design changes. Verify that a design review decision is documented.
(f) Review procedures related to labeling and label controls. Ensure that the
procedures include the UDI requirements and that the appropriate UDI is included
on any labels or labeling and is in conformance with the Device Master Record
(DMR).
(g) Verify that the Device History Record (DHR) includes, or refers, to the location
of any UDI used in manufacturing each batch, lot, or unit of a device.

PROGRAM
7382.845
Page 28 of 59
(h) Verify that complaint-handling procedures include provisions for capturing and
documenting any UDI when performing assessment of the firm’s complaint
investigations.
(i) When servicing is applicable, verify that servicing procedures include provisions
for documenting in the service reports any UDI.
D. Sampling Records
Sampling of records is an important tool that can reduce the time spent reviewing
records. Sampling of records should be used for evaluating the firm’s adherence to
requirements and its procedures, not for performing data verification or analysis. The
QSIT guide includes a binomial sampling table that should be used for sampling
records when trying to make a decision about an endpoint that only has two potential
outcomes (e.g., The device history record is compliant, or the device history record is
noncompliant).
Computer- aided techniques may also be useful tools to efficiently evaluate
electronic records (for example, a large volume of complaint files) or to accomplish
assignment specific objectives (for example, evaluating for trends in product specific
complaint or failure data).
E. Special Instructions Concerning Design Controls
The inspectional authority for review of design control records is derived from
Section 704(e) of the Act. Such authority applies only after the establishment has
manufactured the device for which the design has been under development or has
taken an action that precludes the argument that the product under development is
not a device. Such actions include:
• Submitting to an Institutional Review Board plans for clinical investigation
of the device.
• Submitting to FDA a Product Development Protocol (PDP).
• Submitting to FDA an IDE, 510(k), PMA, Humanitarian Device Exemption
(HDE) or Premarket Report (PMR).
• Changes to an already marketed device.
FDA has inspectional authority to review design control records when the device
has been placed on the market, or when any of the four actions above have
occurred.
Review of design controls should cover any design processes performed after June 1,
1997. The manufacturer is not required to retrospectively apply design controls to any
stages in the design process that it had completed prior to June 1, 1997, unless changes
have been made to the design (including changes in ownership or where the designed
device will be manufactured) after June 1, 1997.

PROGRAM
7382.845
Page 29 of 59
If a manufacturer normally designs its own devices but has not initiated any design
changes to current devices since June 1, 1997 or does not have a design project
underway that is reviewable by FDA given the limitation discussed above,
investigators should limit their coverage to a review of the design change control
procedures that the manufacturer must have defined and documented.
Some manufacturers have their devices designed under contract. These
manufacturers must comply with the requirements for using contractors or service
suppliers, as found under 21 CFR § 820.50 and 21 CFR § 820.30. The manufacturer
must maintain or have reasonable accessibility to copies of a Design History File for
any device that is in its production.
Do not place observations on the FDA-483 that concern the adequacy, safety, or
efficacy of a particular design. Observations relating to Design Controls placed on
the FDA-483 should be limited to the adequacy of, and adherence to, the procedures
and/or controls established by the firm.
F. Inspection of Radiation-Emitting Devices
Medical devices that emit electronic product radiation (for example, diagnostic x-ray
systems and their major components) are subject to both Electronic Product
Radiation Control (EPRC) and Medical Device provisions of the FD&C Act. These
devices have additional Radiological Health requirements meant to protect the public
from unnecessary radiation. Such requirements include affixing certification labeling,
verifying safety features, reporting and record keeping, and the continual testing to
verify product conformance with applicable Federal Performance Standards
promulgated under 21 CFR 1010 – 1050. QS inspections should be performed jointly
with EPRC inspections whenever possible. Medical device inspection and
enforcement activities described in the relevant radiological health compliance
programs (for example, CP 7386.003a, Diagnostic Medical X-Ray Equipment)
should be adhered to, jointly with this Compliance Program. For joint QS/EPRC
inspections, the firm should be informed that EPRC requirements will be evaluated
during the inspection. There are also specific International Electrotechnical
Commission (IEC) standards that cover these products that many manufacturers
conform to as well.
A firm may manufacture medical devices that are capable of emitting electronic
product radiation. If so, when conducting QS inspections, you should also assess the
firm’s devices against any applicable standards promulgated under Chapter V,
Subchapter C - Electronic Product Radiation Control of the FD&C Act. This
assessment is not a QS activity and should not be reported as a QS activity. Instead,
report any Radiological Health time under the appropriate Radiological Health PAC.
For Inspection and Field Testing of Radiation-Emitting Electronic Products use CP
7386.001.

PROGRAM
7382.845
Page 30 of 59
For Field Compliance Testing of Diagnostic Medical X-Ray Equipment use CP
7386.003.
Device manufacturers subject to existing FDA performance standards (21 CFR Parts
1010 – 1050) should include in their device master and history records those
procedures and records demonstrating compliance with the applicable standard, self-
certification (21 CFR Part 1010), and reporting (21 CFR 1002 – 1005).
G. Field Exams
See IOM Chapter 5 regarding field exams (e.g., sterile device packaging).
Take into consideration your sample size based on the lot size and the sample
tables found in QSIT.
If defective packaging is found during a visual field examination, consider collecting
a sample, and contact your supervisor.
H. Sample Collection
Physical samples should not be routinely collected to support QS cases. Generally,
samples are not necessary to support a warning letter for QS, MDR, Tracking,
UDI, and Correction and Removals violations. Discuss whether a sample is needed
with the Division Compliance Branch when discussing the potential for any
administrative or judicial actions.
(1) Physical Sampling
When considering physical sample collections, sampling specifics and
coordination should be completed in conjunction with your supervisor through
consult with the Office of Regulatory Science’s (ORS) Winchester Engineering
and Analytical Center (WEAC). See Part IV Analytical for additional
guidance.
(2) Counterfeit Sampling
The investigator should work with their supervisor and ORS’s Forensic Chemistry
Center (FCC) for guidance on collecting samples for potential counterfeit product.
2. ADDITIONAL CONSIDERATIONS
A. Registration and Listing
Registration and Listing should be reviewed as part of the pre-inspectional activities for

PROGRAM
7382.845
Page 31 of 59
both domestic and foreign inspections. During the inspection, review a sample of device
listings to determine whether listings are accurate.
If a firm failed to register or has not registered accurately, this needs to be discussed
with firm management. If the firm is required to list, and yet has not listed all their
devices, this also needs to be discussed with management.
B. Imports
No import field examinations or sample collections are scheduled under this program.
C. Export-Only Firms
Investigators that are assigned inspections of firms that are export-only should perform a
QSIT inspection and confirm that the device(s) substantially meet the requirements of the
Quality System Regulation, are not adulterated other than due to lack of U.S. marketing
approval and are not mislabeled other than possessing labeling in the language of the
recipient country.
A medical device which would be considered to be adulterated or misbranded, may be
exported under Section 801 or 802 of the FD&C Act provided the device is intended
solely for export. See Exporting Medical Devices | FDA. Although such a device would
not meet the requirements of the FD&C Act to be sold domestically for commercial
distribution, it may be exported legally if certain requirements are met. Records must
clearly demonstrate compliance to the requirements.
See section 5 Guidance for Industry: Exports Under the FDA Export Reform and
Enhancement Act of 1996 | FDA
Investigators should determine if the firm has obtained any export certificate(s) for the
covered devices. See CDRH Export Certificate Validation (CECV) (fda.gov) or CBER’s
Biologics Export Certification Application and Tracking System (BECATS) Export
Certificate.
• If a Certificate of Exportability (COE) Section 801 has been issued, verify subject
device is listed, confirm the firm’s intentions to operate as export only and
discontinue the inspection.
• If no COE Section 801 (COE 801), then proceed to verify all requirements of
801(e)(1).
• If COE Section 802 (COE 802) has been issued, verify subject device is covered
and then conduct QSIT level 1 inspection.
• If no COE Section 802, then verify 801(e)(1) and 802(f) and proceed with a QSIT
level 1 inspection.

