
PATIENT-FOCUSED DRUG DEVELOPMENT
GUIDANCE PUBLIC WORKSHOP
Incorporating Clinical Outcome
Assessments into Endpoints for
Regulatory Decision-Making
Workshop Date: December 6, 2019
Discussion Document for Patient-Focused Drug Development Public Workshop on Guidance 4:
INCORPORATING CLINICAL OUTCOME ASSESSMENTS INTO ENDPOINTS FOR
REGULATORY DECISION-MAKING

TABLE OF CONTENTS
I. INTRODUCTION............................................................................................................. 1
A. Guidance Series ................................................................................................................. 1
B. Document Summary ......................................................................................................... 2
II. ESTIMAND FRAMEWORK OVERVIEW .................................................................. 3
A. COA Research Objective: Foundation for Your Work ................................................ 4
B. Target Study Population: In Whom Are You Going to Do the Research and Which
Subject Records Are in the Analysis? ............................................................................. 5
C. Endpoint of Interest: What Are You Testing or Measuring in the Target Study
Population? ........................................................................................................................ 6
1. Endpoint Definition(s) ............................................................................................... 6
2. Pooling Different Tools and/or Different Concepts to Construct the Endpoint ........ 6
Correlation of Subcomponents and Effect on Power and Type 1 Error ................ 6
Multidomain Responder Index .............................................................................. 7
Personalized Endpoints ......................................................................................... 8
Pooling Scores Across Reporters .......................................................................... 8
Pooling Across Delivery Modes (Same Tool, Same Reporter) ............................. 8
3. Timing of Assessments ............................................................................................... 9
4. Defining Improvement and Worsening .................................................................... 10
5. Clinical Trial Duration and COA-Based Endpoints ................................................ 10
D. Intercurrent Events: What Can Affect Your Measurement’s Interpretation? ........ 11
1. Use of Assistive Devices, Concomitant Medications, and Other Therapies ............ 11
2. Impact of Disease/Condition Progression, Treatment, and Potential Intercurrent
Events ....................................................................................................................... 12
3. Practice Effects ........................................................................................................ 12
4. Participant Burden................................................................................................... 14
5. Mode of Administration ........................................................................................... 15
6. Missing Data and Event-Driven COA Reporting .................................................... 15
7. Missing Scale-Level Data ........................................................................................ 15
E. Population-Level Summary: What Is the Final Way All Data Are Summarized and
Analyzed?......................................................................................................................... 16
1. Landmark Analysis................................................................................................... 16
2. Analyzing Ordinal Data ........................................................................................... 16
3. Time-to-Event Analysis ............................................................................................ 16
4. Responder Analyses and Percent Change From Baseline ....................................... 17
III. MEANINGFUL WITHIN-PATIENT CHANGE ........................................................ 18
A. Anchor-Based Methods to Establish Meaningful Within-Patient Change ................ 19
B. Using Empirical Cumulative Distribution Function and Probability Density
Function Curves to Supplement Anchor-Based Methods ........................................... 20
C. Other Methods ................................................................................................................ 22
1. Potentially Useful Emerging Methods ..................................................................... 22
2. Distribution-Based Methods .................................................................................... 22
3. Receiver Operator Characteristic Curve Analysis .................................................. 22
D. Applying Within-Patient Change to Clinical Trial Data............................................. 22
IV. ADDITIONAL CONSIDERATIONS ........................................................................... 25
A. Other Study Design Considerations .............................................................................. 26
B. Formatting and Submission Considerations ................................................................ 27
APPENDIX 1: CASE STUDY OF ESTIMAND FRAMEWORK ......................................... 29
A. Example Research Objective ......................................................................................... 29
1. Define COA Scientific Research Question A Priori ................................................ 30
2. Define Target Study Population Based on the Research Question A Priori ........... 30
3. Define Endpoint of Interest Based on the Research Question A Priori ................... 31
4. Address Intercurrent Events in Alignment with the Research Question .................. 32
5. Define Population-Level Summary Based on Research Question A Priori ............. 33
6. Prespecify Statistical Analysis Plan......................................................................... 34
B. Summary of Decisions Made in This Case Study ........................................................ 35
APPENDIX 2: EXAMPLE FROM GENE THERAPY .......................................................... 36
APPENDIX 3: REFERENCES .................................................................................................. 42
APPENDIX 4: GLOSSARY ...................................................................................................... 44
TABLE OF FIGURES
Figure 1: Attributes of an Estimand Placed in Context .................................................................. 4
Figure 2: Example of Empirical Cumulative Distribution Function Curves of Change in COA
Score from Baseline to Primary Time Point by Change in PGIS Score ....................................... 21
Figure 3: Example of Density Function Curves of Change in COA Score from Baseline to
Primary Time Point by Change in PGIS Score ............................................................................. 21
Figure 4: An eCDF Curve by Treatment Arm Showing Consistent Separation Between Two
Treatment Arms ............................................................................................................................ 23
Figure 5: An eCDF Curve Where Treatment Effect Is Not in Range Considered Clinically
Meaningful by Patients ................................................................................................................. 24
Figure 6: MLMT Scores in Phase 3 Trial ..................................................................................... 40
TABLE OF TABLES
Table 1: Considerations of Defining a COA Target Study Population .......................................... 5
Table 2: Considerations When Defining a COA-Based Endpoint ................................................ 31
Table 3: Considerations When Addressing Intercurrent Events ................................................... 33
Table 4: Considerations When Defining a COA Population-Level Summary ............................. 33
Table 5: Summary of Estimand Decisions Made ......................................................................... 35
Table 6: MLMT Illuminance Level, Score Code, and Real-World Examples ............................. 37

1
I. INTRODUCTION 1
A. Guidance Series 2
The Food and Drug Administration (FDA) recognizes the need to obtain meaningful patient 3
experience data
1
to understand patients’ experience with their disease and its treatment. This can 4
help inform development of endpoint measures to assess clinical outcomes of importance to 5
patients and caregivers in medical product development. To ensure a patient-focused approach 6
to medical product
2
development and regulation, FDA is developing guidance on methods to 7
identify what matters most to patients for measurement in clinical trials; specifically, how to 8
design and implement studies to capture the patient’s voice in a robust manner. FDA created this 9
Discussion Document to facilitate discussions at the December 6, 2019, public meeting that will 10
inform FDA’s development of a patient-focused drug development (PFDD)
3
guidance on 11
incorporating clinical outcome assessments (COAs) into endpoints for regulatory decision-12
making. 13
14
This public workshop will inform the development of the fourth in a series of four 15
methodological PFDD guidance documents
4
that FDA is developing to describe in a stepwise 16
manner how stakeholders (patients, researchers, medical product developers, and others) can 17
collect and submit patient experience data and other relevant information from patients and 18
caregivers for medical product development and regulatory decision-making. The topics that 19
each guidance document will address are: 20
21
• Methods to collect patient experience data that are accurate and representative of the 22
intended patient population (Guidance 1)
5
23
1
The Glossary defines many of the terms used in this Discussion Document. Words or phrases found in the Glossary
appear in bold italics at first mention.
2
A drug, biological product, or medical device.
3
See https://www.fda.gov/Drugs/NewsEvents/ucm607276.htm.
4
The four guidance documents that will be developed correspond to commitments under section I.J.1 associated with
the sixth authorization of the Prescription Drug User Fee Amendments (PDUFA VI) under Title I of FDA
Reauthorization Act of 2017. The projected time frames for public workshops and guidance publication reflect
FDA’s published plan aligning the PDUFA VI commitments with some of the guidance requirements under section
3002 of the 21st
Century Cures Act (available at
https://www.fda.gov/downloads/forindustry/userfees/prescriptiondruguserfee/ucm563618.pdf
).
5
See draft guidance for industry, FDA staff, and other stakeholders Patient-Focused Drug Development: Collecting
Comprehensive and Representative Input (June 2018). When final, this guidance will represent FDA’s current
thinking on this topic. For the most recent version of a guidance, check the FDA guidance web page at
https://www.fda.gov/RegulatoryInformation/Guidances/default.htm
.

2
• Approaches to identify what is most important to patients with respect to their experience 24
as it relates to disease burden and treatment burden (Guidance 2)
6
25
• Approaches to identify and develop methods to measure impacts in clinical trials 26
(Guidance 3) 27
• Methods, standards, and technologies to collect and analyze COA data for regulatory 28
decision-making (Guidance 4) 29
30
All documents in the series encourage stakeholders to obtain feedback from FDA during the 31
study and trial development period when considering collection of patient experience data. FDA 32
encourages engagement of broader disciplines during clinical development (e.g., qualitative 33
researchers, survey methodologists, statisticians, psychometricians, patient preference 34
researchers, data managers) when designing and implementing studies because the logistics in 35
some cases can be daunting for a seemingly simple piece of patient data to address a simple 36
research objective. 37
38
B. Document Summary 39
The purpose of this Discussion Document is to help stakeholders understand what FDA 40
considers when a COA in a clinical study will be used to eventually support medical product 41
regulatory decision-making. 42
43
The document first lays out a framework that aims to align the clinical study objective with the 44
study design, endpoint, and analysis to improve study planning and the interpretation of analyses. 45
Several examples are provided to help illustrate the framework. 46
47
The document then describes methods to aid in the interpretation of study results to evaluate 48
what constitutes a meaningful within-patient change (i.e., improvement and deterioration from 49
the patients’ perspective) in the concepts assessed by COAs. This information is important 50
because statistical significance can be achieved for small differences between comparator 51
groups, but this finding does not indicate whether individual patients have experienced 52
meaningful clinical benefit. 53
54
A list of considerations when developing an endpoint from a COA is included in Section IV of 55
this Discussion Document. 56
57
6
See draft guidance for industry, FDA staff, and other stakeholders Patient-Focused Drug Development: Methods to
Identify What Is Important to Patients https://www.fda.gov/media/131230/download
. When final, this guidance will
represent FDA’s current thinking on this topic.

3
II. ESTIMAND FRAMEWORK OVERVIEW 58
Section Summary
An estimand is a quantity used to define a treatment effect in a clinical study. The estimand framework
aims to align the clinical study objective with the study design, endpoint, and analysis to improve study
planning and the interpretation of analyses. The attributes of an estimand include specifically defining:
• Who is the target population for the study?
• What is the endpoint (e.g., what variables will be used including which time points)?
• How will events precluding observation or affecting interpretation be accounted for in the
analyses, e.g., dropouts, use of rescue medication, not following prescribed regimen?
• What is the population level summary (e.g., comparing means, hazard ratios)?
Decisions for all the attributes, implicitly or explicitly, are currently present in every data analysis that
is performed. The choices made strongly impact interpretation of the analysis, power, and data
collected.
59
Technical Summary: Key Messages in This Section
To develop endpoints from COAs, a fundamental issue must first be addressed: What is the clinical
question or research objective that the clinical study should be designed to answer? The estimand
framework based on International Council on Harmonisation (ICH) E9(R1) aims to improve clinical
studies by putting the focus on a set of attributes to ensure they align with the study research objectives.
This section discusses four attributes:
Target Study Population
• Patients who are targeted by the scientific question; who will be included in the analysis
• A different population may be appropriate for each scientific question
Endpoint of Interest
• Outcome obtained for each patient that will be statistically analyzed to address the scientific
question; this may include data from multiple variables
• Research protocols should define the concept, COA instrument, score/summary score, type of
endpoint, and thresholds/estimates for clinical interpretation
Intercurrent Events
• Events that occur after randomization/treatment initiation/or study start that could preclude
observation of the variable or affect its interpretation
• An example of an intercurrent event: taking subsequent therapy beyond treatment
discontinuation with an endpoint of physical function measured at a time point several
weeks after treatment discontinuation
• For nonrandomized trials or trials borrowing data, intercurrent events could occur at any
time a subject is considered ‘on study’
• Protocols should specify intercurrent events and how they will be accounted for in analyses to
address the scientific question of interest
4
Population-Level Summary
• Basis for comparison between treatment arms, treatment conditions, other groups, or otherwise
summarizes information
• Examples include a) mean physical function score at baseline for everyone in an
observational research study and b) difference compared to control of a new medical
product’s median time to pain resolution
60
The attributes listed above should be clearly defined prior to developing a protocol and included 61
in both the protocol and Statistical Analysis Plan (SAP). They will determine the data collected, 62
procedures, and other sections of the protocol beyond statistical methods. The attributes also 63
drive the SAP and communication of trial results, as highlighted in Figure 1. 64
65
Figure 1: Attributes of an Estimand Placed in Context 66
67
A. COA Research Objective: Foundation for Your Work 68
The essence of clinical research is to ask important questions and answer them with appropriate 69
studies (ICH E8(R1)). The research objective should be clearly and explicitly stated. To develop 70
the objective, both the natural history of the disease and the treatment goal for the intended 71
product must be considered. For example, the choice of an endpoint will likely be very different 72
between a product intended to treat an acute disease, where the symptoms of many patients will 73
likely resolve within several weeks, versus a product intended to be used for patients living with 74
a chronic disease. Even for a chronic disease, endpoint selection could vary depending on 75
whether the disease is degenerative/progressive, relapsing and remitting, episodic, or relatively 76
stable. Heterogeneity of symptoms or functional status of patients with the disease is also a 77
crucial issue. As an example, relating to the intended goal of the treatment, a product intended to 78
cure a disease is likely to have a different research objective from a product designed to decrease 79
the symptom severity of a chronic disease. 80
81
Research
Objective
Estimand
→ T
arget Study Population
→
Endpoint of Interest
→ I
ntercurrent Events
→
Population-Level Summary
Statistical
Analysis Plan
Communication
of Results

