Affinity Chromatography – Vol. 3: Specific Groups of Biomolecules
www.gelifesciences.com
GE, GE monogram, Amersham, ÄKTA, Biacore, BioProcess, Capto, Cy, CyDye, ECL, ECL Plex, ECL Select,
ExcelGel, GSTrap, HiPrep, HiScale, HiScreen, HiTrap, ImageQuant, Hybond, MiniTrap, MabSelect, MiniTrap,
MultiTrap, PhastGel, PhastSystem, PrimeView, Protran, Sephadex, Sepharose, SPARTAN, SpinTrap,
Superdex, Typhoon, Tricorn, UNICORN, and Whatman are trademarks of General Electric Company.
Coomassie is a trademark of Thermo Fisher Scientific LLC. Neutravidin is a trademark of Pierce
Biotechnology, Inc. Nonidet is a trademark of Air Products and Chemicals, Inc. Pefabloc is a trademark
of DSM IP Assets B.V. Tween is a trademark of Croda Group of Companies. All other third-party
trademarks are the property of their respective owners.
The purchase of CyDye products includes a limited license to use the CyDye products for internal
research and development but not for any commercial purposes. A license to use the Cy and CyDye
trademarks for commercial purposes is subject to a separate license agreement with GE Healthcare.
Commercial use shall include:
1. Sale, lease, license or other transfer of the material or any material derived or produced from it.
2. Sale, lease, license or other grant of rights to use this material or any material derived or produced from it.
3. Use of this material to perform services for a fee for third parties, including contract research and
drug screening.
If you require a commercial license to use the Cy and CyDye trademarks please contact: [email protected].
VIIISelect incorporates BAC BV’s proprietary ligand technology, which has been exclusively licensed to
GE Healthcare in the field of purification of beta domain depleted recombinant factor VIII. Other use of
this product may require a separate license from BAC BV, Huizerstraatweg 28, 1411 GP Naarden, The
Netherlands.
IXSelect incorporates BAC BV’s proprietary ligand technology, which has been exclusively licensed to
GE Healthcare for use in chromatography separation.
GCSFSelect incorporates BAC BV’s proprietary ligand technology, which has been exclusively licensed
to GE Healthcare for use in chromatography separation.
© 1988–2016 General Electric Company. First published 1988.
All goods and services are sold subject to the terms and conditions of sale of the company within
GE Healthcare that supplies them. A copy of these terms and conditions is available on request.
Contact your local GE Healthcare representative for the most current information.
GE Healthcare UK Ltd, Amersham Place, Little Chalfont, Buckinghamshire, HP7 9NA, UK
GE Healthcare Bio-Sciences Corp. 100 Results Way, Marlborough, MA 01752, USA
GE Healthcare Dharmacon, Inc., 2650 Crescent Dr., Lafayette, CO 80026, USA
HyClone Laboratories, Inc., 925 W 1800 S, Logan, UT 84321, USA
GE Healthcare Europe GmbH, Munzinger Strasse 5, D-79111 Freiburg, Germany
GE Healthcare Japan Corporation, Sanken Bldg. 3-25-1, Hyakunincho, Shinjuku-ku, Tokyo 169-0073, Japan
For local office contact information, visit www.gelifesciences.com/contact
18102229 AF 03/2016
GE Healthcare Bio-Sciences AB
Björkgatan 30
751 84 Uppsala
Sweden
Affinity Chromatography
Vol. 3: Specific Groups of Biomolecules
GE Healthcare
imagination at work
imagination at work
imagination at work
Handbooks from GE Healthcare Life Sciences
For more information refer to www.gelifesciences.com/handbooks
Affinity Chromatography
Vol. 1: Antibodies
GE Healthcare
Affinity Chromatography
Vol. 1: Antibodies
18103746
Affinity Chromatography
Vol. 2: Tagged Proteins
GE Healthcare
Affinity Chromatography
Vol. 2: Tagged Proteins
18114275
Affinity Chromatography
Vol. 3: Specific Groups of Biomolecules
GE Healthcare
Affinity Chromatography
Vol. 3: Specific Groups of
Biomolecules
18102229
GE Healthcare
Life Sciences
ÄKTA
™
Laboratory-scale
Chromatography Systems
Instrument Management Handbook
ÄKTA Laboratory-scale
Chromatography
Systems
Instrument Management
Handbook
29010831
GE Healthcare
Life Sciences
Biacore
™
Assay Handbook
Biacore Assay
Handbook
29019400
GE Healthcare
Life Sciences
Biacore
Sensor Surface Handbook
Biacore Sensor Surface
Handbook
BR100571
GE Healthcare
Life Sciences
Cell Separation Media
Methodology and applications
Cell Separation Media
Methodology and
Applications
18111569
Size Exclusion Chromatography
Principles and Methods
GE Healthcare
Life Sciences
Size Exclusion
Chromatography
Principles and Methods
18102218
GE Healthcare
Life Sciences
GST Gene
Fusion System
Handbook
GST Gene Fusion System
Handbook
18115758
GE Healthcare
Life Sciences
High-throughput
Process Development
with PreDictor
™
Plates
Principles and Methods
High-throughput Process
Development with
PreDictor Plates
Principles and Methods
28940358
Hydrophobic Interaction
and Reversed Phase
Chromatography
Principles and Methods
GE Healthcare
Life Sciences
Hydrophobic Interaction
and Reversed Phase
Chromatography
Principles and Methods
11001269
GE Healthcare
Life Sciences
Imaging
Principles and Methods
Laser
CCD
IRUV
IRUV
trans
epi
630
710
520
W
460365
312
473 532 635650 685
785
epi
Imaging
Principles and Methods
29020301
Ion Exchange
Chromatography
Principles and Methods
GE Healthcare
Ion Exchange
Chromatography
Principles and Methods
11000421
GE Healthcare
Life Sciences
Isolation of
mononuclear cells
Methodology and applications
Isolation of
Mononuclear Cells
Methodology and
Applications
18115269
GE Healthcare
Life Sciences
Microcarrier
Cell Culture
Principles and Methods
Microcarrier Cell Culture
Principles and Methods
18114062
imagination at work
GE Healthcare
Life Sciences
Multimodal
Chromatography
Handbook
Multimodal
Chromatography
Handbook
29054808
GE Healthcare
Life Sciences
Nucleic Acid Sample
Preparation for
Downstream Analyses
Principles and Methods
Nucleic Acid Sample
Preparation for
Downstream Analyses
Principles and Methods
28962400
GE Healthcare
Life Sciences
Protein Sample
Preparation
Handbook
Protein Sample
Preparation
Handbook
28988741
GE Healthcare
Life Sciences
Purifying
Challenging Proteins
Principles and Methods
Purifying Challenging
Proteins
Principles and Methods
28909531
GE Healthcare
Life Sciences
Spectrophotometry
Handbook
Spectrophotometry
Handbook
29033182
GE Healthcare
Life Sciences
Strategies for
Protein Purif ication
Handbook
Strategies for Protein
Purification
Handbook
28983331
GE Healthcare
Life Sciences
Western Blotting
Principles and Methods
Western Blotting
Principles and Methods
28999897
2-D Electrophoresis
Principles and Methods
GE Healthcare
Life Sciences
2-D Electrophoresis using
Immobilized pH Gradients
Principles and Methods
80642960

Affinity Chromatography
Vol. 3: Specific Groups of Biomolecules
2 18102229 AF
Content
Introduction.........................................................................................................................................9
Symbols .............................................................................................................................................................................10
Common acronyms and abbreviations .............................................................................................................10
Chapter 1
Principles of affinity chromatography ........................................................................................ 13
Components of an affinity chromatography medium ...............................................................................15
Matrix ..........................................................................................................................................................................15
Ligand .........................................................................................................................................................................16
Spacer arms ............................................................................................................................................................17
Chapter 2
Affinity chromatography in practice ........................................................................................... 19
Selection of chromatography media .................................................................................................................19
Selection of format ......................................................................................................................................................19
Selection of equipment ...........................................................................................................................................20
Selection of purification method ..........................................................................................................................21
Preparation of sample and buffers ..............................................................................................................21
Flow rates .................................................................................................................................................................21
Equilibration .............................................................................................................................................................21
Sample application and wash ........................................................................................................................21
Elution .........................................................................................................................................................................22
Re-equilibration .....................................................................................................................................................23
Analysis of results and further steps ..........................................................................................................24
Troubleshooting .............................................................................................................................................................24
Chapter 3
Purification of specific groups of molecules ............................................................................. 27
Purification or removal of albumin ......................................................................................................................27
Blue Sepharose High Performance, Blue Sepharose 6 Fast Flow, Capto Blue,
Capto Blue (high sub)...........................................................................................................................................27
Chromatography media characteristics ...................................................................................................28
Purification options .............................................................................................................................................28
Purification examples .........................................................................................................................................29
Performing a separation ...................................................................................................................................30
Cleaning .....................................................................................................................................................................30
Chemical stability .................................................................................................................................................30
Storage .......................................................................................................................................................................30
Purification or removal of albumin and IgG ....................................................................................................31
Albumin & IgG Depletion Sepharose High Performance...................................................................31
Chromatography medium characteristics ..............................................................................................31
Purification options ..............................................................................................................................................31
Purification examples .........................................................................................................................................32
Performing a separation ..................................................................................................................................32
Storage .......................................................................................................................................................................33
18102229 AF 3
Purification or removal of biotin and biotinylated substances ..............................................................34
Streptavidin Sepharose High Performance .............................................................................................34
Chromatography medium characteristics ..............................................................................................34
Purification options ..............................................................................................................................................34
Purification example ...........................................................................................................................................35
Performing a separation ...................................................................................................................................36
Storage ......................................................................................................................................................................38
Purification or removal of calmodulin-binding proteins:
ATPases, adenylate cyclases, protein kinases, phosphodiesterases, neurotransmitters ........39
Calmodulin Sepharose 4B ................................................................................................................................39
Chromatography medium characteristics ..............................................................................................39
Purification options ..............................................................................................................................................39
Performing a separation ...................................................................................................................................40
Cleaning .....................................................................................................................................................................40
Chemical stability .................................................................................................................................................40
Storage .......................................................................................................................................................................40
Purification or removal of coagulation factors ..............................................................................................41
VIISelect, VIIISelect, IXSelect, Heparin Sepharose High Performance,
Heparin Sepharose 6 Fast Flow, Capto Heparin ...................................................................................41
Chromatography media characteristics ...................................................................................................41
Purification options ..............................................................................................................................................42
Storage .......................................................................................................................................................................44
Performing a separation ...................................................................................................................................44
Cleaning ...................................................................................................................................................................45
Purification or removal of DNA-binding proteins .........................................................................................46
Heparin Sepharose High Performance, Heparin Sepharose 6 Fast Flow, Capto Heparin 46
Chromatography media characteristics ...................................................................................................47
Purification options ..............................................................................................................................................47
Purification examples .........................................................................................................................................48
Performing a separation ...................................................................................................................................49
Cleaning ...................................................................................................................................................................49
Chemical stability .................................................................................................................................................50
Storage .......................................................................................................................................................................50
Cleaning .....................................................................................................................................................................50
Storage .......................................................................................................................................................................50
Purification or removal of fibronectin .................................................................................................................51
Gelatin Sepharose 4B .........................................................................................................................................51
Chromatography medium characteristics ..............................................................................................51
Purification option ................................................................................................................................................51
Performing a separation ...................................................................................................................................51
Cleaning ...................................................................................................................................................................51
Chemical stability .................................................................................................................................................51
Storage ......................................................................................................................................................................51
Purification or removal of glycoproteins and polysaccharides .............................................................52
4 18102229 AF
Con A Sepharose 4B, Lentil Lectin Sepharose 4B, Capto Lentil Lectin .......................................52
Chromatography media screening ..............................................................................................................52
Chromatography media characteristics ...................................................................................................53
Purification options ..............................................................................................................................................53
Purification example ...........................................................................................................................................54
Performing a separation ...................................................................................................................................54
Cleaning ...................................................................................................................................................................55
Chemical stability .................................................................................................................................................55
Storage ......................................................................................................................................................................55
Performing a separation ...................................................................................................................................55
Cleaning ...................................................................................................................................................................56
Chemical stability .................................................................................................................................................56
Storage ......................................................................................................................................................................56
Purification or removal of granulocyte-colony stimulating factor ......................................................57
GCSFSelect ...............................................................................................................................................................57
Chromatography medium characteristics ..............................................................................................57
Purification options ..............................................................................................................................................57
Purification examples .........................................................................................................................................58
Performing a separation ..................................................................................................................................58
Cleaning ...................................................................................................................................................................58
Storage .......................................................................................................................................................................58
Purification or removal of NAD+-dependent dehydrogenases and ATP-dependent kinases 59
Blue Sepharose 6 Fast Flow, Capto Blue, Capto Blue (high sub) ....................................................59
Performing a separation ...................................................................................................................................59
Purification or removal of NADP+-dependent dehydrogenases and other enzymes with affinity
for NADP+ ..........................................................................................................................................................................60
2’5’ ADP Sepharose 4B ......................................................................................................................................60
Chromatography medium characteristics .............................................................................................60
Purification options ..............................................................................................................................................61
Purification example ...........................................................................................................................................61
Performing a separation ...................................................................................................................................61
Cleaning .....................................................................................................................................................................62
Chemical stability .................................................................................................................................................62
Storage ......................................................................................................................................................................62
Purification or removal of proteins and peptides with exposed amino acids: His,
Cys, Trp, and/or with affinity for metal ions .....................................................................................................63
Chelating Sepharose High Performance, Chelating Sepharose Fast Flow,
Capto Chelating .....................................................................................................................................................63
Chromatography media characteristics .................................................................................................63
Purification options ..............................................................................................................................................64
Performing a separation ...................................................................................................................................65
Scaling up .................................................................................................................................................................66
Cleaning ...................................................................................................................................................................66
Chemical stability .................................................................................................................................................66
Storage ......................................................................................................................................................................66
Purification or removal of serine proteases, such as thrombin and trypsin, and zymogens ...........67
18102229 AF 5
Benzamidine Sepharose 4 Fast Flow (low sub),
Benzamidine Sepharose 4 Fast Flow (high sub) ...................................................................................67
Chromatography media characteristics .................................................................................................68
Purification options ..............................................................................................................................................68
Purification examples .........................................................................................................................................68
Cleaning .....................................................................................................................................................................70
Chemical stability .................................................................................................................................................70
Storage ......................................................................................................................................................................70
Purification or removal of viruses including adeno-associated virus ................................................71
Capto DeVirS, AVB Sepharose High Performance ................................................................................71
Chromatography media characteristics ...................................................................................................71
Purification options ..............................................................................................................................................72
Purification examples .........................................................................................................................................72
Performing a separation ...................................................................................................................................73
Cleaning ...................................................................................................................................................................74
Storage .......................................................................................................................................................................74
BioProcess chromatography media (resins) for AC......................................................................................74
Custom Designed Media ..................................................................................................................................74
Chapter 4
Designing affinity chromatography media using preactivated matrices ........................... 75
Choosing the matrix ....................................................................................................................................................75
Choosing the ligand and spacer arm .................................................................................................................75
Choosing the coupling method .............................................................................................................................76
Coupling the ligand ......................................................................................................................................................77
Binding capacity, ligand density, and coupling efficiency .......................................................................78
Binding and elution conditions ..............................................................................................................................79
Coupling through the primary amine of a ligand .........................................................................................80
NHS-activated Sepharose High Performance, NHS-activated Sepharose 4 Fast Flow ........80
Chromatography media characteristics ...................................................................................................81
Purification options ..............................................................................................................................................81
Purification example ..........................................................................................................................................82
Performing a purification ..................................................................................................................................82
Buffer preparation ................................................................................................................................................82
Ligand and column preparation ...................................................................................................................82
Ligand coupling .....................................................................................................................................................83
Washing and deactivation ...............................................................................................................................83
Storage .......................................................................................................................................................................83
CNBr-activated Sepharose...............................................................................................................................84
Chromatography media characteristics ...................................................................................................84
Purification options ..............................................................................................................................................84
Purification example ..........................................................................................................................................85
Performing a separation ...................................................................................................................................85
Ligand preparation ..............................................................................................................................................86
Ligand coupling .....................................................................................................................................................86
Storage .......................................................................................................................................................................87
Immunoaffinity chromatography .........................................................................................................................88
6 18102229 AF
Coupling small ligands through carboxyl groups via a spacer arm ...................................................89
EAH Sepharose 4B ................................................................................................................................................89
Chromatography medium characteristics ..............................................................................................89
Purification options ..............................................................................................................................................90
Preparation of coupling reagent ...................................................................................................................90
Preparation of EAH Sepharose 4B ................................................................................................................90
Ligand preparation ..............................................................................................................................................91
Ligand coupling ....................................................................................................................................................91
Storage .......................................................................................................................................................................91
Performing a separation ...................................................................................................................................91
Coupling through hydroxy, amino, or thiol groups via a 12-carbon spacer arm ..........................92
Epoxy-activated Sepharose 6B .....................................................................................................................92
Chromatography medium characteristics ..............................................................................................92
Purification options ..............................................................................................................................................92
Purification example ...........................................................................................................................................93
Alternative coupling solutions ........................................................................................................................93
Coupling procedure .............................................................................................................................................93
Storage .......................................................................................................................................................................94
Coupling other functional groups ........................................................................................................................95
Chapter 5
Magnetic beads for affinity chromatography ........................................................................... 97
General magnetic bead separation steps ................................................................................................99
Dispensing the medium slurry .......................................................................................................................99
Handling liquids .....................................................................................................................................................99
Incubation ................................................................................................................................................................99
Purification or removal of biotin and biotinylated biomolecules with magnetic beads ............. 100
Streptavidin Mag Sepharose, Sera-Mag Streptavidin coated,
Sera-Mag SpeedBeads Streptavidin-Coated, Sera-Mag SpeedBeads
Streptavidin-Blocked, Sera-Mag SpeedBeads Neutravidin-Coated .........................................100
Bead characteristics ........................................................................................................................................100
Purification examples ......................................................................................................................................102
Performing a separation: Streptavidin Mag Sepharose ................................................................. 102
Sample preparation .......................................................................................................................................... 103
Performing a separation: Sera-Mag SpeedBeads Streptavidin-Blocked ...............................105
Sample preparation .......................................................................................................................................... 105
Purification or removal of phosphorylated biomolecules .....................................................................106
TiO
2
Mag Sepharose .........................................................................................................................................106
Bead characteristics ........................................................................................................................................ 106
Purification examples ......................................................................................................................................106
Performing a separation ................................................................................................................................ 107
MS analysis ...........................................................................................................................................................108
Preactivated magnetic beads .............................................................................................................................109
NHS Mag Sepharose, Sera-Mag Carboxylate-Modified,
Sera-Mag SpeedBeads Carboxylate-Modified ....................................................................................109
Bead characteristics ........................................................................................................................................ 110
Purification example ........................................................................................................................................ 110
Performing a separation ................................................................................................................................ 111
Chapter 6
18102229 AF 7
Affinity chromatography in a purification strategy (CIPP) ..................................................... 117
Applying C
IPP ................................................................................................................................................................. 118
Selection and combination of purification techniques ...........................................................................118
Appendix 1
Sample preparation ...................................................................................................................... 123
Sample stability ..........................................................................................................................................................123
Sample clarification .................................................................................................................................................. 123
Centrifugation ..................................................................................................................................................... 124
Filtration ................................................................................................................................................................. 124
Desalting ............................................................................................................................................................... 125
Specific sample preparation steps ................................................................................................................... 125
Fractional precipitation .......................................................................................................................................... 125
Ammonium sulfate precipitation ...............................................................................................................126
Resolubilization of protein precipitates ..................................................................................................128
Buffer exchange and desalting .......................................................................................................................... 128
Removal of lipoproteins ..........................................................................................................................................130
Removal of phenol red ............................................................................................................................................130
Removal of LMW contaminants .........................................................................................................................130
Appendix 2
Selection of purification equipment .......................................................................................... 131
Appendix 3
Column packing and preparation ............................................................................................. 132
Column packing and efficiency ..........................................................................................................................134
Custom column packing ........................................................................................................................................ 135
Appendix 4 ...................................................................................................................................... 136
Converting from flow velocity to volumetric flow rates ....................................................... 136
From flow velocity (cm/h) to volumetric flow rate (ml/min) .................................................................136
From volumetric flow rate (ml/min) to flow velocity (cm/h) ...................................................................136
From volumetric flow rate (ml/min) to using a syringe ...........................................................................136
Appendix 5
Conversion data: proteins, column pressures ......................................................................... 137
Proteins ...........................................................................................................................................................................137
Column pressures ...................................................................................................................................................... 137
Appendix 6 ...................................................................................................................................... 138
Table of amino acids ..................................................................................................................... 138
Appendix 7
Analytical assays during purification ....................................................................................... 140
Total protein determination .................................................................................................................................. 140
Purity determination ................................................................................................................................................140
Functional assays .......................................................................................................................................................141
Detection and assay of tagged proteins ........................................................................................................ 142
Appendix 8
8 18102229 AF
Storage of biological samples ..................................................................................................... 143
General recommendations ................................................................................................................................... 143
Specific recommendations for purified proteins........................................................................................ 143
Related literature .......................................................................................................................... 144
Ordering information ................................................................................................................... 145
Product index ................................................................................................................................. 148
18102229 AF 9
Introduction
Biomolecules are purified using purification techniques that separate according to differences
in specific properties, as shown in Figure I.1.
Property Technique
Biorecognition (ligand specificity) Affinity chromatography (AC)
Charge Ion exchange chromatography (IEX)
Size Size exclusion chromatography (SEC),
also called gel filtration (GF)
Hydrophobicity Hydrophobic interaction chromatography (HIC)
Reversed phase chromatography (RPC)
Fig I.1. Separation principles in chromatographic purification.
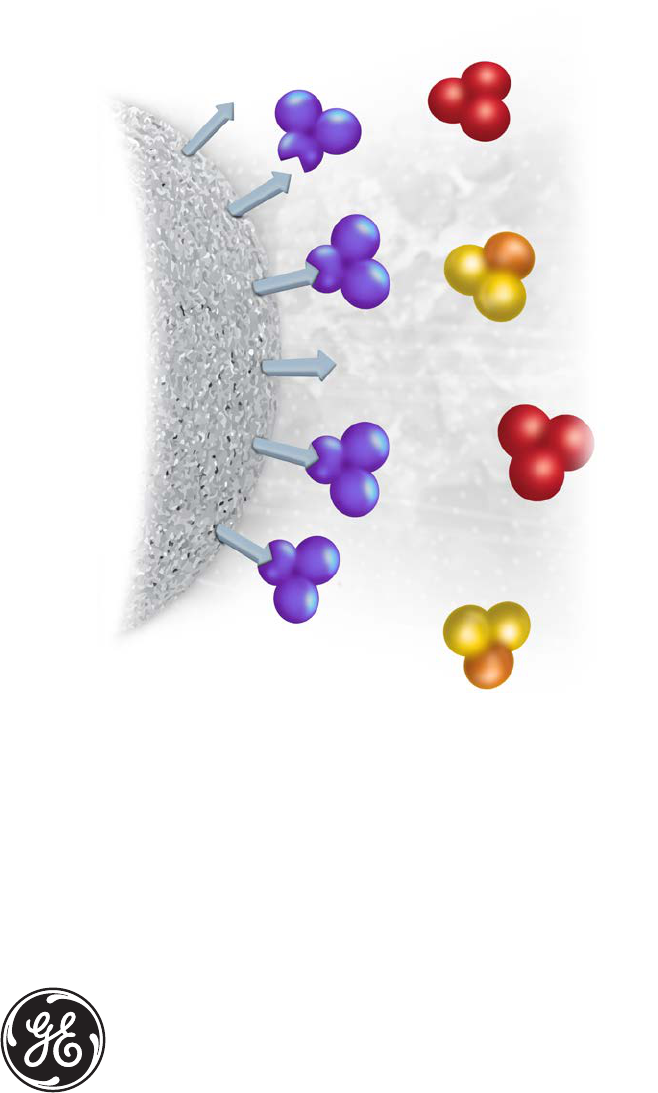
Affinity chromatography (AC) separates proteins on the basis of a reversible interaction
between a protein (or group of proteins) and a specific ligand coupled to a chromatography
matrix. The technique offers high selectivity, hence high resolution, and usually high capacity
for the protein(s) of interest. Purification can be in the order of several thousand-fold and
recoveries of active material are generally very high.
AC is the only chromatography technique that enables the purification of a biomolecule on
the basis of its biological function or individual chemical structure. Purification that would
otherwise be time-consuming, difficult, or even impossible using other techniques can often
be easily achieved with AC. The technique can be used to separate active biomolecules from
denatured or functionally different forms, to isolate pure substances present at low concentration
in large volumes of crude sample and also to remove specific contaminants.
GE Healthcare’s Life Sciences business offers a wide variety of prepacked columns, ready-to-use
chromatography media, and preactivated media for ligand coupling.
The Affinity Chromatography handbook is divided into three volumes:
Affinity Chromatography, Vol. 1: Antibodies
Affinity Chromatography, Vol. 2: Tagged Proteins
Affinity Chromatography, Vol. 3: Specific Groups of Biomolecules
Size exclusion Hydrophobic
interaction
Ion exchange Affinity Reversed phase