PROGRAM
7382.845
Page 32 of 59
Contact the CDRH Exports team at [email protected] (or the CBER Exports
team at [email protected] for CBER regulated medical devices) if you have
questions.
D. Electronic Records and Electronic Signatures
Follow agency policy when inspecting electronic records and signatures, see Part VI.
3. REMARKETED DEVICES
A. Remanufacturers of Used Devices
Remanufacturers are persons who process, condition, renovate, repackage, restore, or do
any other act to a finished device that significantly changes the finished device’s
performance or safety specifications or intended use (21 CFR 820.3(w)).
Remanufacturers are considered to be manufacturers and are subject to all applicable
requirements of the QSR, MDR requirements, device tracking requirements, registration
and listing, and premarket approval or clearance requirements. If an establishment
disputes its regulatory status, the division should refer the EIR to the Office of
Regulatory Programs (ORP) at CDRH (or CBER OCBQ for CBER regulated medical
devices) for assistance in interpreting the definition of a remanufacturer.
NOTE: For a discussion of the above issues, see Federal Register Notice: December 23,
1997 (Volume 62, Number 246), pages 67011 – 67013.
B. Third Party Refurbishers/Reconditioners/Servicers of Used Devices
Third party refurbishers, reconditioners, servicers, and "as is" resellers are currently not
subject to the requirements of the Quality System regulation. Refurbishers should only be
inspected as directed.
C. Reprocessors of Single Use Devices
Third party reprocessors of single use devices are considered to be manufacturers, and so
are subject to those requirements of the Quality System regulation that apply to the
operations they perform.
D. Hospital Reprocessors
Hospital reprocessors are to be only inspected under CDRH assignment.

PROGRAM
7382.845
Page 33 of 59
Self-described refurbishers/reconditioners/servicers should only be inspected as directed,
or if there is reasonable evidence that the entity is actually remanufacturing and there is a
risk to public health.
4. REPORTING
A. Establishment Information Updates
Update establishment information (for example., a firm’s legal name and address) in
eNSpect prior to the issuance of the FDA 483 as needed. Per the IOM, Official
Establishment Inventory (OEI) updates should also be conducted as appropriate.
B. Observation and Discussion Item Reporting Requirements
If there are significant observed violations of the QS requirements (21 CFR 820), you
should follow the IOM instruction and document as necessary on the Form FDA-483.
The QSIT Guide provides instruction concerning major QS requirements and the
identification of significant deviations.
Observations indicating nonconformity with MDR (21 CFR 803), Medical Device
Tracking, (21 CFR 821) and UDI (21 CFR 801 subpart B and 21 CFR 830)
requirements should be discussed and/or cited per the IOM.
Observations relating to violations of Reports of Corrections and Removals (21 CFR
806) should be discussed with division management prior to including on an FDA-
483. See Part V Corrections and Removals Regulatory/Administrative Follow-up.
Observations indicating non-compliance with medical device pre-market notification
requirements and pre-market approval (FD&C Act sections 510(k) and 515) require
confirmation from CDRH or CBER prior to including on an FDA-483.
Violations that are not documented on the Form FDA-483 should be discussed with
management and described in the report with supporting evidence.
C. Registration Listing Discrepancies
All discrepancies in registration listing should be discussed with management during
the inspection close-out meeting. Report any management responses and
commitments to update registration and listing information online.
D. Annotations and Firm Commitments Related to Inspectional Observations and
Discussion Items
Encourage the firm to respond in writing to any observations and violations discussed
during the inspection. See IOM for further instruction.

PROGRAM
7382.845
Page 34 of 59
Report any offers to annotate FDA 483 Inspectional Observations you have issued.
Any commitments made by the firm to your verbal observations or discussion items
should be captured in your report.
E. Establishment Inspection Report Writing
Refer to the IOM for general reporting requirements for writing an establishment
inspection report.
Report the FEI, name, and address of significantly related firms, such as if the
manufacturer contracts a sterilization process or contracts the manufacturing of
significant components, subassemblies, or processes, in the EIR.
F. eNSPECT Profiles
Document device Profiles in eNSpect. Clearly identify what manufacturing processes
and products were covered in this inspection. See IOM for further instruction.
PROGRAM
7382.845
Page 35 of 59
PART IV - ANALYTICAL
1. ANALYZING LABORATORIES
The division will make all the necessary arrangements for proper handling of samples with
the following designated testing facilities:
TYPES OF
DEVICES
ANALYZING LABORATORIES
All General Medical
Devices
Winchester Engineering and Analytical Center
(WEAC)
109 Holton Street
Winchester, MA 01890-1197
ANALYSES
PERFORMED
ANALYZING LABORATORIES
Testing for sterility
of finished devices,
package integrity,
bioburden,
endotoxins, and In
Vitro diagnostics
WEAC – Micro and Chem
Performance Testing
WEAC - Engineering
See PART VI regarding WEAC contact information.
SPECIAL NOTE: For all questions concerning laboratory testing capabilities, contact the
ORA’s Office of Regulatory Science (ORS) medical product program coordination team at
2. ANALYSES TO BE CONDUCTED
Sample collection and analysis will be determined on a case-by-case basis through
consideration of inspectional findings, compliance concerns, and scientific capabilities and
expertise. Full collaboration between investigations and analytical personnel is essential.
See Part III for additional information.
3. METHODOLOGY
A. Testing Finished Device Samples for Sterility
(1) Visually examine each unit to ascertain if its packaging is intact. Report all
observed defects by describing the size, type, and location of the defects. Units
with defective packaging do not need to be examined for sterility.