5
B. Target Study Population: In Whom Are You Going to Do the Research and Which 82
Subject Records Are in the Analysis? 83
The target study population used to address a COA research objective (the COA ‘analysis 84
population’) may vary based on the COA-derived endpoint and scientific research question. 85
There may be multiple COA analysis populations in a single trial. The COA analysis 86
population(s) should be defined a priori in the protocol and SAP, with clear justification made 87
for each COA analysis population. The choice of COA analysis population will affect the COA-88
related estimand and interpretation of patient experience. 89
90
Table 1 presents COA target study population examples. There may be other target study 91
populations of interest depending on your research objective. 92
93
Table 1: Considerations of Defining a COA Target Study Population 94
Target Study Population (Examples)
• ITT: All patients randomized according to
assigned treatment arm, regardless of
adherence
• Safety: All patients who received at least
one dose of product, regardless of
randomization
• Analysis populations are often defined based on
their availability of COA data
• All patients who are eligible for the COA
• Completed the COA at baseline
• Completed baseline and at least one
postbaseline assessment
• COA data are available at any trial
timepoint
Abbreviations: ITT = intent-to-treat; COA = clinical outcome assessment 95
96
For sponsors considering an effectiveness claim from a COA-derived endpoint in a randomized 97
trial, the intent-to-treat (ITT) population generally should be used to preserve the benefits of 98
randomization. Justification should be provided if treatment comparisons are made using a COA 99
analysis population different from the ITT population. Any justification should incorporate 100
discussion of trial blinding
7
procedures and their potential impact on data interpretation. If the 101
COA objective is safety or tolerability, including patients who received at least one dose of the 102
investigational product, regardless of randomization, may be more appropriate. Consider how 103
interpretation of the COA-derived endpoint changes if all patients in a trial are not eligible for 104
the COA. For example, generalizability of the results may be narrowed if some patients do not 105
have access to a COA because it is unavailable in a language in which they are fluent. 106
Additionally, depending on the trial protocol, eligibility to complete a COA may change over 107
time. 108
109
Every effort should be made to have high completion rates throughout the study. At baseline, this 110
is important otherwise all postbaseline assessments will be difficult to put into context without a 111
reference variable. Because there is the potential for patients to have missing assessments, 112
sponsors should clearly specify in the SAP how missing observations will be dealt with for clear 113
7
Blinding is sometimes referred to as “masking”; for purposes of this document, we will use blinding.

6
interpretation. Removing subjects without a baseline measurement is common but depending on 114
the research question it may not be the better option. 115
116
C. Endpoint of Interest: What Are You Testing or Measuring in the Target Study 117
Population? 118
1. Endpoint Definition(s) 119
An endpoint is a precisely defined variable intended to reflect an outcome of interest that is 120
statistically analyzed to address a specific research question. An endpoint definition typically 121
specifies the type of assessments made, the timing of those assessments, the tools used, and 122
possibly other details, as applicable, such as how multiple assessments within an individual are 123
to be combined. Measurement properties remain crucial in the context of developing useful 124
endpoints, as endpoints (as well as COAs) should be understood as imperfectly measuring 125
concepts. Hence, assessment of an endpoint’s reliability, content validity, construct validity, as 126
well as ability to detect change are important (refer to FDA PFDD G3 Public Workshop 127
Discussion Document for details). Within the protocol, the specific COA concept(s) should be 128
assessed by fit-for-purpose COA(s) and should be incorporated into a corresponding clinical trial 129
objective or hypothesis and reflected in the endpoint definition and positioning in the testing 130
hierarchy. 131
132
A multidomain COA may successfully support claims based on one or a subset of the domains 133
measured if an analysis plan prespecifies (1) the domains that will be targeted for supporting 134
endpoints and (2) the method of analysis that will adjust for the multiplicity of tests for the 135
specific claim. The use of domain subsets to support clinical trial endpoints assumes the COA 136
was adequately developed and validated to measure the subset of domains independently from 137
the other domains. A complex, multidomain claim cannot be substantiated by instruments that 138
do not adequately measure the individual components of the domain. 139
140
2. Pooling Different Tools and/or Different Concepts to Construct the Endpoint 141
Correlation of Subcomponents and Effect on Power and Type 1 Error 142
Since most diseases have more than one relevant clinical outcome, trials can be designed to 143
examine the effect of a medical product on more than one endpoint (i.e., multiple endpoints). For 144
example, a COA with multiple domains may be used in a clinical trial to assess the most relevant 145
and meaningful clinical outcomes (i.e., each domain corresponds to one clinical outcome) to 146
patients. In such a case, a multiple endpoint approach would be of clinical interest, specifically a 147
multicomponent endpoint approach (refer to FDA draft guidance Multiple Endpoints in Clinical 148
Trials (January 2017)). 149
150
Other analytical methods, such as global tests, could potentially be used to pool scores from 151
different tools of a similar type, e.g., patient-reported outcomes (PROs). The use of these 152
methods should be discussed with the FDA. 153
154

7
Some researchers have considered combining different scores from different measurement tools 155
that evaluate different parts of the latent construct to create a new endpoint using item response 156
theory and other methods to take in to account the different potential dimensions of the COAs. 157
FDA is open to discussion of well-defined and reliable endpoints. 158
159
For a COA composed of multiple domains, with each domain measured using either an ordinal 160
or a continuous response scale, a within-patient combination (e.g., sum or average) of all the 161
individual domain (i.e., component) scores to calculate a single overall rating creates a type of 162
multicomponent endpoint. Other types of multicomponent endpoints may include a dichotomous 163
(event) endpoint corresponding to an individual patient achieving prespecified criteria on each 164
individual component. Careful considerations should be made regarding the choice of individual 165
components and whether all components will have reasonably similar clinical importance or 166
whether the algorithm combining the scores uses differential weighting and how those weights 167
are determined. Since multicomponent endpoints are constructed as a single endpoint, no 168
multiplicity adjustment is necessary to control Type I error. In addition, multicomponent 169
endpoints may provide gains in efficiency if different components are not that highly correlated. 170
Regardless of how a multicomponent endpoint is constructed, the choice of the endpoint needs to 171
be clinically relevant and interpretable. 172
173
Multidomain Responder Index 174
For some rare diseases with heterogeneous patient populations and variable disease 175
manifestations, it may be challenging to assess a single concept of interest across all patients. 176
Stakeholders occasionally propose combining multiple measurement concepts (e.g., a variety of 177
individual COA-based endpoints) into a single dichotomous (event) endpoint. While FDA 178
regulations allow for flexibility and judgement in considering multicomponent endpoints, the 179
selection of these endpoints faces similar challenges as those described under responder 180
endpoints—responder analyses (refer to Section II.E.4). In addition, the choice of the individual 181
components relies on the requirement that all components are of reasonably similar clinical 182
importance (Multiple Endpoints in Clinical Trials (January 2017)). An example dichotomous 183
(event) endpoint is the multidomain responder index (MDRI) approach, which thus far has not 184
been demonstrated as a viable approach based on evidence submitted to FDA. 185
186
In general, the MDRI approach combines multiple individual domains or endpoints with a 187
prespecified responder threshold for each endpoint. Various methods have been proposed to 188
construct an MDRI endpoint. For example, each domain score is presented as +1 for 189
improvement, 0 for no change, and -1 for decline, and an overall MDRI response for a patient is 190
defined based on the individual scores (e.g., if any domain shows improvement). It is important 191
to note that successful creation of an MDRI requires clearly defined and clinically relevant 192
endpoints with appropriate responder thresholds (i.e., what constitutes a clinically meaningful 193
within-patient change score) established a priori for those endpoints. In practice, these responder 194
thresholds are often hard to establish, especially for rare diseases with small patient populations 195
and limited natural history data. Additionally, defining an endpoint score as +1, 0, or -1 relies on 196
the assumption that the degree of improvement and deterioration in a concept of interest is 197
symmetric, which often is not a valid assumption. Another important consideration for the MDRI 198

8
approach is the amount of missing data for each domain, component, or individual endpoint of 199
the MDRI. Large amounts of missing data will impede the interpretation of the endpoint results. 200
201
Personalized Endpoints 202
Similar concerns exist with personalized or individualized endpoints, which often are analyzed 203
descriptively as exploratory endpoints. The process to construct a personalized endpoint should 204
be standardized, and the criteria for selecting the outcome assessments should be consistent 205
across sites and patients. The same set of outcome assessments should be assessed for all 206
patients, regardless of their own personalized endpoint, to allow for an assessment of any new or 207
worsening symptoms and/or functional limitation(s) during the trial duration. Certain outcome 208
assessments may not be applicable to all trial patients. However, if an outcome is not assessed in 209
a patient at a given time point, the reason for the assessment not being performed should be 210
noted, included in the analysis data set, and used as part of the analysis. 211
212
Pooling Scores Across Reporters 213
To evaluate the treatment benefit of a medical product, sometimes it may be necessary to use 214
different types of COAs to assess the same construct(s) in the same clinical trial (e.g., in a 215
pediatric trial in which a PRO measure is used for older children who can reliably self-report and 216
an observer-reported outcome (ObsRO) measure is used by caregivers to report signs and 217
behaviors of younger children who are unable to reliably self-report). In general, scores 218
generated by different types of COAs, (i.e., PROs, ObsROs, clinician-reported outcomes 219
(ClinROs), and performance outcomes (PerfOs)), cannot be pooled to form a single clinical trial 220
endpoint, even if they are developed to assess the same construct(s), in analyses submitted to 221
FDA to support a medical product application. Because these different types of COAs are 222
developed for different contexts of use (e.g., PRO measures to report direct experiences of 223
symptoms by the patients themselves and ObsRO measures to report observable signs and 224
behaviors of the patients by their caregivers), they are distinct outcome assessments, and it is 225
therefore inappropriate to pool the resulting sets of scores. 226
227
Scores generated by different types of COAs should be analyzed separately with—where 228
feasible—enough reporters included in each group to support any subsequent inferences to the 229
target population. Simply because each tool has the same score range (e.g., 0 to 10) does not 230
mean data can be pooled. 231
232
Pooling Across Delivery Modes (Same Tool, Same Reporter) 233
Scores generated by the same tool administered (“delivered”) via different modes (e.g., 234
interactive voice response; interview; paper-based; electronic device) may be pooled under very 235
specific and limited conditions. Although scores yielded by different modes are generally 236
considered to be comparable when there is no difference between modes in terms of the wording 237
of item stems and response options, item formats, the appearance and usage of graphics or other 238
visuals, or order of the items (see FDA PFDD G3 Public Workshop Discussion Document for 239
9
further discussion), administering a tool using more than one mode or method per study can 240
introduce noise (i.e., construct-irrelevant variance in COA score) that may not be completely 241
random and may make it more difficult to discern treatment effects. 242
243
3. Timing of Assessments 244
Clinical trials using COAs should include a schedule of COA administration as part of the 245
overall study assessment schedule in the protocol. The timing of assessments plays a vital role in 246
gaining reliable and meaningful information on the concept(s) of interest and should be selected 247
carefully and supported by adequate rationale for the choice of assessment time points. The COA 248
schedule should correspond directly with the natural course of the disease or condition (i.e., 249
acute, chronic, or episodic), research questions to be addressed, trial duration, disease stage of 250
the target patient population, and current treatment of patients, and be administered within the 251
expected time frame for observing changes in the concept(s) of interest. Other important 252
considerations for determining the most appropriate timing of assessments for COA-based 253
endpoints include, but are not limited to, the following: 254
255
• Recall period: A COA should not be administered more frequently than the recall period 256
allows (refer to FDA PFDD G3 Public Workshop Discussion Document Section VI.B.7 257
for in-depth discussion of considerations regarding an instrument’s recall period). For 258
example, an instrument with a 1-week recall period should be administered no more 259
frequently than 1 week (7 days) after the previous administration. If the recall period 260
implies assessment at a specific time of day (e.g., in the morning, at night) or at a specific 261
time relative to treatment (e.g., since last dose) or relative to some other event (e.g., since 262
waking up today, since going to bed last night, since last bowel movement), assessments 263
should be timed accordingly. This issue also arises in wording of COAs administered 264
using ecological momentary assessment. 265
• Anticipated rate of change in the underlying construct to be measured: The timing of 266
assessments should align with the anticipated rate of change in the underlying construct 267
to be measured (but, as mentioned above, should be no more frequent than what the 268
instrument’s recall period allows). For example, if the construct to be measured is 269
expected to change rapidly over the course of the study period, assessments should be 270
placed closer together. If the construct is expected to change slowly, one might place 271
assessments further apart. Note that rate and direction of change in the underlying 272
construct is linked to the rate and direction of change in the underlying disease/condition 273
to be treated (i.e., linked to the pace of improvement or deterioration/progression in the 274
underlying disease), but the two may not move together in lock-step (i.e., they probably 275
would move in the same direction but may move at different rates). 276
• COA administration burden: The length and frequency of COA administration should 277
take into consideration patient burden which may result in patient fatigue and lead to an 278
increase of missing data, as well as impact data quality. 279
• COA administration schedule: The schedule of COA administration should align with 280
the administration of other prespecified endpoints (i.e., primary and secondary) and 281
proposed SAP. 282
10
• Collect COA data at baseline: The COAs should be administered at baseline. If the trial 283
includes a run-in period during which the effect on the COA might be expected to change 284
(e.g., medication washout, patient behavior modification), this should be considered 285
when considering the timing of assessments. Note that some diseases, conditions, or 286
clinical trial designs may necessitate more than one baseline assessment and several COA 287
administrations during treatment. 288
• Align anchor administration time: The timing of anchor scale administration should 289
align with both the recall period and the administration of the corresponding COA (e.g., 290
patient global impression of severity (PGIS) with PRO timing; clinician global 291
impression of severity with ClinRO timing). 292
• Use same COA administration order: The order of COA administration should be 293
standardized to help reduce measurement error. 294
• Timing of treatment administration: If treatment is administered repeatedly over the 295
clinical trial period and change in the target construct(s) is to be assessed repeatedly over 296
the trial period, it may be sensible to measure the construct at the same time relative to 297
treatment administration throughout the trial—unless treatment considerations dictate 298
otherwise. 299
300
4. Defining Improvement and Worsening 301
Clinically relevant within-patient thresholds for improvement and worsening should be 302
predefined and justified. A few suitable supplementary analyses may be conducted to evaluate a 303
range of thresholds when appropriate. See Section III of this Discussion Document for additional 304
information. 305
306
Superiority versus noninferiority or equivalence testing of a COA-based endpoint must be 307
predefined in the SAP. It is inappropriate to conclude “no worsening” when there is a 308
nonsignificant test of superiority (e.g., p > 0.05). Trials with small sample sizes lead to wide 309
confidence intervals of the treatment effect of the COA-based endpoint, which will likely not 310
demonstrate superiority. 311
312
5. Clinical Trial Duration and COA-Based Endpoints 313
Generally, the duration a COA is collected should be the same duration as indicated for other 314
measures of effectiveness or safety in the clinical trial protocol. It is important to consider 315
whether the clinical trial’s duration is of adequate length to assess a durable COA-based endpoint 316
in the disease or condition being studied. Determination of the clinical trial duration should be 317
driven by the disease course as well as treatment and endpoint objectives outlined in the clinical 318
trial protocol. 319
320
11
D. Intercurrent Events: What Can Affect Your Measurement’s Interpretation? 321
Intercurrent events are events that occur after randomization/treatment initiation/or trial start that 322
either preclude observation of the variable (and potentially subsequently the endpoint) of interest 323
or affect its interpretation (e.g., taking rescue medication). While missing data is a part of the 324
definition, it is not the only definition. 325
326
1. Use of Assistive Devices, Concomitant Medications, and Other Therapies 327
It is important to consider what other activities may impact the COA score and endpoint value, 328
such as use of assistive devices (e.g., walkers), concomitant medications including rescue 329
therapies (e.g., bronchodilators or pain medication), and other therapies (e.g., physical therapy). 330
For example: 331
332
• Use of assistive devices may particularly impact PerfO assessment of mobility and can 333
impact other types of COAs 334
• If a specific published administrator’s manual is selected for a performance-based test, it 335
is important to conduct the test in accordance with the selected manual, including the use 336
of standardized assistive devices, if allowed 337
• If study procedures are not aligned with the instrument’s user manual, changes should be 338
detailed in the study documents and training should occur specific to the changes 339
• Case report forms (CRFs) for data collection should include information on whether an 340
assistive device (and what type) was used during the test 341
342
For diseases where patients’ underlying disease status is expected to change during the trial, with 343
corresponding changes in the use and the type of assistive device, it would be informative to 344
incorporate the information on the assistive device into the COA-based endpoint construction, as 345
the change in assistive device may reflect either an improvement or a deterioration in the 346
patient’s disease status. 347
348
Two other examples of intercurrent events: 349
350
• If an item assesses difficulty buying groceries and wording does not account for use of a 351
food delivery service, an intercurrent event could occur. 352
• If a patient in a trial breaks their leg in a car accident, that likely impacts the physical 353
function PRO instrument’s score. 354
355
Use of other supportive therapies that may impact the interpretation of the endpoint should be 356
assessed consistently. Data should be collected and recorded in a standardized manner, and 357
incorporated into the endpoint model and supplementary analyses. A discussion with study 358
coordinators, statisticians, clinicians, and patients will result in a list of likely intercurrent events 359
to include in study planning. 360
361