10 18102229 AF
This handbook describes the role of AC in the purification of specific groups of biomolecules,
the principle of the technique, the chromatography media available and how to select them,
application examples, and detailed instructions for the most commonly performed procedures.
Practical information is given as a guide towards obtaining the desired results.
The illustration on the inside cover shows the range of handbooks that have been produced
by GE to ensure that purification with any chromatographic technique becomes a simple and
efficient procedure at any scale and in any laboratory.
Symbols
This symbol indicates general advice on how to improve procedures or recommends
measures to take in specific situations
This symbol indicates where special care should be taken
Highlights chemicals, buffers, and equipment
Outline of experimental protocol
Common acronyms and abbreviations
A
280
UV absorbance at specified wavelength (in this example, 280 nm)
AC affinity chromatography
AIEX anion exchange chromatography
APMSF 4-aminophenyl-methylsulfonyl fluoride
AU absorbance units
BSA bovine serum albumin
cGMP current good manufacturing practice
CF chromatofocusing
CHO Chinese hamster ovary
CIEX cation exchange chromatography
CIP cleaning-in-place
CIPP capture, intermediate purification, polishing
CV column volume
Dab domain antibody, the smallest functional entity of an antibody
DNA deoxyribonucleic acid
DNAse deoxyribonuclease
DOC deoxycholate
DoE design of experiments
DS desalting (group separation by size exclusion chromatography;
buffer exchange)
EDAC 1-ethyl-(3-dimethylaminopropyl)carboiimide
EDTA ethylene diaminetetraacetic acid
EGTA ethylene glycol-O,O’-bis-[2-amino-ethyl]-N,N,N’,N’,-tetraacetic acid
ELISA enzyme-linked immunosorbent assay
F(ab’)
2
fragment
fragment with two antigen binding sites, obtained by pepsin digestion
Fab fragment antigen binding fragment obtained by papain digestion
Fc fragment crystallizable fragment obtained by papain digestion
18102229 AF 11
Fv fragment unstable fragment containing the antigen binding domain
GF gel filtration; also called size exclusion chromatography
GST glutathione S-transferase
HCP host cell protein
HIC hydrophobic interaction chromatography
HMW high molecular weight
HSA human serum albumin
IEF isoelectric focusing
IEX ion exchange chromatography
IgA, IgG etc. different classes of immunoglobulin
IMAC Immobilized metal ion affinity chromatography
LC-MS liquid chromatography–mass spectrometry
LMW low molecular weight
MAb monoclonal antibody
MALDI-ToF Matrix-assisted laser desorption/ionization time-of-flight
mo month
MPa megaPascal
M
r
relative molecular weight
MS mass spectrometry
n native, as in nProtein A
NC nitrocellulose
NHS N-hydroxysuccinimide
PAGE polyacrylamide gel electrophoresis
PBS phosphate buffered saline
PEG polyethylene glycol
pI isoelectric point, the pH at which a protein has zero net surface charge
PMSF phenylmethylsulfonyl fluoride
psi pounds per square inch
PVDF polyvinylidene fluoride
PVP polyvinylpyrrolidine
r recombinant, as in rProtein A
RNA ribonucleic acid
RNAse ribonuclease
RPC reversed phase chromatography
scFv single chain Fv fragment
SDS sodium dodecyl sulfate
SDS-PAGE sodium dodecyl sulfate polyacrylamide gel electrophoresis
SEC size exclusion chromatography
TCEP tris(2-carboxyethyl) phosphine hydrochloride
TFA Trifluoroacetic acid
Tris tris-(hydroxymethyl)-aminomethane
UV ultraviolet
v/v volume to volume
w week
w/v weight to volume
12 18102229 AF
18102229 AF 13
Chapter 1
Principles of affinity chromatography
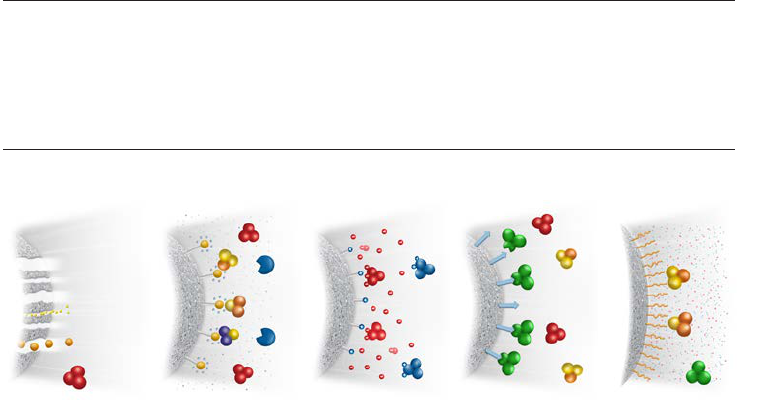
Affinity chromatography (AC) separates biomolecules on the basis of a reversible interaction
between a biomolecule (or group of biomolecules) and a specific ligand coupled to a
chromatography matrix. Figure 1.1 shows the key stages in an affinity purification. The
technique is an excellent choice for a capture or intermediate step in a purification protocol
and can be used whenever a suitable ligand is available for the target molecule(s) of interest.
With high selectivity, hence high resolution, and high capacity, purification levels in the
order of several thousand-fold with high recovery of active material are achievable. Target
biomolecule(s) is collected in a purified, concentrated form.
Column volumes (CV)
begin sample
application
change to
elution buffer
equilibration
adsorption of
sample and
elution of
unbound material
wash
away
unbound
material
elute
bound
protein(s)
Absorbance
re-equilibration
4. Re-equilibration
AC medium is re-equilibrated with binding buffer.
3. Elution
Target protein is recovered by changing conditions
to favor elution of the bound molecules. Target protein
is collected in a purified, concentrated form.
2. Sample application and wash
Sample is applied under conditions that favor specific
binding of the target molecule(s) to a complementary
binding substance (the ligand). Target substances bind
specifically, but reversibly, to the ligand and unbound
material washes through the column.
1. Equilibration
AC medium is equilibrated in binding buffer.
Fig 1.1. Principles of affinity purification.
14 18102229 AF
Biological interactions between ligand and target molecule can be a result of electrostatic or
hydrophobic interactions, van der Waals’ forces, and/or hydrogen bonding. To elute the target
molecule from the AC medium, the interaction can be reversed, either specifically using a
competitive ligand, or nonspecifically, by changing the pH, ionic strength, or polarity.
In a single step, affinity purification can offer immense time-saving over less selective
multistep procedures. The concentrating effect enables large volumes to be processed. Target
molecules can be purified from complex biological mixtures, native forms can be separated
from denatured forms of the same substance and small amounts of biological material can be
purified from high levels of contaminating substances.
Any component can be used as a ligand to purify its respective binding partner. Some typical
biological interactions, frequently used in AC, are listed below:
• Antibody antigen, virus, cell (see the handbook Affinity Chromatography, Vol. 1: Antibodies,
18103746).
• Metal ions Histidine- (his)-tagged proteins (see the handbook Affinity Chromatography,
Vol. 2: Tagged Proteins, 18114275).
• Glutathione glutathione-S-transferase or GST-tagged proteins (see Affinity
Chromatography, Vol. 2: Tagged Proteins).
• Enzyme substrate analog, inhibitor, cofactor (this handbook).
• Lectin polysaccharide, glycoprotein (this handbook).
AC is also used to remove specific contaminants, for example Benzamidine Sepharose™ 6
Fast Flow can remove serine proteases, such as thrombin and Factor Xa.

18102229 AF 15
Components of an affinity chromatography medium
Matrix: for ligand attachment. Matrix should be chemically and
physically inert.
Spacer arm: used to improve binding between ligand and target
molecule by overcoming any effects of steric hindrance.
Ligand: molecule that binds reversibly to a specific target
molecule or group of target molecules.
Fig 1.2. The three components of an AC medium.
Matrix
The matrix is an inert support to which a ligand can be directly or indirectly coupled (Fig 1.2).
The list below highlights many of the properties required for an efficient and effective
chromatography matrix.
• Extremely low nonspecific adsorption, essential since AC relies on specific interactions.
• Easily derivatized groups for covalent attachment of a ligand.
• An open pore structure to ensure high capacity binding even for large biomolecules, since
the interior of the matrix is available for ligand attachment.
• Good flow properties for rapid separation.
• Stability under a range of experimental conditions such as high and low pH, detergents, and
dissociating agents.
Sepharose, a bead-form of agarose (Fig 1.3), provides many of these properties.
CH OH
2
HO
O
HO
O
O
D
-galactose
O
HO
O
3,6 anhydro
L
-galactose
O
Agarose Structure of cross-linked agarose
chromatography media
Fig 1.3. Partial structure of agarose chromatography media (Sepharose).

16 18102229 AF
Sepharose has been modified and developed to further enhance these excellent properties,
resulting in a selection of matrices chosen to suit the particular requirements for each
application (see Table 1.1). A more rigid agarose matrix is used for Capto™ chromatography
media. The average particle sizes of Capto matrices used for AC media are either 75 or 90 µm.
Table 1.1. Sepharose and Capto matrices used with GE affinity chromatography media
Chromatography medium Base matrix Average particle size (µm)
Sepharose High Performance 6% highly cross-linked agarose 34
Sepharose 6 Fast Flow 6% highly cross-linked agarose 90
Sepharose 4 Fast Flow 4% highly cross-linked agarose 90
Sepharose 6B 6% agarose 90
Sepharose 4B 4% agarose 90
Capto Highly cross-linked high-flow agarose 75 and 90
In AC, the particle size and porosity are designed to maximize the surface area available for
coupling a ligand and binding the target molecule. A small average particle size with high
porosity increases the surface area. Increasing the degree of cross-linking of the matrix
improves the chemical stability, in order to tolerate potentially harsh elution and wash
conditions, and creates a rigid matrix that can withstand high flow rates. These high flow
rates, although not always used during a separation, save considerable time during column
equilibration and cleaning procedures.
Ligand
The ligand is the molecule that binds reversibly to a specific molecule or group of molecules,
enabling purification by AC.
The selection of the ligand for AC is influenced by two factors: the ligand must exhibit specific
and reversible binding affinity for the target substance(s) and it must have chemically
modifiable groups that allow it to be attached to the matrix without destroying binding activity.
The dissociation constant (k
D
) for the ligand-target complex should ideally be in the
range 10
-4
to 10
-8
M in free solution. If the dissociation constant is outside the useful
range, changing elution methods can help to promote successful AC.
If no information on the strength of the binding complex is available, a trial-and-error approach
should be used.
For purification of specific molecules or groups of molecules, many ligands are
available coupled to an appropriate matrix (see Chapter 3). Ligands can also be isolated
and purified to prepare a specific AC medium for a specific target molecule. Coupling
of ligands to preactivated matrices is described in Chapter 4.
18102229 AF 17
Spacer arms
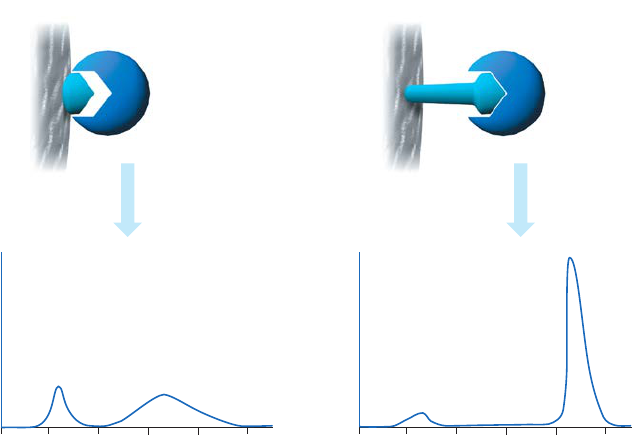
The binding site of a target protein is often located deep within the molecule and an AC medium
prepared by coupling small ligands, such as enzyme cofactors, directly to Sepharose may exhibit
low binding capacity due to steric interference i.e. the ligand is unable to access the binding
site of the target molecule, as shown in Figure 1.4 (A). In these circumstances a “spacer arm” is
interposed between the matrix and the ligand to facilitate effective binding. Spacer arms must
be designed to maximize binding, but to avoid nonspecific binding effects. Figure 1.4 (B) shows
the improvement that can be seen in a purification as the spacer arm creates a more effective
environment for binding.
Volume (ml)
Inefficient binding
Target protein elutes during
binding and elution
0 5 10 15 20 25
Volume (ml)
0 5 10 15 20 25
A
280
A
280
Efficient binding
Target protein elutes
in a single peak
(A) (B)
Fig 1.4. The effect of spacer arms. (A) Ligand attached directly to the matrix. (B) Ligand attached to the
matrix via a spacer arm.
18 18102229 AF

18102229 AF 19
Chapter 2
Affinity chromatography in practice
This chapter provides guidance and advice that is generally applicable to any AC purification.
The first steps towards a successful purification starts with a number of selections aiming for the
most suitable chromatography medium, format, equipment, and purification method (Fig 2.1).
The choices depend on factors such as the purpose of the purification, the purification scale, and
the required purity and yield.
A
280
Binding
buffer
Elution
buffer
Eluted
target
Flowthrough
(unbound material)
Volume (ml)
Media Format Equipment Method
Fig 2.1. Successful AC purification requires making the right initial choices.
Selection of chromatography media
A suitable AC medium has a ligand which interacts reversibly with the target molecule or group
of molecules. Media with ligands for purification of, for example, enzymes, coagulation factors,
and proteases, are described in Chapters 3 and 5. The media can be used immediately for
purification without any prepreparation, simply following the supplied purification protocol.
Preactivated chromatography media are useful when no ready to use media are available for the
purpose. This requires a specific biomolecule (often an antibody) directed towards the target protein.
The specific biomolecule is used as a ligand and covalently coupled to the preactivated media.
The media can then be used for affinity purification of the target protein (see Chapters 4 and 5).
In addition to the ligand, the matrix of the chromatography medium affects the purification
(see Chapter 1). The most suitable matrix can be selected according to the degree of resolution,
binding capacity, and the scale desired for the separation. For example, performing gradient
elution on Sepharose High Performance (34 µm) will result in high-resolution separations.
Media with larger particles such as Sepharose Fast Flow and Capto have better pressure/flow
properties and are suitable for small-scale purification as well as for scaling up.
Selection of format
A number of prepacked formats are available from GE to facilitate and speed up the affinity
purification. Prepacked HiTrap™ (1 and 5 ml media, bed height 2.5 cm) and HiScreen™ (4.7 ml
medium, bed height 10 cm) columns provide flexibility as they can be operated using a syringe,
pump, or chromatography system. The columns are useful for fast method development before
scaling up as well as for small-scale purification. The prepacked HiPrep™ column (20 ml) is
suitable for preparative purification, and chromatography media can also be packed in XK,
Tricorn™, or HiScale™ columns for larger scale purification.
Figure 2.2 shows the simple procedure to perform a typical affinity purification using prepacked
HiTrap columns. The different method steps are discussed more in detail later in this chapter.

20 18102229 AF
Equilibrate column
with binding buffer
Apply sample.
Wash with binding buffer
W
aste
Collect
elution buffer
Collect fractions
5 min 5 to 15 min
5 min
Elute with
Fig 2.2. The purification procedure consists of equilibration, sample application, wash, and elution.
In addition, some AC media are available in other formats, such as small-scale SpinTrap™
columns and MultiTrap™ 96-well plates (Fig 2.3). Prepacked SpinTrap columns are used
together with a microcentrifuge and can be an alternative to screening in MultiTrap 96-well
plate format when fewer samples are to be screened.
Fig 2.3. Examples of different prepacked formats. MultiTrap 96-well plates, SpinTrap columns, and HiTrap
columns are designed for fast and convenient screening and small-scale AC purification.
Purification can also be performed in batch mode, where the loose chromatography medium is
used directly in a container or test tube together with buffers and sample. This allows for increased
binding time, for example, the sample can be incubated with the chromatography medium
overnight. After binding, the chromatography medium can be poured into an empty gravity-flow
column before wash and elution.
Avoid using magnetic stirrers when the medium is used in batch mode as they can
damage the chromatography beads. Use mild rotation or end-over-end stirring.
Another example of batch mode purification is using magnetic beads in combination with a
magnetic device. This approach is discussed in detail in Chapter 5.
Selection of equipment
The selection of equipment depends on the purpose of the purification. A simple stepwise
purification can for example be performed using a HiTrap column and a syringe for the buffers
and sample. More advanced methods require a chromatography system; Appendix 2 provides
a guide for the selection of ÄKTA™ chromatography systems.

18102229 AF 21
Selection of purification method
AC media are supplied with purification methods for the specific media. These methods
are often sufficient for a successful purification, but in some cases additional optimization
of the method might be required. A purification method consists of several different steps:
equilibration, sample application, wash, elution and re-equilibration (see Fig 1.1, Chapter 1). As
each step has an impact on the final results, the different steps are described in detail below.
Preparation of sample and buffers
Adjust the sample to the composition and pH of the binding buffer. This will promote efficient
binding and can be done by performing a buffer exchange with a desalting column or simply
by dilution in the binding buffer. Samples should also be clear and free from particulate matter
in order to avoid clogging the column and reduce the need for stringent washing procedures.
Appendix 1 contains an overview of sample preparation techniques.
Binding and elution buffers are specific for each AC medium since their influence on the
interaction between the target molecule and the ligand affects the affinity-based separation.
The instructions supplied with the AC medium contain suggested binding and elution buffers.
Use high-quality water and chemicals. Solutions should be filtered through 0.22 or
0.45 µm filters.
Flow rates
The optimal flow rate in AC depends on the dissociation rates of ligand/target molecule
interactions and varies widely. For ready-to-use AC media, follow the supplied instructions and,
if required, optimize:
• the flow rate to achieve efficient binding
• the flow rate for elution to maximize recovery
To obtain sharp elution curves and maximum recovery with minimum dilution of separated
molecules, use the lowest acceptable flow rate.
Equilibration
Equilibration of the AC medium with binding buffer is necessary since any remaining storage
solution might disturb the binding of the target protein. Wash away the storage solution
thoroughly according to the instructions. If the medium is supplied as a freeze-dried powder,
reswell the medium in the correct buffer according to the instructions.
Sample application and wash
The column must be equilibrated in binding buffer before beginning sample application.
The sample volume is not critical and does not affect the separation since AC is a binding
technique. For interactions with weak affinity and/or slow equilibrium, a lower flow rate might
be required; alternatively the purification can be performed in batch mode with increased time
for binding.
Wash the column/medium thoroughly after sample application until all unbound material has
been washed away, as determined by UV absorbance at 280 nm. This will improve the purity of
the eluted target protein.
If possible, test the affinity of the ligand-target molecule interaction. Too low affinity will
result in poor yields since the target protein can wash through or leak from the column
during sample application. Too high affinity will result in low yields since the target
molecule might not dissociate from the ligand during elution.
22 18102229 AF
Elution
AC media from GE are supplied with recommendations for the most suitable elution buffer to
reverse the interaction between the ligand and target protein. Elution methods may be either
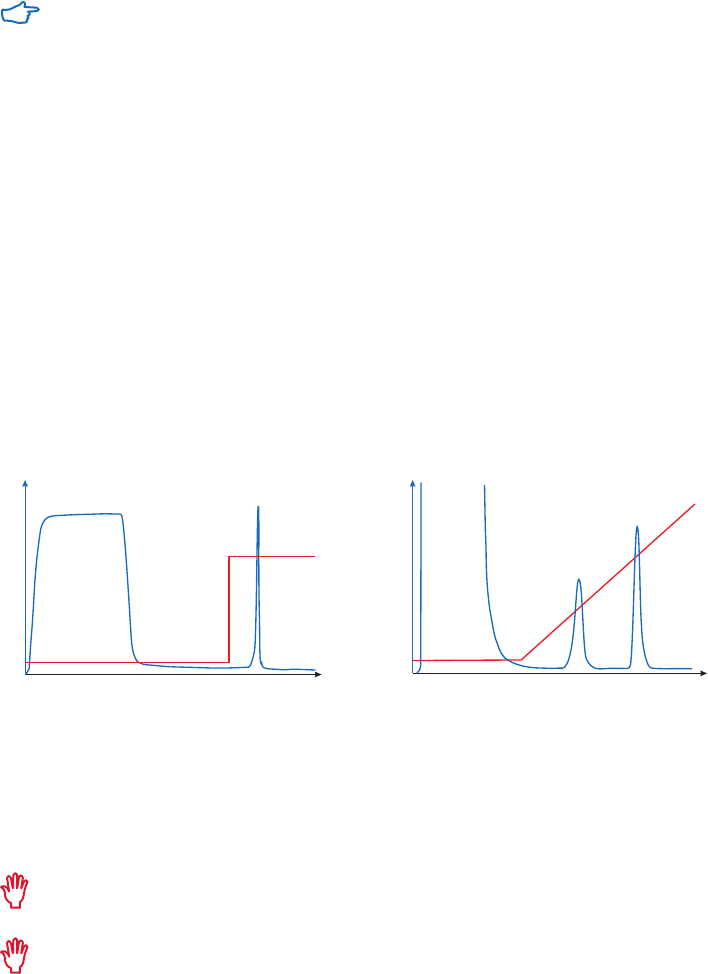
selective or nonselective, as shown in Figure 2.4.
Method 1
The simplest case. A change of buffer
composition elutes the bound substance
without harming either it or the ligand.
Method 2
Extremes of pH or high concentrations of
chaotropic agents are required for elution,
but these can cause permanent or
temporary damage.
Methods 3 and 4
Specific elution by addition of a substance
that competes for binding. These methods
can enhance the specificity of media that
use group-specific ligands.
1
2
3
4
Fig 2.4. Elution methods in AC.
Ionic-strength elution
The exact mechanism for elution by changes in ionic strength will depend upon the specific
interaction between the ligand and target protein. This is a mild elution using a buffer with
increased ionic strength (usually NaCl), applied as a linear gradient or in steps.
pH elution
A change in pH alters the degree of ionization of charged groups on the ligand and/or the
bound protein. This change can affect the binding sites directly reducing their affinity, or cause
indirect changes in affinity by alterations in conformation.
If low pH must be used, collect fractions into neutralization buffer such as 1 M Tris-HCl, pH 9.0
(60 to 200 µl/ml eluted fraction) to return the fraction to a neutral pH. The column should also
be re-equilibrated to neutral pH immediately.
Competitive elution
Selective eluents are often used to separate substances on a group-specific chromatography
medium or when the binding affinity of the ligand/target protein interaction is relatively high.
The eluting agent competes either for binding to the target protein or for binding to the ligand.
Substances may be eluted either by a gradient or step elution (see below).
For elution, it is common to use a concentration 10-fold higher than that of the ligand.
Other elution methods
Substances that reduce the polarity of the buffer can facilitate elution without affecting protein
activity, such as dioxane (up to 10%) and ethylene glycol (up to 50%).
If other elution methods fail, buffers which alter the structure of proteins can be used, for
example, chaotropic agents such as guanidine hydrochloride or urea. Chaotropes should be
avoided whenever possible since they are likely to denature the eluted protein.
18102229 AF 23
When substances are very tightly bound to the AC medium, it can be useful to stop
the flow for some time after applying eluent (10 min to 2 h is commonly used) before
continuing elution. This gives more time for dissociation to take place and thus helps to
improve recoveries of bound substances.
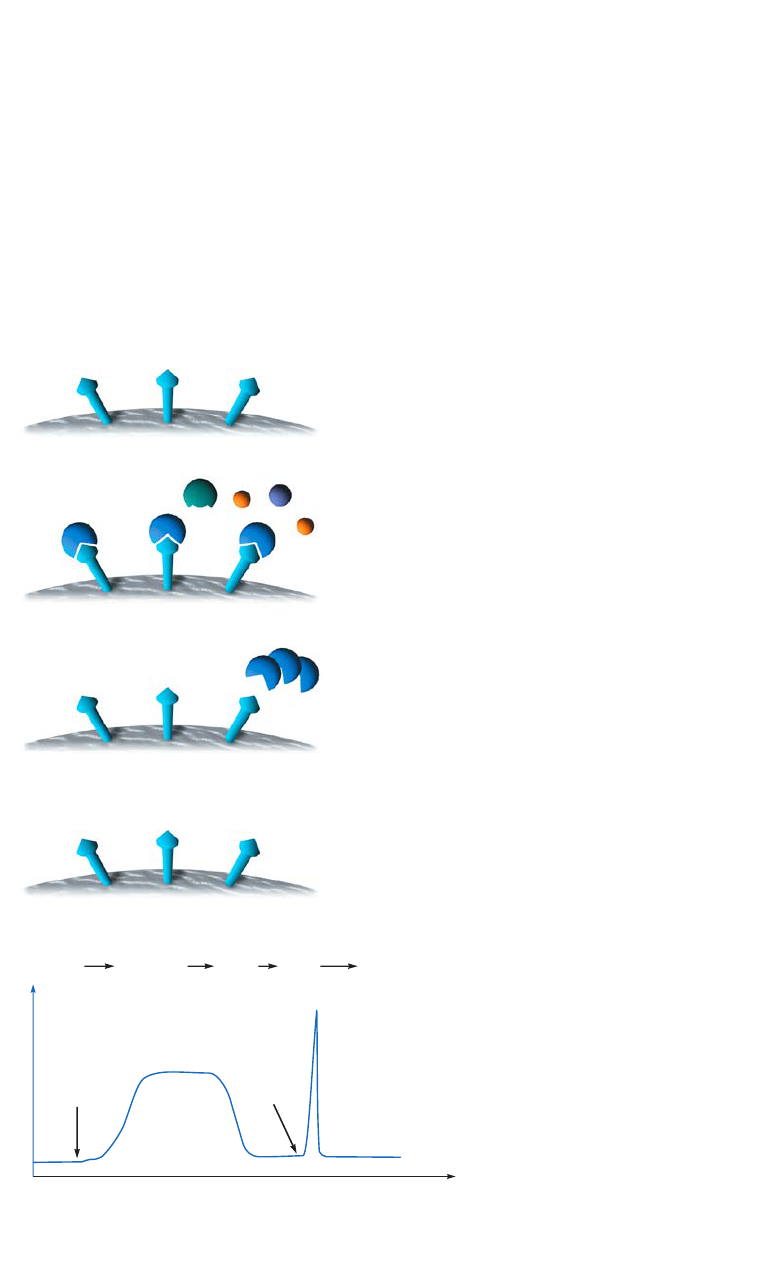
Gradient and step elution
The figures below illustrate the principle of separations in which proteins are eluted using step
elution or linear gradient elution (Fig 2.5).
Step elution can be used for less complex samples or after optimizing using gradient elution.
Changing to a step elution speeds up separation time and reduces buffer consumption. Step
elution can also be used for group separation in order to concentrate the proteins of interest
and rapidly remove them from unwanted substances.
Gradient elution is often used when starting from an unknown sample (the components
are bound to the column and eluted differentially to give a total protein profile) and for
development of a purification method. The position of the eluted peaks can give information
about the optimal binding and elution conditions to be used in step elution. A chromatography
system is essential when gradient elution is performed.
A
280
A
280
Time/volume
Time/volume
Binding
conditions
Elution
conditions
Binding
conditions
Linear
change
in elution
conditions
(A) (B)
A
280
A
280
Time/volume
Time/volume
Binding
conditions
Elution
conditions
Binding
conditions
Linear
change
in elution
conditions
Fig 2.5. Typical conditions for (A) step and (B) gradient elution in AC.
Re-equilibration
After elution, the AC medium needs to be re-equilibrated before the next purification run.
Depending on sample, it might also be necessary to perform additional cleaning, for example if
pressure has increased or if color change is noted.
Reuse of AC media depends on the nature of the sample and should only be considered
when processing identical samples to avoid cross-contamination.
If an AC medium is to be reused routinely, care must be taken to ensure that any
contaminants from the applied sample can be removed by procedures that do not
damage the ligand.