PROGRAM
7382.845
Page 36 of 59
(2) Finished device samples are to be tested in accordance with the requirements of
current USP methodology for Sterility Tests, and FDA Pharmaceutical
Microbiology Manual.
(3) Device samples are to consist of 60 units.
NOTE: Some medical devices may naturally contain antimicrobial properties or be
impregnated or packaged with antimicrobials or preservatives that inhibit microbes.
Common antimicrobial-containing products include:
• Bandages or gauze pads containing Ag (silver), alcohol, chlorohexidine,
honey, beeswax, ointments, or gels.
• Latex gloves.
• Isoprene gloves.
Examine the sample for claims of antimicrobial properties. If antimicrobial
ingredients are suspected, collect a sample size of 100 units, rather than 60 units.
When the required number of units are not available because of lot size or cost,
contact the analyzing lab to discuss a sampling strategy.
See PART VI regarding contact information for WEAC.
(4) Positive subsamples
During incubation, check cultures for growth at regular intervals. If any growth is
detected, you should begin qualitative analysis of that growth f ollowing
subculturing procedures in the FDA Pharmaceutical Microbiology Manual.
All isolates from sterility tests must be maintained until the sample disposition
authorization is completed in FACTS.
B. Pre-sterilization Microbial Contamination (Bioburden)
Bioburden testing is to be performed in accordance with the guidance provided in ISO
11737-1, Sterilization of health care products - Microbiological methods - Part I:
Estimation of population of microorganisms on products. The methodology used for
estimating the bioburden must be validated. Twenty units are to be tested.
NOTE: the above refenced ISO method must be purchased or one must have a
membership to access.
C. Analysis of Packaging Defects

PROGRAM
7382.845
Page 37 of 59
Perform a visual, nondestructive inspection of the package noting the existence and
location of any seal or material defects. Normally, 20 packaged devices should be
collected to ensure adequate analysis. Further testing may be performed using
consensus standards such as those identified in the Part VI. A.1 references for the
American Society for Testing and Materials (ASTM). Selection of the test will depend
on the materials and construction of the package, and on the nature of the noted or
suspected problem.
D. Analysis of Endotoxins
Samples will be analyzed using the Bacterial Endotoxins Test found in the current USP
and the FDA Pharmaceutical Microbiology Manual. Ten units are required for
endotoxin testing.
E. Antimicrobial Effectiveness Testing
Samples will be analyzed using the Antimicrobial Effectiveness Test found in the
current USP and FDA Pharmaceutical Microbiology Manual. Ten units are required
for testing.

PROGRAM
7382.845
Page 38 of 59
PART V - REGULATORY/ADMINISTRATIVE STRATEGY
Voluntary corrections are often the most effective and expedient means by which to protect
public health, support the availability of quality medical devices to patients, and maintain
compliance. As such, FDA’s goal, as part of this regulatory strategy, is to obtain prompt
voluntary correction of violations by industry, to reduce risks to public health safety and ensure
compliance with regulatory requirements. However, when voluntary corrections to address
significant violations are not forthcoming, and/or do not adequately mitigate risk to the user,
regulatory action may be taken, when significant violations present a threat to public health. The
potential adverse effect of the violations on the safety and intended use of the finished device
will be considered when determining the appropriate level of regulatory significance and
resulting actions and/or communications with the firm.
Traditionally, domestic cases are primarily handled by ORA’s OMDRHO under direct reference,
while foreign cases are handled by CDRH’s OPEQ. For the purposes of this document, the
compliance role can be performed by either organization.
Domestic and foreign cases for CBER regulated devices are initially handled by ORA’s OBPO
and referred to CBER OCBQ. For the purposes of this document, the compliance role can be
performed by either organization.
Note: If a combination product and/or there are violations for products regulated by other centers
(e.g. CDER or CBER), coordinate appropriately.
1. REGULATORY SIGNIFICANCE FOR COMPLIANCE DECISIONS
The evidence in its totality, and the associated risk to users and patients, should be assessed
when determining regulatory significance. Regulatory significance can be determined by
evaluating the inspectional observations and findings (Section A below) and factors
considered by compliance (Section B below). By assessing this full complement of
information, you can determine the following: the overall criticality of the inspectional
findings, how the current inspection should be classified, and what regulatory action(s) are
most appropriate.
A. Inspectional Observations/Findings
When the Investigations Branch is deciding the initial type of action to recommend, the
initial decision should be based on the seriousness and/or frequency of the deficiencies.
The following are examples of such deficiencies.
(1) Situation 1
Evidence that supports a significant deficiency, and/or a pattern of deficiencies,
within one or more systems covered, should result in an Official Action Indicated
(OAI) recommendation. Examples to consider include:

PROGRAM
7382.845
Page 39 of 59
(a) A firm’s total failure to define, document, or implement all or part of its quality
system, particularly where there is evidence of an adverse impact from this
failure. The following is a list of examples and is not all inclusive:
i. Failure to establish CAPA procedures paired with, for example,
uninvestigated quality data sources, such as uninvestigated complaints and
unreported MDRs.
ii. Failure to establish design procedures, or design change procedures for
devices where significant changes have been made that were not reviewed
appropriately.
iii. Inadequate or undocumented process validation for process(es) where
results cannot be fully verified that have either resulted in or are likely to
result in nonconforming product.
(b) Overall inspectional findings reveal a pattern of serious deviations from the
regulation, or other factors that have or may result in a significant public health
risk. Particular attention should be paid to the relationships of requirements. The
following list only provides examples and is not all inclusive:
i. Distribution of nonconforming product(s) that may have caused injury or
death without effective mitigation, and/or adequate corrective actions by
the firm.
ii. Failure to investigate, evaluate, and act upon quality data sources that have
demonstrated an adverse impact(s) to the finished product and/or to patient
safety.
iii. Failure to adequately analyze quality data sources, and/or failure to utilize
current risk information that results in a decision not to proceed with
formal investigations and/or corrective actions, leading to unmitigated
adverse health consequences or nonconformances. This may result from
underestimated or inadequate risk analysis, or outdated risk information
used to make decisions (for example, on risk analysis documents and
appropriate corrections and/or removal decisions).
iv. A deficiency in one or more element(s) of the subsystems. For example,
deficiencies in both purchasing controls and acceptance activities can
indicate a major deficiency, because adequate control of components and
suppliers depends on both activities. If there are problems with one or
both subsystems product quality may be diminished.
v. Failure to correct, or inadequate correction of, significant deficiencies
from previous inspection(s). Observation of same or similar significant
deficiencies from previous inspection(s).
NOTE: If a firm fails to evaluate changes for new clearance (510(k)) or approval
(PMA) in instances when such changes appear to warrant a new clearance or
approval, compliance branch will consult with CDRH (or CBER if CBER
regulated medical device).