12
2. Impact of Disease/Condition Progression, Treatment, and Potential Intercurrent Events 362
In the planning stages of a clinical study, it is important to consider how both the 363
disease/condition and treatment may impact a patient’s ability to function cognitively and 364
physically over the course of the study as the disease/condition progresses or as treatment side 365
effects manifest, including ability to communicate, follow instructions (verbal and written), 366
receive and understand information, and complete the assessment.
8
Missed or incomplete 367
assessments due to disease progression or treatment side effects “may provide meaningful 368
information on the effect of a treatment and hence may be incorporated into a variable [(or 369
endpoint)], with appropriate summary measure, that describes a meaningful treatment effect” 370
(ICH E9(R1)). 371
372
Since model-based estimates generally tend to be “very sensitive” to model misspecification, it is 373
recommended that supplementary and sensitivity analyses be conducted to examine how much 374
the results/findings change under various assumptions about the missing data mechanism 375
(National Research Council, 2010). Principles and methods for sensitivity analyses are discussed 376
further in ICH E9(R1) and Chapter 5 of the National Research Council’s 2010 report on The 377
Prevention and Treatment of Missing Data in Clinical Trials (National Research Council, 2010). 378
379
Changes in physical or cognitive function due to disease/condition progression and/or treatment 380
effects are important outcomes to be measured and either incorporated into the study endpoint 381
structure or reported as safety findings. 382
383
For some risk factors of cognitive or physical change unrelated to disease or treatment (such as 384
advancing age), the chances of a patient’s cognitive or physical function changing over the 385
course of the study may increase with study duration. Use of appropriate inclusion and exclusion 386
criteria may help mitigate some potential causes of cognitive and physical change. However, 387
restrictive criteria can impact the ability to recruit and the generalizability of study results. 388
Because changes in cognitive and physical function may still occur during the study, it is 389
important to note sources of competing risks and other intercurrent events in the SAP and Study 390
Report. 391
392
3. Practice Effects 393
A practice effect
9
(sometimes also called a learning effect) is any change that results from 394
practice or repetition of completing particular tasks or activities including repeated exposure to 395
an instrument. A simple example is taking a math test. After completing the same test three times 396
8
When disease progression or treatment side effects result in missed or incomplete assessments, those missing COA
data are considered to be informatively missing or missing not at random (MNAR). Missing observations (e.g.,
missing COA data) are considered to be informatively missing or MNAR “when there is some association between
whether or not an observation is missing (or observed) and the status of the patient’s underlying disease” (Lachin,
1999). Failing to incorporate both observed and unobserved (i.e., missing but potentially observable) COA data from
the entire ITT population in analyses involving the COA-based endpoint will likely yield biased (erroneous;
misleading) results.
9
Note that practice effects may be referred to using different terminology in different disciplines.
13
your speed (and maybe accuracy in answering) likely will improve because you recognize the 397
questions and have ‘learned’ the test. While potentially an issue for any COA, practice effects 398
may be of particular concern in studies utilizing PerfOs with within-subject designs in which 399
repeated measurements are taken over time, i.e., over the course of the study period (American 400
Psychological Association, 2018; Shadish, Cook, & Campbell, 2002). 401
402
Practice effects may be problematic for studies conducted to support a medical product 403
regulatory application. Practice effects, by definition, lead to improvement in the score of the 404
assessment. This score improvement confounds score changes attributable to the clinical trial 405
intervention. In randomized controlled trials, if practice effects are constant across trial arms, 406
they will not bias the difference of the outcomes between arms. However, if practice effects 407
interact with clinical trial intervention such that the magnitude and direction of practice effects 408
differ by trial arm, the treatment effects may be deflated or inflated (Song & Ward, 2015). 409
Deflation of treatment effects may result in delayed patient access to effective treatment options, 410
and inflation of treatment effects may expose patients to risk due to wasted time and resources 411
spent pursuing ineffective treatments. Whether the practice effects are constant or differ across 412
clinical trial arms is generally unknown. Therefore, the best strategy is to minimize the potential 413
for practice effects in clinical studies. 414
415
Currently, approaches exist for attenuating, but not eliminating, practice effects (Jones, 2015). In 416
addition, no consensus on best practices for attenuating practice effects has yet been reached 417
(Jones, 2015). Some general strategies for mitigating practice effects are summarized below. 418
These strategies may be used in isolation but may be more effective when used in combination. 419
420
• Consider available evidence on practice effects when identifying an instrument: 421
Some instruments may be more robust to practice effects than others. When selecting an 422
instrument, one may wish to consider available evidence of the candidate instruments’ 423
robustness (or vulnerability) to practice effects. Such evidence may be obtained through, 424
for example, a thorough review of the literature. 425
• Increase length of time (spacing) between assessments: In general—and all else being 426
equal—the magnitude of practice effects is expected to decrease as time between 427
assessments increases (Shadish, Cook, & Campbell, 2002). Decisions regarding the 428
length of time (spacing) to place between assessments should take into consideration both 429
how rapidly (or slowly) change in the underlying construct is expected to occur and the 430
recall period utilized by the instrument. Refer to Section II.C.3 of this Discussion 431
Document for more detailed considerations regarding timing of assessments. 432
• Increase the length of the run-in period: In general, the magnitude of practice effects is 433
largest at the beginning of a study and gradually levels off or decreases as the number of 434
assessments increases. Having a long run-in period allows large practice effects to occur 435
for the first few assessments until its magnitude does not significantly increase such that 436
the baseline and postbaseline score are minimally affected by practice effects. 437
• Use alternate forms (sometimes also referred to as parallel forms or equivalent 438
forms): Alternate forms are different versions of an instrument “that are considered 439
interchangeable, in that they measure the same constructs in the same ways, are built to 440
the same content and statistical specifications, and are administered under the same 441