24 18102229 AF
Troubleshooting
This section focuses on practical problems that can occur when running an AC column.
Situation Cause Remedy
Poor binding of the protein. Sample has not been filtered properly. Clean the column, filter the sample, and repeat.
Sample has altered during storage. Prepare fresh samples.
Sample has wrong pH or buffer
conditions are incorrect.
Use a desalting column to transfer sample
into the correct buffer (see Buffer exchange and
desalting in Appendix 1).
Solutions have wrong pH. Calibrate pH meter, prepare new solutions.
The column is not equilibrated
sufficiently in the buffer.
Repeat or prolong the equilibration step.
Column is overloaded with sample. Decrease the sample load.
Microbial growth has occurred in
the column.
Store in 20% ethanol when possible.
Low yield. Protein is still attached to ligand If using competitive elution, increase the
concentration of the competitor in the elution
buffer.
Protein has been degraded by
proteases.
Add protease inhibitors to the sample and
buffers to prevent proteolytic digestion. Run
sample through a medium such as Benzamidine
Sepharose 4 Fast Flow (high sub) to remove
serine proteases.
Adsorption to filter during sample
preparation.
Use another type of filter.
Sample precipitates. Can be caused by removal of salts or unsuitable
buffer conditions.
Hydrophobic proteins. Protein is
still attached to ligand.
Use chaotropic agents, polarity reducing
agents, or detergents.
Analysis of results and further steps
The analysis of the eluted sample can indicate if the purification method needs to be optimized
to increase the yield or achieve higher purity. Commonly used assays are outlined in Appendix 7.
AC offers high selectivity and is often the first and sometimes the only step required. The target
molecule is concentrated into a small volume and purity levels are often above 95%. However,
to achieve satisfactory sample homogeneity, a further polishing step, often size exclusion
chromatography (SEC), might be required to remove any aggregates. SEC is used to separate
molecules according to differences in size, and to transfer the sample into storage buffer, removing
excess salt and other small molecules. The chromatogram will also give an indication of the
homogeneity of the purified sample, see the Size Exclusion Chromatography Handbook, 18102218
from GE. Alternatively, a desalting column that gives low resolution, but high sample capacity, can
be used to quickly transfer the sample into storage buffer and remove excess salt (see Appendix 1).
18102229 AF 25
Situation Cause Remedy
Low recovery of activity, but
normal recovery of protein.
Protein is unstable or inactive in the
elution buffer.
Determine the pH and salt stability of the protein.
Collect fractions into neutralization buffer such
as 1 M Tris-HCl, pH 9.0 (60 to 200 µl per fraction)
if elution is performed at low pH.
Enzyme separated from cofactor or
similar.
Test by pooling aliquots from the fractions and
repeating the assay.
More activity is recovered
than was applied to the
column.
Different assay conditions have
been used before and after the
chromatographic step.
Use the same assay conditions for all the
assays in the purification scheme.
Reduced or poor flow
through the column and/or
too high back pressure.
Presence of lipoproteins or protein
aggregates.
Remove lipoproteins and aggregrates during
sample preparation (see Appendix 1).
Protein precipitation in the column
caused by removal of stabilizing
agents during fractionation.
Modify the eluent to maintain stability.
Clogged column filter. Replace the filter or use a new column. Always
filter samples and buffer before use.
Clogged end-piece, adapter,
or tubing.
Remove and clean or use a new column.
Precipitated proteins. Clean the column using recommended methods or
use a new column.
Bed is too compressed. Repack the column, if possible, or use a new column.
Microbial growth. Store in 20% ethanol when possible.
Back pressure increases
during a run or during
successive runs.
Turbid sample. Improve sample preparation (see Appendix 1).
Improve sample solubility by the addition of
ethylene glycol, detergents, or organic solvents.
Precipitation of protein in the
column filter and/or at the top
of the bed.
Clean using recommended methods. Exchange
or clean filter or use a new column.
Include any additives that were used for initial
sample solubilization in the solutions used for
chromatography.
Bubbles in the bed. Column packed or stored at cool
temperature and then warmed up.
Remove small bubbles by passing degassed
buffer upwards through the column. Take
special care if buffers are used after storage
in a fridge or cold-room. Do not allow column
to warm up in direct sunlight or by placement
in close proximity to heating system. Repack
column if possible (see Appendix 3).
Buffers not properly degassed. Degas buffers thoroughly.
Cracks in the bed. Large air leak in column. Check all connections for leaks. Repack the
column if possible (see Appendix 3).
26 18102229 AF
18102229 AF 27
Chapter 3
Purification of specific groups of molecules
This chapter describes the affinity chromatography media and prepacked formats available
from GE for purification of specific groups of molecules, such as glycoproteins and coagulation
factors. Advice on handling of the different formats is provided and purification protocols
for each format are described. For purification of antibodies and tagged proteins, see the
handbooks Affinity Chromatography, Vol. 1: Antibodies, 18103746 and Vol. 2: Tagged Proteins,
18114275, respectively. A group-specific AC medium has an affinity for a group of related
substances rather than for a single type of molecule. The same general ligand can be used
to purify several substances (for example members of a class of enzymes) without the need
to prepare a new medium for each different substance in the group. Within each group there
is either structural or functional similarity. The specificity of the AC medium derives from the
selectivity of the ligand and the use of selective elution conditions.
AC media can be used either for purification or removal of the target substance. In the case of
removal, the depleted sample is collected during sample application and wash.
Purification or removal of albumin
Blue Sepharose High Performance, Blue Sepharose 6 Fast Flow, Capto Blue,
Capto Blue (high sub)
Albumin binds to Cibacron Blue F3G-A, a synthetic polycyclic dye that acts as an aromatic
anionic ligand binding the albumin via electrostatic and/or hydrophobic interactions. Similar
interactions are seen with coagulation factors, lipoproteins and interferon. Cibacron Blue
F3G-A is linked to Sepharose to create Blue Sepharose AC media (Fig 3.1).
NH
NH
NH
N
N
N
O
NH
2
O
O
SO
3
Na
SO
3
Na
SO
3
Na
Sepharose
Fig 3.1. Partial structure of Blue Sepharose Fast Flow and Blue Sepharose High Performance.
Capto Blue and Capto Blue (high sub) have a more rigid agarose base matrix compared with
Blue Sepharose 6 Fast Flow, which results in improved pressure/flow properties, optimized pore
structure, and high chemical stability to support cleaning-in-place (CIP) procedures.
Use Blue Sepharose or Capto Blue to remove host albumin from mammalian expression
systems, or when the sample is known to contain high levels of albumin that can mask
the visualization of other protein peaks seen by UV absorption.
Advice on the selection of techniques for the removal of albumin during antibody purification
is given in the handbook Affinity Chromatography, Vol. 1: Antibodies, 18103746 from GE.

28 18102229 AF
Cibacron Blue F3G-A also shows certain structural similarities to naturally occurring
molecules, such as the cofactor NAD
+
, that enable it to bind strongly and specifically to
a wide range of proteins including kinases, dehydrogenases, and most other enzymes
requiring adenylyl-containing cofactors.
Chromatography media characteristics
Characteristics of Blue Sepharose and Capto Blue chromatography media are summarized
in Table 3.1.
Table 3.1. Characteristics of Blue Sepharose and Capto Blue chromatography media
Ligand density Composition pH stability
1
Average
particle size
(µm)
Blue Sepharose
High Performance
4 mg/ml Cibacron Blue F3G-A
coupled to Sepharose
High Performance using
the triazine method.
Short term: 3 to 13
Long term: 4 to 12
34
Blue Sepharose 6
Fast Flow
6.7 to 7.9 µmol/ml Cibacron Blue F3G-A
coupled to Sepharose
6 Fast Flow using the
triazine method.
Short term: 3 to 13
Long term: 4 to 12
90
Capto Blue 13 µmol/ml Cibacron Blue F3G-A
coupled to Capto.
Short term: 3 to 13
Long term: 2 to 13.5
75
Capto Blue
(high sub)
18 µmol/ml Cibacron Blue F3G-A
coupled to Capto.
Short term: 3 to 13
Long term: 2 to 13.5
75
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Long term refers
to the pH interval over which the medium is stable over a long period of time without adverse effects on its subsequent
chromatographic performance.
Purification options
Blue Sepharose and Capto Blue are available in chromatography media packs for packing
into empty columns. The media are also available in prepacked columns for convenience.
Purification options for the media and prepacked columns are shown in Table 3.2.
Table 3.2. Purification options for Blue Sepharose and Capto Blue chromatography media and
prepacked columns
Binding capacity
Maximum
operating flow Comments
Blue Sepharose
6 Fast Flow
1
Human serum albumin
(HSA), 18 mg/ml medium
> 750 cm/h
2
Supplied as a suspension ready
for column packing.
HiTrap Blue HP, 1 ml
HiTrap Blue HP, 5 ml
HSA, 20 mg/column
HSA, 100 mg/column
4 ml/min
20 ml/min
Prepacked 1 ml column.
Prepacked 5 ml column.
HiScreen Blue FF HSA, 85 mg/column 4.6 ml/min Prepacked 4.7 ml column.
Capto Blue HSA, 24 mg/ml medium At least 600 cm/h
1
Supplied as suspension ready
for packing.
HiScreen Capto Blue HSA, 118 mg/column 4.6 ml/min Prepacked 4.7 ml column.
Capto Blue (high sub) HSA, 30 mg/ml medium At least 600 cm/h
1
Supplied as suspension ready
for packing.
1
In a 1 m column with 20 cm bed height at 20°C using process buffers with the same viscosity as water.
2
See Appendix 4 to convert flow velocity (cm/h) to volumetric flow rate (ml/min) and vice versa. Maximum operating flow is
calculated from measurement in a packed column with a bed height of 10 cm and i.d. of 5 cm.
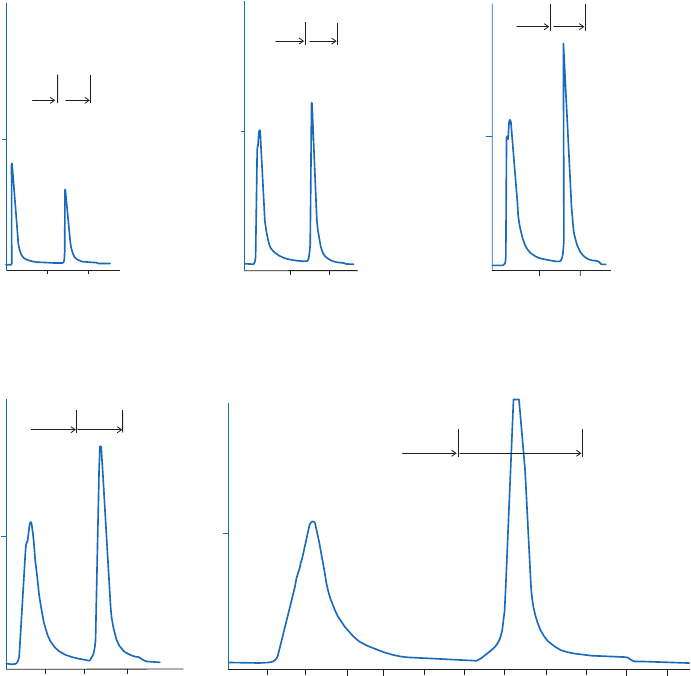
18102229 AF 29
Purification examples
Figure 3.2 shows the use of HiTrap Blue HP for purification of increasing amounts of human
serum albumin. The process is easily scaled up by connecting several 1 ml or 5 ml HiTrap
columns in series.
Figure 3.3 shows the use of Blue Sepharose 6 Fast Flow for the separation of HSA from interferon b.
A
280 nm
A
280 nm
A
280 nm
A
280 nm
A
280 nm
1.0
10
20
Binding
buffer
Elution
buffer
Volume (ml)
Volume (ml) Volume (ml)
Volume (ml) Volume (ml)
1.0
10
20
Binding
buffer
Elution
buffer
10
20
1.0
Binding
buffer
Elution
buffer
10
20
30
Binding
buffer
Elution
buffer
1.0
Binding
buffer
Elution
buffer
1.0
10 20
30
40 50 60 70
80
90
100
110
Column: HiTrap Blue HP, 1 ml or 5 ml
Sample: Human serum buffer exchanged on a PD-10 Desalting column
to binding buffer. Filtered on a 0.45 µm filter
Binding buffer: 50 mM potassium dihydrogen phosphate (KH
2
PO
4
), pH 7.0
Elution buffer: 50 mM KH
2
PO
4
, 1.5 M KCl, pH 7.0
Flow rate: 2 ml/min (1 ml column), 4 ml/min (5 ml column)
Configuration: 1 × 1 ml column
Sample vol.: 0.7 ml human serum
Yield: 16.7 mg HSA
Configuration: 3 × 5 ml columns connected in series
Sample vol.: 10.5 ml human serum
Yield: 286.9 mg HSA
Configuration: 1 × 5 ml column
Sample vol.: 3.5 ml human serum
Yield: 98.5 mg HSA
Configuration: 3 × 1 ml column
Sample vol.: 2.1 ml human serum
Yield: 52.0 mg HSA
Configuration: 2 × 1 ml column
Sample vol.: 1.4 ml human serum
Yield: 33.2 mg HSA
Fig. 3.2. Scaling up on HiTrap Blue HP gives predictable separations and quantitatively reproducible yields.
30 18102229 AF
Column: Blue Sepharose 6 Fast Flow (0.5 ml)
in packed column
Sample: 0.5 ml of interferon b (1 000 000 U/ml)
in 100 mM phosphate, pH 7.4, with
1 mg/ml of human serum albumin
Binding buffer: 20 mM phosphate, 150 mM NaCl, pH 7.2
Elution buffer 1: 20 mM phosphate, 2 M NaCl, pH 7.2
Elution buffer 2: 20 mM phosphate, 2 M NaCl,
50% ethylene glycol, pH 7.2
Flow rate: Gravity feed
0
0
0
Elution
buffer 1
Elution
buffer 2
5 10
Volume (ml)
A
280nm
IFN-
activity
IFN-act. units
0.02
50
0.04
100
0.06
150
0.08
200
0.10
250
0.12
A
280 nm
Fig 3.3. Purification of human serum albumin and interferon b on Blue Sepharose 6 Fast Flow.
In these examples elution is achieved by increasing the ionic strength of the buffer or changing
the polarity of the buffer. Changing the pH of the buffer can also work, but the correct cofactor
is preferable for the elution of specifically bound proteins.
Performing a separation
Binding buffer: 50 mM potassium dihydrogen phosphate (KH
2
PO
4
), pH 7.0 or
20 mM sodium phosphate, pH 7.0
Elution buffer: 50 mM KH
2
PO
4
, 1.5 M KCl, pH 7.0 or 20 mM sodium phosphate,
2 M NaCl, pH 7.0
1. Equilibrate the column with 5 CV of binding buffer.
2. Adjust the sample to starting conditions and apply to the column, using a syringe or a pump.
3. Wash with 10 CV of binding buffer or until no material appears in the eluent (monitored
by absorption at A
280 nm
).
4. Elute with 5 CV of elution buffer (step elution) or with 0% to 100% elution buffer in
binding buffer (gradient elution).
Cleaning
Wash with 5 CV of high pH (100 mM Tris-HCl, 500 mM NaCl, pH 8.5) followed by low pH
(100 mM sodium acetate, 500 mM NaCl, pH 4.5). Repeat four to five times. Re-equilibrate
immediately with binding buffer.
Remove precipitated proteins with 4 CV of 100 mM NaOH at a low flow rate, followed by washing
with 3 to 4 CV of 70% ethanol or 2 M potassium thiocyanate. Alternatively, wash with 2 CV of 6 M
guanidine hydrochloride. Re-equilibrate immediately with binding buffer.
Remove strongly hydrophobic proteins, lipoproteins and lipids by washing with 3 to 4 CV of up to
70% ethanol or 30% isopropanol. Alternatively, wash with 2 CV of detergent in a basic or acidic
solution, e.g. 0.1% nonionic detergent in 1 M acetic acid at a low flow rate, followed by 5 CV of
70% ethanol to remove residual detergent. Re-equilibrate immediately in binding buffer.
Chemical stability
Stable in all commonly used aqueous buffers, 70% ethanol, 8 M urea, and 6 M guanidine hydrochloride.
Storage
Wash chromatography media and columns with 20% ethanol (use approximately 5 CV for
packed media) and store at 4°C to 8°C.

18102229 AF 31
Purification or removal of albumin and IgG
Albumin & IgG Depletion Sepharose High Performance
Albumin and IgG are the most abundant proteins in plasma which tend to obscure the signals
of less abundant proteins, preventing accurate detection. The high abundance of albumin and
IgG also interferes with the detection of other proteins by preventing a sufficient amount of
less abundant proteins from being included in the analysis. By depleting samples of albumin
and IgG, the quality and depth of the analysis can be greatly enhanced. Depletion of the two
removes more than 60% of the total protein content in human plasma, allowing proteins
normally obscured by albumin and IgG to be visualized.
Albumin & IgG Depletion Sepharose High Performance is available prepacked in in HiTrap and
SpinTrap formats for removal of human serum albumin (HSA) and IgG. Both column types are
prepacked with a mixture of antiHSA Sepharose High Performance and Protein G Sepharose
High Performance. The ligand of antiHSA Sepharose High Performance is based on a single
domain antibody fragment with high specificity and capacity for HSA. The ligand of Protein G
Sepharose High Performance is derived from the IgG binding regions of Protein G, a cell surface
protein of Streptococcus bacteria. The protein G ligand binds human IgG
1
, IgG
2
, IgG
3
, and IgG
4
.
The primary use of the products is small-scale preparation of protein samples prior to
downstream analyses such as 1-D or 2-D gel electrophoresis and mass spectrometry (MS).
A lower sample volume should be used when applying plasma containing albumin and
IgG above the normal levels of human plasma (40 mg albumin/ml and 15 mg IgG/ml).
Chromatography medium characteristics
Table 3.3 shows the characteristics of the chromatography medium.
Table 3.3. Characteristics of Alumin & IgG Depletion Sepharose High Performance medium
Product Ligand Composition pH stability
1
Average
particle size
(µm)
Albumin &
IgG Depletion
Sepharose High
Performance
Recombinant protein G
fragment and recombinant
protein binding HSA.
Ligand coupled to
Sepharose High
Performance.
Short term: 2 to 9
Long term: 3 to 9
34
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Long term refers
to the pH interval over which the medium is stable over a long period of time without adverse effects on its subsequent
chromatographic performance.
Purification options
The purification options for HiTrap Albumin & IgG Depletion and Albumin & IgG Depletion
SpinTrap column are shown in Table 3.4.
Table 3.4. Purification options: HiTrap Albumin & IgG Depletion as well as Albumin & IgG Depletion SpinTrap
Binding capacity
Maximum operating
flow rate (ml/min) Comments
HiTrap Albumin & IgG
Depletion
Human plasma, ~ 150 µl
1
4 Prepacked 1 ml
column.
Albumin & IgG Depletion
SpinTrap
Human plasma, ~ 50 µl
1
Not applicable To be used with a
benchtop centrifuge.
1
Human plasma containing ~ 40 mg albumin/ml and ~ 15 mg IgG/ml. Results according to ELISA: > 95% albumin depletion
and > 90% IgG depletion.
32 18102229 AF
Purification examples
HiTrap Albumin & IgG Depletion can be used for depletion of human plasma without dilution
of the sample before loading. A volume of 150 µl human plasma was applied to the 1 ml
column, and the unbound fraction containing the depleted sample was collected. The depletion
of albumin and IgG is shown by SDS-PAGE analysis (Fig 3.4). The depletion level was also
determined by ELISA, and the result for the unbound fraction was 99% albumin depletion and
98% IgG depletion.
IgG
albumin
M
r
97 000
66 000
45 000
30 000
123 4
Lanes
1. LMW marker
2. Human plasma
3. Depleted fraction
4. Bound fraction containing albumin and IgG
Fig 3.4. Deep Purple stained SDS-PAGE analysis (nonreducing conditions) of fractions from the depletion of
human plasma using HiTrap Albumin & IgG Depletion.
Performing a separation
Binding buffer: 20 mM sodium phosphate, 150 mM NaCl, pH 7.4.
Elution buffer: 100 mM glycine-HCl, pH 2.7
Sample preparation: Dilution of the human plasma is not required. Filter the human
plasma through a 0.22 or 0.45 µm filter shortly before applying it to the column.
HiTrap Albumin and IgG Depletion
A flow rate of 1 ml/min is recommended for the entire depletion procedure.
1. Fill the pump tubing with binding buffer. Remove the stopper and the snap-off end
from the column and connect it to the pump tubing ‘drop-to-drop’ to avoid introducing
air into the system.
2. Wash the column with 5 ml binding buffer to remove the 20% ethanol storage solution.
3. Equilibrate with 10 ml of binding buffer.
4. Apply 150 µl filtrated human plasma and wash with at least 5 ml binding buffer until
the absorbance reaches a steady baseline. Collect the sample flowthrough during
sample application and wash. The flowthrough contains the depleted sample.
5. Optional: Elute and collect the bound proteins (albumin and IgG) with 10 ml of elution buffer.
Note: Step 5 should be performed if the column is to be reused or if the bound
albumin and IgG fraction is to be analyzed.
For the manual depletion procedure (without using a pump), the syringe is connected to the
column by the provided Luer connector. Be sure to use a flow rate of approximately 1 ml/min.

18102229 AF 33
Note that too high a flow rate will damage the packing of the chromatography medium
and result in high back pressure.
Albumin & IgG Depletion SpinTrap
1. Remove storage solution
A. Invert and shake the SpinTrap column repeatedly to resuspend the medium.
B. Twist off the bottom cap from the SpinTrap column and loosen the top cap
one-quarter of a turn.
C. Place the column in a 2 ml microcentrifuge tube and centrifuge for 30 s at 70 to
100 × g. Discard the collected liquid.
D. Remove and discard the top cap.
2. Column equilibration
A. Add 400 µl binding buffer and centrifuge for 30 s at 800 × g. Discard the collected liquid.
B. Add 400 µl binding buffer a second time and centrifuge for 30 s at 800 × g. Discard
the collected liquid.
3. Sample application and incubation
A. Place the column in a new 2 ml tube.
B. Dilute the 50 µl plasma sample with binding buffer to a final volume of 100 µl and
apply to the column.
C. Incubate for 5 min without mixing.
4. Collection of depleted sample
A. Centrifuge for 30 s at 800 × g. Collect the eluate.
B. Add 100 µl binding buffer and centrifuge for 30 s at 800 × g. Collect the eluate.
C. Add 100 µl binding buffer a second time and centrifuge for 30 s at 800 × g.
Collect the eluate.
Note: All eluates can be collected in the same 2 ml tube.
5. Optional: elution of albumin and IgG
A. Bound albumin and IgG can be eluted by 100 mM glycine-HCl, pH 2.7.
Storage
Store at 4°C to 8°C in 20% ethanol.

34 18102229 AF
Purification or removal of biotin and biotinylated substances
Streptavidin Sepharose High Performance
Biotin and biotinylated substances bind to streptavidin, a molecule isolated from Streptomyces
avidinii. The binding of streptavidin to biotin is one of the strongest known noncovalent
biological interactions. Hence, denaturing conditions are generally required for the efficient
elution of biotinylated biomolecules. Alternatively, biotinylated biomolecules bound to
streptavidin chromatography media can be used to capture interacting target substances such
as proteins. Impurities are removed by washing, and the enriched target protein is eluted using
relatively mild elution conditions.
Chromatography medium characteristics
Characteristics of Streptavidin Sepharose High Performance AC medium are given in Table 3.5
Table 3.5. Characteristics of Streptavidin Sepharose High Performance chromatography medium
Composition pH stability
1
Average particle size (µm)
Streptavidin Sepharose
High Performance
Streptavidin is coupled
to Sepharose High
Performance using a
N-hydroxysuccinimide
coupling method.
Short term: 2 to 10.5
Long term: 4 to 9
34
1
Short term refers to the pH interval for regeneration, cleaning-in-place and sanitization procedures. Long term refers
to the pH interval over which the medium is stable over a long period of time without adverse effects on its subsequent
chromatographic performance.
Purification options
Streptavidin Sepharose High Performance is available in chromatography media packs and
is prepacked in HiTrap 1 ml columns, MultiTrap 96-well plates, and SpinTrap minicolumns
(Table 3.6). These different formats can be used for protein purification and enrichment, where
a biotinylated antibody (or other biotinylated molecule) is attached to the Streptavidin and
the protein of interest is enriched through the affinity interaction with the antibody/target
molecule.
Streptavidin Mag Sepharose magnetic beads are also available for small-scale
immunoprecipitation and purification of biotinylated molecules, see Chapter 5.
Table 3.6. Purification options for Streptavidin High Performance chromatography media and
prepacked columns
Binding capacity
Maximum
operating flow Comments
HiTrap Streptavidin HP,
1 ml
Biotin, > 300 nmol/column
Biotinylated BSA, 6 mg/ml medium
4 ml/min Prepacked 1 ml column.
Streptavidin Sepharose
High Performance
Biotin, > 300 nmol/medium
Biotinylated BSA, 6 mg/ml medium
150 cm/h
1
Supplied as a suspension
ready for column packing.
Streptavidin HP
MultiTrap
Biotin, > 15 nmol/well
Biotinylated BSA, 0.3 mg/well
Not applicable 96-well filter plate.
Streptavidin HP
SpinTrap
Biotin, > 30 nmol/column
Biotinylated BSA, 0.6 mg/column
Not applicable To be used with a
benchtop centrifuge.
1
See Appendix 4 to convert flow velocity (cm/h) to volumetric flow rate (ml/min). Maximum operating flow is calculated
from measurement in a packed column with a bed height of 10 cm and i.d. of 5 cm.
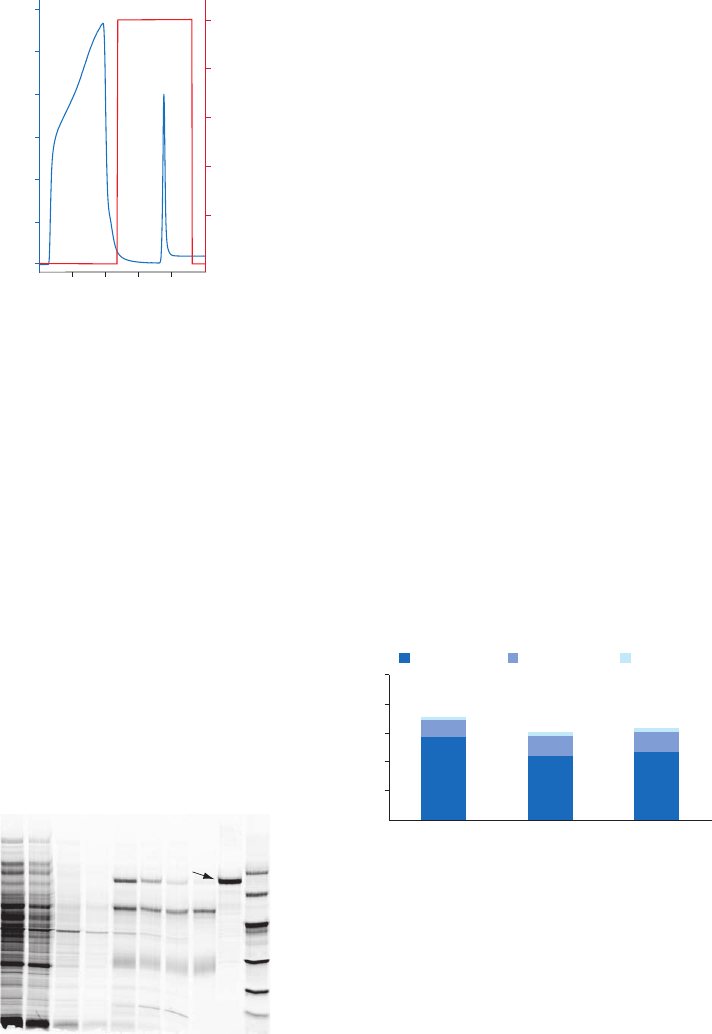
18102229 AF 35
Purification example
An alternative to labeling the biomolecule, for example the antibody, with biotin is to use
2-iminobiotin that binds to streptavidin above pH 9.5 and can be eluted at pH 4.0 (Fig 3.5).
Column: HiTrap Streptavidin HP, 1 ml
Sample: 9.0 ml of a mixture of BSA and iminobiotinylated
BSA, filtered through a 0.45 µm filter
Binding buffer: 50 mM ammonium carbonate buffer,
500 mM NaCl, pH 10.0
Elution buffer: 50 mM ammonium acetate buffer,
500 mM NaCl, pH 4.0
Flow rate: 1 ml/min (0.3 ml/min during
sample application)
System: ÄKTA system
0 5 10 15 20
Volume (ml)
0
0.4
0.8
1.2
0
20
40
60
80
100
Elution buffer (%)
A
280 nm
Fig 3.5. Purification of iminobiotinylated BSA on HiTrap Streptavidin HP, 1 ml.
Enrichment of a particular protein is often desired to increase its signal in subsequent
analysis steps. In this example Streptavidin HP SpinTrap was used for enrichment of human
transferrin from E. coli sample. The concentration of the protein of interest was 0.15% of
the total E. coli protein content, which approximately corresponds to the concentration of a
medium-abundance protein. Capture of transferrin was achieved using a biotinylated antibody
(polyclonal rabbit antihuman transferrin immobilized on the medium).
Analysis by SDS-PAGE of the collected fractions from the runs revealed a significant enrichment
of transferrin (Fig 3.6). Recovery of the start material was 60% to 70% with the majority of
the protein eluted in the first elution step. The enrichment of transferrin relative to the start
material was approximately 100-fold with Streptavidin HP SpinTrap.
Second elution
Third elution
First elution
1
2
3 4
5
6
7 8
100
80
60
40
20
0
Replicate 1 Replicate 2
Replicate 3
Percentage of start material
9 10
97 000
66 000
45 000
30 000
20 100
14 400
M
r
Transferrin
Column: Streptavidin HP SpinTrap
Sample: 5 mg/ml E. coli protein containing
7.5 µg/ml human transferrin
Sample volume: 200 µl
Binding buffer: Tris buffered saline (TBS: 50 mM Tris,
150 mM NaCl, pH 7.5)
Wash buffer: TBS, 2 M urea, pH 7.5
Elution buffer: 100 mM glycine/HCl, 2 M urea, pH 3.0
Antibody: Polyclonal rabbit antihuman transferrin
(biotinylated)
Lanes
1. Flowthrough (diluted 1:30)
2. First wash (diluted 1:10)
3. Third wash
4. Fifth wash
5. First elution
6. Second elution
7. Third elution
8. Antibody
9. Transferrin, M
r
77 000
10. LMW markers
(B)(A)
Second elution
Third elution
First elution
1
2
3 4
5
6
7 8
100
80
60
40
20
0
Replicate 1 Replicate 2
Replicate 3
Percenta
ge of start material
9 10
97 000
66 000
45 000
30 000
20 100
14 400
M
r
Transferrin
Fig 3.6. Enrichment of transferrin from E. coli cell lysate. (A) Analysis by SDS-PAGE (wash steps 2 and 4 have
been omitted from the gel). The gel was post-stained with Deep Purple Total Protein Stain and scanned.
(B) All three elution steps were analyzed using ImageQuant™ TL software. Recovery (percentage of start
material) of three replicates is shown.