PROGRAM
7382.845
Page 40 of 59
(2) Situation 2
Overall inspectional findings reveal a pattern of less significant deviations that may
have minimal or no public health impact. These findings will typically be classified
Voluntary Action Indicated (VAI).
(a) The inspection documents QS deficiencies of a quantity and/or type to conclude
that there is low probability, considering the relationship between quality system
deficiencies observed and the device and manufacturing processes involved, that
the establishment will produce nonconforming and/or defective finished devices.
The Form FDA-483, Inspectional Observations, will serve to inform the
establishment of any objectionable findings.
(b) Definitions of classifications NAI, VAI and OAI are found in Field Management
Directives, FMD-86 Establishment Inspection Report Conclusions and
Decisions, and described below in section B. When Investigations Branch (IB)
determines an inspection classification of NAI or VAI, they will typically finalize
the classification of an inspection. When IB has determined that significant
objectionable conditions are found, they will usually make an initial classification
of OAI and send the corresponding case package for review by Compliance. See
the Classification section below for more information.
B. Factors Considered by Compliance
Compliance Branch will review a case sent from Investigations Branch along with the
below factors to evaluate the overall evidence and make a regulatory decision. Factors to
consider include, but are not limited to:
(1) Quality Data
Quality data is related to the quality and/or performance of the device(s) as observed
during the current inspection, or otherwise available to FDA. The quality data could
include significant quality issues, or signals with the product or across the devices
manufactured, as observed by FDA or the firm. Examples could include CAPA data
sources, recalls, post-approval study data, MDR data, MDSAP audit results, RRA
results, or the firm’s analysis reports that give insight into the product’s performance.
NOTE: If covering a combination product, please ensure all necessary ORA
counterparts and Centers are included on review and case decisions.
(2) Firm’s Inspection & Compliance History
A firm’s inspection history may include aspects of their previous compliance history,
such as the severity of repetitive issues, correction of deficiencies in a timely manner,
and the firm’s timely communications to provide evidence of their corrective actions.

PROGRAM
7382.845
Page 41 of 59
Inspection history may also include examples of whether or not a firm has
demonstrated sustained quality system control.
(3) Firm’s Commitments
The firm’s responsiveness to address regulatory deficiencies include their willingness
and ability to make corrective actions, and/or put effective mitigations in place to
decrease the risk to public health. Responsiveness encompasses firm management’s
oral and written communications during and after the inspection. The firm
demonstrates commitment through their ability to identify and execute a thorough
corrective action plan, including commitment from senior leadership, and allocation
of adequate resources to ensure the firm’s quality system is operating in a state of
control. For example, evidence is submitted to:
• Provide an adequate corrective action plan with appropriate depth and an
adequate timeline for corrective actions and/or mitigations.
• Consider retrospective reviews or other mechanisms to identify the impact of
deficiencies across the quality system.
• Provide evidence in updates that demonstrates timely progress towards
corrective action plan, and any changes or delays to the plan are
communicated and indicate continued commitment to quality.
• Consider preventive measures that demonstrate a state of control, such as the
implementation of consistent processes to identify and correct issues.
• Demonstrate that communications received from the firm are timely,
complete, and transparent.
2. CLASSIFICATION
Inspection classifications are based on the public health significance of the deficiencies
observed during the inspection and the firm’s response, including corrective actions.
Classifications are as follows:
• No Action Indicated – NAI. No objectionable conditions or practices were found
during the inspection (or the significance of the documented objectionable conditions
found does not justify further action).
• Voluntary Action Indicated - VAI. Objectional conditions were found and
documented but the Division and/or Center is not prepared to take or recommend any
of the following regulatory actions (warning letter, administrative, or judicial) since
the objectionable conditions do not meet the threshold for these actions.
• Official Action Indicated - OAI. Objectionable conditions were found, and
regulatory action should be recommended. This includes inspections resulting in
recalls where the Division has decided conditions warrant regulatory (advisory,

PROGRAM
7382.845
Page 42 of 59
administrative, or judicial) action. An OAI classification is most often made if an
FDA 483, or state equivalent, has been issued and the documented evidence supports
the action recommended, unless the only significant observations are non-reportable.
An OAI classification may also be made when a state contract inspection is
determined to be violative and the Investigations Branch management, in consultation
with the Compliance Branch and/or the State Contract Agency, determines that FDA
follow up or action is required.
The procedures for developing recommendations and determining the need for regulatory
action following an inspection are conducted consistent with the established processes within
and between ORA and CDRH (or CBER if CBER regulated device). (See also PART VII-
CENTER RESPONSIBILITIES and FMD-86 Establishment Inspection Report Conclusions
and Decisions for further guidance.)
3. REGULATORY/ADMINISTRATIVE FOLLOW-UP
All reasonable efforts to achieve voluntary compliance will be considered before initiating
regulatory action, if received in a timely manner after an inspection closes. Corrections and
corrective action proposals and plans, including evidence of corrections implemented, should
be submitted by the firm in writing within 15 business days of the inspection close, detailing
the action(s) taken or to correct the deviations within a specified time frame. Voluntary
correction does not preclude the initiation of advisory, administrative, and/or judicial action.
The decision on the type of action to recommend should be based on the seriousness of the
documented deficiencies, while taking into consideration the most effective way to protect
public health. As listed above in the Regulatory Significance section, Compliance Officers
and CDRH (or CBER, if CBER regulated device) Reviewers take into consideration the
criticality of inspectional findings and additional factors when determining the best course of
action utilizing the RPM. Final classification of inspections should align with FMD-86.
A. Considerations in Determining Regulatory Action
Other considerations may cause the overall level of regulatory significance to increase or
decrease when evaluating the inspectional outcome and proposed action type by the
Compliance Officer or CDRH (or CBER, if CBER regulated device) reviewer while a
case is under consult. These considerations include:
(1) Benefit and risk to public health: Consideration of the benefits and risks related to the
likelihood of nonconforming product release that may cause patient harm or result in
adverse events for a significant and/or at-risk population. ORA Compliance Officers
should consider a consult to CDRH (or CBER if CBER regulated device) to
determine the health hazard(s) for situations in which there are risks associated with
either continued use of, or lack of availability, of the product that would significantly
and adversely affect public health. Additionally, if a potential recall or shortage
situation exists, ORA and CDRH should discuss strategy considerations for products

PROGRAM
7382.845
Page 43 of 59
remaining on the market including, but not limited to, the firm’s plan for: coming into
compliance, risk mitigation, and communication plan. When appropriate, FDA will
use the Factors to Consider Regarding Benefit-Risk in Medical Device Product
Availability, Compliance, and Enforcement Decisions | FDA issued on December 27,
2016, to inform decisions related to product availability, compliance, and
enforcement.
(2) Emergency provisions, allowances, and exceptions resulting from a public health
emergency.
(3) Requirements, exceptions, or priorities resulting from executive orders, across agency
programs, or policies, such as, combination products or small business guidance.
(4) Relevant agency initiatives that impact device quality and safety outcomes.
B. Regulatory Actions
(1) Advisory Action
(a) Options
i. Untitled Letter: Untitled letters are used for violations that may not meet the
threshold of regulatory significance for a Warning Letter, and request
correction of the violations. (See RPM Section 4-2 Untitled Letters).
ii. Warning Letter: The Warning Letter is the Agency’s principal means of
notifying regulated industry of violations and achieving prompt voluntary
correction.
a. Issuance of all Warning Letters should follow RPM Section 4-1. Consult
the Office of Policy, Compliance and Enforcement’s (OPCE) Enforcement
Resources page on ORA’s intranet website for current instructions
regarding obtaining Office of Chief Counsel (OCC) clearance and current
approved Warning Letter templates.
b. See below for policy on recidivist Warning Letters.
(b) ORA (OMDRHO) has DIRECT REFERENCE AUTHORITY for Warning
Letters and Untitled Letters in certain areas described in Chapter 4 of the RPM.
When the investigator identifies that firms have not reported a Correction and
Removal, Warning Letters may be issued once CDRH has reviewed and classified
the recall as Class I or II. Circumstances in which ORA should obtain CDRH
concurrence before FDA issues Warning Letters or Untitled Letters are described
in Chapter 4 of the RPM (4-1-4, All Centers and 4-1-4, section 4, CDRH).
(2) Regulatory Meeting
A Regulatory Meeting is a meeting requested by FDA management at its discretion,
to inform responsible individuals or facilities about how one or more products,
practices, processes, or other activities are determined to be in violation of the law.