14
conditions using the same directions” (Test Design and Development, 2014). 442
Administering different forms comprised of distinct sets of items may make practice 443
effects less likely to occur. 444
For the use of alternate forms to attenuate practice effects without introducing additional 445
bias: (1) alternate forms must be truly psychometrically equivalent;
10
and (2) alternate 446
forms must be administered in a random order that differs by study arm (i.e., a 447
counterbalanced, randomized order) (Jones, 2015; Goldberg, Harvey, Wesnes, Snyder, & 448
Schneider, 2015). 449
450
4. Participant Burden 451
The possibility of participant burden compromising the validity of the endpoint should be 452
assessed. Burden may lead to missing data or inaccurate data (e.g. answering the first response to 453
every item). When an endpoint is derived from multiple administrations of a COA, attention 454
should be paid to whether study subject fatigue or patient burden might diminish the validity of 455
COA scale scores. This, in turn, could compromise the validity of the endpoint itself, leading to 456
biased estimates of treatment effects and inaccurate hypothesis tests. Study subject fatigue is less 457
likely to occur if an endpoint is based on a small number of widely spaced administrations, and 458
more likely to occur if an endpoint is based on a larger number of administrations over a limited 459
period of time. The effort required for the subject to complete the COA also influences the 460
probability that subject fatigue will compromise scale and endpoint validity. 461
462
For the sake of illustration, suppose subjects are expected to complete a 25-item PRO for seven 463
consecutive days, with the endpoint being the average of the seven daily scores. Some study 464
subjects may grow fatigued at needing to complete the PRO for seven consecutive days, and 465
such fatigue could manifest itself in a variety of ways: 466
467
• Subjects stop completing the PRO at some point after the initial administration and/or 468
choose not to respond to some items at a given administration. 469
• Subjects recall item responses they made the previous day and repeat prior item 470
responses rather than carefully considering how to respond to each item. 471
• Subjects tend to give the same rote response to each item rather than carefully 472
considering how to respond to each item. 473
474
The first type of fatigue response will increase endpoint missingness. While this is not, strictly 475
speaking, an issue of reliability or validity, it clearly compromises the use of the endpoint for 476
assessing its construct. The second and third types of fatigue responses compromise the validity 477
10
For two different instruments to be considered parallel, they must have matching content (i.e., each instrument
must measure the same symptom, function, or impact); estimated item parameters and corresponding standard errors
must not significantly differ; estimated score reliability and corresponding standard errors must not significantly
differ; and score means and standard deviations (surrogates for the distributions of the two sets of scores) in the
target population must not significantly differ (Test Design and Development, 2014).
15
of the endpoint, as the validity of at least some of the PRO administrations per fatigued subject 478
are compromised. 479
480
5. Mode of Administration 481
Changes or disruptions to standardized instrument administration procedures should be 482
documented and may need to be included in the data analyses. Depending on the construct being 483
measured, the assessment environment should provide the reporter with reasonable comfort and 484
minimal distractions to avoid introducing construct-irrelevant variance into the resulting COA 485
scores (American Educational Research Association; American Psychological Association; 486
National Council on Measurement in Education, 2014). 487
488
COA data collection modes can include paper-based and/or electronic-based approaches. Types 489
of COA administration can include self-administration, interviewer-administration (e.g. face-to-490
face, via telephone or electronic means), clinician-administration, and/or trained administrator-491
administration (FDA PFDD G3 Discussion Document). To help ensure the instrument’s 492
established psychometric measurement properties hold in the study at hand, the COA must be 493
administered in accordance with standardized administration and scoring algorithm specified by 494
the instrument developer (such as in the instrument’s user manual or website). For modes of data 495
collection that do not include a date and time stamp (e.g., paper diaries), it is difficult to ensure 496
that patients enter data at the protocol-specified time. 497
498
6. Missing Data and Event-Driven COA Reporting 499
Programming errors can result in significant amounts of missing data which impedes 500
interpretation of analysis results. For example, a COA may be designed to give patients the 501
option to report additional events and event-related symptoms not reported during the day; 502
however, a potential programming error could cause the additional questions to not be 503
administered at the end of the day. Large amounts of missing data would be generated, resulting 504
in underreporting of the event and the study endpoint itself being unreliable and uninterpretable. 505
506
7. Missing Scale-Level Data 507
Missing data should be distinguished from data that do not exist or data that are not considered 508
meaningful due to an intercurrent event. The protocol and the SAP should address plans for how 509
the statistical analyses will handle missing COA data when evaluating clinical benefit and when 510
considering patient success or patient response. 511
512
In cases where patient-level COA data are missing for the entire domain(s) or the entire 513
measurement(s), sponsors should clearly define missing data and propose statistical methods that 514
properly account for such data with respect to a particular estimand. Methods to handle the 515
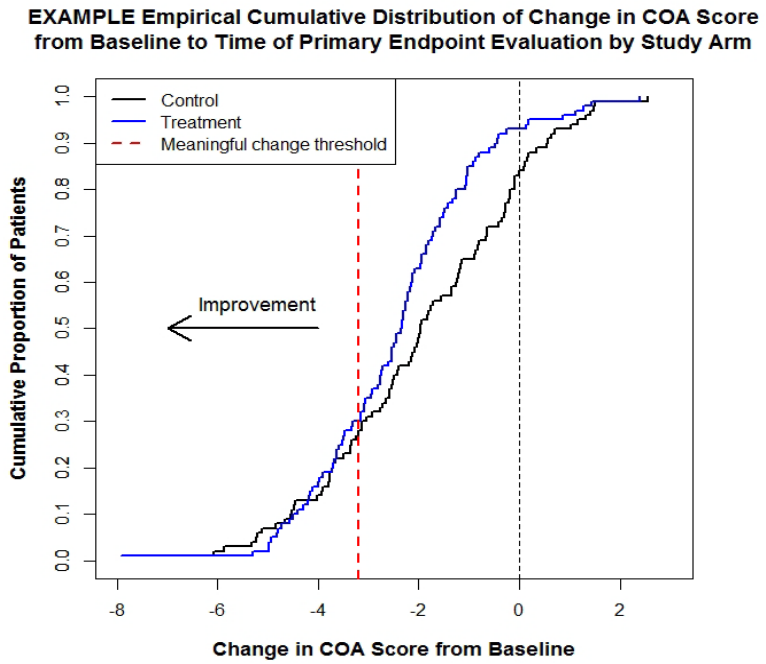
missing data for a COA-based endpoint and any related supportive endpoints should be 516
addressed in the protocol and the SAP. In addition, the supplementary and sensitivity analyses of 517
the COA-based endpoints should be prospectively proposed in the protocol and the SAP. These 518
16
analyses investigate assumptions used in the statistical model for the main analytic approach, 519
with the objective of verifying that inferences based on an estimand are robust to limitations in 520
the data and deviations from the assumptions. 521
522
E. Population-Level Summary: What Is the Final Way All Data Are Summarized and 523
Analyzed? 524
The population-level summary serves as the basis for comparison between treatment arms, 525
treatment conditions, other groups, or otherwise summarizes information. Examples include a) 526
mean physical function score at baseline for everyone in an observational research study and b) 527
difference compared to control of a new medical product’s median time to pain resolution. 528
529
The statistical analysis considerations for COA-based endpoints are similar to the statistical 530
considerations for any other endpoint used in medical product development. This section briefly 531
discusses several considerations that commonly arise when analyzing COA-based endpoints. 532
533
1. Landmark Analysis 534
Sponsors should justify the use of and time in which a landmark analysis (an analysis at a fixed 535
time point, e.g. 12 weeks) is to be performed. If a COA-based endpoint is collected repeatedly, 536
information may be lost in conducting a landmark analysis. However, even when conducting a 537
landmark analysis at a fixed time point, data from intermediate time points (i.e., measurements 538
taken prior to the fixed time point) can still be included in the model. Interpretation of an 539
analysis of overall COA score over time may be difficult in the presence of missing data. The 540
interpretation of potential analyses when COA data collection is truncated due to death or other 541
events should be carefully discussed within the research team. 542
543
2. Analyzing Ordinal Data 544
When an ordinal endpoint has a limited number (e.g., 3 to 7) of categories, you should describe, 545
analyze, and interpret the study result on this endpoint using methods appropriate for ordinal 546
variables, e.g., ordinal regression. For descriptive statistics, mean and standard deviation should 547
not be used on an ordinal endpoint. Percentiles and bar graphs can be informative. 548
549
3. Time-to-Event Analysis 550
Defining and identifying an event is an issue for time-to-event analysis of COA-based endpoints 551
and responder analyses of ordinal or continuous COA data. A clinically relevant threshold for 552
deterioration, maintenance, or improvement must be predefined and justified. Relevant 553
information on intercurrent events, censoring rules, and defining an event should be prespecified 554
in the protocol. It is important to explicitly state how to handle intercurrent events. For example, 555
estimates may differ if death is considered a deterioration event versus censored. Censoring rules 556
in the presence of missing COA data should be prespecified in the SAP. Furthermore, analyses to 557
17
evaluate assumptions of the primary time-to-event analysis should be performed under differing 558
censoring rules. 559
560
4. Responder Analyses and Percent Change From Baseline 561
As previously mentioned, COA data often are ordinal or continuous in nature. Sponsors should 562
consider analyzing COA-based endpoints as continuous or ordinal variables rather than as a 563
responder (i.e., dichotomized from either ordinal or continuous COA data) to avoid 564
misclassification errors and potential loss of statistical power. There tends to be more precision 565
in the evaluation of medical product effects on continuous variables (i.e., based on a comparison 566
of means), especially when sample size is of concern. Alternative approaches for analysis (e.g., 567
analyses based on ranks) should be included, if appropriate, in the SAP to account for occurrence 568
of extreme outliers. 569
570
If a responder endpoint is deemed appropriate for a trial and the endpoint is proposed based on 571
dichotomization from either ordinal or continuous data, it is prudent for the sponsor to prespecify 572
a single responder threshold and provide evidence to justify that the proposed responder 573
threshold constitutes a clinically meaningful within-patient change prior to the initiation of the 574
trial. Proposed responder threshold(s) should be discussed with FDA prior to the initiation of the 575
trial as it is crucial for sample size planning and to appropriately power the study. 576
577
In general, for COA-based endpoints FDA does not recommend a responder analysis endpoint or 578
a percent change from baseline endpoint unless the targeted response is complete resolution of 579
signs and symptoms. While percent change from baseline is popular in other contexts, the 580
statistical measurement properties are poor. Strange occurrences arise, for example in 581
randomized withdrawal studies we have seen subjects needing to reach a percent change from 582
baseline threshold who end up needing significantly higher symptom burden to go back on 583
treatment compared to symptom levels needed to enter the trial based on inclusion criteria. 584
Extreme caution should be exercised, and all potential endpoint situations explored especially 585
near the floor and ceiling of the COA or COA-based endpoint’s values, before using percent 586
change from baseline as the population-level summary. 587
588
The Appendix contains a case study that illustrates several of these concepts and guides us to 589
using the estimand framework to better develop the SAP and ultimately more transparently 590
communicate study results. 591
592
593
594

18
III. MEANINGFUL WITHIN-PATIENT CHANGE 595
Section Summary
To aid in the interpretation of study results, FDA is interested in what constitutes a meaningful within-
patient change (i.e., improvement and deterioration from the patients’ perspective) in the concepts
assessed by COAs. Statistical significance can be achieved for small differences between comparator
groups, but this finding does not indicate whether individual patients have experienced meaningful
clinical benefit.
596
Technical Summary: Key Messages in This Section
• What constitutes, from a patient perspective, a meaningful within-patient change in the concepts
evaluated by COAs.
• FDA recommends the use of anchor-based methods to establish meaningful within-patient changes,
although there are other methods that can be used.
• Anchors selected for the trial should be plainly understood in context, easier to interpret than the
clinical outcome itself, and sufficiently associated with the target COA and/or endpoint.
• Anchor-based methods should be supplemented by the use of empirical cumulative distribution
function (eCDF) curves and probability density function (PDF) curves.
597
Interpretation of Within-Patient Meaningful Change 598
599
To holistically determine what is a meaningful change, both benefit and risk, improvement and 600
deterioration, may need to be accounted for. This document is not directly addressing this 601
integration of benefit and risk, but the methods described can be used to help interpret benefit or 602
risk. As such, special consideration should be given by the sponsor to assess how meaningful the 603
observed differences are likely to be. To aid in the interpretation of the COA-based endpoint 604
results, sponsors should propose an appropriate threshold(s) (e.g., a range of score change) that 605
would constitute a clinically meaningful within-patient change in scores in the target patient 606
population for FDA review. 607
608
In addition, if the selected threshold(s) are based on transformed scores (e.g., linear 609
transformation of a 0-4 raw score scale to a 0-100 score scale), it is important to consider score 610
interpretability of the meaningful change threshold(s) for both transformed scores and raw 611
scores. Depending on the proposed score transformation, selected threshold(s) based on 612
transformed scores may reflect less than one category change on the raw score scale, which is 613
not useful for the evaluation and interpretation of clinically meaningful change. 614
615

19
Meaningful Within-Patient Change Versus Between-Group Difference 616
617
It is important to recognize that individual within-patient change is different from between-618
group difference. From a regulatory standpoint, FDA is more interested in what constitutes a 619
meaningful within-patient change in scores from the patient perspective (i.e., individual 620
patient level). The between-group difference is the difference in the score endpoint between 621
two trial arms that is commonly used to evaluate treatment difference. Between-group 622
differences do not address the individual within-patient change that is used to evaluate 623
whether a meaningful score change is observed. A treatment effect is different from a 624
meaningful within-patient change. The terms minimally clinically important difference 625
(MCID) and minimum important difference (MID) do not define meaningful within-patient 626
change if derived from group-level data and therefore should be avoided. Additionally, the 627
minimum change may not be sufficient to serve as a basis for regulatory decisions. 628
629
A. Anchor-Based Methods to Establish Meaningful Within-Patient Change 630
Anchor-based methods utilize the associations between the concept of interest assessed by the 631
target COA and the concept measured by separate measure(s), referred to as anchoring 632
measure(s), often other COAs. FDA recommends the use of anchor-based methods 633
supplemented with both empirical cumulative distribution function (eCDF) and PDF curves to 634
establish a threshold(s), or a range of thresholds, that would constitute a meaningful within-635
patient change score of the target COA or the derived endpoint for the target patient population. 636
The anchor measure(s) are used as external criteria to define patients who have or have not 637
experienced a meaningful change in their condition, with the change in COA score evaluated in 638
these sets of patients. Sponsors should provide evidence for what constitutes a meaningful 639
change on the anchor scale by specifying and justifying the anchor response category that 640
represents a clinically meaningful change to patients on the anchor scale, e.g., a 2-category 641
decrease on a 5-category patient global impression of severity scale. 642
643
Considerations for Anchor Measures 644
645
• Selected anchors should be plainly understood in context, easier to interpret than the 646
COA itself, and sufficiently associated with the target COA or COA endpoint 647
• Multiple anchors should be explored to provide an accumulation of evidence to help 648
interpret a clinically meaningful within-patient score change (can also be a range) in the 649
clinical outcome endpoint score 650
• Selected anchors should be assessed at comparable time points as the target COA but 651
completed after the target COA 652
20
• The following anchors are sometimes recommended to generate appropriate threshold(s) 653
that represent a meaningful within-patient change in the target patient population: 654
– Static, current-state global impression of severity scale (e.g., PGIS) 655
– Global impression of change scale (e.g., patient global impression of change or 656
PGIC) 657
– Well-established clinical outcomes (if relevant) 658
• A static, current state global impression of severity scale is recommended at minimum, 659
when appropriate, since these scales are less likely to be subject to recall error than global 660
impression of change scales; they also can be used to assess change from baseline. 661
662
B. Using Empirical Cumulative Distribution Function and Probability Density 663
Function Curves to Supplement Anchor-Based Methods 664
The eCDF curves and PDF curves can be used to supplement anchor-based methods. The eCDF 665
curves display a continuous view of the score change (both positive and negative) in the COA-666
based endpoint score from baseline to the proposed time point on the horizontal axis, with the 667
vertical axis representing the cumulative proportion of patients experiencing up to that level of 668
score change. An eCDF curve should be plotted for each distinct anchor category as defined and 669
identified by the anchor measure(s) (e.g., much worse, worse, no change, improved, much 670
improved). 671
672
As a reference, Figure 2 provides an example of eCDF curves. Note that the median change is 673
indicated by the red line in this example. The number of PGIS category increases and decreases 674
defines the example’s curves. In some instances, not all two (or 1 or 0) category changes are the 675
same. This should be considered when choosing an anchor summary and interpreting these 676
figures and data. 677
678

21
Figure 2: Example of Empirical Cumulative Distribution Function Curves of Change in 679
COA Score from Baseline to Primary Time Point by Change in PGIS Score 680
681
Abbreviations: PGIS = patient global impression of severity; COA = clinical outcome assessment 682
683
The PDF curves are useful in aiding the interpretation of eCDF curves. Compared with eCDF 684
curves, PDF curves may provide a more intuitive overview of the shape, dispersion, and 685
skewness of the distribution of the change from baseline in the endpoint of interest across 686
various anchor categories. Figure 3 provides an example of PDF curves. 687
688
Figure 3: Example of Density Function Curves of Change in COA Score from Baseline to 689
Primary Time Point by Change in PGIS Score 690
691
Abbreviations: PGIS = patient global impression of severity; COA = clinical outcome assessment 692
22
C. Other Methods 693
1. Potentially Useful Emerging Methods 694
Other methods may be explored to complement the anchor-based methods or when anchor-based 695
methods are not feasible (i.e., when no adequate anchor measure(s) are available). For example, 696
mixed methods may be used to triangulate and interpret COA-based endpoint results. The 697
qualitative research methods in the PFDD Guidance 1 and Guidance 2 documents are frequently 698
used, including cognitive interviews, exit interviews, or surveys to help inform the improvement 699
threshold. In addition, patient preference studies, typically surveys or interviews, may be utilized 700
to help interpret and support clinical trial results. 701
702
There are several methods emerging in the health sector as potential ways to derive and interpret 703
clinically meaningful change (Duke Margolis meeting summary, 2017), including scale-704
judgement and bookmarking/standard-setting. These methods are relatively new in the regulatory 705
setting. 706
707
2. Distribution-Based Methods 708
Distribution-based methods (e.g., effect sizes, certain proportions of the standard deviation 709
and/or standard error of measurement) do not directly take into account the patient voice and as 710
such cannot be the primary evidence for within-patient clinical meaningfulness. Distribution-711
based methods can provide information about measurement variability. 712
713
3. Receiver Operator Characteristic Curve Analysis 714
Unless there is significant knowledge about how a COA performs in a specific context of use, 715
FDA does not recommend using receiver operator characteristic (ROC) curve analysis as a 716
primary method to determine the thresholds for within-patient meaningful change score. The 717
ROC curve method is a model-based approach, such that different models may yield different 718
threshold values. Additionally, the ROC curve method is partially a distributional-based 719
approach, such that the distribution of the change scores of the two groups will determine the 720
location of the threshold. The most sensitive threshold identified by ROC may not actually be the 721
most clinically meaningful threshold to patients. 722
723
The ROC curve method is appropriate for evaluating the performance (e.g., sensitivity and 724
specificity) of the proposed responder thresholds derived from the anchor-based methods. 725
726
D. Applying Within-Patient Change to Clinical Trial Data 727
Clinical trials compare groups. To help evaluate what constitutes a meaningful within-patient 728
change (i.e., improvement and deterioration from the patients’ perspective), you should examine 729
whether treatment arms show separation in the range of clinically meaningful within-patient 730
change thresholds evaluated using methodologies described in other parts of this document. 731
732