36 18102229 AF
Performing a separation
HiTrap Streptavidin HP
The following protocol describes AC using a HiTrap Streptavidin HP 1 ml column by syringe,
using a pump, or a chromatography system.
Biotinylated substances
Binding buffer: 20 mM sodium phosphate, 150 mM NaCl, pH 7.5
Elution buffer: 8 M guanidine-HCl, pH 1.5
Iminobiotinylated substances
Binding buffer: 50 mM ammonium carbonate, 500 mM NaCl, pH 10.0
Elution buffer: 50 mM ammonium acetate, 500 mM NaCl, pH 4.0
1. Equilibrate the column with 10 CV of binding buffer.
2. Apply the sample. For optimal results, use a low flow rate of 0.1 to 0.5 ml/min during
sample application.
3. Wash with at least 10 CV of binding buffer or until no material appears in the eluent
(monitored by UV absorption at A
280 nm
).
4. Elute with 10 to 20 CV of elution buffer.
1
1
Since elution conditions can be quite harsh, collect fractions into neutralization buffer (100 to 200 µl 1 M Tris-HCl, pH 9.0
per ml of fraction), so that the final pH of the fractions will be approximately neutral or perform a rapid buffer exchange
on a desalting column (see Buffer exchange and desalting, Appendix 1).
The harsh conditions required to break the streptavidin-biotin bond can affect both the
sample and the ligand. Streptavidin Sepharose columns cannot be reused after elution
under these conditions.
Antigen purification
Antigens can be purified from biotinylated biomolecule (often antibody)-antigen complexes
bound to streptavidin. The following method is adapted for HiTrap Streptavidin HP.
Solubilization buffer: 20 mM sodium phosphate, 150 mM NaCl, pH 7.5 with 0.1% SDS,
1.0% Nonidet™-P-40, 0.5% sodium deoxycholate
Elution buffer: 100 mM glycine-HCl, pH 2.2
1. Solubilize the antigen with an appropriate amount of solubilization buffer, clear the
sample by centrifuging at 12 000 × g for 15 min.
2. Add the biotinylated antibody and adjust the volume to 1 ml.
3. Incubate with end-over-end mixing, for at least 1 h or overnight.
4. Equilibrate the column with 10 CV of solubilization buffer.
6. Apply antibody-antigen solution to the column at a low flow rate such as 0.2 ml/min. If
the sample volume is less than 1 ml, apply the sample, and leave for a few minutes to
allow binding to take place.
7. Wash out unbound sample with 10 CV of solubilization buffer or until no material is
found in eluent (monitored by UV absorption at A
280 nm
).
8. Elute with 5 to 10 CV of elution buffer
1
.
1
Since elution conditions are quite harsh, it is recommended to collect fractions into neutralization buffer (100 to 200 µl of 1 M
Tris-HCl, pH 9.0 per ml of fraction), so that the final pH of the fractions will be approximately neutral or perform a rapid
buffer exchange on a desalting column (see Buffer exchange and desalting, Appendix 1).

18102229 AF 37
Streptavidin HP MultiTrap 96-well plate
The protocol is designed for enrichment of target proteins by using immobilized antibodies.
Centrifuge the MultiTrap 96-well plates at 700 × g or use vacuum. If vacuum is used, apply
-0.15 bar until the wells are empty, then slowly increase the vacuum to -0.3 bar (do not apply
more vacuum than -0.5 bar). Turn off the vacuum after approximately 5 s.
Mix briefly before removal of liquid in the equilibration, wash, and elution steps to increase the
efficiency of the step. Incubating on a plate shaker is recommended. Remember to change or
empty the collection plate between steps.
Binding buffer: TBS (50 mM Tris, 150 mM NaCl, pH 7.5)
Washing buffer: TBS with 2 M urea, pH 7.5
Elution buffer: 100 mM glycine with 2 M urea, pH 3.0
Blocking buffer: 2 mM biotin in TBS
1. Remove storage solution
A. Suspend the medium by gently shaking the plate upside down.
B. Remove top and bottom seals and place plate on the collection plate.
C. Remove the storage solution by centrifugation for 1 min at 700 × g.
2. Equilibration for immobilization (perform this step three times)
A. Add 400 µl binding buffer per well, mix briefly and centrifuge for 1 min at 700 × g to
equilibrate the medium.
3. Binding of biotinylated antibody
A. Immediately after equilibration, add 200 µl of the biotinylated antibody solution per
well (0.1 to 1.0 mg/ml in binding buffer).
B. Incubate on shaker for 20 min.
C. Centrifuge for 1 min at 700 × g to remove unbound antibody.
4. Blocking (perform this step twice)
A. Add 400 µl blocking buffer per well and incubate on shaker for 5 min to block free
biotin binding sites.
B. Centrifuge for 1 min at 700 × g.
5. Washing (perform this step three times)
A. Add 400 µl binding buffer per well and mix briefly.
B. Centrifuge for 1 min at 700 × g.
6. Binding of target protein
A. Add 200 µl clarified sample in binding buffer per well and incubate on shaker for
60 min.
B. Centrifuge for 1 min at 700 × g to wash out unbound protein. Collect flowthrough.
7. Washing
A. Add 400 µl binding/wash buffer per well and mix briefly.
B. Centrifuge for 1 min at 700 × g. Perform this step five times in total. (Collect and save
washes in case troubleshooting is needed).
8. Elution (perform this step three times)
A. Add 200 µl of desired elution buffer and shake for 1 min.
B. Centrifuge for 1 min at 700 × g.
C. Collect the eluates in separate collection plates.

38 18102229 AF
Streptavidin HP SpinTrap columns
This protocol is designed for enrichment of target proteins by using immobilized antibodies. In
each step, place the SpinTrap column in a fresh 2 ml microcentrifuge tube for liquid collection.
Lids and bottom caps of Streptavidin HP SpinTrap are used during the incubation and elution
but not during equilibration and washing. Before centrifugation, remove the bottom cap and
slightly open the screw cap lid (twist the cap lid ~ 90º counterclockwise).
Binding buffer: TBS (50 mM Tris, 150 mM NaCl, pH 7.5)
Washing buffer: TBS with 2 M urea, pH 7.5
Elution buffer: 100 mM glycine with 2 M urea, pH 2.9
Blocking buffer: 2 mM biotin in TBS
1. Remove storage solution
A. Break off the bottom cap from the spin column. Save the bottom cap.
B. Remove the storage solution by centrifugation for 1 min at 150 × g.
2. Equilibrate for immobilization (perform this step three times)
A. Add 400 µl binding buffer and centrifuge for1 min at 150 × g to equilibrate the medium.
B. Remove the binding buffer.
3. Binding biotinylated antibody
A. Immediately after the equilibration, add 200 µl of the biotinylated antibody
(0.1 to 1.0 mg/ml).
B. Fully suspend the medium by manual inversion and incubate with slow, end-over-
end mixing for 20 min at room temperature.
C. Centrifuge for 1 min at 150 × g to remove unbound antibody.
4. Blocking (perform this step twice)
A. Add 400 µl blocking buffer.
B. Mix by manual inversion and incubate with end-over-end mixing for 5 min to block
free biotin binding sites.
C. Centrifuge for 1 min at 150 × g.
5. Washing (perform this step three times)
A. Add 400 µl binding buffer and centrifuge for 1 min at 150 × g.
6. Binding of target protein
A. Add 200 µl clarified sample in binding buffer.
B. Mix by manual inversion and incubate with slow, end-over-end mixing for 60 min at
room temperature.
C. Centrifuge for 1 min at 150 × g to wash out unbound protein. Collect flowthrough.
7. Washing (perform this step five times)
A. Add 400 µl wash buffer and centrifuge for 1 min at 150 × g.
B. During optimization/troubleshooting: Collect flowthrough.
8. Elution (perform this step three times)
A. Add 200 µl of desired elution buffer and mix by inversion.
B. Centrifuge for 1 min at 1000 × g.
C. Collect the eluates in individual tubes.
Storage
Wash chromatography media and HiTrap columns with 20% ethanol (use approximately 5 CV for
packed media) and store at 4°C to 8°C. Streptavidin MultiTrap and Streptavidin SpinTrap are for
single-use only.

18102229 AF 39
Purification or removal of calmodulin-binding proteins:
ATPases, adenylate cyclases, protein kinases, phosphodiesterases,
neurotransmitters
Calmodulin Sepharose 4B
Calmodulin is a highly conserved regulatory protein found in all eukaryotic cells. This protein
is involved in many cellular processes such as glycogen metabolism, cytoskeletal control,
neurotransmission, phosphate activity, and control of NAD
+
/NADP
+
ratios. Calmodulin
Sepharose 4B provides a convenient method for the isolation of many of the calmodulin-
binding proteins involved in these pathways.
Calmodulin binds proteins principally through interactions with hydrophobic sites on its
surface. These sites are exposed after a conformational change induced by the action of
Ca
2+
on separate Ca
2+
-binding sites. The binding of enzymes can be enhanced if the enzyme
substrate is present and enzyme-substrate-calmodulin-Ca
2+
complexes are particularly stable.
Chromatography medium characteristics
The charactersistics of Calmodulin Sepharose 4B chromatography medium are shown in Table 3.7.
Table 3.7. Characteristics of Calmodulin Sepharose 4B chromatography medium
Ligand density
(mg/ml) Composition pH stability
1
Average
particle size
(µm)
Calmodulin Sepharose 4B 0.9 to 1.3 Bovine testicular
calmodulin coupled to
Sepharose 4B by the
CNBr method.
Short term: 4 to 9
Long term: 4 to 9
90
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Long term refers
to the pH interval over which the medium is stable over a long period of time without adverse effects on its subsequent
chromatographic performance.
Purification options
Calmodulin Sepharose 4B chromatography medium is available in chromatography media
packs for packing in columns, Table 3.8.
Table 3.8. Purification options for Calmodulin Sepharose 4B
Binding capacity
Maximum operating
flow velocity (cm/h
1
) Comments
Calmodulin Sepharose 4B No data available 75 Supplied as a suspension
ready for column packing.
1
See Appendix 4 to convert flow velocity (cm/h) to volumetric flow rate (ml/min). Maximum operating flow is calculated
from measurement in a packed column with a bed height of 10 cm and i.d. of 5 cm.

40 18102229 AF
Performing a separation
Binding buffer: 50 mM Tris-HCl, 50 to 200 mM NaCl, 2 mM CaCl
2
, pH 7.5
Elution buffer: 50 mM Tris-HCl, 50 to 200 mM NaCl, 2 mM EGTA, pH 7.5
1. Pack the column (see Appendix 3) and wash with at least 10 CV of binding buffer to
remove preservative.
2. Equilibrate the column with 10 CV of binding buffer.
3. Apply the sample, using a low flow from 15 cm/h, during sample application (flow rate
is the most significant factor for maximum binding).
4. Wash with 5 to 10 CV of binding buffer or until no material appears in the eluent
(monitored by UV absorption at A
280 nm
).
5. Elute with 5 CV of elution buffer.
Remove proteases as quickly as possible from the sample as the calmodulin-binding
sites on proteins are frequently very susceptible to protease action (see Purification or
removal of serine proteases in this chapter).
Remove free calmodulin from the sample by HIC in the presence of Ca
2+
on HiTrap
Phenyl FF (high sub) or by IEX on HiTrap Q FF.
Since some nonspecific ionic interactions can occur, a low salt concentration
(50 to 200 mM NaCl) is recommended to promote binding to the ligand while eliminating
any nonspecific binding.
Use chelating agents to elute the proteins. Chelating agents strip Ca
2+
from the
calmodulin, reversing the conformational change that exposed the protein binding
sites. Calcium ions can also be displaced by a high salt concentration, 1 M NaCl.
Cleaning
Alternative 1
Wash with 3 CV of 50 mM Tris-HCl, 1 M NaCl, 2 mM EGTA, pH 7.5 and reequilibrate immediately
with 5 to 10 CV of binding buffer.
Alternative 2
Wash with 3 CV of 100 mM ammonium carbonate buffer, 2 mM EGTA, pH 8.6 followed by 3 CV
of 1 M NaCl, 2 mM CaCl
2
. Continue washing with 3 CV of 100 mM sodium acetate buffer, 2 mM
CaCl
2
, pH 4.4 followed by 3 CV of binding buffer.
Remove severe contamination by washing with nonionic detergent such as 0.1% Tween™ 20 at
37°C for 1 min.
Chemical stability
Stable in all commonly used aqueous solutions.
Storage
Wash chromatography media and columns with 20% ethanol (use approximately 5 CV for
packed media) and store at 4°C to 8°C.

18102229 AF 41
Purification or removal of coagulation factors
VIISelect, VIIISelect, IXSelect, Heparin Sepharose High Performance,
Heparin Sepharose 6 Fast Flow, Capto Heparin
Blood coagulation factors form an extremely important group of proteins for research, medical
and clinical applications.
Different coagulation factors involved in bleeding disorders are selectively purified using
VIISelect, VIIISelect, and IXSelect. Hemophilia type A is caused by a deficiency or defect in factor
VIII (FVIII) while hemophilia type B is known as factor IX (FIX) deficiency. Factor VII (FVII) is used
for hemophilia patients with FVIII or FIX deficiencies who have developed inhibitors against the
replacement coagulation factors.
In addition, coagulation factors obtained from plasma or expressed as recombinant proteins
in various cell types can be purified by Heparin Sepharose and Capto Heparin chromatography
media. For details about Heparin Sepharose High Performance, Heparin Sepharose 6 Fast Flow
and Capto Heparin, see Purification or removal of DNA-binding proteins in this chapter. The protocols
are also applicable for coagulation factors.
Chromatography media characteristics
The characteristics of VIISelect, VIIISelect, and IXSelect chromatography media for purification
of coagulation factors are shown in Table 3.9.
Table 3.9. Characteristics of VIISelect, VIIISelect, and IXSelect chromatography media
Product Ligand Composition pH stability
1
Average
particle size
(µm)
VIISelect Recombinant protein
(M
r
14 080) produced
in Saccharomyces
cerevisiae.
Ligand coupled to
Capto.
Short term: 2 to 12
Long term: 3 to 10
75
VIIISelect Recombinant protein
(M
r
13 000) produced
in S. cerevisiae.
Ligand coupled to
Capto.
Short term: 2 to 12
Long term: 3 to 10
75
IXSelect Single-chain antibody
fragment (M
r
13 151)
directed against
FIX and produced
in Saccharomyces
cerevisiae.
Ligand coupled to
Capto.
Short term: 2 to 12
Long term: 3 to 10
75
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Long term refers
to the pH interval over which the medium is stable over a long period of time without adverse effects on its subsequent
chromatographic performance.
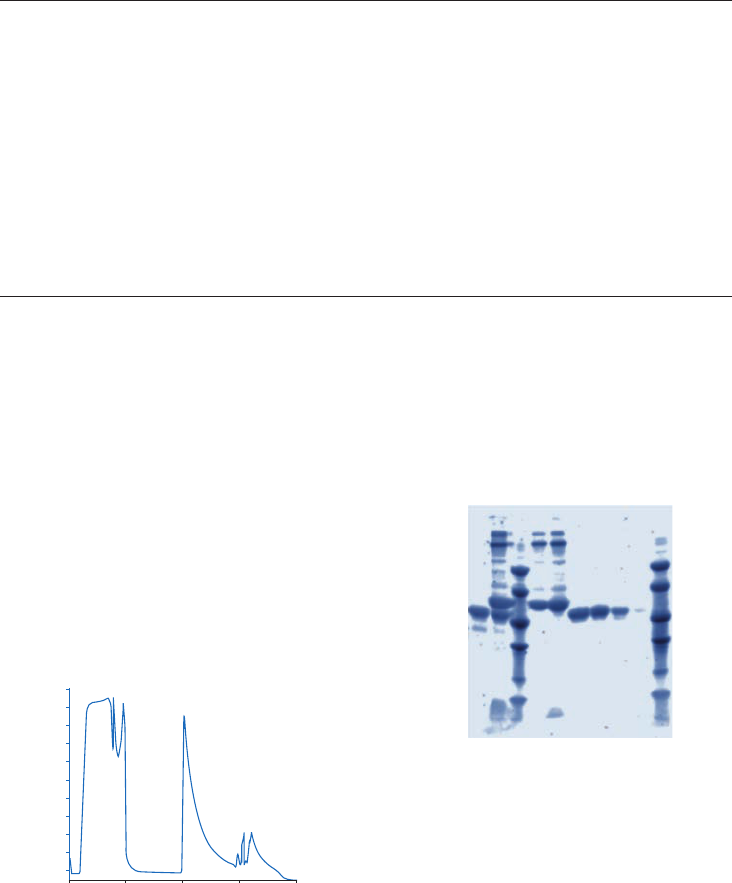
42 18102229 AF
Purification options
A wide range of chromatography media packs and prepacked columns for purification of
coagulation factors is available (Table 3.10).
Table 3.10. Chromatography media and prepacked columns for purification of coagulation factors
Product Binding capacity
Maximum
operating flow Comments
VIISelect FVII, ~ 8 mg/ml medium At least 600 cm/h
in a 1 m diameter
column with 20 cm
bed height
1
Supplied as suspension ready
for column packing.
VIIISelect Typically 20 000 IU/ml
medium
Up to 300 cm/h at
30 cm bed height
1
Supplied as suspension ready
for column packing.
IXSelect FIX, ~ 6 mg/ml medium At least 600 cm/h
in a 1 m diameter
column, with 20 cm
bed height
1
Supplied as suspension ready
for column packing.
HiTrap IXSelect, 1 ml FIX, ~ 6 mg/column 4 ml/min Prepacked 1 ml column.
HiTrap IXSelect, 5 ml FIX, ~ 30 mg/column 20 ml/min Prepacked 5 ml column.
HiScreen IXSelect FIX, ~ 28 mg/column 4.6 ml/min Prepacked 4.7 ml column.
1
20°C using buffers with the same viscosity as water at < 0.3 MPa (3 bar, 43.5 psi).
Purification of FVII
A commercially available drug approved for infusion therapy was spiked in human plasma, and
FVII was purified (Fig 3.7A). Gel electrophoresis was run on SDS-PAGE gradient gels, 8% to 16%,
under nonreducing conditions. Figure 3.7B shows the high purity of FVII in the eluted fractions
obtained using VIISelect.
350
300
250
200
150
100
50
0
mAU
0510
Sample
Elution
Strip
15
20
Volume (ml)
Lanes
1. Commercial FVII
2. Commercial FVII spiked in plasma
3. LMW markers
4. Flowthrough fraction
5. Wash after sample loading
6. Elution fraction 5
7. Elution fraction 6
8. Elution fraction 1 to 12 (pooled)
9. Strip after elution
10. LMW markers
Column: Tricorn™ 5/20 packed with 0.45 ml of VIISelect
Sample: Registered pharmaceutical FVII drug
diluted with solution for injection and spiked in
human plasma
Sample load: 7 mg/ml of chromatography VIISelect
(below the maximum capacity)
Binding/equilibration buffer: 10 CV of 50 mM Tris, 150 mM NaCl, pH 7.5.
Elution buffer: 12 CV of 50 mM Tris, 1.5 M NaCl,
50% (v/v) propylene glycol, pH 7.5
Wash: 12 CV binding/equilibration buffer
Flow rate (flow velocity): Sample load, 0.2 ml/min (61 cm/h); wash and
elution, 0.5 ml/min (153 cm/h)
97 000
66 000
45 000
30 000
20 100
14 400
M
r
12345678910
(A) (B)
Fig 3.7. (A) UV280 absorbance curve for purification of FVII from spiked plasma using VIISelect for the initial
capture step. (B) SDS-PAGE of the FVII drug before and after purification on VIISelect. The gel was stained
with Deep Purple total protein stain and scanned in a Typhoon™ scanner.
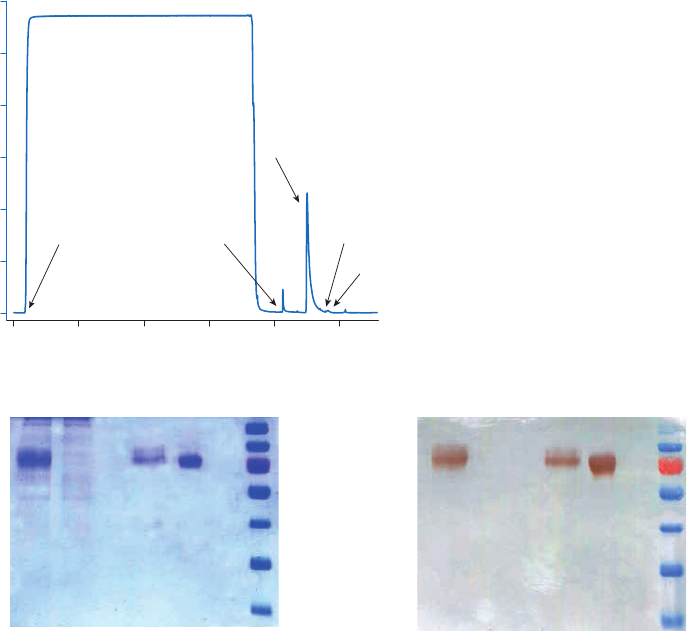
18102229 AF 43
Capture of FIX
Capture of FIX from a Chinese hamster ovary (CHO) cell lysate was performed using IXSelect
chromatography medium (Fig 3.8A). Fractions from the FIX capture step were analyzed by
SDS-PAGE using a FIX reference preparation as standard (Fig 3.8B). The identity of the target
protein was confirmed by Western blot analysis (Fig 3.8C).
0
200
400
600
800
1000
1200
A
280
(mAU)
0 500 1000 1500 2000 2500
Volume (ml)
RegenerationLoad Wash
Elution
CIP
M
r
130 000
100 000
70 000
55 000
40 000
35 000
25 000
1 2 3 4 5
M
r
130 000
100 000
70 000
55 000
40 000
35 000
25 000
1 2 3 4 5
Lanes
1. Cell culture medium
2. Flowthrough
3. Elution peak
4. FIX standard
5. LMW markers
Lanes
1. Cell culture supernatant
2. Flowthrough
3. Elution peak
4. FIX standard
5. LMW markers
Column: HiScale 16/20 packed with
13 ml of IXSelect medium
Sample: FIX-containing CHO cell lysate
Equilibration: 6 CV of 20 mM Tris-HCl,
150 mM NaCl, pH 7.4
Sample load: 1500 ml sample feed loaded at 100 cm/h
Wash: 10 CV of equilibration buffer
10 CV of 20 mM Tris-HCl, 500 mM NaCl,
0.01% Tween 80, pH 7.4
3 CV of equilibration buffer
Elution: 20 mM Tris-HCl, 2 M MgCl
2
, pH 7.4
Regeneration: 100 mM glycine buffer
CIP: 10 mM NaOH
(A)
(B) (C)
Fig 3.8. (A) FIX capture using IXSelect. (B) SDS-PAGE analysis of fractions from FIX capture step. (C) Western
blot analysis of fractions from FIX capture step. Data from customer evaluation.
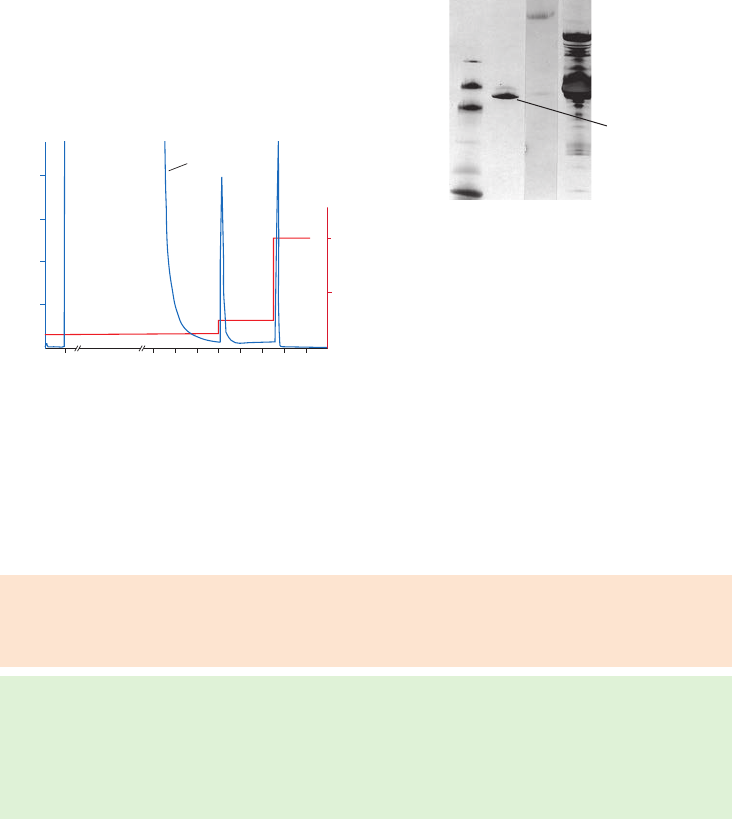
44 18102229 AF
Purification of antithrombin III from bovine plasma
Figure 3.9 shows the result from purification of antithrombin III from bovine plasma using
HiTrap Heparin HP. The antithrombin III eluted in the seond peak.
A
280 nm
A
280
0.8
0
10
0.6
0.4
0.2
50 60
Volume (ml)
70 80 90 100 110 120 130
50
100
Elution buffer (%)
Flowthrough peak I peak II
1 2 3 4
M
r
97 000
20 100
30 000
45 000
14 400
66 000
Lanes
1. LMW-SDS Marker Kit, reduced
2. Peak II, reduced, diluted two-fold
3. Peak I, reduced, diluted two-fold
4. Unbound material, reduced, diluted 15-fold
Antithrombin III
Column: HiTrap Heparin HP, 1 ml
Sample: 30 ml bovine plasma diluted with 15 ml 100 mM Tris,
10 mM citrate, 225 mM NaCl, pH 7.4
Binding buffer: 100 mM Tris, 10 mM citric acid, 225 mM NaCl, pH 7.4
Elution buffer: 100 mM Tris, 10 mM citric acid, 2 M NaCl, pH 7.4
2.9 mg antithrombin-III was eluted in peak II
Flow rate: 1.0 ml/min
Electrophoresis: SDS-PAGE, PhastSystem™ electrophoresis unit,
PhastGel™ Gradient 8–25 SDS gel, 1 µl sample, silver stained
A
280 nm
A
280
0.8
0
10
0.6
0.4
0.2
50 60
Volume (ml)
70 80 90 100 110 120 130
50
100
Elution buffer (%)
Flowthrough peak I peak II
1 2 3 4
M
r
97 000
20 100
30 000
45 000
14 400
66 000
Fig 3.9. Purification of antithrombin III from bovine plasma on HiTrap Heparin HP, 1 ml.
Storage
Store at 4°C to 8°C in 20% ethanol.
Performing a separation
VIISelect
Binding buffer: 50 mM Tris-HCl, 150 mM NaCl, pH 7.5
Elution buffer: 50 mM Tris, 1.5 M NaCl, 50% (v/v) propylene glycol, pH 7.5
Regeneration buffer: 100 mM glycine
1. Equilibrate with 6 to 10 CV of binding buffer.
2. Load the sample.
3. Wash with 10 to 12 CV of binding buffer.
4. Elute with 6 to 12 CV elution buffer.
5. Regenerate the column with regeneration buffer.