PROGRAM
7382.845
Page 44 of 59
Regulatory Meetings can be an effective enforcement tool to obtain prompt voluntary
compliance and have been used successfully in a variety of different situations,
including, but not limited to: (1) in conjunction with another advisory action (e.g.,
untitled or warning letter), (2) as a follow-up to other advisory actions, or (3) to
communicate violations that would not warrant another type of advisory action.
Regulatory meetings provide the benefit of a real time, two-way discussion of the
violations and the appropriate corrective actions. (See RPM Section 10-3).
(3) Administrative Actions
(a) Civil Money Penalty: Section 303(f)(1)(B)(i) of the FD&C Act states that civil
money penalties shall not apply to QS violations “unless such violation
constitutes (I) a significant or knowing departure from such requirements, or (II) a
risk to public health.” Section 303(f)(1)(B)(iii) further stipulates those civil
penalties shall not apply to violations of “section 501(a)(2)(A) which involve one
or more devices which are not defective.” (See RPM Section 5-9-1, item 4).
(b) Administrative Detention: The intent of administrative detention is to protect the
public by preventing distribution or use of violative devices until FDA has had
time to consider the appropriate action to take and, where appropriate, to initiate a
regulatory action. Prior to invoking an administrative detention, for a period of 20
or 30 days, the Division Director should have reason to believe: (1) the device is
misbranded or adulterated; (2) the establishment holding the device is likely to
quickly distribute or otherwise dispose of the device; and (3) detention is
necessary to prevent use of the device by the public until appropriate regulatory
action may be taken by the Agency. (See RPM Section 5-6)
i. Division Directors should consult with the appropriate Office of Health
Technology (OHT) at CDRH (or CBER OCBQ if CBER regulated
device), OPCE/DCE, and the Office of Chief Counsel (OCC) concerning
administrative detention. Concurrence should be given by the OHT in
CDRH, based on a recommendation by both CDRH and OCC staff.
ii. The Division should consider immediately recommending seizure of the
detained devices to assure continued control of the violative device after
the 20/30 days of administrative detention expire.
(c) Citations: A citation should be recommended, if appropriate, as stated in the RPM
Section 5-1.
(d) 518 (e)Recall Authority: If the Division believes that prompt removal of a
violative device from channels of commerce is necessary per Section 518(e) of
the Act, it should proceed in accordance with the requirements of 21 CFR Part
806 and the established recall procedures found in Chapter 7 of the RPM and 21
CFR Part 7 (Enforcement Policy), Subpart C (Recalls). In the event of serious
adverse health consequences or a death, CDRH may order a firm to discontinue
further distribution and advise customers of the problem and may subsequently
order the recall of a device to the user level in accordance with Section 518(e) of

PROGRAM
7382.845
Page 45 of 59
the Act. Refer to Section 7-5-3 in RPM.
(e) 518 (a) Notification Order
(f) 518 (b) Repair, Replacement or Refund Order:
(4) Judicial Actions
(a) Seizure: A seizure is an action that is intended to take quick control over the
violative product and put it under the possession or custody of the court. (See
Section 304 of the Act and RPM Chapter 6-1)
(b) Injunction: If an establishment has a continuing pattern of significant deviations,
despite past warnings, an injunction will usually be the recommended action of
choice. If a serious health hazard exists, the recommendation should include a
request for a temporary restraining order (TRO) to prevent the distribution of
devices that have been manufactured under the violative conditions documented
by the inspection report. (See Section 302 of the Act and RPM Section 6-2.)
(c) Prosecution: The criteria for consideration of prosecution of individuals in
violation of applicable requirements is described in RPM Section 6-5.
(5) Other Communications and Compliance Activities
(a) Withhold PMA approval: Refer to Compliance Program 7383.001.
(b) Compliance actions and communications resulting from Agency evaluation of
Regulatory Audit Reports (RARs) submitted by recognized third-party Auditing
Organizations under the Medical Device Single Audit Program (MDSAP): See
Medical Device Single Audit Program (MDSAP) | FDA for more information or
contact [email protected] with questions.
(c) Engage with the firm in other written or verbal communications as deemed
necessary by the Agency, with or without an inspection, to address potential
safety issues or promote voluntary corrective actions.
C. Violative Compliance Follow-Up Inspections
After issuance of a Warning Letter, the next inspection should be a compliance follow up
inspection to advise manufacturers it is their responsibility to investigate, identify, and
correct systemic problems. Refer to Part III for compliance follow up inspection
coverage. When investigators identify the same or additional conditions that meet the
criteria for OAI classification, the Division should consider subsequent enforcement
actions, such as seizure, injunction, prosecution, or civil penalties. During compliance
follow-up inspections, the investigator should work closely with the Division Compliance
Officer and, where appropriate, CDRH to assure that appropriate coverage is provided,

PROGRAM
7382.845
Page 46 of 59
and that deviations are properly documented.
(1) The Recidivist Policy -- Enforcement Strategy for Establishments with Repeated
Violative Inspections
(a) Some establishments have a high rate of recidivism. They have repeated
occurrences of correcting violative conditions in response to a Warning Letter or
other advisory or administrative action, and usually maintain those corrections
long enough to result in a follow-up inspection with no subsequent compliance
action. When FDA next inspects the establishment (sometimes, as a follow-up to
a recall), the investigator identifies similar conditions that again meet the criteria
of OAI classification. This tendency toward recidivism is often due to the failure
of the establishment to have an effectively established quality management
system implemented.
(b) When dealing with another violative inspection for such an establishment, the
Division should consider using the following strategy:
i. Issue a Warning Letter that follows the Recidivist Warning Letter
approved template found on Office of Policy, Compliance and
Enforcement’s (OPCE) Enforcement Resources page on ORA’s intranet
website. This Recidivist Warning Letter requests the manufacturer to
submit to the Division (for up to two years if the Division believes that it
is necessary) an annual certification by an outside expert consultant stating
that it has conducted a complete audit of the establishment's quality
management system relative to the requirements of the Quality System
regulation. The manufacturer should submit a copy of the consultant's
report
7
and certification by the establishment's CEO stating that they
personally have received and reviewed the consultant's report and that the
establishment has made, or taken, all corrections and corrective actions
identified in the report. To keep the process on track, schedules,
milestones, update reports and other similar activities should be
established between the firm and FDA, or by the firm after issuance of the
Recidivist Warning Letter.
ii. Compliance Officers have the option of limiting the review of the
certification only to the extent necessary to confirm that the consultant and
the establishment have met the requirements set forth in the Recidivist
Warning Letter. Compliance Officers may also request a technical
evaluation of the consultant's report by the appropriate Division within the
OHT at CDRH (or CBER OCBQ if CBER regulated device). Compliance
Officers have no obligations, however, to send to the establishment
comments regarding the adequacy of the consultant's report, or the
establishment's corrections.
7
Establishments may be asked to release consultant’s reports as part of their voluntary agreement with FDA.
Because of its voluntary nature, the request is not in conflict with 21 CFR 820.180(c).