23
When analyzing a COA-based endpoint as either a continuous or an ordinal variable, it is 733
important to evaluate and justify the clinical relevance of any observed treatment effect. 734
Sponsors should plan to evaluate the meaningfulness of within-patient changes to aid in the 735
interpretation of the COA-based endpoint results by submitting a supportive graph (i.e., eCDF) 736
of within-patient changes in scores from baseline with separate curves for each treatment arm. 737
The graph will be used to assess whether the treatment effect occurs in the range that patients 738
consider to be clinically meaningful. 739
740
Figure 4 provides an example of an eCDF curve by treatment arm, where there is consistent 741
separation between the treatment arms. The treatment effect occurs in the range patients consider 742
to be clinically meaningful. 743
744
Figure 4: An eCDF Curve by Treatment Arm Showing Consistent Separation Between 745
Two Treatment Arms 746
747
Abbreviation: eCDF = empirical cumulative distribution function 748
749
Figure 5 provides an example of an eCDF curve where the treatment effect does not occur in the 750
range patients consider to be clinically meaningful. Of note, the eCDF does not take in to 751
account estimation uncertainty and is not a test. 752
753
24
Figure 5: An eCDF Curve Where Treatment Effect Is Not in Range Considered Clinically 754
Meaningful by Patients 755
756
Abbreviations: eCDF = empirical cumulative distribution function; COA = clinical outcome assessment 757
758
759

25
IV. ADDITIONAL CONSIDERATIONS 760
Key Messages in This Section
• Appropriately positioned COAs intended to support approval and/or labeling claims are in the
endpoint testing hierarchy.
• A trial’s protocol and SAP should state each COA-based endpoint as a specific clinical trial
objective.
• Address multiplicity concerns and plans for handling missing data at both the instrument and
patient level.
• Short list of formatting and submission considerations applicable to COA data.
761
When planning a study, confirm the following: 762
763
1. Each COA-based endpoint is stated as part of a specific clinical trial objective 764
2. COAs intended to support meaningful outcomes to patients (i.e., labeling claims or other 765
communications) are fit-for-purpose and sensitive to detect clinically meaningful changes 766
3. Clinical trial duration is adequate to support COA objectives 767
4. Frequency and timing of COA administration is appropriate given patient population, clinical 768
trial design and objectives, and demonstrated COA measurement properties 769
5. How blinding or masking will be implemented (e.g. assessor blinding) 770
6. Plans for instrument administration are consistent with instrument’s user manual, or if 771
different are well-developed, communicated to all study sites, and documented 772
7. Procedures for training are well-described 773
8. Content and scoring information are clearly delineated in the clinical trial protocol 774
9. Plans for COA scoring are consistent with those used during instrument development 775
10. COA-based endpoints intended to support approval and/or labeling claims are appropriately 776
positioned in the endpoint testing hierarchy 777
11. Plans for multiplicity adjustment 778
12. Plans for handling missing data at both the instrument (e.g. person skips an item but answers 779
other items on a PRO) and patient (e.g. patient does not provide any responses for a PRO at a 780
study visit) level 781
13. Procedures include assessment of COA-based endpoint before or shortly after a patient 782
withdraws from the clinical trial 783
26
14. Plans for COA measurement after discontinuation from treatment are driven by the research 784
questions 785
15. Description of how between-group differences will be portrayed (e.g., cumulative 786
distribution function) 787
16. Data collection, data storage, and data handling and transmission of procedures, including 788
electronic COAs, are specified 789
790
Both SPIRIT (Calvert et al, 2018) and CONSORT (Calvert et al, 2013) consensus documents 791
have been published with extensive details on what PRO information should be included in trial 792
protocols and manuscripts. This information is extensible for most COAs. 793
794
A. Other Study Design Considerations 795
Blinding: Patients’ and/or clinicians’
knowledge of treatment assignment may lead to changes in 796
how they report information on a COA, or how they engage with PerfO tasks (e.g., amount of 797
encouragement given to patients when measuring walking distance). The protocol should specify 798
who will evaluate the COA-based endpoints, outcomes, or measurements in relation to the 799
subjects (e.g., the investigator or an independent evaluator/rater) as well as who the intended 800
reporter of patient information will be (e.g., clinicians, patients, caregivers) and to what extent 801
blinding will be maintained among the investigators, evaluators/raters and reporters (e.g., 802
clinicians, patients, caregivers). 803
804
Considerations When Using a Nonrandomized or Nonconcurrent Control: When 805
considering the use of COAs to support endpoints in an externally controlled trial, it is important 806
to establish comparability of the COAs both within each of the treatment and external control 807
groups and between the treatment and external control groups. It will be essential to use well-808
defined and reliable COAs across comparator arms, in conjunction with standardized rater 809
training and instructions for administration within each comparator arm and across comparator 810
arms. Every effort should be made to ensure comparability in the assessment methods and timing 811
of COA administration, together with the use of standardized data collection methods (e.g., 812
standardized case report forms), to allow meaningful comparison of changes over time. 813
814
These considerations apply to clinical trials, as well as natural history studies (see FDA draft 815
guidances Rare Diseases: Natural History Studies for Drug Development (FDA, 2019) and Rare 816
Diseases: Common Issues in Drug Development (FDA, 2019), and FDA final guidance Use of 817
Real-World Evidence to Support Regulatory Decision-Making for Medical Devices (FDA, 818
2017)), disease registries, baseline-controlled trials, and trials with a more complicated 819
sequential on-off-on (medical product-control-medical product) designs. Considerations for the 820
various types of control groups are discussed at length in the ICH guidance for industry E10 821
Choice of Control Group and Related Issues in Clinical Trials (ICH E10). 822
823

27
Computerized Adaptive Testing (CAT): We are asked questions about the use of CAT during 824
trials. We encourage people to submit to the docket content they would like to see in the 825
guidance. 826
827
B. Formatting and Submission Considerations 828
Regardless of how a COA is administered in a given study, COA data collected and submitted to 829
FDA to support a regulatory medical product application are subject to all the same regulations 830
and submission requirements as other types of study data, such as, but not limited to, the 831
following: 832
833
• ICH guidelines, such as M8 Electronic Common Technical Document (eCTD) 834
• The Electronic Code of Federal Regulations, Title 21, Chapter 1 (21 eCFR, Chapter 1)—835
with particular attention given to Parts 11, 21, 312.57, and 312.62(b, c) 836
• FDA guidance Use of Electronic Records and Electronic Signatures in Clinical 837
Investigations Under 21 CFR Part 11 – Questions and Answers (June 2017) 838
• FDA guidance Computerized Systems Used in Clinical Investigations (May 2007) 839
• FDA guidance Electronic Source Data in Clinical Investigations (September 2013) 840
• FDA guidance Providing Regulatory Submissions in Electronic Format—Standardized 841
Study Data (December 2014) 842
• FDA guidance Providing Regulatory Submissions in Electronic Format — Submissions 843
Under Section 745A(a) of the Federal Food, Drug, and Cosmetic Act (December 2014) 844
• FDA guidance Providing Regulatory Submissions in Electronic Format – Certain Human 845
Pharmaceutical Product Applications and Related Submissions Using the eCTD 846
Specifications
(January 2019) 847
• The FDA Data Standards Catalog and other data standards, accessible here 848
849
Electronic devices used to administer COAs in studies conducted to support a regulatory medical 850
product application have special development, testing, and deployment considerations like any 851
digital health technology, including a need for usability studies. The following FDA guidances 852
and related discussion documents have more information: 853
854
• FDA PFDD G3 Discussion Document 855
• FDA guidance Contents of a Complete Submission for Threshold Analyses and Human 856
Factors Submissions to Drug and Biologic Applications (September 2018) 857
• FDA guidance Comparative Analyses and Related Comparative Use Human Factors 858
Studies for a Drug-Device Combination Product Submitted in an ANDA (January 2017) 859
• FDA guidance Applying Human Factors and Usability Engineering to Medical Devices 860
(February 2016) 861


29
APPENDIX 1: CASE STUDY OF ESTIMAND FRAMEWORK 866
Key Messages in This Section
• Example of a randomized, concurrently controlled trial in which patients are randomized to one of
two treatment arms: the current standard of care plus the investigational medical product; or the
current standard of care plus placebo.
• The trial goal is to collect and analyze data to provide compelling evidence of the investigational
product’s efficacy in improving progression-free survival (PFS) and on a secondary endpoint of
physical function measured using a PRO to support a labeling claim.
• The Case Study shows how the trial’s research objective and scientific research question drive the
approaches used within the attributes of the estimand framework presented earlier in this document:
Target Study Population, Endpoint of Interest, Intercurrent Events, and Population-Level Summary.
• It also shows how the previous considerations drive the nature of the trial’s SAP.
867
This Case Study exemplifies employment of the estimand framework when considering physical 868
function in certain breast cancer patients that have progression on first line (standard of care) 869
therapy. Breast cancer has heterogeneous disease symptoms and many women will be 870
asymptomatic at baseline even in the second line setting. For this example, second line prior 871
studies have shown a median overall survival (OS) time of 2-2.5 years with second line hormone 872
therapy alone and a median PFS time of approximately 10-12 months. OS is defined as the time 873
from randomization to date of death. PFS is defined as the time from randomization to date of 874
first progression of disease or death due to any cause. This is a randomized controlled trial where 875
patients are randomized in a 1:1 ratio to the following treatment arms: 876
877
• Treatment: Standard of care + oral targeted investigational agent 878
• Control: Standard of care + placebo 879
880
The primary efficacy endpoint is PFS, which is expected to show 6- to 8-month benefit with the 881
addition of targeted therapy. OS may be impacted due to crossover. Symptomatic toxicities 882
including diarrhea, fatigue, and rash are expected to be greater in the investigational arm. The 883
population is generally high functioning (Eastern Cooperative Oncology Group (ECOG) 0 or 1) 884
and is generally asymptomatic from disease at baseline. 885
886
A. Example Research Objective 887
The secondary endpoint’s focused research objective is to use physical function assessed using a 888
PRO to support a labeling claim. In this case, we would like to make conclusions by comparing 889
the treatment arms. Therefore, a hypothesis test should be prespecified, and a correction for 890
multiple testing is needed to control for Type I error. 891
892

30
1. Define COA Scientific Research Question A Priori 893
In the broad research objective, we prespecify that we intend to look at a superior benefit in 894
physical function for the investigational arm. Based on this, we define our scientific research 895
question as follows: 896
897
898
899
We would like to treat PRO endpoints with the same rigor as we would see with efficacy 900
endpoints in oncology such as OS and PFS, particularly when we want to support a labeling 901
claim. Often in oncology trials the sample size may be very small, leading to wide confidence 902
intervals that do not demonstrate superiority. In addition, the COA may not be sensitive to 903
change. 904
905
2. Define Target Study Population Based on the Research Question A Priori 906
Since we are aiming to compare the two treatment arms for a labeling claim, we are defining our 907
study population based on inclusion/exclusion criteria to reflect the targeted patient population 908
for medical product approval. 909
910
911
912
Scientific Research Question
Is the average change in physical function from baseline to Week 28 better (superior) in the
investigational arm compared to the control arm?
Broad COA Research Objective
Evaluate efficacy
Target Study Population
Defined through inclusion/exclusion criteria to reflect the targeted patient population for
medical product approval.
Scientific Research Question
Is the average change in physical function from baseline to Week 28 better (superior) in the
investigational arm compared to the control arm?
31
If assessing tolerability (not efficacy) of the product while a patient is on treatment is of interest, 913
we may want to include only patients who received at least one dose of the product, regardless of 914
randomization. 915
916
3. Define Endpoint of Interest Based on the Research Question A Priori 917
Based on our scientific research question, we aim to collect change from baseline in physical 918
function score at Week 28 assuming we already have a well-defined measurement tool. 919
920
921
922
We are looking at Week 28, which is around a 6-month time point in which the cumulative 923
effects of the product in terms of both efficacy and toxicity have equilibrated. 924
925
Table 2 presents considerations in defining the COA-based endpoint of interest. 926
927
Table 2: Considerations When Defining a COA-Based Endpoint 928
Concepts
(Examples)
Measurement Tool
Qualities
Endpoint Type
Analysis Time
Point
• Physical
function
• Pain
• Well-defined
• Reliable
• Validated
• Sensitive
• Time-to-event
• Proportion with event at time t
• Continuous summary score at time t
• Overall PRO score over time
• Response patterns/profiles
(longitudinal)
• Specific time
point
• Over time
(specify time
frame)
Abbreviations: COA = clinical outcome assessment; PRO = patient-reported outcome 929
930
Considerations for defining the endpoint include the concepts of interest. We chose physical 931
function as an example, but another concept we might be interested in is pain. For each PRO 932
endpoint, we look at all the measurement properties to check the instrument well-defined. Since 933
our variable is change from baseline, statisticians may refer to this as a “continuous summary 934
score at time t.” If we were interested in time-to-deterioration, the endpoint type would have 935
Endpoint of Interest
Change from baseline in physical function score using a well-defined measurement tool. Use
measurements at baseline and at Week 28.
Scientific Research Question
Is the average change in physical function from baseline to Week 28 better (superior) in the
investigational arm compared to the control arm?