18102229 AF 45
VIIISelect
Binding buffer: 10 mM histidine, 20 mM calcium chloride, 300 mM sodium chloride,
0.02% Tween 80, pH 7.0
Wash buffer 1: 20 mM histidine, 20 mM calcium chloride, 300 mM sodium chloride,
0.02% Tween 80, pH 6.5
Wash buffer 2: 20 mM histidine, 20 mM calcium chloride, 1 M sodium chloride,
0.02% Tween 80, pH 6.5
Elution buffer: 20 mM histidine, 20 mM calcium chloride, 1.5 M sodium chloride,
0.02% Tween 80 dissolved in 50% ethylene glycol at pH 6.5
1. Equilibrate with 10 CV of binding buffer.
2. Load the sample in loading buffer.
3. Wash with 5 CV of wash buffer 1.
4. Wash with 5 CV of wash buffer 2.
5. Elute with 5 to 10 CV of elution buffer.
IXSelect
Binding buffer: 20 mM Tris-HCl, 150 mM NaCl, pH 7.4
Wash buffer: 20 mM Tris-HCl, 500 mM NaCl, 0.01% Tween 80, pH 7.4
Elution buffer: 20 mM Tris-HCl, 2 M MgCl
2
, pH 7.4
Regeneration buffer: 100 mM glycine, 100 mM NaCl, pH 2.0
1. Equilibrate with 6 to 10 CV binding buffer.
2. Load the sample.
3. Wash with 10 CV of binding buffer.
4. Wash with 10 CV washing buffer.
5. Wash with 3 CV of binding buffer.
6. Elute with 5 to 10 CV elution buffer.
7. Regenerate the column with regeneration buffer.
Cleaning
The following are suggestions for solutions to be used during cleaning. Prolonged exposure
to pH < 2.0 and pH > 12.0 should be avoided. The required cleaning is strongly dependent on
the sample used, number of runs, conditions of the chromatography media etc. and has to be
designed for each application.
• PAB (120 mM phosphoric acid, 167 mM acetic acid, 2.2% benzyl alcohol). Store in dark.
• 10 mM sodium hydroxide
• 100 mM citric acid
46 18102229 AF
Purification or removal of DNA-binding proteins
Heparin Sepharose High Performance, Heparin Sepharose 6 Fast Flow, Capto Heparin
DNA-binding proteins form an extremely diverse class of proteins sharing a single
characteristic, their ability to bind to DNA. Functionally the group can be divided into those
responsible for the replication and orientation of the DNA such as histones, nucleosomes and
replicases and those involved in transcription such as RNA/DNA polymerases, transcriptional
activators and repressors and restriction enzymes. They can be produced as tagged proteins
to enable more specific purification but their ability to bind DNA also enables group specific
affinity purification using heparin as a ligand. Heparin is a highly sufonated glycosaminoglycan
with the ability to bind a very wide range of biomolecules including:
• DNA binding proteins such as initiation factors, elongation factors, restriction
endonucleases, DNA ligase, DNA, and RNA polymerases.
• Serine protease inhibitors such as antithrombin III, protease nexins.
• Enzymes such as mast cell proteases, lipoprotein lipase, coagulation enzymes, superoxide
dismutase.
• Growth factors such as fibroblast growth factor, Schwann cell growth factor, endothelial cell
growth factor.
• Extracellular matrix proteins such as fibronectin, vitronectin, laminin, thrombospondin, collagens.
• Hormone receptors such as estrogen and androgen receptors.
• Lipoproteins.
The structure of heparin is shown in Figure 3.10. Heparin has two modes of interaction with
proteins and, in both cases, the interaction can be weakened by increases in ionic strength.
1. In its interaction with DNA binding proteins heparin mimics the polyanionic structure of the
nucleic acid.
2. In its interaction with coagulation factors such as antithrombin III, heparin acts as an
affinity ligand.
O
O
O
O
O
O
O O
COO
–
COO
–
OH
OH
OH
OR
1
H
2
COR
1
OH
HNR
2
O
Fig 3.10. Structure of a heparin polysaccharide consisting of alternating hexuronic acid (A) and
D-glucosamine residues (B). The hexuronic acid can either be D-glucuronic acid (top) or its C-5 epimer,
L-iduronic acid (bottom). R
1
= -H or -SO
3
–
, R
2
= -SO
3
–
or -COCH
3
.

18102229 AF 47
Chromatography media characteristics
Characteristics of chromatography media for purification or removal of DNA-binding proteins
are shown in Table 3.11.
Table 3.11. Characteristics of Heparin Sepharose and Capto Heparin chromatography media
Ligand
density
(mg/ml) Composition pH stability
1
Average
particle size
(µm)
Heparin Sepharose
High Performance
10 Heparin coupled to Sepharose
High Performance by reductive
amination to give a stable
attachment even in alkaline
conditions.
Short term: 5 to 10
Long term: 5 to 10
34
Heparin Sepharose 6
Fast Flow
5.0 Heparin coupled to Sepharose 6
Fast Flow by reductive amination
to give a stable attachment even
in alkaline conditions.
Short term: 4 to 13
Long term: 4 to 12
90
Capto Heparin 1.8 Heparin coupled to Capto. Short term: 4 to 13
Long term: 4 to 12
90
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Long term refers
to the pH interval over which the medium is stable over a long period of time without adverse effects on its subsequent
chromatographic performance.
Purification options
Purification options for Heparin Sepharose 6 Fast Flow chromatography medium and
prepacked columns as well as Capto Heparin are shown in Table 3.12.
Table 3.12. Purification options for purification of DNA-binding proteins
Binding capacity
Maximum
operating flow Comments
HiTrap Heparin HP, 1 ml
HiTrap Heparin HP, 5 ml
Bovine antithrombin III, 3 mg/column
Bovine antithrombin III, 15 mg/column
4 ml/min
20 ml/min
Prepacked 1 ml column.
Prepacked 5 ml column.
HiPrep Heparin FF 16/10 Bovine antithrombin III, 40 mg/column 10 ml/min Prepacked 20 ml column.
Heparin Sepharose 6
Fast Flow
Bovine antithrombin III, 2 mg/ml medium 400 cm/h
1
Supplied as a suspension
ready for column packing.
Capto Heparin Antithrombin III, 1.4 mg/ml medium 700 cm/h
2
Supplied as suspension
ready for column packing.
1
See Appendix 4 to convert flow velocity (cm/h) to volumetric flow rate (ml/min). Maximum operating flow is calculated
from measurement in a packed column with a bed height of 10 cm and i.d. of 5 cm.
2
1 m diameter column, 20 cm bed height.
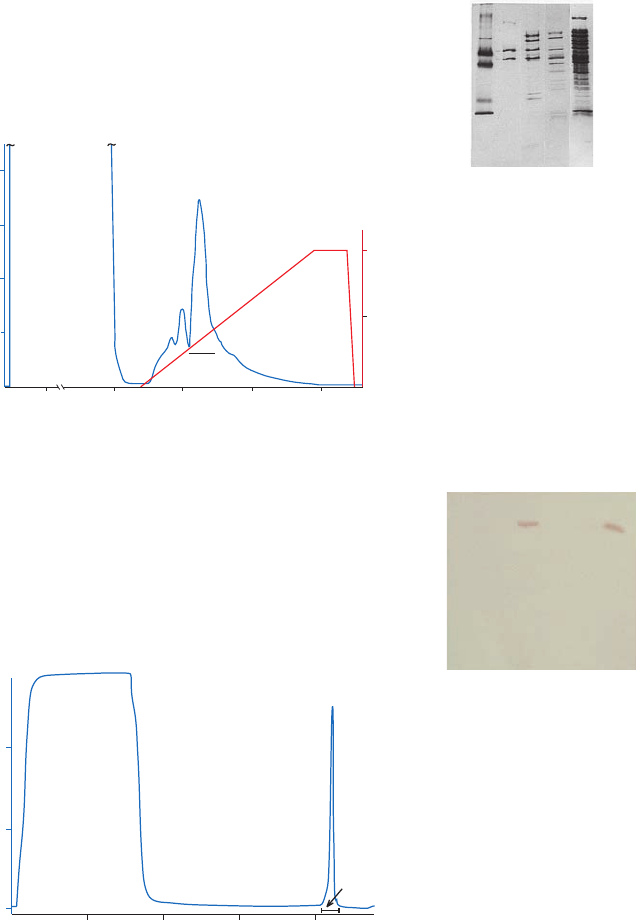
48 18102229 AF
Purification examples
Figures 3.11 to 3.13 show examples of conditions used for the purification of different DNA
binding proteins.
50
0.1
0.2
70
60
6
0
pool I
0.3
0.4
80
50
100
Elution buffer (%)
A
280 nm
1 2 3 4 5
M
r
97 000
66 000
20 100
30 000
45 000
14 400
Volume (ml)
Column: HiTrap Heparin HP, 1 ml
Sample: 49 ml E. coli lysate (= 1 g cells) after passage through a
5 ml DEAE Sepharose Fast Flow packed in column
Binding buffer: 20 mM Tris-HCl, 1 mM EDTA, 1 mM 2-mercaptoethanol,
2% glycerol, pH 8.0
Elution buffer: Binding buffer + 1 M NaCl
Elution conditions: 25 ml elution buffer, linear gradient 0% to 100%
Flow rate: 1.0 ml/min
Lanes
SDS-PAGE, Gradient 8–25, 1 ml sample, silver stained.
1. LMW-SDS Marker Kit, reduced
2. Reverse transciptase, reduced
3. Pool I from HiTrap Heparin HP, 1 ml reduced
4. Unbound material from
DEAE Sepharose Fast Flow, reduced
5. Cell lysate, reduced
50
0.1
0.2
70
60
6
0
pool I
0.3
0.4
80
50
100
Elution buffer (%)
A
280 nm
1 2 3 4 5
M
r
97 000
66 000
20 100
30 000
45 000
14 400
Volume (ml)
Fig 3.11. Partial purification of recombinant HIV-reverse transcriptase on HiTrap Heparin HP.
Column: HiTrap Heparin HP, 5 ml
Sample: 30 ml extract containing Oct-1, filtered (0.45 µm) and
transferred to binding buffer using a PD-10 desalting column
Binding buffer: 20 mM Tris-HCl, 5% (v/v) glycerol, 0.1 mM EDTA,
10 mM 2-mercaptoethanol 0.1 mM Pefabloc™,
500 mM NaCl, pH 8
Elution buffer: 20 mM Tris-HCl, 5% (v/v) glycerol,
0.1 mM EDTA,
10 mM 2-mercaptoethanol,
0.1 mM Pefabloc, 2 M NaCl, pH 8.0
Flow rate (flow velocity): 1 ml/min (30 cm/h)
A
280nm
1.0
0.5
0.0
Pool
0.0 20.0 40.0 60.0 80.0
Volume (ml)
Flowthrough
Eluate
4 3 2 1
M
r
97 000
20 100
30 000
45 000
14 400
66 000
Lanes
Western blot of the electrophoresis gel using rabbit
anti-Oct human-1 and alkaline phosphatase
1. Starting material
2. Flowthrough
3. LMW-SDS Marker Kit
4. Eluate pool
A
280nm
1.0
0.5
0.0
Pool
0.0 20.0 40.0 60.0 80.0
Volume (ml)
Flowthrough
Eluate
4 3 2 1
M
r
97 000
20 100
30 000
45 000
14 400
66 000
Fig 3.12. Partial purification of the recombinant DNA binding Oct-1 protein (courtesy of Dr Gunnar Westin,
University Hospital, Uppsala, Sweden) using HiTrap Heparin HP, 5 ml.
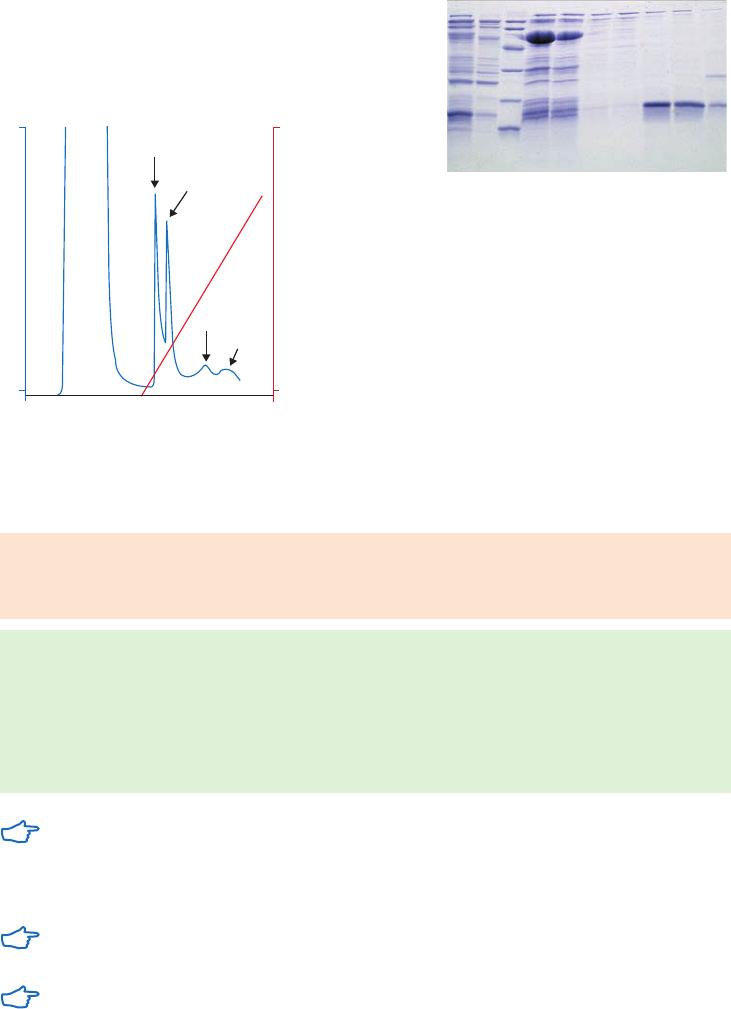
18102229 AF 49
Column: HiPrep Heparin FF 16/10
Sample: 2000 ml partially purified sample from
DEAE Sepharose CL-4B flowthrough, pH 7.0
Binding buffer: 50 mM sodium phosphate, pH 7.5
Elution buffer: 50 mM sodium phosphate,
1 M sodium chloride, pH 7.5
Flow rate (flow velocity): 1.5 ml/min (45 cm/h)
Lanes
Electrophoresis: SDS-PAGE, 12% gel, Coomassie™ Blue staining
1. Pool from HiPrep 26/10 Desalting
2. Flowthrough pool from DEAE Sepharose CL-4B
3. LMW-SDS Marker Kit
4–10. Eluted fractions from HiPrep Heparin FF 16/10
4. Fraction 13
5. Fraction 14
6. Fraction 23
7. Fraction 24
8. Fraction 54
9. Fraction 55
10. Fraction 69
1
1
fr. 23–24
fr. 13–14
A
NaCl concentration (M)
0 0
scCro8, fr . 54–55
fr. 69
280
1 2 3 4 5 6 7 8 9 10
M
r
97 000
20 100
30 000
45 000
66 000
14 400
1
1
fr. 23–24
fr. 13–14
A
NaCl concentration (M)
0 0
scCro8, fr . 54–55
fr. 69
280
1 2 3 4 5 6 7 8 9 10
M
r
97 000
20 100
30 000
45 000
66 000
14 400
Fig 3.13. scCro8 purification on HiPrep Heparin FF 16/10.
Performing a separation
Heparin Sepharose 6 Fast Flow, Heparin Sepharose High Performance
Binding buffers: 20 mM Tris-HCl, pH 8.0 or 10 mM sodium phosphate, pH 7.0
Elution buffer: 20 mM Tris-HCl, 1 to 2 M NaCl, pH 8.0 or 10 mM sodium phosphate,
1 to 2 M NaCl, pH 7.0
1. Equilibrate the column with 10 CV of binding buffer.
2. Apply the sample.
3. Wash with 5 to 10 CV of binding buffer or until no material appears in the eluent
(monitored by UV absorption at A
280 nm
).
4. Elute with 5 to 10 CV of elution buffer using a continuous or step gradient from 0% to
100% elution buffer.
Modify the selectivity of heparin by altering pH or ionic strength of the buffers. Elute
using a continuous or step gradient with NaCl, KCl or (NH
4
)
2
SO
4
up to 2 M.
If used for purification or removal of coagulation factors:
Since the heparin acts as an affinity ligand for coagulation factors, it is advisable to
include a minimum concentration of 150 mM NaCl in the binding buffer.
If an increasing salt gradient gives unsatisfactory results, use heparin (1 to 5 mg/ml) as
a competing agent in the elution buffer.
Cleaning
Remove ionically bound proteins by washing with 0.5 CV of 2 M NaCl for 10 to 15 min.
Remove precipitated or denatured proteins by washing with 4 CV of 100 mM NaOH for 1 to 2 h;
or 2 CV of 6 M guanidine hydrochloride for 30 to 60 min; or 2 CV of 8 M urea for 30 to 60 min.
Remove hydrophobically bound proteins by washing with 4 CV of 0.1% to 0.5% Tween 20 for 1 to 2 h.

50 18102229 AF
Chemical stability
100 mM NaOH (1 w at 20°C), 50 mM sodium acetate, pH 4.0, 4 M NaCl, 8 M urea,
6 M guanidine hydrochloride.
Storage
Wash chromatography media and columns with 50 mM sodium acetate containing 20% ethanol
(use approximately 5 CV for packed media) and store at 4°C to 8°C.
Capto Heparin
Binding buffer: 100 mM Tris-HCl, 10 mM trisodium citrate, 225 mM NaCl, pH 7.4
Wash buffer: 100 mM Tris-HCl, 10 mM trisodium citrate, 330 mM NaCl, pH 7.4
Elution buffer: 100 mM Tris-HCl, 10 mM trisodium citrate, 2 M NaCl, pH 7.4
1. Equilibrate with 5 CV of binding buffer.
2. Apply the sample.
3. Wash step 1: wash with 40 CV of binding buffer.
4. Wash step 2: wash with 15 CV of wash buffer.
5. Elute with 9.5 CV of elution buffer.
A flow rate of 0.5 ml/min is recommended for a 1 ml column.
Cleaning
Substances such as denatured proteins that do not elute during regeneration can be removed
by cleaning-in-place (CIP) procedures. A recommended CIP procedure for Capto Heparin is 4 CV
of 100 mM NaOH with a contact time of 1 to 2 h.
Storage
Store unused chromatography media at 4ºC to 30ºC in 20% ethanol and 50 mM of sodium acetate.
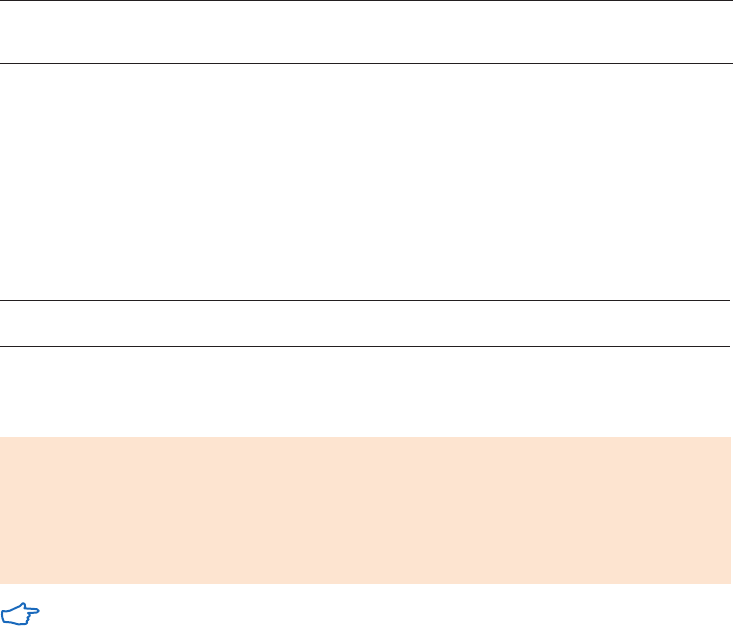
18102229 AF 51
Purification or removal of fibronectin
Gelatin Sepharose 4B
Fibronectin is a high-molecular weight glycoprotein found on the surfaces of many cell types and
present in many extracellular fluids including plasma. Fibronectin binds specifically to gelatin
at or around physiological pH and ionic strength.
Chromatography medium characteristics
The characteristics os Gelatin Sepharose 4B chromatography medium are shown in Table 3.13.
Table 3.13. Characteristics of Gelatin Sepharose 4B chromatography medium
Ligand density
(mg/ml) Composition pH stability
1
Average
particle size
(µm)
Gelatin Sepharose 4B 4.5 to 8.0 Gelatin coupled to
Sepharose 4B using
the CNBr method.
Short term: 3 to 10
Long term: 3 to 10
90
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Long term refers
to the pH interval over which the medium is stable over a long period of time without adverse effects on its subsequent
chromatographic performance.
Purification option
Gelatin Sepharose 4B is available in chromatography media packs for packing into columns of
your choice (Table 3.14).
Table 3.14. Purification option for Gelatin Sepharose 4B
Binding capacity/
ml medium
Maximum operating
flow (cm/h)
1
Comments
Gelatin Sepharose 4B Human plasma
fibronectin, 1 mg
75 Supplied as a suspension
ready for column packing.
1
See Appendix 4 to convert flow velocity (cm/h) to volumetric flow rate (ml/min). Maximum operating flow is calculated
from measurement in a packed column with a bed height of 10 cm and i.d. of 5 cm.
Performing a separation
Binding buffer: PBS: 140 mM NaCl, 2.7 mM KCl, 10 mM Na
2
HPO
4
,
1.8 mM KH
2
PO
4
, pH 7.4
Elution buffer alternatives: 50 mM sodium acetate, 1 M sodium bromide
(or potassium bromide), pH 5.0
Binding buffer + 8 M urea
Binding buffer + arginine
Fibronectin has a tendency to bind to glass. Use siliconized glass to prevent adsorption.
Cleaning
Wash three times with 2 to 3 CV of buffer, alternating between high pH (100 mM Tris-HCl, 500 mM NaCl,
pH 8.5) and low pH (100 mM sodium acetate, 500 mM NaCl, pH 4.5). Re-equilbrate immediately
with 3 to 5 CV of binding buffer. Remove denatured proteins or lipids by washing the column
with 0.1% Tween 20 at 37°C for 1 min. Re-equilibrate immediately with 5 CV of binding buffer.
Chemical stability
Stable in all commonly used aqueous buffers.
Storage
Wash chromatography media and columns with 20% ethanol at neutral pH (use approximately
5 CV for packed media) and store at 4°C to 8°C.

52 18102229 AF
Purification or removal of glycoproteins and polysaccharides
Con A Sepharose 4B, Lentil Lectin Sepharose 4B, Capto Lentil Lectin
Glycoproteins and polysaccharides react reversibly, via specific sugar residues, with a group of
proteins known as lectins.
As ligands for purification media, lectins are used to isolate and separate glycoproteins,
glycolipids, polysaccharides, subcellular particles and cells, and to purify detergent-solubilized
cell membrane components. Substances bound to the lectin are resolved by using a gradient of
ionic strength or of a competitive binding substance.
Chromatography media screening
To select the optimum lectin for purification, it may be necessary to screen different
chromatography media. The ligands, Concanavalin A (Con A) and Lentil Lectin provide a
spectrum of parameters for the separation of glycoproteins. Table 3.15 gives their specificity.
Table 3.15. Specificity of lectins
Lectin Specificity
Mannose/glucose binding lectins
Con A, Canavalia ensiformis Branched mannoses, carbohydrates with terminal mannose or
glucose (aMan > aGlc > GlcNAc).
Lentil Lectin, Lens culinaris Branched mannoses with fucose linked a(1,6) to N-acetyl-
glucosamine, (aMan > aGlc > GlcNAc).
Con A is a tetrameric metalloprotein isolated from Canavalia ensiformis (jack bean). Con A binds
molecules containing a-
D-mannopyranosyl, a-D-glucopyranosyl and sterically related residues.
The binding sugar requires the presence of C-3, C-4, and C-5 hydroxyl groups for reaction with
Con A. Con A can be used for applications such as:
• Separation and purification of glycoproteins, polysaccharides, and glycolipids.
• Detection of changes in composition of carbohydrate-containing substances,
for example, during development.
• Isolation of cell surface glycoproteins from detergent-solubilized membranes.
• Separation of membrane vesicles into “inside out” and “right side out” fractions.
Lentil lectin binds a-
D-glucose and a-D-mannose residues and is an affinity ligand used for
the purification of glycoproteins including detergent-solubilized membrane glycoproteins, cell
surface antigens and viral glycoproteins. Lentil lectin is the hemagglutinin from the common
lentil, Lens culinaris. When compared to Con A, it distinguishes less sharply between glucosyl
and mannosyl residues and binds simple sugars less strongly. It also retains its binding ability
in the presence of 1% sodium deoxycholate. For these reasons Lentil Lectin Sepharose 4B is
useful for the purification of detergent-solubilized membrane proteins, giving high capacities
and extremely high recoveries.