PROGRAM
7382.845
Page 47 of 59
iii. Follow-up inspections will normally be conducted after the establishment
certifies that it has completed all corrections and corrective actions to the
Warning Letter violations.
iv. If the follow-up inspection indicates that the corrections and corrective
actions are satisfactory, the Division should notify the establishment that it
has no objections. The ORA Division should then update the profile data
appropriately. The Division should also remind the establishment that it
should continue to submit to the Division, in accordance with the schedule
specified in the Recidivist Warning Letter, certification by an outside
expert consultant. This certification should state it has conducted an
updated audit; has certification by the establishment's CEO that any
corrections and corrective actions noted to be necessary by the consultant
have been made; and remains in compliance with the requirements of the
Quality System regulation. The establishment should continue to submit
copies of the audit results.
(c) If conditions identified by the immediate follow-up inspection or subsequent
inspections meet the criteria of OAI classification, the Division should consider
administrative or judicial action.
(d) If the evidence indicates that the consultant's or establishment's certifications are
fraudulent, the Division is encouraged to advise and seek assistance from the
Office of Criminal Investigations. When there is clear evidence that the
establishment falsified its status report to the Division, the Division should initiate
appropriate action under 18 USC 1001.
D. Facilitating Review of Regulatory Recommendations for Judicial Actions
(1) Division responsibilities are listed in Section 6-1B of the RPM. It is recommended
that ORA work with CDRH (or CBER if CBER regulated device) “up front” (during
the inspection) to review inspectional findings and discuss potential observations, so
that both sets of staff are fully aware of the situation. At the discretion of the
Division, notification to CDRH (or CBER if CBER regulated device) may occur prior
to an inspection, while the inspection is ongoing, or after issuance of the Form FDA-
483.
(2) Center responsibilities for judicial actions are found in Section 6-2-9B of the RPM. It
is recommended that CDRH (or CBER if CBER regulated device) reviewers discuss
the case with ORA, prior to the Preliminary Assessment Call (PAC,) if contacted in
advance.

PROGRAM
7382.845
Page 48 of 59
(3) OPCE Division of Compliance and Enforcement (DCE) responsibilities for judicial
actions are found in Section 6-2-9C of the RPM. ORA compliance may also provide
“up front” information to OPOP/OPCE/DCE prior to the PAC.
E. Deciding Responsibility When Taking Regulatory Action - Contract Sterilizers,
Contract Device Manufacturers, and Finished Device Manufacturers
(1) The following is provided as instruction for deciding which party is to be held
responsible when a finished device manufacturer uses a contract sterilizer to perform
terminal sterilization on its devices or uses a contract device manufacturer.
(a) Contract sterilization and contract manufacturing are considered an extension of
the finished device manufacturer's process. The finished device manufacturer is
ultimately responsible for assuring that validations, operations, process controls,
quality assurance checks, etc., are appropriate, adequately documented, and
correctly performed.
(b) Contract sterilizers and contract manufacturers of finished devices are considered
manufacturers for the purpose of applying the Quality System regulation in that
they meet the definitions as described in 21 CFR § 820.3(l) (“finished device”)
and 21 CFR § 820.3(o) (“manufacturer”). Contract sterilizers and contract
manufacturers of finished devices are subject to those parts of the Quality System
regulation that apply to the operations that are performed.
(c) The finished device manufacturer bears overall responsibility for the safety and
effectiveness of the finished device and must control all contractors under
purchasing and receiving, in-process, and finished device acceptance. However, a
contract sterilizer/contract manufacturer of finished devices and the finished
device manufacturer are all legally responsible for compliance with the Quality
System regulation and for assuring the safety and effectiveness of the finished
device.
(d) Contract manufacturers, including contract testing or contract laboratories, that
are not manufacturing a device, meeting the definition of a finished device in 21
CFR § 820.3(l), are not required to meet the Quality System regulation. These
contractors, even though they may meet the definition of a “manufacturer,” are
controlled by the finished device manufacturer under purchasing and receiving,
in-process, and finished device acceptance.

PROGRAM
7382.845
Page 49 of 59
(e) For contract sterilization, the written agreement between the manufacturer and
contract sterilizer required by 21 CFR 801.150(e), may be referenced to determine
how the parties have defined their respective responsibilities. For other contract
manufacturers, any written agreements used as part of supplier controls under
Purchasing controls, may be referenced to determine how the parties have defined
their activities and respective responsibilities.
(1) When deviations are observed, proposed regulatory actions should reflect and identify
the shared responsibilities between the contractor and finished device manufacturer.
In some situations, it may be appropriate to initiate regulatory action against both the
contractor and the device manufacturer.
(a) Appropriate action should be considered against the contract sterilizer or contract
manufacturer of finished devices in areas for which either party has the prime
responsibility under any written agreement. It may be necessary to inspect more
than one customer to develop supporting documentation to demonstrate the
contractor does not appear to have adequate controls.
(b) When an inspection of a contractor finds violations in areas that are the
responsibility of the finished device manufacturer (such as validation, biological
indicators, package seal testing, etc.), these deviations are reported to the FDA
home Program Division of the finished device manufacturer. Regulatory action
consistent with the action of choice for the contractor should be considered for the
finished device manufacturer.
(c) Because the finished device manufacturer is ultimately responsible for the safety
and effectiveness of the device and, therefore, the contractor's activities, serious
deficiencies found at a contractor’s establishment should lead to consideration of
regulatory action against the finished device manufacturer. Copies of
appropriately redacted Warning Letters issued to a contract sterilizer or contract
manufacturer of finished devices should be sent to the finished device
manufacturer. A copy should also be sent to the FDA home Program Division of
the finished device manufacturer. These documents should be used as a basis for
the next scheduled inspection of the finished device manufacturer.
(d) When a possible health hazard situation exists due to the contractor’s operation,
or an administrative or legal action is contemplated against a contract sterilizer or
contract manufacturer of finished devices, the FDA home Program Division(s) of
all finished device manufacturers utilizing that contractor should schedule an
immediate follow-up inspection(s) at all affected device manufacturers.