32
been time-to-event. Other possibilities include proportions, overall PRO score over time, and 936
response profiles. 937
938
We specified our analysis time point at Week 28. In addition, we might be interested in 939
analyzing data over a specific time frame. For example, it may be of interest to analyze data at 940
each PRO assessment time point while a patient is on treatment. 941
942
4. Address Intercurrent Events in Alignment with the Research Question 943
Based on our research question, some examples of intercurrent events that may impact 944
interpretation include death, progression, and discontinuation. These events and the way they are 945
handled will impact the estimate of the treatment effect. We specify that after date of death, we 946
cannot collect or include physical function assessments in our analyses. We do not expect a high 947
proportion of death to occur at the time of the analysis. For patients who discontinue treatment, 948
progress, start physical therapy, initiate subsequent therapy or experience any other intercurrent 949
event, we continue to collect physical function assessments regardless of these intercurrent 950
events and will include them in our analysis. 951
952
953
954
Table 3 presents a list of additional intercurrent events that may impact interpretation of physical 955
function. It is crucial to list intercurrent events and how they are handled in the analysis so that 956
there is a clear understanding between regulators and sponsors of what is being estimated. 957
958
Scientific Research Question
Is the average change in physical function from baseline to Week 28 better (superior) in the
investigational arm compared to the control arm?

33
Table 3: Considerations When Addressing Intercurrent Events 959
Intercurrent Events (Examples)
Handling Intercurrent Events
• Death
• Progression
• Discontinuation due to adverse event
• Taking subsequent therapy beyond discontinuation
• Use of rescue medication or therapy
• Analgesic use
• Hospitalization
• Nonadherence
• Prespecify handling of intercurrent
events in alignment with research
question
• There are multiple ways to handle
intercurrent events
960
5. Define Population-Level Summary Based on Research Question A Priori 961
Since our research question is looking at mean change from baseline between the two treatment 962
arms, we chose the population-level summary to be the difference between the two arms in mean 963
change from baseline to Week 28. 964
965
966
967
Table 4 presents considerations when defining the COA population-level summary. 968
969
Table 4: Considerations When Defining a COA Population-Level Summary 970
Population-Level Summary (Examples)
Clinical Relevance
• Median time to event, hazard ratio
• Proportion of patients with event at time t
• Mean change at time t
• Mean overall PRO score over time (e.g., mean
area under the curve)
• Mean longitudinal profile
• Clinically relevant thresholds
• Within-patient change
• Estimate
• Within-group mean change
• Between-group difference
Population Level Summary
Difference between treatment arms in mean change from baseline in physical function score
using baseline and Week 28 measurements.
Scientific Research Question
Is the average change in physical function from baseline to Week 28 better (superior) in the
investigational arm compared to the control arm?

34
Abbreviations: COA = clinical outcome assessment; PRO = patient-reported outcome 971
Since we are looking at a magnitude, we chose mean change from baseline as the population 972
level summary. If we were doing a time-to-event analysis, perhaps a hazard ratio and median 973
time to event summary measure would have been used. 974
975
Next, we want to evaluate clinical relevance of our estimates. Once we have results, we want to 976
know what these numbers mean and how they apply to the patient perspective. FDA is interested 977
in evaluating within-patient change to interpret clinical meaningfulness, a topic in Section III of 978
this Discussion Document. Other estimates that are important in interpretation of clinical 979
relevance include the within-group and between-group difference of mean change from baseline. 980
981
6. Prespecify Statistical Analysis Plan 982
Our primary endpoint is PFS with a secondary endpoint of mean change from baseline in 983
physical function score at Week 28. We will analyze this endpoint using a mixed model for 984
repeated measurements (MMRM) in the ITT population to obtain a least squares (LS) mean 985
change from baseline in physical function score at Week 28 for each treatment arm, difference 986
from control arm and their associated 95% confidence intervals. 987
988
989
990
We defined how we handle intercurrent events where patients are assumed missing after death, 991
progression, or treatment discontinuation. Note that the MMRM assumes patients who drop out 992
behave similarly to other patients in the same treatment group, who had similar covariate and 993
COA data prior to dropping out. If, for example, a patient discontinues because of toxicity, this 994
assumption may not be reasonable. The estimated treatment effect may be biased, leading to 995
Scientific Research Question
Is the average change in physical function from baseline to Week 28 better (superior) in the
investigational arm compared to the control arm?

35
uncertainty regarding information. Similar issues arise when death occurs prior to the landmark 996
analysis date. 997
998
In this example trial, MMRM may be reasonable because we do not expect a high proportion of 999
death to occur and will continue to collect physical function assessments regardless of 1000
progression or treatment discontinuation. Suitable supplementary analyses should be performed 1001
to challenge the assumptions of the prespecified analysis by incorporating reasons for 1002
missingness in the analysis. 1003
1004
B. Summary of Decisions Made in This Case Study 1005
We applied the estimand framework to an example research objective to support a labeling claim 1006
in a second line advanced cancer trial. A summary of decisions made for the estimand based on 1007
the research question is given in Table 5. 1008
1009
Table 5: Summary of Estimand Decisions Made 1010
Estimand Attributes
Decisions Based on Research Question
Target population
Defined through inclusion/exclusion criteria to reflect the targeted
patient population for approval.
Endpoint of interest
Change from baseline in physical function score using well-defined
measurement tool. Use measurements at baseline and at Week 28.
Handling of intercurrent events
Death
Physical function data not collected after this intercurrent event
occurs.
Disease progression;
Treatment discontinuation;
Physical therapy;
Initiation of subsequent
therapy
Physical function collected and analyzed regardless of whether
these intercurrent events occur.
Population-level summary
Difference between treatment arms in mean change from baseline
in physical function score using baseline and Week 28
measurements.
Abbreviations: ITT = intent-to-treat; CI = confidence interval; LS = least squares 1011
1012
This case study is not an endorsement of any singular study design, outcome or analysis; rather, 1013
it is meant to demonstrate application of the estimand framework on a COA-based endpoint. 1014
1015
1016
36
APPENDIX 2: EXAMPLE FROM GENE THERAPY 1017
In December 2017, Luxturna (voretigene neparvovec-rzyl), a gene therapy delivered though 1018
subretinal injection, was approved by FDA for the treatment of patients with confirmed biallelic 1019
RPE65 mutation-associated retinal dystrophy (information page for Luxturna BLA (biologics 1020
license application) 125610 (FDA, 2018), and information page for the October 12, 2017, 1021
Cellular, Tissue and Gene Therapies Advisory Committee Meeting (FDA, 2018)), a condition 1022
that leads to visual function decline with age, resulting in total blindness in young adulthood. 1023
There is no approved pharmacological treatment for this condition, which affects approximately 1024
1,000 to 3,000 patients in the United States. The phase 3 trial that provided the primary evidence 1025
of efficacy of Luxturna received the 2017 David Sackett Trial of the Year Award from the 1026
Society of Clinical Trials (Evans, 2018). 1027
1028
The open-label, two-center trial randomized 31 eligible subjects in a 2:1 ratio to the Luxturna 1029
intervention group or the control (nonintervention) group. Primary and key secondary efficacy 1030
endpoints were measured after one year for both groups. The control group was then crossed 1031
over to receive the Luxturna intervention. After another year of follow-up, the same efficacy 1032
outcomes were collected for crossed-over control subjects and subjects in the original 1033
intervention group. 1034
1035
This trial used a novel performance outcome assessment (PerfO) as the primary efficacy 1036
endpoint. Biallelic mutations in the RPE65 gene cause a progressive retinal dystrophy 1037
characterized by decreased light sensitivity, constricted visual fields, and impaired visual acuity, 1038
resulting in poor functional vision, defined as the ability to conduct vision dependent activities of 1039
daily living independently. Because traditional mobility metrics do not address the effects of 1040
illumination on speed and accuracy of navigation in a standardized and quantitative manner, to 1041
evaluate the effect of Luxturna on functional vision the sponsor developed and validated a novel 1042
PerfO of mobility, the multiluminance mobility test (MLMT) (Chung et al, 2018), targeted 1043
specifically at the treatment effect on retinal dystrophy. 1044
1045
In MLMT, a patient navigated a marked path along a 5-foot by 10-foot obstacle course relying 1046
on vision, in varying environmental illuminations, including very low light levels. There were 1047
seven light levels, ranging from 1 Lux to 400 Lux, each assigned a score code going from 6 to 0, 1048
respectively (Table 6). The patient’s MLMT score corresponded to the lowest light level at 1049
which the patient completed the course accurately and at a reasonable pace. A score of -1 was 1050
assigned to patients who could not pass MLMT at a light level of 400 lux, the highest light level 1051
tested. 1052
1053

37
Table 6: MLMT Illuminance Level, Score Code, and Real-World Examples 1054
Illuminance
(Lux)
Score
Code
Corresponding Environment
1
6
Moonless summer night; or indoor nightlight
4
5
Cloudless summer night with half-moon; or outdoor parking lot at night
10
4
60 min after sunset in a city setting; or a bus stop at night
50
3
Outdoor train station at night; or inside of illuminated office building stairwell
125
2
30 min before cloudless sunrise; or interior of shopping mall, train or bus at night
250
1
Interior of elevator, library or office hallway
400
0
Office environment; or food court
Adapted from Chung et al, 2018. 1055
Abbreviation: MLMT = multiluminance mobility test 1056
1057
The primary efficacy endpoint was the MLMT score change from the Baseline visit to the Year 1 1058
visit. A positive score change indicated that the patient was able to complete the MLMT at a 1059
lower light level. The trial showed that Luxturna treatment led to a clinically meaningful and 1060
statistically significant improvement in the ability to navigate independently in lower light 1061
conditions compared with control (see Figure 6). 1062
1063
In many aspects, this trial reflects the challenges and opportunities with using a novel PerfO 1064
endpoint in a registration trial (Richardson et al, 2019), especially in the context of a rare disease 1065
and a new therapeutic class. These challenges and opportunities are also addressed in two recent 1066
FDA draft guidance documents on human gene therapies for rare diseases (Human Gene 1067
Therapy for Rare Diseases (FDA, 2018), and retinal disorders (Human Gene Therapy for Retinal 1068
Disorders (FDA, 2018)), respectively. In what follows, we summarize the salient features of the 1069
Luxturna trial with regard to the use of the MLMT endpoint to demonstrate the efficacy of 1070
Luxturna, and general considerations on using a novel PerfO as the primary efficacy endpoint. 1071
1072
• After a phase 1 trial, the sponsor identified the need to develop a novel clinically 1073
meaningful PerfO endpoint specific to the treatment effect of Luxturna on the target 1074
patient population, and went on to develop and validate the MLMT, in discussion with 1075
FDA. This illustrates the importance of carefully designed and conducted early-phase 1076
trials in informing the design of late-phase trials. 1077
• The phase 3 trial used a randomized concurrent control group, instead of a single-arm 1078
design, despite the limited number of patients potentially eligible for the trial. In general, 1079
a lack of adequate information on the natural history of a condition, coupled with a high 1080
diversity in clinical manifestations and rates of progression, would call for a randomized 1081
concurrent-controlled trial, instead of a single-arm trial, to provide the primary evidence 1082
of efficacy. Using a novel PerfO endpoint further adds to the importance of using a 1083
randomized concurrent control for comparison with the investigational product. 1084
38
• The cross-over component, together with a 2:1 randomization ratio, not only potentially 1085
increased enrollment, but also provided additional data to strengthen the efficacy 1086
conclusion, which was primarily based on the primary analysis comparing the MLMT 1087
score change between the two groups one year after randomization. The MLMT score 1088
change in the control group one year after crossing-over to receive the intervention 1089
showed similar improvement to that observed in the original intervention group one year 1090
after randomization (Russell, Bennett, Wellman, et al, 2017a). This design also allowed 1091
the observation of the maintenance of the treatment effect in the original intervention 1092
group two years after randomization. 1093
• The trial was designed to be open-label, due to various considerations, some of which are 1094
listed in FDA draft guidance of gene therapy for retinal disorders (Human Gene Therapy 1095
for Retinal Disorders (FDA, 2018)). However, it was also designed with considerable 1096
focus on mitigating potential biases on endpoint evaluation. 1097
– MLMT evaluation was masked to the evaluator. Audio and video recordings of 1098
MLMT were independently graded by two trained reviewers and an adjudicator, if 1099
needed, at a separate time and location from the testing. The reviewers were affiliated 1100
with an independent reading center and were masked to treatment group by receiving 1101
coded video files that did not reference date or group assignment. 1102
– To mitigate learning effects, the MLMT used 12 different configurations of the 1103
obstacle course of comparable difficulties. Each test was randomly assigned one of 1104
the 12 configurations. 1105
• The Package Insert (see information page for Luxturna BLA 125610 (FDA, 2018)) states 1106
that “An MLMT score change of two or greater is considered a clinically meaningful 1107
benefit in functional vision.” In this trial, this threshold of 2 seems to refer to both the 1108
difference in the medians between the two trial groups and the within-patient change. In 1109
general, however, the between-group difference and a meaningful within-patient change 1110
are two distinct concepts. It may be challenging to reach a consensus on the threshold for 1111
a meaningful within-patient change, especially for an endpoint based on an ordinal scale. 1112
– Patients entering the trial with a MLMT score of 5 at most could improve by one light 1113
level to 6, the highest attainable light score. This ceiling effect precludes a 1114
demonstration of attaining the meaningful within-patient change of at least 2, but it is 1115
also important to include these patients to provide data for a broad target population. 1116
In the Luxturna trial, all four (4) patients in the Luxturna group with a baseline 1117
MLMT score of 5 improved to a score of 6 at the Year 1 visit, consistent with the 1118
efficacy result in other patients. 1119
• In the evaluation of treatment efficacy, FDA, applicant, and Advisory Committee also 1120
considered supportive evidence from the secondary endpoints (information page for 1121
Luxturna BLA 125610 (FDA, 2018), and information page for the October 12, 2017, 1122
Cellular, Tissue and Gene Therapies Advisory Committee Meeting (FDA, 2018)). 1123
Russell and colleagues (Russell, Bennett, Wellman, et al, 2017b) considered the 1124
unavailability of traditional bilateral best-corrected visual acuity data to be a limitation of 1125
the trial. In the case of a primary endpoint that is novel to most clinicians, and/or when 1126
the primary endpoint does not comprehensively capture the potential impact of a 1127
treatment on the disease, it is important to include secondary endpoints that are directly 1128
39
interpretable to clinicians and that characterize the treatment effect more fully. Care 1129
should be taken in the study design and conduct to collect good quality data on the 1130
secondary endpoints, ideally with the same care as afforded the primary efficacy 1131
endpoint. 1132
• The primary endpoint, score change in the MLMT test, is on an ordinal scale, because the 1133
log-unit illuminance levels are not evenly spaced (Table 6) (information page for 1134
Luxturna BLA 125610 (FDA, 2018), and information page for the October 12, 2017, 1135
Cellular, Tissue and Gene Therapies Advisory Committee Meeting (FDA, 2018)). Some 1136
of the statistical analyses used for this endpoint have an interpretation only for variables 1137
of an interval scale, e.g., a mean difference between the two trial groups and the 1138
corresponding confidence interval. Other analyses have an interpretation for ordinal 1139
variables as well, e.g., median and Wilcoxon rank sum tests. For this particular example, 1140
it is unclear whether treating the primary endpoint as an interval-scale variable is 1141
reasonable. While the endpoint scores do not correspond to evenly spaced log-1142
illuminance level, they are aligned to real-world ambiance illumination that one can 1143
relate to. In general, statistical methods, including choice of effect parameters and effect 1144
size estimators, should correspond to the scale of the endpoint. 1145
1146