18102229 AF 53
Chromatography media characteristics
Characteristics of Con A and Lentil Lectin chromatography media are shown in Table 3.16.
Table 3.16. Characteristics of Con A and Lentil Lectin chromatography media
Ligand density
(mg/ml) Composition pH stability
1
Average
particle size
(µm)
Con A Sepharose 4B 10 to 16 Con A coupled to
Sepharose 4B by
CNBr method.
Short term: 4 to 9
Long term: 4 to 9
90
Lentil Lectin Sepharose 4B 2.5 Lentil lectin coupled
to Sepharose 4B by
CNBr method.
Short term: 3 to 10
Long term: 3 to 10
90
Capto Lentil Lectin 3 Lentil lectin coupled
to Capto matrix by
NHS-method.
Short term: 3 to 10
Long term: 3 to 10
75
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Long term refers
to the pH interval over which the medium is stable over a long period of time without adverse effects on its subsequent
chromatographic performance.
Purification options
Purification options for Con A and Lentil Lectin chromatography media and prepacked columns
are shown in Table 3.17.
Table 3.17. Con A and Lentil Lectin chromatography media and prepacked columns
Binding capacity/ml
medium
Maximum
operating flow Comments
Con A Sepharose 4B Porcine thyroglobulin,
20 to 45 mg
75 cm/h
2
Supplied as a suspension
ready for column packing
1
.
Lentil Lectin
Sepharose 4B
Porcine thyroglobulin,
16 to 35 mg
75 cm/h
1
Supplied as a suspension
ready for column packing.
Capto Lentil Lectin Porcine thyroglobulin
~ 15 mg
100 to 300 cm/h
2
Supplied as suspension
ready for column packing.
HiTrap Con A 4B, 1 ml Porcine thyroglobulin,
20 to 45 mg
4 ml/min Prepacked 1 ml column.
HiTrap Con A 4B, 5 ml 100 to 225 mg Porcine
thyroglobulin
20 ml/min Prepacked 5 ml column.
HiTrap Capto
Lentil Lectin, 1 ml
Porcine thyroglobulin
~ 15 mg
2 ml/min Prepacked 1 ml column.
HiTrap Capto
Lentil Lectin, 5 ml
Porcine thyroglobulin
~ 75 mg
10 ml/min Prepacked 5 ml column.
HiScreen Capto
Lentil Lectin
Porcine thyroglobulin
~ 70 mg
2.3 ml/min Prepacked 4.7 ml column.
1
See Appendix 4 to convert flow velocity (cm/h) to volumetric flow rate (ml/min). Maximum operating flow is calculated
from measurement in a packed column with a bed height of 10 cm and i.d. of 5 cm.
2
Supplied in acetate buffer solution (100 mM, pH 6.0) containing 1 M NaCl, 1 mM CaCl
2
, 1 mM MgCl
2
, 1 mM MnCl
2
, 20% ethanol.
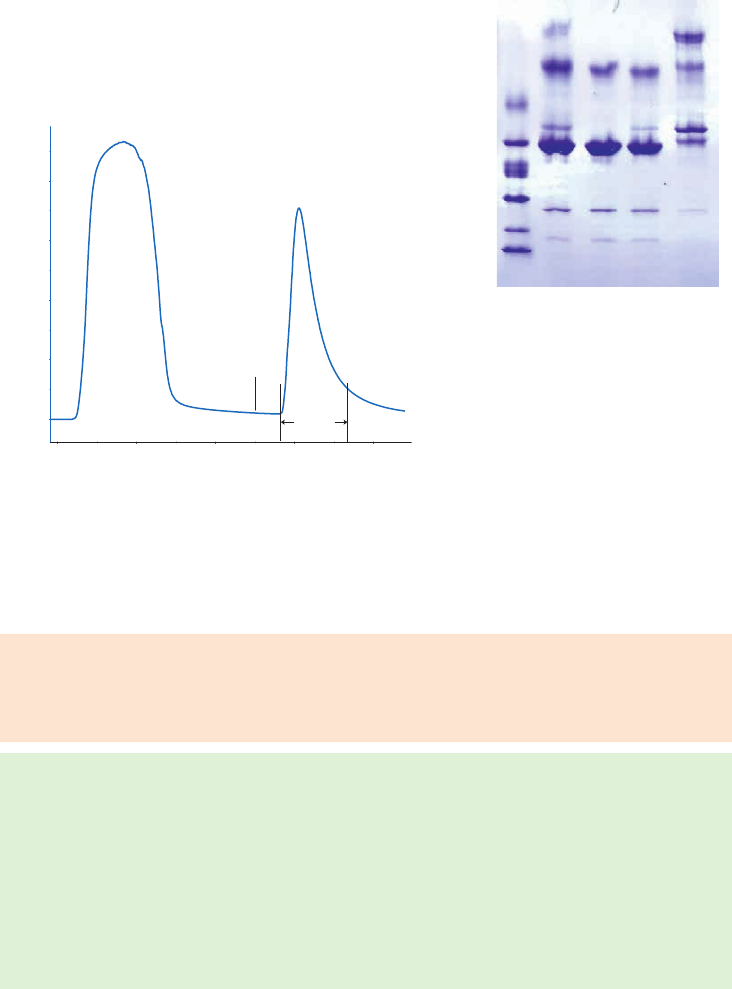
54 18102229 AF
Purification example
Enrichment of glycoproteins from human plasma
Glycoproteins from human plasma were enriched on a HiTrap Con A 4B 1 ml column
(Fig 3.15A). Analysis by Coomassie stained SDS-PAGE (nonreducing conditions), showed
that unfractionated plasma (start material), flowthrough, and wash fractions all had band
corresponding to molecular weight of 67 000 (Fig 3.15B). This corresponds to high-abundance,
nonglycosylated serum albumin, which was removed from the sample and not detected in the
eluate containing the glycoproteins.
Lanes
1. LMW markers
2. Start material
3. Flowthrough
4. Wash
5. Eluate
Column: HiTrap Con A 4B, 1 ml
Sample: 0.5 ml human plasma diluted to 5 ml with binding buffer
Binding buffer: 20 mM Tris, 500 mM NaCl, 1 mM MnCl
2
, 1 mM CaCl
2
, pH 7.4
Elution buffer: 20 mM Tris, 500 mM NaCl, 300 mM methyl-D-glucoside, pH 7.4
Flow rates: 0.2 ml/min (sample load)
0.1 ml/min with pause 4 min (elution)
System: ÄKTA system
20
Wash
0
200
400
600
800
A
280
(mAU)
0 5 10 15
Volume (ml)
Elution start
Eluate
Flowthrough
M
r
97 000
66 000
45 000
30 000
20 100
14 400
1 2 3 4 5
20
Wash
0
200
400
600
800
A
280
(mAU)
0 5 10 15
Volume (ml)
Elution start
Eluate
Flowthrough
M
r
97 000
66 000
45 000
30 000
20 100
14 400
1 2 3 4 5
(A) (B)
Fig 3.15. (A) Chromatographic enrichment of glycoproteins from human plasma using HiTrap Con A 4B,
1 ml. (B) SDS-PAGE anaysis with Coomassie stained ExcelGel™ 8–18 Gradient gel (nonreducing conditions)
of fractions from enrichment of glycoproteins from human plasma using HiTrap Con A 4B, 1 ml.
Performing a separation
Con A Sepharose 4B
Binding buffer: 20 mM Tris-HCl, 500 mM NaCl, 1 mM MnCl
2
, 1 mM CaCl
2
, pH 7.4
Elution buffer: 100 to 500 mM methyl-a-
D-glucopyranoside (methyl-a-D-glucoside)
or methyl-a-
D-mannopyranoside (methyl-a-D-mannoside),
20 mM Tris-HCl, 500 mM NaCl, pH 7.4
1. Pack the column (see Appendix 3) and wash with at least 10 CV of binding buffer to
remove preservative.
2. Equilibrate the column with 10 CV of binding buffer.
3. Apply the sample, using a low flow velocity from 15 cm/h, during sample application
(flow velocity is the most significant factor to obtain maximum binding).
4. Wash with 5 to 10 CV of binding buffer or until no material appears in the eluent
(monitored by UV absorption at A
280 nm
).
5. Elute with 5 CV of elution buffer.

18102229 AF 55
Recovery from Con A Sepharose 4B is decreased in the presence of detergents. If the
glycoprotein of interest needs the presence of detergent and has affinity for lentil lectin,
the Lentil Lectin Sepharose 4B chromatography medium provides a suitable alternative
to improve recovery.
For complex samples containing glycoproteins with different affinities for the lectin, a
continuous gradient or step elution can improve resolution. Recovery can sometimes be
improved by pausing the flow for a few minutes during elution.
Elute tightly bound substances by lowering the pH. Note that elution below pH 4.0 is not
recommended and that below pH 5.0, manganese ions (Mn
2+
) will begin to dissociate
from the Con A and the column will need to be reloaded with Mn
2+
before reuse.
Cleaning
Wash with 10 CV of 500 mM NaCl, 20 mM Tris-HCl, pH 8.5, followed by 500 mM NaCl,
20 mM acetate, pH 4.5. Repeat three times before re-equilibrating with binding buffer.
Remove strongly bound substances by:
• washing with 100 mM borate, pH 6.5 at a low flow rate.
• washing with 20% ethanol or up to 50% ethylene glycol.
• washing with 0.1% Tween 20 at 37°C for 1 min.
Re-equilibrate immediately with 5 CV of binding buffer after any of these wash steps.
Chemical stability
Stable to all commonly used aqueous buffers. Avoid 8 M urea, high concentrations of guanidine
hydrochloride, chelating agents such as EDTA, or solutions with pH < 4.0 as these remove the
Mn
2+
from the lectin or dissociate Con A, resulting in loss of activity.
Storage
Wash chromatography media and columns with 20% ethanol in 100 mM acetate, 1 M NaCl,
1 mM CaCl
2
, 1 mM MnCl
2
, 1 mM MgCl
2
, pH 6.0 (use approximately 5 CV for packed media) and
store at 4°C to 8°C.
Performing a separation
Lentil Lectin Sepharose
Binding buffer: 20 mM Tris-HCl, 500 mM NaCl, 1 mM MnCl
2
, 1 mM CaCl
2
, pH 7.4
Elution buffer: 100 to 500 mM methyl-a-
D-glucopyranoside (methyl-a-D-glucoside),
20 mM Tris-HCl, 500 mM NaCl, pH 7.4
Buffers for soluble glycoproteins:
Binding buffer: 20 mM Tris-HCl, 500 mM NaCl, 1 mM MnCl
2
, 1 mM CaCl
2
, pH 7.4
Elution buffer: 300 mM methyl-a-
D-mannopyranoside (methyl-a-D-mannoside),
20 mM Tris-HCl, 500 mM NaCl, pH 7.4
Buffers for detergent-solubilized proteins:
Equilibrate column with the buffer 20 mM Tris-HCl, 500 mM NaCl, 1 mM MnCl
2
, 1 mM CaCl
2
,
pH 7.4, to ensure saturation with Mn
2+
and Ca
2+
.
Binding buffer: 20 mM Tris-HCl, 500 mM NaCl, 0.5% sodium deoxycholate, pH 8.3
Elution buffer: 300 mM methyl-a-
D-mannopyranoside, 20 mM Tris-HCl, 500 mM NaCl,
0.5% sodium deoxycholate, pH 8.3

56 18102229 AF
1. Pack the column (see Appendix 3) and wash with at least 10 CV of binding buffer to
remove preservative.
2. Equilibrate the column with 10 CV of binding buffer.
3. Apply the sample, using a low flow velocity from 15 cm/h, during sample application
(flow velocity is the most significant factor to obtain maximum binding).
4. Wash with 5 to 10 CV of binding buffer or until no material appears in the eluent
(monitored by UV absorption at A
280 nm
).
5. Elute with 5 CV of elution buffer using a step or gradient elution.
Below pH 5.0, excess Mn
2+
and Ca
2+
(1 mM) are essential to preserve binding activity. It
is not necessary to include excess Ca
2+
or Mn
2+
in buffers if conditions that lead to their
removal from the coupled lectin can be avoided.
For complex samples containing glycoproteins with different affinities for the lectin,
a continuous gradient or multistep elution can improve resolution. Recovery can
sometimes be improved by pausing the flow for a few minutes during elution
Elute tightly bound substances by lowering pH, but not below pH 3.0. In some
cases, strongly bound substances can be eluted with detergent, for example 1.0%
deoxycholate.
Cleaning
Wash with 10 CV of 500 mM NaCl, 20 mM Tris-HCl, pH 8.5, followed by 500 mM NaCl,
20 mM acetate, pH 4.5. Repeat three times before re-equilibrating with binding buffer.
Remove strongly bound substances by:
• washing with 100 mM borate, pH 6.5 at a low flow rate.
• washing with 20% ethanol or up to 50% ethylene glycol.
• washing with 0.1% Tween 20 at 37°C for 1 min.
Re-equilibrate immediately with 5 CV of binding buffer after any of these wash steps.
Chemical stability
To avoid loss of activity of the coupled lectin, avoid solutions having a pH below 3.0 or above 10.0,
buffers that contain metal chelating agents such as EDTA, high concentrations of guanidine
hydrochloride, or high concentrations of urea.
Storage
Wash chromatography media and columns with 20% ethanol (use approximately 5 CV for
packed media) and store at 4°C to 8°C.

18102229 AF 57
Purification or removal of granulocyte-colony stimulating factor
GCSFSelect
Granulocyte-colony stimulating factor (G-CSF) is a hormone that stimulates production
of white blood cells in bone marrow. GSCFSelect is specifically designed for purification of
recombinant G-CSF and is based on a highly rigid agarose base matrix that allows for high flow
rates at large production scales. For a highly selective purification step, the affinity ligand is
based on a single-chain antibody fragment directed against G-CSF. To facilitate binding of the
target molecule, the ligand is attached to the base matrix through a hydrophilic spacer arm
(Fig 3.16). The ligand is produced in a yeast expression system, where fermentation, subsequent
purification, and formulation are performed in the absence of animal-derived components. The
rigid agarose base matrix of GCSFSelect allows for processing of large sample volumes.
OH
O
O
Ligand
H
N
H
N
Fig 3.16. Structure of GCSFSelect.
Chromatography medium characteristics
Characteristics of GCSFSelect chromatography medium are shown in Table 3.18.
Table 3.18. Characteristics of GCSFSelect chromatography medium
Product Ligand Composition pH stability
1
Average
particle size
(µm)
GCSFSelect Single-chain antibody
fragment (M
r
14 400)
directed against G-CSF
and produced in
Saccharomyces cerevisiae
Ligand coupled to Capto
via stable amide bonds
Short term: 2 to 12
Long term: 3 to 10
75
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Long term refers
to the pH interval over which the medium is stable over a long period of time without adverse effects on its subsequent
chromatographic performance.
Purification options
GCSFSelect is available in chromatography media packs for packing in columns and in
prepacked HiTrap columns, see Table 3.19.
Table 3.19. Purification options for GCSFSelect chromatography medium and prepacked columns
Binding capacity
Maximum
operating flow Comments
GCSFSelect G-CSF, 3.9 mg/ml medium 600 cm/h
1
Media suspension ready
for column packing.
HiTrap GCSFSelect, 1 ml G-CSF, 3.9 mg/column 4 ml/min Prepacked 1 ml column.
HiTrap GCSFSelect, 5 ml G-CSF, 19.5 mg/column 20 ml/min Prepacked 5 ml column.
1
Flow velocity measured in a 1 m diameter column, 20 cm bed height at 20°C; buffers used had same viscosity as water at
< 0.3 MPa (3 bar, 43.5 psi).
58 18102229 AF
Purification examples
Figure 3.17A shows an example of a purification of G-CSF using GCSFSelect. The sample was
E. coli lysate spiked with recombinant G-CSF and elution was performed using bis-Tris buffer
containing 0.08% Tween and 1 M MgCl
2
(pH 7.0). Fractions from the purification were further
analyzed by SDS-PAGE (Fig 3.17B). The single eluted peak with high purity demonstrates the
high selectivity for G-CSF of the GCSFSelect medium.
A
280
(mAU)
Volume (ml)
0
400
200
800
600
1000
1200
0510 15
Flowthrough
G-CSF
M
r
97 000
66 000
45 000
30 000
20 100
14 400
1 2 3 4 65
Lanes
1. G-CSF registered drug (M
r
19 900)
2. E. coli lysate spiked with G-CSF registered drug
3. Flowthrough
4. Wash
5. Eluate
6. LMW markers
Column: Tricorn 5/20 packed with 1 ml of GCSFSelect
Sample: 1.65 ml homogenized and clarified E. coli lysate
spiked with 2.5 mg/ml G-CSF registered drug
Binding buffer: PBS, pH 7.4
Elution buffer: 20 mM bis-Tris, 0.08% Tween 20, 1 M MgCl
2
, pH 7.0
CIP: 100 mM glycine, pH 2.0
System: ÄKTA system
A
280
(mAU)
Volume (ml)
0
400
200
800
600
1000
1200
0510 15
Flowthrough
G-CSF
M
r
97 000
66 000
45 000
30 000
20 100
14 400
1 2 3 4 65
(A) (B)
Fig 3.17. (A) Purification of G-CSF from a G-CSF spiked E. coli lysate. (B) SDS-PAGE of the different fractions
collected during purification of G-CSF using GCSFSelect.
Performing a separation
Binding buffer: PBS (10 mM sodium phosphate, 140 mM NaCl), pH 7.4
Elution buffer: 20 mM bis-Tris, 0.08% Tween 20, 1 M MgCl
2
, pH 7.0
1. Equilibrate with 10 CV of binding buffer.
2. Load the sample.
3. Wash with binding buffer until no material appears in the eluent (monitored by UV
absorption at A
280 nm
).
4. Elute with 5 to 10 CV of elution buffer.
Cleaning
Solutions such as PAB (120 mM phosphoric acid, 167 mM acetic acid, 2.2% benzyl alcohol) in
cleaning protocols are suggested. Cleaning and sanitization protocols should be designed for
each process as the efficiency of the protocol is strongly associated with the sample and other
related operating conditions.
Storage
Store at 4°C to 8°C in 20% ethanol.

18102229 AF 59
Purification or removal of NAD
+
-dependent dehydrogenases and
ATP-dependent kinases
Blue Sepharose 6 Fast Flow, Capto Blue, Capto Blue (high sub)
NAD
+
-dependent dehydrogenases and ATP-dependent kinases are members of a group of
proteins that will interact with Cibacron Blue F3G-A, a synthetic polycyclic dye that shows
certain structural similarities to the cofactor NAD
+
. When used as an affinity ligand, Cibacron
Blue F3G-A will bind strongly and specifically to a wide range of proteins. Some proteins bind
specifically due to their requirement for nucleotide cofactors, while others such as albumin,
lipoproteins, blood coagulation factors, and interferon, bind in a less specific manner by
electrostatic and/or hydrophobic interactions with the aromatic anionic ligand. For details
about Blue Sepharose 6 Fast Flow, Capto Blue, and Capto Blue (high sub), see Purification or
removal of albumin in this chapter.
Performing a separation
Blue Sepharose 6 Fast Flow, Capto Blue, Capto Blue (high sub)
The information supplied in Purification or removal of albumin earlier in this chapter is applicable
also to the purification of enzymes with an affinity for NAD
+
, but note the following:
For elution, use low concentrations of the free cofactor, NAD
+
or NADP
+
(1 to 20 mM), or
increase ionic strength (up to 2 M NaCl or KCl, 1 M is usually sufficient).
For less specifically bound proteins: use higher concentrations of cofactor or salt or
more severe eluents such as urea or potassium isothiocyanate. Polarity reducing
agents such as dioxane (up to 10%) or ethylene glycol (up to 50%) may be used.
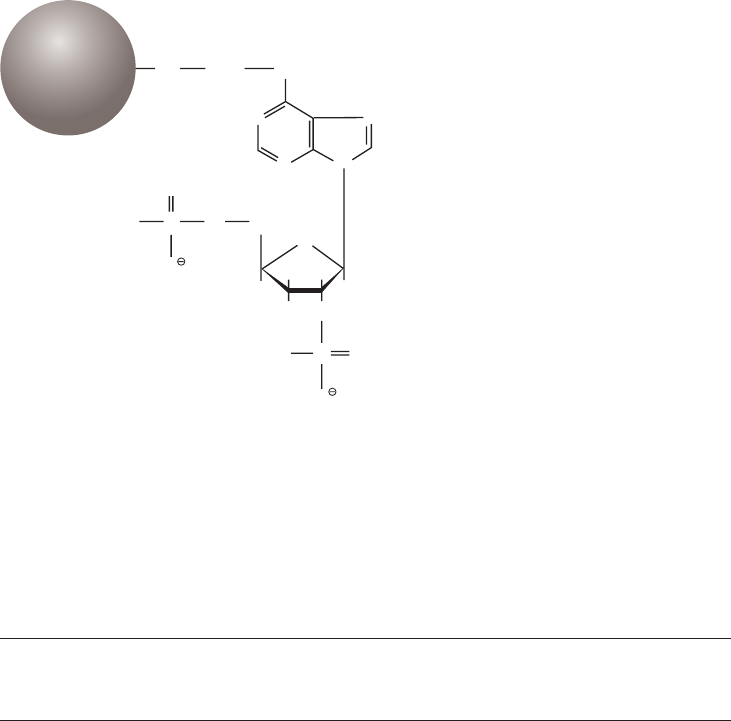
60 18102229 AF
Purification or removal of NADP
+
-dependent dehydrogenases and
other enzymes with affinity for NADP
+
2’5’ ADP Sepharose 4B
NADP
+
-dependent dehydrogenases interact strongly with 2’5’ ADP. Selective elution with
gradients of NAD
+
or NADP
+
has allowed the resolution of complex mixtures of dehydrogenase
isoenzymes using 2’5’ ADP Sepharose 4B.
Synthesis of the medium takes place in several steps. Diaminohexane is linked to 2’5’ ADP
via the N6 of the purine ring. The derivatized ADP is then coupled to Sepharose 4B via the
aminohexane spacer. Figure 3.18 shows the partial structure of 2’5’ ADP Sepharose 4B.
N
N N
O
O
O
P CH
(CH )
2
2
n
OH O
HO
HO O
O
P
N
NH
NH
O
Sepharose
Fig 3.18. Partial structure of 2’5’ ADP Sepharose 4B.
Chromatography medium characteristics
Characteristics of 2’5’ ADP Sepharose 4B are shown in Table 3.21.
Table 3.21. Characteristics of 2’5’ ADP Sepharose 4B chromatography medium
Ligand
density
(µmol/ml) Composition pH stability
1
Average
particle size
(µm)
2’5’ ADP Sepharose 4B 2 N6-(6-aminohexyl)adenosine
2’5’ bisphosphate coupled
to Sepharose 4B by CNBr
method
2
.
Short term: 4 to 10
Long term: 4 to 10
90
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Long term refers
to the pH interval over which the medium is stable over a long period of time without adverse effects on its subsequent
chromatographic performance.
2
Coupling via the N
6
position of the NADP
+
-analog, adenosine 2’5’ bisphosphate, gives a ligand that is stereochemically
acceptable to most NADP
+
-dependent enzymes.
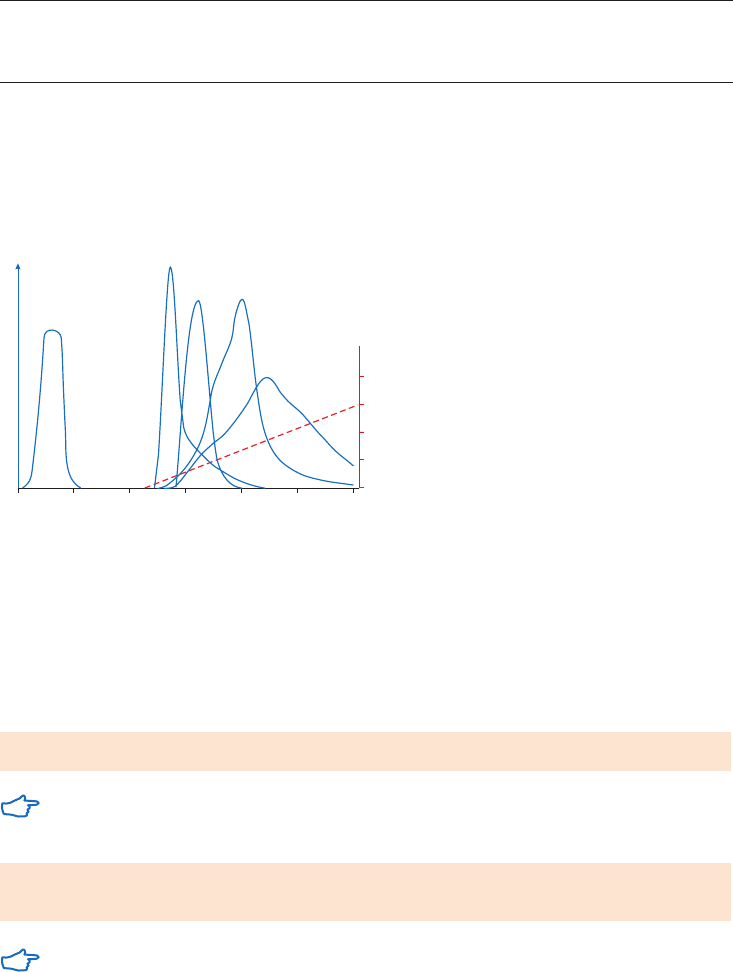
18102229 AF 61
Purification options
2’5’ ADP Sepharose 4B is available in chromatography media packs for packing in columns,
see Table 3.22.
Table 3.22. Purification options for 2’5’ ADP Sepharose 4B
Binding capacity/ml medium
Maximum operating
flow velocity (cm/h)
1
Comments
2’5’ ADP
Sepharose 4B
Glucose-6-phosphate,
dehydrogenase, 0.4 mg (100 mM
Tris-HCl, 5 mM EDTA, 1 mM
2-mercaptoethanol buffer, pH 7.6)
75 Supplied as a
freeze-dried powder,
rehydration required.
1
See Appendix 4 to convert flow velocity (cm/h) to volumetric flow rate (ml/min). Maximum operating flow is calculated
from measurement in a packed column with a bed height of 10 cm and i.d. of 5 cm.
Purification example
Figure 3.19 shows a linear gradient elution used for the initial separation of NADP
+
-dependent
enzymes from a crude extract of Candida utilis.
0.2
0.4
0.6
0.8
Fraction number
ytivitca emyznE
)Mm( noitartnecnocPDAN
+
20
(A)
(B)
(C)
(D)
(E)
0 40 60 80 100 120
Fig 3.19. Gradient elution with 0 to 0.6 mM NADP
+
. (A) noninteracting protein, (B) glucose-6-phosphate
dehydrogenase, (C) glutamate dehydrogenase, (D) glutathione reductase, (E) 6-phosphogluconate
dehydrogenase.
Performing a separation
Swell the required amount of powder for 15 min. in 100 mM phosphate buffer, pH 7.3 (100 ml
per gram dry powder) and wash on a sintered glass filter (porosity G3). Pack the column (see
Appendix 3).
Binding buffer: 10 mM phosphate, 150 mM NaCl, pH 7.3
If the protein of interest binds to the medium via ionic forces, it might be necessary to
reduce the concentration of NaCl in the binding buffer.
Elution buffers: use low concentrations of the free cofactor, NAD
+
or NADP
+
(up to 20 mM) with step or gradient elution
If detergent or denaturing agents have been used during purification, these can also be
used in the low and high pH wash buffers.
62 18102229 AF
Cleaning
Wash three times with 2 to 3 CV of buffers, alternating between high pH (100 mM Tris-HCl,
500 mM NaCl, pH 8.5) and low pH (100 mM sodium acetate, 500 mM NaCl, pH 4.5).
Re-equilibrate immediately with 3 to 5 CV of binding buffer.
Remove denatured proteins or lipids by washing the column with 2 CV of detergent, for
example, 0.1% Tween 20 for 1 min. Re-equilibrate immediately with 5 CV of binding buffer.
Chemical stability
Stable in all commonly used aqueous buffers and additives such as detergents. Avoid high
concentrations of EDTA, urea, guanidine hydrochloride, chaotropic salts, and strong oxidizing
agents. Exposure to pH > 10.0 can cause loss of phosphate groups.
Storage
Store freeze-dried product below 8°C under dry conditions.
Wash chromatography media and columns with 20% ethanol at neutral pH (use approximately
5 CV for packed media) and store at 4°C to 8°C.