PROGRAM
7382.845
Page 50 of 59
F. Violative Devices Sold to Government Agencies
It is agency policy to treat devices sold to the federal government in the same manner as
devices sold to commercial accounts. Consequently, when FDA recommends against
acceptance of a device by a government agency because that device, or its manufacturer,
is in violation of the FD&C Act, FDA should also recommend appropriate
regulatory/administrative action against the same or similar device sold to commercial
accounts.
If an establishment has shipped a violative device to a government agency, appropriate
regulatory action consistent with the nature of the violation(s) may be taken even though
there have been no shipments to commercial customers. Formal regulatory action in
connection with a violative shipment may not be necessary in some cases. (For example,
the establishment promptly corrects the violative condition, and the Agency would not
require further action if the matter involved a device shipped to a non-government
customer.) However, where corrections are not made, or cannot be made, promptly, the
main concern is preventing the subsequent shipment of the device to another customer.
When the device has been shipped solely to a government agency and is under control of
that agency and there is no threat to the public, the Division Compliance Officer will
notify ORA’s Division of Compliance Information and Quality Assurance (DCIQA) staff
and the ORA DCIQA should ascertain the intention of the agency holding the goods (for
example, determining whether the agency will return or destroy the goods; whether it will
request FDA to initiate seizure, etc.). If the procuring agency requests FDA action, ORA
DCIQA staff will refer the matter to the responsible Division Compliance Branch for its
consideration of an appropriate recommendation. Questions on this subject can be
directed to gw[email protected].
Refer to RPM 4-1-2 for “Warning Letters to Government Agencies” for additional
information.
4. MDR REGULATORY/ADMINISTRATIVE FOLLOW UP
The Division should consider an action (see above list) when any of the following 21
CFR Part 803 MDR violations were found during the inspection along with other
deficiencies. This list only provides examples and is not all-inclusive:
• Firm fails to report, within five workdays, after becoming aware that a reportable
MDR-event necessitates remedial action to prevent an unreasonable risk of
substantial harm to the public health.
• Firm fails to submit an MDR death report.
• Firm fails to submit multiple MDR serious injury reports.
• Firm fails to submit multiple MDR malfunction reports.

PROGRAM
7382.845
Page 51 of 59
• Firm fails to develop, maintain, and implement written MDR procedures.
All failures to comply with MDR should be listed on the FDA-483. See Questions and
Answers about eMDR - Electronic Medical Device Reporting - Guidance for Industry,
User Facilities and FDA Staff | FDA for more information.
IMPORTANT NOTE: Warning Letters based on failure to report malfunctions should
have CDRH (or CBER if CBER regulated device) review/concurrence, per instructions in
Chapter 4 of the RPM.
5. TRACKING REGULATORY/ADMINISTRATIVE FOLLOW-UP
The Division should consider an action (see above list) when any of the following 21
CFR Part 821 tracking violations were found during the inspection along with other
deficiencies. This list only provides examples and is not all-inclusive:
• Firm distributes tracked device and does not have a tracking system.
• Firm does not have written standard operating procedures for collection,
maintenance, and auditing of the data for its tracked device(s).
• Firm's tracking system is ineffective in locating tracked devices during
recall/notification.
• Firm does not perform audits of its tracking system.
All failures to comply with the tracking regulation should be listed on the FDA-483. See
Medical Device Tracking | FDA for further information.
IMPORTANT NOTE: CDRH concurrence is required for a Warning Letter for any
violation of device tracking regulation requirements other than failure of the firm to
implement any form of tracking system per the instructions in Chapter 4 of the RPM.
6. CORRECTIONS AND REMOVALS REGULATORY/ADMINISTRATIVE
FOLLOW-UP
The Division should consider an action (see above list) for Corrections and Removals
regulation violation(s) found during an inspection along with other deficiencies, while
also considering the firm’s response to the 21 CFR Part 806 deficiencies. This is only an
example and is not all-inclusive:

PROGRAM
7382.845
Page 52 of 59
• Firm fails to submit a Corrections and Removals report to the Division after its
initiation of a medical device correction or removal action which meets the
definitions of a Class I or II recall situation, along with other deficiencies.
When the firm has already received a Warning Letter for Corrections and Removals
violations and still fails to comply with the Corrections and Removals regulation along
with other deficiencies, then the Division should consider recommending an
administrative or judicial action.
All failures to comply with the Corrections and Removals regulation should be listed on
the FDA-483 once the investigator has confirmed with their Division Recall Coordinator
whether the situation would likely be classified as a Class I or II recall.
7. REGISTRATION AND LISTING REGULATORY/ADMINISTRATIVE
FOLLOW-UP
Chapter 4 of the RPM states agency policy is that Warning Letters should only be issued
for violations of regulatory significance. Generally, 21 CFR Part 807 registration and
listing violations, as a sole finding, should not be the basis of a Warning Letter.
However, when those violations are found in combination with other deficiencies, they
should be included on the Warning Letter, after CDRH (or CBER if CBER regulated
device) concurrence.
8. UNIQUE DEVICE IDENTIFIER (UDI) REGULATORY/ADMINISTRATIVE
FOLLOW-UP
The Division should consider an action (see above list) when any of the following UDI
violation(s) were disclosed during the inspection. This list only provides examples and is
not all-inclusive:
• Firm fails to ensure that the label of every medical device required to bear a UDI,
pursuant to 21 CFR 801.20, contains a UDI.
• Firm fails to provide required information to Global Unique Device Identification
Database (GUDID) pursuant to 21 CFR 830.300.
When the firm has already received a Warning Letter for UDI violations and still fails to
comply with the UDI regulation, then the Division should consider recommending an
administrative or judicial action.
IMPORTANT NOTE: CDRH (or CBER if CBER regulated device) concurrence is
required for a Warning Letter for any violation of device UDI regulatory requirements.

PROGRAM
7382.845
Page 53 of 59
See UDI Rule and Guidance’s, Training, Resources, and Dockets | FDA for more
information.
9. RADIATION EMITTING DEVICE REGULATORY/ADMINISTRATIVE
FOLLOW-UP
Refer to Part V in Compliance Programs found at Center for Devices and Radiological
Health (CDRH) Compliance Programs for instruction on regulatory actions related to
radiation emitting devices.
10. IMPORTS REGULATORY/ADMINISTRATIVE FOLLOW-UP
Foreign establishment inspection reports are provided to CDRH (or CBER if CBER
regulated device) from the Office of Medical Products and Tobacco Operations
(OMPTO). CDRH (or CBER if CBER regulated device) reviews the establishment
inspection findings. When review of inspectional findings reveals conditions or practices
warranting Detention Without Physical Examination (DWPE), CDRH (or CBER if
CBER regulated device) should submit a recommendation to DIO Import Compliance
Branch that the articles offered for import from such firm be subject to DWPE.
Recommendations for addition to DWPE based on establishment inspections follow the
instruction in Regulatory Procedures Manual (RPM), Chapter 9-8-12,
“RECOMMENDATIONS BASED ON ESTABLISHMENT INSPECTION”. Violations
may include deviations from Good Manufacturing Practices (GMPs), insanitary
conditions, or other practices that may cause articles to be misbranded, adulterated, or
otherwise in violation of the FD&C Act per Section 801(a). Import Alert 89-04 (IA 89-
04) "Detention Without Physical Examination of Devices from Firms that Have not met
Device Quality System Requirements". Additionally, if a foreign establishment or
foreign government refuses a foreign inspection, the firm and its products may be subject
to DWPE and could be added to Import Alert 89-16 (IA 89-16),“Detention Without
Physical Examination of Products from Medical Device Firms Refusing FDA Foreign
Establishment Inspection”.
To remove a firm's product from an import alert, information should be provided to the
Agency to adequately demonstrate that the firm has resolved the condition that gave rise
to the appearance of the violation. Firms should provide adequate documentation
(evidence) to the Agency to establish a higher level of confidence to ensure future entries
will be in compliance with the Federal Food Drug and Cosmetic Act (FD&C Act).
General requirements for removal from DWPE can be found in the FDA’s Regulatory
Procedure Manual, Chapter 9, Subchapter: Detention Without Physical Examination
(DWPE). Additionally, the “Guidance” section within an import alert may provide
specific requirements and contact information.
For more information visit: Removal from Import Alert | FDA.