40
Figure 6: MLMT Scores in Phase 3 Trial 1147
1148
Excerpted from FDA’s presentation at the advisory committee meeting (2). 1149
Abbreviations: MLMT = multiluminance mobility test; ITT = intent-to-treat 1150
1151
1152
References Specific to Appendix 2 1153
Chung DC, McCague S, Yu ZF, et al. Novel mobility test to assess functional vision in patients 1154
with inherited retinal dystrophies. Clin Exp Ophthalmol. 2018;46(3):247-259. 1155
Evans S. The 2017 David Sackett Trial of the Year Award. Society of Clinical Trials Newsletter, 1156
June 2018, Volume 29, #1. 1157
FDA. Information page for Luxturna BLA 125610, including package insert and approval 1158
supporting documents. Available at: https://www.fda.gov/vaccines-blood-biologics/cellular-1159
gene-therapy-products/luxturna. Content current as of 07/26/2018. Accessed 11/1/2019. 1160
FDA. Information page for the Cellular, Tissue and Gene Therapies Advisory Committee 1161
meeting, October 12, 2017, on the Luxturna BLA discussion. Includes package insert, briefing 1162
documents, presentations, and summary minutes. Available at: https://www.fda.gov/advisory-1163
committees/cellular-tissue-and-gene-therapies-advisory-committee/2017-meeting-materials-1164

41
cellular-tissue-and-gene-therapies-advisory-committee. Content current as of 03/23/2018. 1165
Accessed 11/1/2019. 1166
FDA Guidance for Industry. Human Gene Therapy for Rare Diseases. Draft, July 2018. 1167
Available at: https://www.fda.gov/media/113807/download. Accessed 11/1/2019.* 1168
FDA Guidance for Industry. Human Gene Therapy for Retinal Disorders. Draft, July 2018. 1169
Available at: https://www.fda.gov/media/124641/download. Accessed 11/1/2019.* 1170
Richardson E, Burnell J, HR Adams HR, et al. Developing and implementing performance 1171
outcome assessments: evidentiary, methodologic, and operational considerations. Ther Innov 1172
Regul Sci. 2019;53(1):146-153. 1173
Russell S, Bennett J, Wellman JA, et al. Phase 3 trial update of voretigene neparvovec in biallelic 1174
RPE65-mediated inherited retinal disease. Presentation given at the American Academy of 1175
Ophthalmology meeting; 2017a; New Orleans. 1176
Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-1177
hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, 1178
controlled, open-label, phase 3 trial. Lancet. 2017b;390(10097):849-860. (Including a 17-page 1179
“Supplementary Appendix” accessed online.) 1180
* When finalized, this guidance will represent FDA’s current thinking on this topic. 1181

42
APPENDIX 3: REFERENCES 1182
American Educational Research Association; American Psychological Association; National 1183
Council on Measurement in Education. Standards for educational and psychological testing. 1184
Washington, DC: American Educational Research Association, 2014. 1185
American Psychological Association (2018). Retrieved July 16, 2019, from APA Dictionary of 1186
Psychology. Available at: https://dictionary.apa.org. 1187
Calvert M, Blazeby J, Altman DG, et al. Reporting of patient-reported outcomes in randomized 1188
trials: the CONSORT PRO Extension. JAMA. 2013;309(8):814-822. 1189
Calvert M, Kyte D, Mercieca-Bebber R, et al: the SPIRIT-PRO Group. Guidelines for inclusion 1190
of patient-reported outcomes in clinical trial protocols: the SPIRIT-PRO Extension. JAMA. 1191
2018;319(5):483-494. 1192
Duke Margolis Center for Health Policy. Report of an event on April 4, 2017. Clinical outcome 1193
assessments: establishing and interpreting meaningful within-patient change. Available at: 1194
https://healthpolicy.duke.edu/events/clinical-outcome-assessments-establishing-and-1195
interpreting-meaningful-within-patient-change. Accessed 11/1/2019. 1196
FDA. Discussion Document for the Patient-Focused Drug Development Public Workshop on 1197
Guidance 3: Select, Develop or Modify Fit-for-Purpose Clinical Outcome Assessments. 1198
Prepared for a Patient-Focused Drug Development Guidance Public Workshop held on 1199
October 15-16, 2018. Available at: https://www.fda.gov/media/116277/download. Accessed 1200
11/1/2019. 1201
FDA. Guidance for Industry. Multiple Endpoints in Clinical Trials. Draft, January 2017. 1202
Available at: https://www.fda.gov/media/102657/download. Accessed 11/1/2019. 1203
FDA. Guidance for Industry. Non-Inferiority Clinical Trials to Establish Effectiveness. 1204
November 2016. Available at: https://www.fda.gov/media/78504/download
. Accessed 1205
11/1/2019. 1206
FDA Guidance for Industry. Rare Diseases: Natural History Studies for Drug Development. 1207
Draft, March 2019. Available at: https://www.fda.gov/media/122425/download. Accessed 1208
11/1/2019. 1209
FDA Guidance for Industry. Rare Diseases: Common Issues in Drug Development. Draft, 1210
January 2019. Available at: https://www.fda.gov/media/119757/download. Accessed 1211
11/1/2019. 1212
FDA Guidance for Industry and Food and Drug Administration Staff. Use of Real-World 1213
Evidence to Support Regulatory Decision-Making for Medical Devices. August 2017. 1214
Available at: https://www.fda.gov/media/99447/download. Accessed 11/1/2019. 1215

43
Goldberg TE, Harvey PD, Wesnes KA, Snyder PJ, Schneider LS. Practice effects due to serial 1216
cognitive assessment: implications for preclinical Alzheimer's disease randomized controlled 1217
trials. Alzheimers Dement (Amst). 2015;1(1):103-111. 1218
ICH Guideline E8(R1). General Considerations for Clinical Studies. May 2019. Available at: 1219
https://database.ich.org/sites/default/files/E8-R1_EWG_Draft_Guideline.pdf. Accessed 1220
11/1/2019. 1221
ICH Guideline E9(R1). Estimands and Sensitivity Analysis in Clinical Trials. June 2017. 1222
Available at: https://database.ich.org/sites/default/files/E9-R1_EWG_Draft_Guideline.pdf. 1223
Accessed 11/1/2019. 1224
ICH E10. Choice of Control Group and Related Issues in Clinical Trials. July 2000. Available at: 1225
https://database.ich.org/sites/default/files/E10_Guideline.pdf. Accessed 11/1/2019. 1226
Jones RN. Practice and retest effects in longitudinal studies of cognitive functioning. Alzheimers 1227
Dement (Amst). 2015;1(1);101-102. 1228
Lachin JM. Worst-rank score analysis with informatively missing observations in clinical trials. 1229
Control Clin Trials. 1999;20(5):408-422. 1230
National Research Council (US) Panel on Handling Missing Data in Clinical Trials. Washington, 1231
DC: National Academies Press (US), 2010. 1232
Shadish WR, Cook TD, Campbell DT. Experimental and quasi-experimental designs for 1233
generalized causal inference. Belmont, CA: Wadsworth Cengage Learning, 2002. 1234
Song MK, Ward SE. Assessment effects in educational and psychosocial intervention trials: an 1235
important but often-overlooked problem. Res Nurs Health. 2015;38(3):241-247. 1236
Test Design and Development. In A. E. Association, A. P. Association, & N. C. Education, 1237
Standards for Educational and Psychological Testing (pp. 75-93). Washington, DC: 1238
American Educational Research Association, 2014. 1239

44
APPENDIX 4: GLOSSARY 1240
This glossary defines terms that will be used in the series of methodological patient-focused drug 1241
development (PFDD) FDA guidance documents that are required by the 21st Century Cures Act, 1242
and part of commitments made by FDA under the sixth authorization of the Prescription Drug 1243
User Fee Act (PDUFA VI). The goal of this glossary is to provide standardized nomenclature 1244
and terminologies related to patient-focused medical product development. As appropriate, 1245
definitions from existing federal resources (e.g., Biomarkers, EndpointS, and Other Tools 1246
(BEST) Resource)
11
have been incorporated into this glossary. External resources were also used 1247
to define terms and are cited. 1248
1249
Ability to Detect Change: Evidence that a COA can identify differences in scores over time 1250
in individuals or groups who have changed with respect to the measurement concept. 1251
1252
Alternate Forms (also referred to as parallel forms or equivalent forms): Different versions 1253
of an instrument “that are considered interchangeable, in that they measure the same constructs 1254
in the same ways, are built to the same content and statistical specifications, and are administered 1255
under the same conditions using the same directions” (Test Design and Development, 2014). 1256
1257
Benefit: Benefits are the favorable effects of a medical product. Types of benefit include clinical 1258
benefit (see clinical benefit). Benefits may also include important characteristics of the medical 1259
product, such as convenience (e.g., a more convenient dosing regimen or route of administration) 1260
that may lead to improved patient compliance, or benefits that affect those other than the patient. 1261
(Source: International Conference on Harmonisation (ICH) guideline Revision of M4E Guideline 1262
on Enhancing the Format and Structure of Benefit-Risk Information in ICH (Efficacy – 1263
M4E(R2)), available at 1264
http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/CTD/M4E_R2_Efficacy/M4E_R1265
2Step_4.pdf; ANSI/AAMI/ ISO 14971: 2007/(R)2016 Medical devices—Application of risk 1266
management to medical devices.) 1267
1268
Caregiver: A person who helps a patient with daily activities, health care, or any other activities 1269
that the patient is unable to perform because of illness or disability, and who understands the 1270
patient’s health-related needs. This person may or may not have decision-making authority for 1271
the patient and is not the patient’s health care provider. 1272
1273
Case Report Form: A form used throughout clinical trials to record data collected from subjects 1274
in the trial. The form captures all the information specified in the trial’s protocol for each subject. 1275
All data recorded on the form must be verifiable from original source documentation. 1276
1277
Ceiling Effect: A ceiling effect can occur at the item level or at the scale score level. An item 1278
level ceiling effect is observed when a large concentration of participants endorses the highest 1279
response category within an item. A scale score level ceiling effect is observed when a large 1280
concentration of participants’ scores fall at or near the upper limit of the scale score of the 1281
instrument. Either situation may occur when the upper extreme of the concept(s) assessed by 1282
11
Available at https://www.ncbi.nlm.nih.gov/books/NBK338448/