18102229 AF 63
Purification or removal of proteins and peptides with exposed amino
acids: His, Cys, Trp, and/or with affinity for metal ions
Chelating Sepharose High Performance, Chelating Sepharose Fast Flow,
Capto Chelating
Proteins and peptides that have an affinity for metal ions can be separated using immobilized
metal-ion affinity chromatography, IMAC. The metals are immobilized onto a chromatographic
medium by chelation. Certain amino acids, for example, histidine and cysteine, form complexes
with the chelated metals around neutral pH (pH 6.0 to 8.0) and it is primarily the histidine-
content of a protein which is responsible for its binding to a chelated metal.
IMAC is excellent for purifying recombinant (his)
6
-tagged proteins (see the handbook Affinity
Chromatography, Vol. 2: Tagged Proteins, 18114275) as well as many natural proteins. Chelating
Sepharose, the medium used for IMAC purification of proteins with exposed His, Cys, and Trp, is
formed by coupling a metal chelate forming ligand (iminodiacetic acid) to Sepharose.
Before use the medium is loaded with a solution of divalent metal ions such as Ni
2+
, Zn
2+
, Cu
2+
, or
Co
2+
. The binding reaction with the target protein is pH dependent and bound sample is eluted
by reducing the pH and increasing the ionic strength of the buffer or by including imidazole in
the buffer. The structure of the ligand, iminodiacetic acid, is shown in Figure 3.20.
OH
OH
2
2
2
2
O OCH HC HC
2
COOH
CH
2
COOH
CH
CH NCH CH
Sepharose
Fig 3.20. Partial structure of Chelating Sepharose High Performance and Chelating Sepharose Fast Flow.
Metalloproteins are not usually suitable candidates for purification by chelating
chromatography since they tend to scavenge the metal ions from the column.
Chromatography media characteristics
Characteristics of Chelating Sepharose and Capto Chelating chromatography media are
given in Table 3.23.
Table 3.23. Characteristics of Chelating Sepharose and Capto Chelating chromatography media
Composition
Metal ion
capacity pH stability
1
Average
particle size
(µm)
Chelating Sepharose
High Performance
Iminodiacetic acid
coupled to Sepharose
High Performance via an
ether bond.
23 µmol Cu
2+
/ml Short term: 2 to 14
Long term: 3 to 13
34
Chelating Sepharose
Fast Flow
Iminodiacetic acid
coupled to Sepharose
Fast Flow via a spacer
arm using epoxy coupling.
22 to 30 µmol
Zn
2+
/ml
Short term: 2 to 14
Long term: 3 to 13
90
Capto Chelating Iminodiacetic acid
coupled to Capto.
22 to 33 µmol
Cu
2+
/ml medium
Short term: 2 to 14
Long term: 3 to 12
75
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Long term refers
to the pH interval over which the medium is stable over a long period of time without adverse effects on its subsequent
chromatographic performance.

64 18102229 AF
Purification options
Options for purification of proteins and peptides with exposed amino acid groups are shown in
Table 3.24.
Table 3.24. Purification options for Chelating Sepharose and Capto Chelating chromatography media
packs and prepacked columns
Binding capacity
Maximum
operating flow Comments
HiTrap Chelating HP, 1 ml 12 mg/column 4 ml/min Prepacked 1 ml column.
HiTrap Chelating HP, 5 ml 60 mg/column 20 ml/min Prepacked 5 ml column.
Chelating Sepharose
Fast Flow
12 mg/ml medium 400 cm/h
1
Supplied as suspension ready
for column packing.
HiTrap Capto Chelating, 1 ml 30 mg green fluorescent
protein (GFP)-his/column
3.8 ml/min Prepacked 1 ml column.
HiTrap Capto Chelating, 5 ml 150 mg GFP-his/column 20 ml/min Prepacked 5 ml column.
HiScreen Capto Chelating 130 mg GFP-his/column 4.6 ml/min Prepacked 4.7 ml column.
Capto Chelating 30 mg GFP-his/ml medium 600 cm/h Supplied as suspension ready
for column packing.
1
See Appendix 4 to convert flow velocity (cm/h) to volumetric flow rate (ml/min). Maximum operating flow is calculated
from measurement in a packed column with a bed height of 10 cm and i.d. of 5 cm.
Selecting the metal ion
The following guidelines may be used for preliminary experiments to select the metal ion that is
most useful for a given separation:
• Cu
2+
gives strong binding and some proteins will only bind to Cu
2+
. Load metal-ion solution
equivalent to 60% of the packed column volume during charging to avoid leakage of metal
ions during sample application. Alternatively, the medium can be saturated and a short
secondary uncharged column of HiTrap Chelating HP or packed Chelating Sepharose Fast
Flow should be connected in series after the main column to collect excess metal ions.
• Zn
2+
gives a weaker binding and this can, in many cases, be exploited to achieve selective
elution of a protein mixture. Load metal-ion solution equivalent to 85% of the packed
column volume to charge the column.
• Ni
2+
is commonly used for his-tagged proteins. Ni
2+
solution equivalent to half the column
volume is usually sufficient to charge the column.
• Co
2+
and Ca
2+
are also alternatives.
Charge the column with metal ions by passing through a solution of the appropriate salt
through the column, for example, ZnCl
2
, NiSO
4
, or CuSO
4
in distilled water. Chloride salts can be
used for other metals.
Several methods can be used to determine when the column is charged. If a solution of metal
salt in distilled water is used during charging, the eluate initially has a low pH and returns to
neutral pH as the medium becomes saturated with metal ions. The progress of charging with
Cu
2+
is easily followed by eye (the column contents become blue). When charging a column with
zinc ions, sodium carbonate can be used to detect the presence of zinc in the eluate. Wash the
medium thoroughly with binding buffer after charging the column.

18102229 AF 65
Choice of binding buffer
A neutral or slightly alkaline pH will favor binding. Tris-acetate (50 mM), sodium phosphate
(20 to 50 mM) and Tris-HCl (20 to 50 mM) are suitable buffers. Tris-HCl tends to reduce binding
and should only be used when metal-protein affinity is fairly high.
High concentrations of salt or detergents in the buffer normally have no effect on
the adsorption of protein and it is good practice to maintain a high ionic strength
(e.g., 500 mM to 1 M NaCl) to avoid unwanted ion exchange effects.
Chelating agents such as EDTA or citrate should not be included as they will strip the
metal ions from the medium.
Choice of elution buffers
Differential elution of bound substances may be obtained using a gradient of an agent that
competes for either the ligand or the target molecules. An increased concentration of imidazole
(0 to 500 mM), ammonium chloride (0 to 150 mM), or substances such as histamine or glycine
with affinity for the chelated metal can be used. The gradient is best run in the binding buffer at
constant pH.
Since pH governs the degree of ionization of charged groups at the binding sites, a gradient
or stepwise reduction in pH can be used for nonspecific elution of bound material. A range of pH
7.0 to 4.0 is normal, most proteins eluting between pH 6.0 and 4.2. Deforming eluents such as
8 M urea or 6 M guanidine hydrochloride can be used.
Elution with EDTA (50 mM) or other strong chelating agents will strip away metal ions
and other material bound. This method does not usually resolve different proteins.
If harsh elution conditions are used, it is recommended to transfer eluted fractions
immediately to milder conditions (either by collecting them in neutralization buffer or by
passing directly onto a desalting column for buffer exchange (see Buffer exchange and
desalting, Appendix 1).
The loss of metal ions is more pronounced at lower pH. The column does not have to
be stripped between consecutive purifications if the same protein is going to be purified.
Although metal-ion leakage is very low, the presence of any free metal in the purified
product can be avoided by connecting an uncharged HiTrap Chelating HP column in
series after the first column and before the protein is eluted. This column will bind any
metal ions removing them from the protein as it passes through the second column.
Performing a separation
This protocol can be used as a base from which to develop purification methods for proteins
and peptides with affinity for metal ions:
Metal-ion solution: 100 mM CuSO
4
Binding buffer: 20 mM sodium phosphate, 500 mM NaCl, 10 mM imidazole, pH 7.4
Elution buffer: 20 mM sodium phosphate, 500 mM NaCl, 500 mM imidazole, pH 7.4

66 18102229 AF
Use water, not buffer, to wash away the column storage solution which contains 20%
ethanol. This avoids the risk of nickel salt precipitation in the next step. If air is trapped
in the column, wash the column with distilled water until the air bubbles are expelled.
1. Wash the column with at least 2 CV of distilled water.
2. Load 0.5 CV of the 100 mM copper solution onto the column.
3. Wash with 5 CV of distilled water.
4. Equilibrate the column with 10 CV of binding buffer.
5. Apply sample at a flow rate of 1 to 4 ml/min (1 ml column) or 5 ml/min (5 ml column).
Collect the flowthrough fraction. A pump is more suitable for application of sample
volumes greater than 15 ml.
6. Wash with 10 CV of binding buffer. Collect wash fraction.
7. Elute with 5 CV of elution buffer. Collect eluted fractions in small fractions such as 1 ml
to avoid dilution of the eluate.
8. Wash with 10 CV of binding buffer. The column is now ready for a new purification and
there is rarely a need to reload with metal if the same (his)6-tagged protein is to be purified.
Reuse of purification columns depends on the nature of the sample and should only be
considered when processing identical samples to avoid cross-contamination.
Scaling up
To increase capacity, use several HiTrap Chelating HP columns (1 ml or 5 ml) in series
(note that back pressure will increase) or, for even larger capacity, pack Chelating
Sepharose Fast Flow into a suitable column (see Appendix 3).
Cleaning
Remove metal ions by washing with 5 CV of 20 mM sodium phosphate, 500 mM NaCl,
50 mM EDTA, pH 7.4.
Remove precipitated proteins by filling the column with 1 M NaOH and incubate for 2 h. Wash out
dissolved proteins with 5 CV of water and a buffer at pH 7.0 until the pH of the flowthrough
reaches pH 7.0.
Alternatively wash with a nonionic detergent such as 0.1% Tween 20 at 37°C for 1 min.
Remove lipid and very hydrophobic proteins by washing with 70% ethanol, or with a gradient
0%–30%–0% isopropanol/water.
Chemical stability
Stable in all commonly used aqueous buffers and denaturants such as 6 M guanidine
hydrochloride, 8 M urea, and other chaotropic agents.
Storage
Wash chromatography media and columns with 20% ethanol at neutral pH (use approximately
5 CV for packed media) and store at 4°C to 8°C.
Before long-term storage, remove metal ions by washing with 5 CV of 20 mM sodium
phosphate, 500 mM NaCl, 50 mM EDTA, pH 7.4.
The column must be recharged with metal ions after long-term storage.
18102229 AF 67
Purification or removal of serine proteases, such as thrombin and
trypsin, and zymogens
Benzamidine Sepharose 4 Fast Flow (low sub),
Benzamidine Sepharose 4 Fast Flow (high sub)
Sample extraction procedures often release proteases into solution, requiring the addition of
protease inhibitors to prevent unwanted proteolysis. An alternative to the addition of inhibitors
is to use a group-specific AC medium to remove the proteases from the sample. The same
procedure can be used to either specifically remove these proteases or purify them.
The synthetic inhibitor para-aminobenzamidine is used as the affinity ligand for trypsin,
trypsin-like serine proteases, and zymogens. Benzamidine Sepharose 4 Fast Flow is frequently
used to remove molecules from cell culture supernatant, bacterial lysate or serum. During the
production of recombinant proteins, tags such as GST are often used to facilitate purification
and detection. Enzyme specific recognition sites are included in the recombinant protein to
allow the removal of the tag by enzymatic cleavage when required. Thrombin is commonly
used for enzymatic cleavage, and must often be removed from the recombinant product.
HiTrap Benzamidine FF (high sub) provides a simple, ready-to-use solution for this process.
Figure 3.21 shows the partial structure of Benzamidine Sepharose 4 Fast Flow and Table 3.25
gives examples of different serine proteases. Benzamidine Sepharose 4 Fast Flow (high sub) is
a medium designed for high capacity. It has a trypsin binding capacity greater than 35 mg/ml
medium. For some applications, Benzamidine Sepharose 4 Fast Flow (high sub) induces strong
interactions between ligand and target molecules, which may lead to reduced purity and
recovery. Benzamidine Sepharose 4 Fast Flow (low sub) is designed to balance good capacity
with high purity and recovery.
O
N
NH
NH
2
O
N
O H
O H
O
H
H
Sepharose
Fig 3.21. Partial structure of Benzamidine Sepharose 4 Fast Flow.
Table 3.25. Examples of different serine proteases
Source M
r
pI
Thrombin Bovine pancreas 37 000 10.5
Trypsin Human plasma chain A
Human plasma chain B
5 700
31 000
7.1
Urokinase Human urine 54 000 8.9
Enterokinase Porcine intestine heavy chain
Porcine intestine light chain
134 000
62 000
4.2
Plasminogen Human plasma 90 000 6.4–8.5
Prekallikrein Human plasma nd nd
Kallikrein Human plasma
Human saliva
86 000
nd
nd (plasma)
4.0 (saliva)

68 18102229 AF
Chromatography media characteristics
Characteristics of Benzamidine Sepharose chromatography media are shown in Table 3.26.
Table 3.26. Characteristics of Benzamidine Sepharose 4 Fast Flow (low sub) and (high sub)
chromatography media
Ligand density
(µmol/ml) Composition pH stability
1
Average particle size
(µm)
Benzamidine
Sepharose 4
Fast Flow
(high sub)
> 12 Amide coupling of
p-aminobenzamidine
ligand via a 14 atom
spacer to Sepharose 4
Fast Flow (high sub).
Short term: 1 to 9
Long term: 2 to 8
90
Benzamidine
Sepharose 4
Fast Flow
(low sub)
6 Amide coupling of
p-aminobenzamidine
ligand via a 14 atom
spacer to Sepharose 4
Fast Flow (high sub).
Short term: 1 to 9
Long term: 2 to 8
90
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Long term refers
to the pH interval over which the medium is stable over a long period of time without adverse effects on its subsequent
chromatographic performance.
Purification options
Benzamidine Sepharose 4 Fast Flow low and high sub chromatography media and prepacked
columns are described in Table 3.27.
Table 3.27. Purification options for Benzamidine Sepharose 4 Fast Flow chromatography media and
prepacked columns
Binding capacity
Maximum
operating flow Comments
HiTrap Benzamidine FF
(high sub)
Trypsin, > 35 mg/column
Trypsin, > 175 mg/column
4 ml/min
20 ml/min
Prepacked: 1 ml column
2
.
Prepacked: 5 ml column
2
.
Benzamidine Sepharose 4
Fast Flow (high sub)
Trypsin, > 35 mg/ml medium 300 cm/h
1
Supplied as a suspension
ready for column packing
2
.
Benzamidine Sepharose 4
Fast Flow (low sub)
Trypsin, 25 mg/ml medium 300 cm/h
1
Supplied as a suspension
ready for column packing
2
.
1
See Appendix 4 to convert flow velocity (cm/h) to volumetric flow rate (ml/min). Maximum operating flow is calculated
from measurement in a packed column with a bed height of 10 cm and i.d. of 5 cm.
2
Supplied in 50 mM acetate, pH 4.0 containing 20% ethanol.
Purification examples
Figure 3.23 shows an example of the removal of trypsin-like proteases from human plasma to
prevent proteolysis of the plasma components, using a low pH elution. The activity test demonstrated
that almost all trypsin-like protease activity is removed from the sample and bound to the column.
Column: HiTrap Benzamidine FF (high sub), 1 ml
Sample: 1 ml human plasma filtered
through a 0.45 µm filter
Binding buffer: 20 mM Tris-HCl, 500 mM NaCl, pH 7.4
Elution buffer: 50 mM glycine, pH 3.0
0% to 100% elution buffer in one step
Flow rate: 1.0 ml/min
Protease activity: S-2288 from Chromogenix,
Heamochrom Diagnostica AB
A
405
measurement. The activity
is presented as the proteolytic
activity/mg protein
System: ÄKTA system
0
0.20
0.40
0.60
0.80
1.00
3.0
2.5
2.0
1.5
1.0
0.5
A
A
A
280
280
405
0.0 5.0 10.0
Volume (ml)
15.0
10 × IU/liter
-3
Fig 3.22. Removal of trypsin-like serine proteases from human plasma using HiTrap Benzamidine FF (high sub), 1 ml.
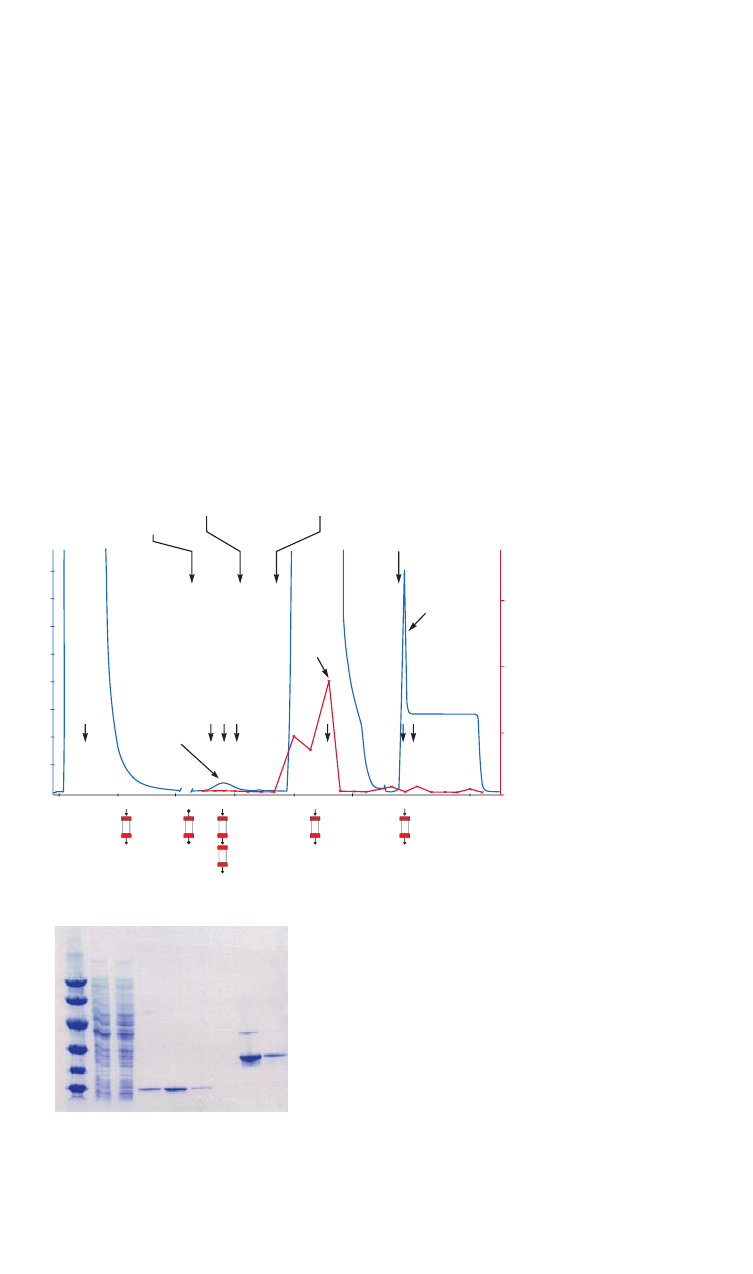
18102229 AF 69
Figure 3.23 shows the effectiveness of using a GSTrap™ FF column with a HiTrap Benzamidine FF
(high sub) for purification of a GST-tagged protein, followed by cleavage of the GST tag via the
thrombin cleavage site and subsequent removal of the thrombin enzyme. The GST-tagged protein
binds to the GSTrap FF column as other proteins wash through the column. Thrombin is applied to
the column and incubated for 2 h.
A HiTrap Benzamidine FF (high sub) column, pre-equilibrated in binding buffer, is attached after
the GSTrap FF column and both columns are washed in binding buffer followed by a high salt
buffer. The cleaved protein and thrombin wash through from the GSTrap FF column, thrombin
binds to the HiTrap Benzamidine FF (high sub) column, and the eluted fractions contain pure
cleaved protein.
0
0
0.10
0.20
M
r
97 000
66 000
20 100
30 000
45 000
14 400
1 2 3 4 5 6 7 8 9
0.20
0.30
Thrombin activity
0.40
0.80
0.60
0
fr.6
Cleaved SH2
protein
Thrombin
Thrombin
High salt
buffer wash
Elution on HiTrap
Benzamidine FF (high sub)
Elution on
GSTrap FF
GST-tag
fr.7
fr.8
fr.14
fr.21
fr.22
fr.2
10 20 50
Volume (ml)
15 25
A
280 nm
A
405 nm
(A) (A) (A)
(B)
(B) (A)
(A)
(B)
GSTrap FF, 1 ml
HiTrap Benzamidine FF (high sub), 1 ml
Columns: GSTrap FF, 1 ml and HiTrap Benzamidine FF (high sub), 1 ml
Sample: 2 ml clarified E. coli homogenate expressing a M
r
37 000 SH2-GST-tagged protein with
a thrombin cleavage site
Binding buffer: 20 mM sodium phosphate, 150 mM NaCl, pH 7.5
High salt wash buffer: 20 mM sodium phosphate, 1 M NaCl, pH 7.5
Benzamidine elution buffer: 20 mM p-aminobenzamidine in binding buffer
GST elution buffer: 20 mM reduced glutathione, 50 mM Tris, pH 8.0
Flow rate: 0.5 ml/min
Protease treatment: 20 units/ml thrombin (GE) for 2 h at room temperature
Thrombin activity: S-2238 (Chromogenix, Haemochrom Diagnostica AB) was used as a substrate and its absorbance at
405 nm was measured
System: ÄKTA system
Lanes
ExcelGel SDS Gradient 8–18, Coomassie Blue staining
1. LMW-SDS Marker Kit
2. Clarified E. coli homogenate expressing SH2-GST-tagged protein
3. Flowthrough from GSTrap FF (Fraction 2)
4. SH2 GST-tag cleaved, washed off with binding buffer through
both columns (Fraction 6)
5. as above (Fraction 7)
6. as above (Fraction 8)
7. Elution of thrombin, HiTrap Benzamidine FF (high sub)
8. Elution of GST-tag and some noncleaved SH2-GST, GSTrap FF (Fraction 21)
9. as above (Fraction 22)
Fig 3.23. On-column cleavage of a GST-tagged protein and removal of thrombin after on-column
cleavage, using GSTrap FF and HiTrap Benzamidine FF (high sub).

70 18102229 AF
Performing a separation
Binding buffer: 50 mM Tris-HCl, 500 mM NaCl, pH 7.4
Elution buffer alternatives:
- pH elution: 50 mM glycine-HCl, pH 3.0 or 10 mM HCl, 50 mM NaCl, pH 2.0
- competitive elution: 20 mM p-aminobenzamidine in binding buffer
- denaturing eluents: 8 M urea or 6 M guanidine hydrochloride
1. Equilibrate the column with 5 CV of binding buffer.
2. Apply the sample.
3. Wash with 5 to 10 CV of binding buffer or until no material appears in the eluent
(monitored by UV absorption at A
280 nm
).
4. Elute with 5 to 10 CV of elution buffer. Collect fractions in neutralization buffer if low
pH elution is used
1
. The purified fractions can be buffer exchanged using desalting
columns (see Buffer exchange and desalting, Appendix 1).
1
Since elution conditions are quite harsh, collect fractions into neutralization buffer (60 to 200 µl of 1 M Tris-HCl, pH 9.0 per
milliliter of fraction), so that the final pH of the fractions will be approximately neutral.
Since Benzamidine Sepharose 4 Fast Flow has some ionic binding characteristics, the
use of 500 mM NaCl and pH elution between 7.4 and 8.0 is recommended. If lower salt
concentrations are used, include a high salt wash step after sample application and
before elution.
The elution buffer used for competitive elution has a high absorbance at 280 nm. The
eluted protein must be detected by other methods, such as an activity assay, total
protein or SDS-PAGE analysis. The advantage with competitive elution is that the pH is
kept constant throughout the purification.
Cleaning
Wash with 3 to 5 CV of 100 mM Tris-HCl, 500 mM NaCl, pH 8.5 followed with 3 to 5 CV of
100 mM sodium acetate, 500 mM NaCl, pH 4.5 and re-equilibrate immediately with 3 to 5 CV
of binding buffer.
Remove severe contamination by washing with nonionic detergent such as 0.1%
Tween 20 at 37°C for 1 min.
Chemical stability
All commonly used aqueous buffers.
Storage
Wash chromatography media and columns with 20% ethanol in 50 mM sodium acetate, pH 4.0
(use approximately 5 CV for packed media) and store at 4°C to 8°C.
18102229 AF 71
Purification or removal of viruses including
adeno-associated virus
Capto DeVirS, AVB Sepharose High Performance
Capto DeVirS is a chromatography medium for capture and intermediate purification of virus.
The dextran sulfate ligand (Fig 3.24) has affinity for several virus types, which makes Capto DeVirS
suitable for different virus applications, for example vaccine manufacturing processes. The
matrix of Capto DeVirS is based on highly cross-linked high-flow agarose that is highly rigid and
offers outstanding pressure/flow properties, enabling rapid processing of large sample volumes.
AVB Sepharose High Performance is designed for the purification of adeno-associated virus
(AAV) of subclasses 1, 2, 3, and 5. Adeno associated viruses are of increasing interest as
potential vectors for gene therapy. The ligand of AVB Sepharose High Performance is a
recombinant protein, M
r
14 000, attached to a highly cross-linked 6% agarose matrix via a long,
hydrophilic spacer arm to make it easily available for binding of the virus (Fig 3.25).
O
O
H
H
OH
H
NaSO
3
O
OSO
3
Na
H
O
O
O
H
H
OH
H
NaSO
3
O
OSO
3
Na
H
OH
O H
H
OH
H
NaSO
3
O
OSO
3
Na
H
CH
2
CH
2
CH
2
Fig 3.24. Capto DeVirS consists of highly cross-linked agarose base matrix coupled to dextran sulfate ligand.
OH
O
Ligand
O
H
N
N
H
Matrix
Fig 3.25. Partial structure of AVB Sepharose High Performance.
Chromatography media characteristics
Characteristics of chromatography media for the purification of viruses are described in Table 3.28.
Table 3.28. Characteristics of Capto DeVirS and AVB Sepharose High Performance chromatography media
Product Ligand Composition pH stability
1
Average
particle
size (µm)
Capto DeVirS Dextran sulfate Ligand coupled to
Capto.
Short term: 6 to 14
Long term: 7 to 13
75
AVB Sepharose
High Performance
M
r
14 000 recombinant
protein produced in
S. cerevisiae. Binds
AAV of subclasses 1, 2,
3, and 5
Ligand coupled
to Sepharose
High Performance.
Short term: 2 to 12
Long term: 3 to 10
34
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Long term refers
to the pH interval over which the medium is stable over a long period of time without adverse effects on its subsequent
chromatographic performance.