PROGRAM
7382.845
Page 54 of 59
Visit Import for Export Overview | FDA for an overview of the import for export (IFE)
provisions of the FD&C Act section 802(d)(3). IFE allows for the importation of a
product that is unapproved or otherwise does not comply with FDA laws and regulations
if it is coming into the U.S. for further processing and ultimately exported out of the U.S.
11. EXPORTS REGULATORY/ADMINISTRATIVE FOLLOW-UP
When violations meet the criteria for OAI classification for those unapproved devices
exported under Section 802 of the FD&C Act, note that fact in the Warning Letter.
Submit a copy of the Warning Letter to CDRH, Office of Product Evaluation and Quality
(OPEQ), Office of Regulatory Policy (ORP), Exports Team (or CBER OCBQ for CBER
regulated devices) with a recommendation to rescind all current or unexpired certificates
of export.

PROGRAM
7382.845
Page 55 of 59
PART VI - REFERENCES AND PROGRAM CONTACTS
1. APPLICABLE REFERENCES
Copies of CDRH QS publications and other FDA-guidance documents are available from the
Division of Industry and Consumer Education (DICE), Telephone: 800-638-2041 or email at:
[email protected].gov.
Many of these publications are also available in the CDRH Good
Guidance Practices (GGP) Database.
A. APPLICABLE REFERENCES – SPECIFIC TO STERILIZATION
The following sources may be referenced for further guidance regarding sterilization
processes.
(1) Food and Drug Administration
(a) Submission and Review of Sterility Information in Premarket Notification
(510(k)) Submissions for Devices Labeled as Sterile
https://www.fda.gov/media/74445/download.
(Supersedes “Updated 510(k) Sterility Review Guidance K90-1” issued on
August 30, 2002)
(b) A searchable database of FDA-recognized standards is available at:
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfstandards/search.cfm.
A list of FDA-recognized standards related to sterilization of medical devices
can be obtained by searching on the category “Sterility.”
(2) United States Pharmacopeia (USP)/National Formulary (NF), current edition:
http://www.usp.orghttp://www.uspnf.com (USP/NF Online)
<61> Microbial Limit Tests
<71> Sterility Tests
<85> Bacterial Endotoxins Test (LAL)
<151> Pyrogen Test (USP Rabbit Test)
<161> Transfusion and Infusion Assemblies and Similar Medical Devices
<1211> Sterilization and Sterility Assurance of Compendial Articles
2. PROGRAM CONTACTS
A. ORA Contacts
(1) For questions regarding inspectional requirements and/or technical assistance,
refer to the following:

PROGRAM
7382.845
Page 56 of 59
Office of Medical Devices and Radiological Health Operations (OMDRHO)
Operations Staff by email: ORA[email protected].
(2) For questions regarding inspectional requirements for CBER regulated devices
and/or technical assistance, refer to the following:
Office of Biological Product Operations (OBPO) Operations Staff by
email: [email protected].
(3) For questions regarding database helpdesk resources, refer to the following:
Contact ERIC Helpdesk
(301) 827-ERIC (3742)
(4) For questions regarding sampling of devices and laboratory capabilities, refer to
the following:
ORA Office of Regulatory Science
(5) For questions regarding WEAC contacts for testing medical devices, refer to the
following:
Engineering Branch Chief
Telephone: (781) 756-9857
Analytical Branch Chief
Telephone: (781) 756-9707
(6) ORA ORS MPT WEAC Sample Communications refer to the following:
Telephone: (781) 756-9701
B. CDRH Contacts
Refer to the Office of Product Evaluation and Quality (OPEQ) Organizational Chart to
identify the office that is responsible for the type of device for which you have a

PROGRAM
7382.845
Page 57 of 59
question or need guidance, including the interpretation and applicability of the device
QS regulation and GMP exemptions.
Refer to the CDRH “Who’s the Lead” list of internal contacts for staff to use when
trying to get assistance on a specific topic.
(1) For questions regarding MDR Regulation Interpretation and Policy Questions
refer to the following:
MDR Team
Division of Regulatory Programs 3
Office of Regulatory Programs
(2) For questions regarding sampling and/or testing of general medical devices refer
to the following:
Office of Science and Engineering Laboratories (OSEL)
(3) Recommendations are electronically submitted to CDRH. Questions can be
directed to:
FDA Regulatory Inspections and Audits Team Inspections Contact:
Medical Device Single Audit Program Contact: [email protected]
(4) For questions regarding Medical Device Tracking refer to the following:
PMA, HDE, Presubmission & Device Tracking Lifecycle Team
Division of Regulatory Programs 1
Office of Regulatory Programs
Stacy Monza
(5) For questions regarding the reprocessing of single-use devices refer to the
following:
OPEQ Compliance-Quality Program
(6) For questions regarding compliance of medical device software, quality system
software, or production/manufacturing equipment software refer to the
following:
OPEQ Compliance-Quality Program

PROGRAM
7382.845
Page 58 of 59
C. CBER Contacts
For CBER regulated devices questions and guidance refer to CBER’s Office of
Compliance and Biologics Quality .
D. Questions regarding COMSTAT (Compliance Status Information System):
Email: [email protected]
Contact: GWQAP Team Leader in ORA OPCE
3. FDA WEB SITES
A. FDA
Home Page: http://www.fda.gov
B. ORA
Home Page: https://www.fda.gov/about-fda/fda-organization/office-regulatory-
affairs
C. ORA Office of Medical Device and Radiological Health (OMDRHO)
Home Page: https://www.fda.gov/about-fda/ora-program-areas/office-medical-
device-and-radiological-health-operations-omdrho
D. CDRH
Home Page: https://www.fda.gov/medical-devices
E. Registration and Listing
Who Must Register, List and Pay the Fee | FDA
F. Medical Device Reporting (MDR)
https://www.fda.gov/regulatory-information/search-fda-guidance-
documents/medical-device-reporting-manufacturers
G. MedWatch
http://www.fda.gov/medwatch
https://www.fda.gov/media/133177/download (Instructions for completing
MedWatch Form 3500A)

PROGRAM
7382.845
Page 59 of 59
H. Medical Device Tracking
https://www.fda.gov/medical-devices/postmarket-requirements-devices/medical-
device-tracking
I. Recalls, Corrections and Removals
https://www.fda.gov/medical-devices/postmarket-requirements-devices/recalls-
corrections-and-removals-devices
J. QSIT Guide
http://www.fda.gov/downloads/ICECI/Inspections/UCM142981.pdf
K. FDA Recognized Standards
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfstandards/search.cfm
NOTE: A list of FDA-recognized standards related to sterilization of medical devices
can be obtained by searching on the above FDA Recognized Standards link under the
category “Sterility.”