45
item response categories or by the scale score of the instrument does not sufficiently match the 1283
level of the upper extreme of the target patient population. 1284
1285
Clinical Benefit: A positive clinically meaningful effect of an intervention (i.e., a positive effect 1286
on how an individual feels, functions, or survives). (Source: BEST (Biomarkers, EndpointS, and 1287
other Tools) Resource) 1288
1289
Clinical Outcome: A positive clinically meaningful effect of an intervention (i.e., a positive 1290
effect on how an individual feels, functions, or survives). (Source: BEST (Biomarkers, 1291
EndpointS, and other Tools) Resource) 1292
1293
Clinical Outcome Assessment (COA): Assessment of a clinical outcome can be made through 1294
report by a clinician, a patient, a nonclinician observer, or through a performance- based 1295
assessment. Types of COAs include: patient-reported outcome (PRO) measures, clinician-1296
reported outcome (ClinRO) measures, observer-reported outcome (ObsRO) measures, and 1297
performance outcome (PerfO) measures. (Source: BEST (Biomarkers, EndpointS, and other 1298
Tools) Resource) 1299
1300
Clinician-Reported Outcome (ClinRo): A measurement based on a report that comes from a 1301
trained health-care professional after observation of a patient’s health condition. Most ClinRO 1302
measures involve a clinical judgment or interpretation of the observable signs, behaviors, or 1303
other manifestations related to a disease or condition. ClinRO measures cannot directly assess 1304
symptoms that are known only to the patient (e.g., pain intensity). (Source: BEST (Biomarkers, 1305
EndpointS, and other Tools) Resource
) 1306
1307
Clinical Study: Research according to a protocol involving one or more human subjects to 1308
evaluate biomedical or health-related outcomes, including interventional studies and 1309
observational research. (Source:
https://www.federalregister.gov/documents/2016/09/21/2016-1310
22129/clinical-trials-registration-and-results-information-submission#p-1195
) 1311
1312
Cognitive Interviews: A qualitative research process used to determine whether concepts and 1313
items are understood by respondents in the same way that instrument developers intend. 1314
Cognitive interviews involve incorporating follow-up questions in a field test interview to gain a 1315
better understanding of how respondents interpret questions/tasks asked of them. In this method, 1316
respondents are often asked to think aloud and describe their thought processes as they answer 1317
the instrument questions. Respondents should reflect the target population who will be 1318
responding to the instrument during the study. 1319
1320
Competing Risks: A competing risk is an event whose occurrence precludes the occurrence of 1321
the primary event of interest. 1322
1323
Concept (also referred to as concept of interest): In a regulatory context, the concept is the 1324
aspect of an individual’s clinical, biological, physical, or functional state, or experience that the 1325
assessment is intended to capture (or reflect). (Source: BEST (Biomarkers, EndpointS, and other 1326
Tools) Resource) 1327
1328

46
Construct Validity: Evidence that relationships among items, domains, and concepts 1329
conform to a priori hypotheses concerning logical relationships that should exist with other 1330
measures or characteristics of patients and patient groups. 1331
1332
Content Validity: Evidence from qualitative research demonstrating that an instrument 1333
measures the concept of interest, including evidence that the items and domains of an 1334
instrument are appropriate and comprehensive relative to its intended measurement concept, 1335
population, and use. Testing other measurement properties will not replace or rectify problems 1336
with content validity. 1337
1338
Context of Use: A statement that fully and clearly describes the way a medical product 1339
development tool is to be used and the medical product development-related purpose of the use. 1340
(Source: BEST (Biomarkers, EndpointS, and other Tools) Resource) 1341
1342
Digital Health Technologies (DHTs): Use of computing platforms, connectivity, software 1343
and/or sensors for healthcare and related uses. These technologies span a range of products, from 1344
general wellness applications to medical devices. These products are also used as diagnostics, 1345
therapeutics or adjuncts to medical products (devices, drugs, and biologics). They may also be 1346
used to develop or study medical products. 1347
1348
Disease Burden (also referred to as burden of disease): The impacts, direct and indirect, of 1349
the patient’s health condition that have a negative effect on his or her health, functioning, and 1350
overall well-being. Disease burden includes but is not limited to the physical and physiologic 1351
impacts of the disease and its symptoms; co-morbidities; emotional and psychological effects of 1352
the disease, its management, or its prognosis; social impacts; effects on relationships; impacts on 1353
the patient’s ability to care for self and others; time and financial impacts of the disease and its 1354
management; and considerations of the impacts on the patient’s family. 1355
1356
Domain: A sub-concept represented by a score of an instrument that measures a larger concept 1357
comprised of multiple domains. For example, psychological function is the larger concept 1358
containing the domains subdivided into items describing emotional function and cognitive 1359
function. 1360
1361
Endpoint: A precisely defined variable intended to reflect an outcome of interest that is 1362
statistically analyzed to address a particular research question. A precise definition of an 1363
endpoint typically specifies the type of assessments made; the timing of those assessments; the 1364
assessment tools used; and possibly other details, as applicable, such as how multiple 1365
assessments within an individual are to be combined. (Source: BEST (Biomarkers, EndpointS, 1366
and other Tools) Resource) 1367
1368
Estimand: A precise description of the treatment effect reflecting the clinical question posed by 1369
the trial objective. It summarizes at a population-level what the outcomes would be in the same 1370
patients under different treatment conditions being compared. (Source: ICH E9(R1)) 1371
1372
Fit-for-Purpose: A conclusion that the level of validation associated with a tool is sufficient to 1373
support its context of use. (Source: BEST (Biomarkers, EndpointS, and other Tools) Resource) 1374

47
1375
Generalizability: The extent to which study findings can be reliably extended to the target 1376
population of interest. 1377
1378
Instrument or Tool: An assessment system comprising three essential components: (1) 1379
materials for measurement; (2) an assay for obtaining the measurement; and (3) method and/or 1380
criteria for interpreting those measurements. (Source: BEST (Biomarkers, EndpointS, and other 1381
Tools) Resource) 1382
1383
Item: An individual question, statement, or task (and its standardized response options) that is 1384
evaluated or performed by the patient to address a particular concept. 1385
1386
Learning Effect: See Practice Effect 1387
1388
Measurement Properties: All the attributes relevant to the application of a COA including the 1389
content validity, construct validity, reliability, and ability to detect change. These attributes are 1390
specific to the measurement application and cannot be assumed to be relevant to all measurement 1391
situations, purposes, populations, or settings in which the instrument is used. 1392
1393
Multicomponent Endpoint: A within-patient combination of two or more components. In some 1394
cases, multiple aspects of a disease may appropriately be combined into a single endpoint, but 1395
subsequent analysis of the aspects or components is generally important for an adequate 1396
understanding of the drug’s effect. In this type of endpoint, an individual patient’s evaluation is 1397
dependent upon observation of all the specified components in that patient. A single overall 1398
rating or status is often determined according to specified rules. 1399
1400
Observer-Reported Outcome (ObsRO): A measurement based on a report of observable signs, 1401
events, or behaviors related to a patient’s health condition by someone other than that patient or a 1402
health professional. Generally, ObsROs are reported by a parent, caregiver, or someone who 1403
observes the patient in daily life, and ObsROs are particularly useful for patients who cannot 1404
report for themselves (e.g., infants or individuals who are cognitively impaired). An ObsRO 1405
measure does not include medical judgment or interpretation. (Source: BEST (Biomarkers, 1406
EndpointS, and other Tools) Resource
) 1407
1408
Observational Research: A type of nonexperimental social science research technique in which 1409
a researcher directly observes ongoing phenomena in a natural setting. In health sciences, this 1410
can include, but is not limited to, observing behaviors and disease signs (tremors) in real-world 1411
settings and in real-time. 1412
1413
Patient: Any individual with or at risk of a specific health condition, whether the individual 1414
currently receives any therapy to prevent or treat that condition. Patients are the individuals who 1415
directly experience the benefits and harms associated with medical products. 1416
1417
Practice Effect: Any change or improvement that results from practice or repetition of task 1418
items or activities, including repeated exposure to an instrument. 1419
1420
48
Patient Experience Data: Defined in Title III, section 3001, of the 21st Century Cures Act of 1421
2016, as amended by section 605 of the Food and Drug Administration Reauthorization Act of 1422
2017, and includes data that are collected by any persons and are intended to provide information 1423
about patients’ experiences with a disease or condition. Patient experience data can be 1424
interpreted as information that captures patients’ experiences, perspectives, needs, and priorities 1425
related to but not limited to (1) the symptoms of their condition and its natural history; (2) the 1426
impact of the conditions on their functioning and quality of life; (3) their experience with 1427
treatments; (4) input on which outcomes are important to them; (5) patient preferences for 1428
outcomes and treatments; and (6) the relative importance of any issue as defined by patients. 1429
1430
Patient-Focused (also referred to as patient-centered): Ensuring that patients’ experiences, 1431
perspectives, needs, and priorities are meaningfully incorporated into decisions and activities 1432
related to their health and well-being. 1433
1434
Patient-Focused Drug Development (also referred to as patient-focused medical product 1435
development): A systematic approach to help ensure that patients’ experiences, perspectives, 1436
needs, and priorities are captured and meaningfully incorporated into the development and 1437
evaluation of medical products throughout the medical product life cycle. 1438
1439
Patient Perspective: A type of patient experience data that specifically relates to patients’ 1440
attitudes or points of view about their condition or its management. Patient perspectives may 1441
include, but are not limited to, perceptions, goals, priorities, concerns, opinions, and preferences. 1442
1443
Patient Preference: A statement of the relative desirability or acceptability to patients of 1444
specified alternatives or choice among outcomes or other attributes that differ among alternative 1445
health interventions. (Source: FDA guidance for industry Patient Preference Information –1446
Voluntary Submission, Review in Premarket Approval Applications, Humanitarian Device 1447
Exemption Applications, and De Novo Requests, and Inclusion in Decision Summaries and
1448
Device Labeling)
1449
1450
Patient-Reported Outcome (PRO): A measurement based on a report that comes directly from 1451
the patient (i.e., study subject) about the status of a patient's health condition without amendment 1452
or interpretation of the patient's response by a clinician or anyone else. A PRO can be measured 1453
by self-report or by interview, provided that the interviewer records only the patient's response. 1454
Symptoms or other unobservable concepts known only to the patient (e.g., pain severity or 1455
nausea) can only be measured by PRO measures. PROs can also assess the patient perspective on 1456
functioning or activities that may also be observable by others. (Source: BEST (Biomarkers, 1457
EndpointS, and other Tools) Resource) 1458
1459
Performance Outcome (PerfO): A measurement based on a standardized task performed by a 1460
patient that is administered and evaluated by an appropriately trained individual or is 1461
independently completed. 1462
1463
Qualitative Research Methods: Methods associated with the gathering, analysis, interpretation, 1464
and presentation of narrative information (e.g., spoken or written accounts of experiences, 1465

49
observations, and events). Qualitative research methods may also include direct observations 1466
(e.g., nonverbal communication and behaviors). 1467
1468
Recall Period: The period of time patients, caregivers, or clinicians are asked to consider in 1469
responding to a COA item or task. Recall can be momentary (real time) or retrospective of 1470
varying lengths. 1471
1472
Reliability: The ability of a COA to yield consistent, reproducible estimates. 1473
1474
Reporter: In research studies designed to collect patient experience data, the reporter is the 1475
individual, group of individuals, or entity providing patient experience data. Reporters may be 1476
patients, parents, sexual/romantic partners, caregivers, physicians, or other healthcare 1477
professionals. Selection of an appropriate reporter in a given research study will depend on the 1478
definition of the target patient population of interest. If a patient in the target population can be 1479
reasonably expected to reliably self-report, then one would expect the patient herself/himself to 1480
be the reporter in that research study. 1481
1482
Research Protocol: A document that describes the background, rationale, objectives, design, 1483
methodology, statistical considerations, and organization of a clinical research project. (Source: 1484
University of California San Francisco, 2017) A research protocol guides the study and 1485
associated data collection and analysis in a productive and standardized manner. 1486
1487
Response Scale: The system of numbers or verbal anchors by which a value or score is derived 1488
for an item. Examples include verbal rating scale (VRS), numeric rating scale (NRS), and visual 1489
analog scale (VAS). 1490
1491
Risks: Risks are adverse events and other unfavorable effects associated with a medical product. 1492
Risks include drug interactions, risks identified in the nonclinical data, risks to those other than 1493
the patient (e.g., fetus, those preparing and administering the medical product), and risks based 1494
on pharmacologic class or current knowledge of the product. Factors such as potential misuse, 1495
abuse, or diversion of the product may also be considered. (Source: ICH guidelines Efficacy 1496
M4E(R2)) 1497
1498
Score: A number derived from a patient’s, caregiver’s, or clinician’s response to items or tasks 1499
in an instrument. A score is computed based on a prespecified, appropriate scoring algorithm and 1500
is subsequently used in statistical analyses of clinical trial results. Scores can be computed for 1501
individual items, domains, or concepts, or as a summary of items, domains, or concepts. 1502
1503
Scoring Algorithm: A set of prespecified rules to assign numerical value or values to quantify 1504
the responses to the instrument. A scoring algorithm may create a single score from a single item 1505
or multiple items (e.g., domain score). 1506
1507
Side Effects (also referred to as adverse reactions): Unwanted or unexpected events or 1508
reactions to a medical product (Source:
https://www.fda.gov/drugs/drug-information-1509
consumers/finding-and-learning-about-side-effects-adverse-reactions) 1510
1511

50
Sign: Any observable evidence of a disease, health condition, or treatment-related effect. Signs 1512
are usually observed and interpreted by the clinician but may be noticed and reported by the 1513
patient. 1514
1515
Symptom: Any experience of a disease, health condition, or treatment-related effect that can be 1516
known and confirmed only by the patient, and therefore is most reliably assessed by direct 1517
patient report. 1518
1519
Target population (also referred to as target patient population, underlying population, or 1520
intended population): The group of individuals (patients) about whom one wishes to make an 1521
inference. 1522
1523
Task: See item 1524
1525
Treatment Burden (also referred to as burden of treatment): The impacts of a specific 1526
treatment or treatment regimen that have a negative impact on a patient’s health, functioning, or 1527
overall well-being. Treatment burden includes but is not limited to side effects, discomfort, 1528
uncertainty about treatment outcomes, dosing and route of administration, requirements, and 1529
financial impacts. 1530
1531
Usability Studies: Studies conducted to demonstrate that the device can be used by the intended 1532
users without serious errors or problems, for the intended uses and under the expected use 1533
conditions.
12
1534
12
See guidance for industry Applying Human Factors and Usability Engineering to Medical Devices. Definition
derived from Human Factors Validation Testing.