72 18102229 AF
Purification options
Purification options for Capto DeVirS and AVB Sepharose High Performance and prepacked
columns are shown in Table 3.29.
Table 3.29. Purification options for Capto DeVirS, AVB Sepharose High Performance and prepacked columns
Binding capacity
Maximum
operating flow Comments
Capto DeVirs Influenza virus: up to 9 log
10
FFU
1
/ml 600 cm/h
2
Media suspension ready
for column packing.
HiTrap AVB
Sepharose HP, 1 ml
> 1012 genome copies/column 1 ml/min Prepacked 1 ml column.
HiTrap AVB
Sepharose HP, 5 ml
> 5060 genome copies/column 5 ml/min Prepacked 5 ml column.
AVB Sepharose
High Performance
> 1012 genome copies/ml medium 150 cm/h
3
Media suspension ready
for column packing.
1
FFU: Fluorescence Focal Unit.
2
1 m diameter column with a 20 cm bed height at 20°C using process buffers with the same viscosity as water.
3
Bed height 30 cm.
Purification examples
Capto DeVirS was used as a capture step in the purification of influenza virus. In order to
establish the optimal purification protocol, Design of Experiments (DoE) was used. The effect
of running parameters on virus binding, recovery, and clearance of host cell protein (HCP) and
DNA was investigated. The evaluation showed that optimum conductivity for the binding of
influenza virus to Capto DeVirS was below 5 mS/cm, while optimum pH for binding and elution
was pH 6.8 and pH 7.8, respectively.
Table 3.30 shows the results for the different strains of influenza virus in optimized Capto DeVirs
purification.
Table 3.30. Purification of different influenza strains on Capto DeVirS in an XK50 column (5 × 17 cm) with
330 ml of medium and a flow velocity of 150 cm/h
Influenza strain A/South Dakota A/Uruguay B/Florida
Loading titer
(log
10
FFU
1
/ml) 9.3 6.6 7.9
Step yield (%) 76 77 84
HCD
2
level (ng/dose) 0.20 N/A 0.93
1
FFU = Fluorescence Focal Unit.
2
HCD = Host-cell DNA
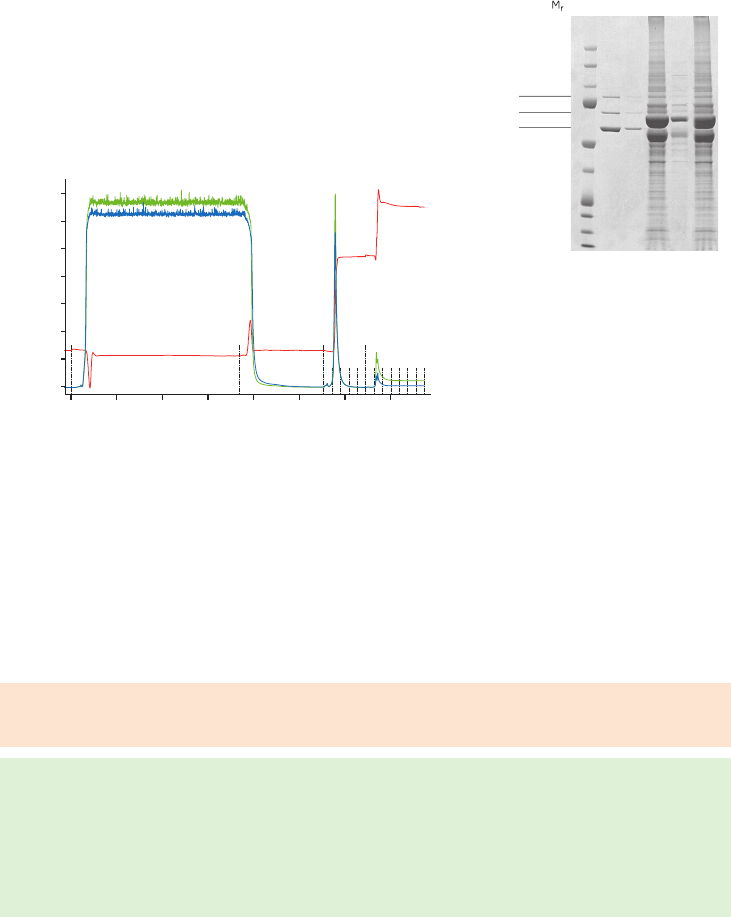
18102229 AF 73
Figure 3.26A shows an example of a purification of adeno-associated virus using AVB
Sepharose High Performance. Recombinant AAV1 was eluted using low pH followed by a
second elution buffer containing arginine (high pH). Eluted virus was detected by ELISA and
SDS-PAGE analysis (Fig 3.26B). SDS-PAGE showed three AAV viral capsid proteins, VP1, VP2,
and VP3 (at M
r
87 000, 73 000, and 62 000, respectively) eluted in the initial low pH elution. An
additional 6% of the bound virus eluted with the second high-pH elution containing arginine.
injection
Wash
Start gradient
High pH + Arginine
3500
3000
2500
2000
1500
1000
500
0
mAU
0.0 5.0 10.0 15.0 20.0 25.0
pH
30.0 35.0
Volume (ml)
1 2 3 4 5 6
225 000
150 000
102 000
76 000
52 000
38 000
31 000
24 000
17 000
12 000
VP1
VP2
VP3
Column: Tricorn 5/50 (1 ml CV)
Chromatography medium: AVB Sepharose High Performance, 1 ml
Sample: rAAV1 (7.0 × 10
10
viral genomes/ml)
Sample load: 20 ml
Wash buffer: 20 mM Tris-HCl, 500 mM NaCl, pH 8.0
First elution buffer: 100 mM sodium acetate, 500 mM NaCl, pH 2.5
Second elution buffer: 20 mM Tris-HCl, 500 mM NaCl, 500 mM arginine, pH 10.5
Flow velocity: 153 cm/h
System: ÄKTA system
Lanes
1. High molecular weight (HMW) markers
2. Fraction from peak eluted at low pH
3. Fraction from peak eluted at high pH
(500 mM arginine)
4. Flowthrough
5. Wash
6. Loaded sample
(A) (B)
injection
Wash
Start gradient
High pH + Arginine
3500
3000
2500
2000
1500
1000
500
0
mAU
0.0 5.0 10.0 15.0 20.0 25.0
pH
30.0 35.0
Volume (ml)
1 2 3 4 5 6
225 000
150 000
102 000
76 000
52 000
38 000
31 000
24 000
17 000
12 000
VP1
VP2
VP3
Fig 3.26. (A) Purification of rAAV on AVB Sepharose High Performance using low pH elution followed by
high pH elution with 500 mM arginine. The absorbance at 260 and 280 nm is shown in green and blue,
respectively. Conductivity is shown in red. (B) SDS-PAGE analysis on fractions collected during purification
of rAAV1 on AVB Sepharose High Performance.
Performing a separation
Capto DeVirS
Sample preparation: Concentrate the clarified feed with ultrafiltration and perform a buffer
exchange to the start buffer
Binding buffer: 20 mM sodium phosphate, pH 6.8
Elution buffer: 20 mM sodium phosphate, 1.5 M NaCl pH 7.4
1. Equilibrate with 10 CV of binding buffer.
2. Load the clarified sample.
3. Wash with binding buffer until no material appears in the eluent (monitored by UV
absorption at A
280 nm
).
4. Elute with 5 to 10 CV of elution buffer.

74 18102229 AF
AVB Sepharose High Performance
Sample preparation: Clarified AAV vector cell lysate can be applied directly to the column.
Binding buffer: 20 mM Tris-HCl, 500 mM NaCl, pH 8.0
Elution buffer: 100 mM sodium acetate, 500 mM NaCl, pH 2.5
1. Equilibrate with 10 CV of binding buffer.
2. Load the clarified sample.
3. Wash with binding buffer until no material appears in the eluent (monitored by UV
absorption at A
280 nm
).
4. Elute with 5 to 10 CV of elution buffer.
Note: Elution at high pH can be performed as an alternative if the virus is sensitive to low
pH. The recommended elution buffer in this case is 20 mM Tris-HCl, 500 mM NaCl,
500 mM arginine, pH 10.8.
Cleaning
Cleaning and sanitization protocols should be designed for each process as the efficiency of
the protocol is strongly associated with the feedstock and other related operating conditions.
Suggested solutions for a contact time of at least 30 min:
Capto DeVirS: 1 M NaOH
AVB Sepharose High Performance: PAB (120 mM phosphoric acid, 167 mM acetic acid,
2.2% benzyl alcohol)
Storage
Store at 4°C to 8°C in 20% ethanol.
BioProcess
™
chromatography media (resins) for AC
BioProcess chromatography media family includes chromatography media widely used by
biopharmaceutical manufacturers. Support for these products includes validated manufacturing
methods, secure long-term medium supply, safe and easy handling, and regulatory support
files (RSF) to assist process validation and submissions to regulatory authorities. In addition, the
Fast Trak Training & Education team provides high-level hands-on training for all key aspects of
bioprocess development and manufacturing. All BioProcess media have high chemical stability
to allow efficient cleaning/sanitization procedures and validated packing methods established
for a wide range of large-scale columns.
The range of BioProcess chromatography media for large-scale purification includes Capto media
such as Capto Blue for removal or purification of albumin, and a large number of Sepharose
Fast Flow media for purification of specific groups of molecules. These AC media can be run at
high flow rates and have high dynamic binding capacities.
Most of the chromatography media are available in HiTrap and HiScreen prepacked columns
for development of efficient and robust purification parameters before scaling up. By using
these small-scale formats in the early stages of process development, valuable time is saved
and buffer and sample consumption reduced.
Custom Designed Media
Custom Designed Media (CDM) can be produced for specific industrial process separations
when suitable chromatography media are not available from the standard range. The
Custom Designed Media group (CDM group) works in close collaboration with the user to
design, manufacture, test, and deliver chromatography media for specialized purification
requirements. Visit www.gelifesciences.com/cdm for more information.
18102229 AF 75
Chapter 4
Designing affinity chromatography media
using preactivated matrices
Chapter 3 in this handbook covers a wide range of ligands that have been coupled to matrices
to provide ready-to-use AC media for specific groups of molecules. However, it is also possible
to design new media for special purposes. When a ready-to-use AC medium is not available,
a medium can be designed for the purification of one or more target molecules by coupling a
specific ligand onto a preactivated chromatography matrix. For example, antibodies, antigens,
enzymes, receptors, small nucleic acids, or peptides can be used as affinity ligands to enable the
purification of their corresponding binding partners.
There are three key steps in the design of an AC medium:
• Choosing the matrix.
• Choosing the ligand and spacer arm.
• Choosing the coupling method.
Choosing the matrix
Sepharose provides a macroporous matrix with high chemical and physical stability and low
nonspecific adsorption to facilitate a high binding capacity and sample recovery and to ensure
resistance to potentially harsh elution and wash conditions. The choice of a preactivated
Sepharose matrix depends on the functional groups available on the ligand and whether or not
a spacer arm is required. Table 4.1 reviews the preactivated matrices available.
Choosing the ligand and spacer arm
The ligand must selectively and reversibly interact with the target molecule(s) and must
be compatible with the anticipated binding and elution conditions. The ligand must carry
chemically modifiable functional groups through which it can be attached to the matrix
without loss of activity (see Table 4.1).
If possible, test the affinity of the ligand: target molecule interaction. Too low affinity will result
in poor yields since the target protein can wash through or leak from the column during sample
application. too high affinity will result in low yields since the target molecule might not dissociate
from the ligand during elution.
Use a ligand with the highest possible purity since the final purity of the target substance
depends on the biospecific interaction.
As discussed in Chapter 1, when using small ligands (M
r
< 5 000) there is a risk of steric
hindrance between the ligand and the matrix that restricts the binding of target molecules.
In this case, select a preactivated matrix with a spacer arm. For ligands with M
r
> 5 000, no
spacer arm is necessary.
76 18102229 AF
Choosing the coupling method
Ligands are coupled via reactive functional groups such as amino, carboxyl, hydroxyl, thiol, and
aldehyde moieties. In the absence of information on the location of binding sites in the ligand, a
systematic trial-and-error approach should be used.
Couple a ligand through the least critical region of the ligand to minimize interference with the
normal binding reaction. For example, an enzyme inhibitor containing amino groups can be
attached to a matrix through its amino groups, provided that the specific binding activity with
the enzyme is retained. However, if the amino groups are involved in the binding reaction, an
alternative, nonessential, functional group must be used.
Avoid using a functional group that is close to a binding site or that plays a role in the
interaction between the ligand and target molecule.
If a suitable functional group does not exist, consider derivatizing the ligand to add a
functional group.
Table 4.1. Chemical groups on ligands and spacer arms for preactivated chromatography media
Chemical group
on ligand
Length of
spacer arm Structure of spacer arm Product
Proteins,
peptides,
amino acids
amino 10-atom
14-atom
NHS-activated High Performance
NHS-activated Sepharose 4 Fast Flow
None – CNBr-activated Sepharose 4B
None – CNBr-activated Sepharose 4 Fast Flow
carboxyl 11-atom EAH Sepharose 4B
9-atom Activated Thiol Sepharose 4B
12-atom Epoxy-activated Sepharose 6B
Sugars
hydroxyl 12-atom Epoxy-activated Sepharose 6B
amino 10-atom NHS-activated High Performance
12-atom Epoxy-activated Sepharose 6B
carboxyl 11-atom EAH Sepharose 4B
Polynucleotides
amino None – CNBr-activated Sepharose 4B
CNBr-activated Sepharose 4 Fast Flow
Coenzymes,
cofactors,
antibiotics,
steroids
amino, carboxyl,
thiol, or hydroxyl
Use matrix with spacer arm (see above)
O
O
O
OH
O
O
N
OH
O
O
N
O
O
O
N
OH
OH
O
O
O
O
OH
O
O
N
OH
NH
2
O
S
OH
S
N
O
N
OH
O
O
N
O
O
O
N
OH
OH
O
O
N
OH
NH
2
O
S
OH
S
N
O
O
O
OH
O
N
N
S
O
S
N
N
O
HO O
OH
O
O
O
O
OH
O
O
N
OH
O
O
N
O
O
O
N
OH
OH
O
O
O
O
OH
O
O
N
OH
NH
2
O
S
OH
S
N
O
N
OH
O
O
N
O
O
O
N
OH
OH
O
O
N
OH
NH
2
O
S
OH
S
N
O
O
O
OH
O
N
N
S
O
S
N
N
O
HO O
OH
O
O
O
O
OH
O
O
N
OH
O
O
N
O
O
O
N
OH
OH
O
O
O
O
OH
O
O
N
OH
NH
2
O
S
OH
S
N
O
N
OH
O
O
N
O
O
O
N
OH
OH
O
O
N
OH
NH
2
O
S
OH
S
N
O
O
O
OH
O
N
N
S
O
S
N
N
O
HO O
OH
O
O
O
O
OH
O
O
N
OH
O
O
N
O
O
O
N
OH
OH
O
O
O
O
OH
O
O
N
OH
NH
2
O
S
OH
S
N
O
N
OH
O
O
N
O
O
O
N
OH
OH
O
O
N
OH
NH
2
O
S
OH
S
N
O
O
O
OH
O
N
N
S
O
S
N
N
O
HO O
OH
O
O
O
O
OH
O
O
N
OH
O
O
N
O
O
O
N
OH
OH
O
O
O
O
OH
O
O
N
OH
NH
2
O
S
OH
S
N
O
N
OH
O
O
N
O
O
O
N
OH
OH
O
O
N
OH
NH
2
O
S
OH
S
N
O
O
O
OH
O
N
N
S
O
S
N
N
O
HO O
OH
O

18102229 AF 77
Coupling the ligand
The principle for coupling of ligand is described in this protocol (see also specific
protocols for each preactivated medium).
1. Prepare the ligand solution in coupling buffer, either by dissolving the ligand in coupling
buffer or exchanging the solubilized ligand into the coupling buffer using a desalting column.
2. Prepare the preactivated matrix according to the supplied instructions.
3. Mix the ligand solution and the matrix in the coupling buffer until the coupling reaction
is completed.
4. Block any remaining active groups.
5. Wash the coupled matrix alternately at high and low pH to remove excess ligand and
reaction by-products.
6. Equilibrate in binding buffer or transfer to storage solution.
It is not usually necessary to couple a large amount of ligand to produce an efficient AC
medium. After coupling, wash the medium thoroughly using buffers of alternating low and
high pH to remove noncovalently bound ligand.
A high concentration of coupled ligand is likely to have adverse effects on AC. The
binding efficiency of the medium may be reduced due to steric hindrance between the
active sites (particularly important when large molecules such as antibodies, antigens
and enzymes interact with small ligands). Target substances can bind more strongly to
the ligand making elution difficult. The extent of nonspecific binding increases at very
high ligand concentrations thus reducing the selectivity of the medium.
Remember that the useful capacity of an AC medium can be significantly affected
by flow rate.
For applications that require operating at high pH, the amide bond formed when using
NHS-activated Sepharose is stable up to pH 13.0.
Figure 4.1 shows the effect of ligand concentration on the final amount of ligand coupled to a matrix.
Chymotrypsinogen
Chymotrypsin
Protein added (mg)
0 10
20
30 40
5
10
15
Protein coupled (mg)
0
Fig 4.1 Effect of protein concentration on amount of protein coupled. Protein was coupled to 2 ml
CNBr-activated Sepharose 4B in NaHCO
3
, NaCl solution, pH 8.0.
Table 4.2 summarizes recommended ligand concentrations according for various ligand types.

78 18102229 AF
Table 4.2. Recommended ligand concentrations for coupling
Experimental condition Recommended concentration for coupling
Readily available ligands 10- to 100-fold molar excess of ligand over available groups
Small ligands 1 to 20 µmol/ml medium (typically 2 µmol/ml medium)
Protein ligands 5 to 10 mg protein/ml medium
Antibodies 5 mg protein/ml medium
Very low affinity systems Maximum possible ligand concentration to increase the binding
For certain preactivated matrices, agents are used to block any activated groups
that remain on the matrix after ligand coupling. These blocking agents such as
ethanolamine and glycine can introduce a small number of charged groups into the
matrix. The effect of these charges is overcome by the use of a relatively high salt
concentration (500 mM NaCl) in the binding buffer for affinity purification. A wash cycle
of low and high pH is essential to ensure that no free ligand remains ionically bound to
the coupled ligand. This wash cycle does not cause loss of covalently bound ligand.
Binding capacity, ligand density, and coupling efficiency
Testing the binding capacity of the medium after coupling will give an indication of the success
of the coupling procedure and establish the usefulness of the new AC medium.
Several different methods can be used to determine the ligand density (µmol/ml
medium) and coupling efficiency.
• The fastest and easiest, but least accurate way to quantitate the free ligand in solution is by
spectrophotometry. Measure the ligand concentration before coupling and compare this
with the concentration of the unbound ligand after coupling. The difference is the amount
that is coupled to the matrix.
• Spectroscopic methods can also be used if the ligand has been suitably prelabeled.
The coupled ligand can be quantitated by direct spectroscopy of the AC medium suspended
in a solution with the same refractive index, such as 50% glycerol or ethylene glycol.
By-products of the coupling reaction, such as N-hydroxysuccinimide in the case of NHS-
activated matrices, can be quantitated by spectroscopy.
• The medium can be titrated to determine ligand concentration. The titrant must be relevant
to the ligand.
• The most accurate method to determine ligand concentration is direct amino acid analysis
or determination of characteristic elements. Note that these are destructive techniques.
If the binding capacity for the target is insufficient there are several ways to try to
increase the coupling efficiency:
• Ensure that the ligand is of high purity; there might be contaminants present that are
preferentially coupled.
• Increase the ligand concentration to increase the ligand density on the matrix, but avoid
overloading the matrix as this may cause steric hindrance and so reduce the binding
capacity again.
• Modify reaction conditions such as pH, temperature, buffers, or contact time. Most pre-
activated matrices are supplied with details of the preferred conditions for a coupling
reaction that can be used as a basis for further optimization.

18102229 AF 79
Binding and elution conditions
Binding and elution conditions will depend on the nature of the interaction between the ligand
and target. As for any affinity purification, the general guidelines outlined in Chapter 2 can be
applied during development.
For the first run, perform a blank run to ensure that any loosely bound ligand is removed.
Immunospecific interactions can be strong and sometimes difficult to reverse. The
specific nature of the interaction determines the elution conditions. Always check the
reversibility of the interaction before coupling a ligand to an affinity matrix. If standard
elution buffers do not reverse the interaction, try alternative elution buffers such as:
• Low pH (below pH 2.5).
• High pH (up to pH 11.0).
• Substances that reduce the polarity of the buffer can facilitate elution without affecting
protein activity such as dioxane (up to 10%), ethylene glycol (up to 50%).
The following protocol can be used as a guideline for a preliminary separation:
1. Prepare the column (blank run)
A. Wash with 2 CV of binding buffer.
B. Wash with 3 CV of elution buffer.
2. Equilibrate with 10 CV of binding buffer.
3. Apply sample. The optimal flow rate is dependent on the binding constant of the
ligand, but a recommended flow rate range is, for example, 0.5 to 1 ml/ min on a
HiTrap NHS-activated HP 1 ml column.
4. Wash with 5 to 10 CV of binding buffer, or until no material appears in the eluent,
as monitored by absorption at A
280 nm
.
5. Elute with 1 to 3 CV of elution buffer (larger volumes might be necessary).
6. If required purified fractions can be desalted and transferred into the buffer of choice
using prepacked desalting columns (see Buffer exchange and desalting, Appendix 1).
7. Re-equilibrate the column immediately by washing with 5 to 10 CV of binding buffer.
Avoid excessive washing if the interaction between the protein of interest and the
ligand is weak since this can decrease the yield.
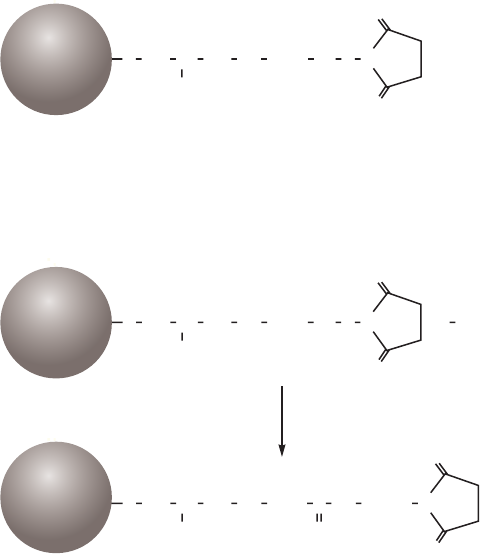
80 18102229 AF
Coupling through the primary amine of a ligand
NHS-activated Sepharose High Performance, NHS-activated Sepharose 4 Fast Flow
NHS-activated Sepharose is designed for the covalent coupling of ligands (often antigens or
antibodies) containing primary amino groups (the most common form of attachment). The
matrix of NHS-activated Sepharose High Performance is based on highly cross-linked agarose
beads with 10-atom spacer arms (6-aminohexanoic acid) attached by epichlorohydrin and
activated by N-hydroxysuccinimide (Fig 4.2). The matrix of NHS-activated Sepharose 4 Fast
Flow is based on highly cross-linked agarose beads with 14-atom spacer arms. Nonspecific
adsorption of proteins to NHS-activated Sepharose (which can reduce binding capacity of the
target protein) is negligible due to the excellent hydrophilic properties of the base matrix. The
matrix is stable at high pH to allow stringent washing procedures (subject to the pH stability of
the coupled ligand).
O
O
OH
+ R
CO
5
2 22 2
CH
(CH )
NH NHOO CH C H N
O
O
OH
C R
5
22 2
CH
(CH )
NHNH + HOO CH CH N
O
O
O
OH
CO
5
2
2 2
CH
(CH )
NH OO CH CH N
Sepharose
Sepharose
Sepharose
Fig 4.2. Partial structure of NHS-activated Sepharose High Performance bearing activated spacer arms.
Ligands containing amino groups couple rapidly and spontaneously by nucleophilic attack at the
ester linkage to give a very stable amide linkage (Fig 4.3). The amide bond is stable up to pH 13.0
making NHS-activated Sepharose suitable for applications that require conditions at high pH.
O
O
OH
+ R
CO
5
2 2
2 2
CH
(CH )
NH NHOO CH C H N
O
O
OH
C R
5
2
2 2
CH
(CH )
NHNH + HOO CH CH N
O
O
O
OH
CO
5
22 2
CH
(CH )
NH OO CH CH N
Sepharose
Sepharose
Sepharose
Fig 4.3. Coupling a ligand to NHS-activated Sepharose High Performance.

18102229 AF 81
Chromatography media characteristics
Characteristics of NHS-activated Sepharose chromatography media are shown in Table 4.3.
Table 4.3. Characteristics of NHS-activated Sepharose chromatography media
Product
Ligand
density
(µmol/ml) Composition pH stability
1
Average
particle size
(µm)
NHS-activated Sepharose
High Performance
10 6-aminohexanoic acid
linked by epoxy coupling
to Sepharose High
Performance, terminal
carboxyl group esterified
with NHS.
Short term: 3 to 12
Long term: 3 to 12
34
NHS-activated
Sepharose 4 Fast Flow
16 to 23 As above. Short term: 3 to 13
Long term: 3 to 13
90
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Stability data refers
to the coupled medium provided that the ligand can withstand the pH. Long term refers to the pH interval over which the
matrix is stable over a long period of time without adverse effects on its subsequent chromatographic performance.
Purification options
NHS-activated Sepharose chromatography media and prepacked columns are described in
Table 4.4.
Table 4.4. Purification options for NHS-activated chromatography media and prepacked columns
Product Spacer arm
Coupling
conditions
Maximum
operating flow Comments
HiTrap NHS-activated HP 10-atom pH 6.5 to 9.0, 15
to 30 min at room
temp. or 4 h at 4°C.
4 ml/min
(1 ml column)
20 ml/min
(5 ml column)
Prepacked 1 ml column.
Prepacked 5 ml column.
NHS-activated Sepharose
4 Fast Flow
14-atom pH 6 to 9, 16 h
4°C to room temp.
300 cm/h
1
Supplied as a suspension
ready for column packing.
1
See Appendix 4 to convert flow velocity (cm/h) to volumetric flow rate (ml/min). Maximum operating flow is calculated
from measurement in a packed column with a bed height of 10 cm and i.d. of 5 cm.
Figure 4.4 shows that over 30 mg IgG can be coupled to a 1 ml HiTrap NHS-activated HP column.
The coupling process takes less than 15 min. The AC medium is then ready to use for antigen
purification.
Protein coupled (mg)
Protein added (mg/1 ml column)
40
30
20
10
0
20 40
600
80
100
Fig 4.4. Ligand coupling to HiTrap NHS-activated HP.

82 18102229 AF
Purification example
AC can be used to produce monospecific antibodies from polyclonal sera. This approach was
taken by the Human Protein Atlas project (www.proteinatlas.org) on a proteome-wide scale.
Polyclonal antibodies were raised in rabbits to all proteins encoded for by the human genome.
Each antibody serum was purified using HiTrap NHS-activated columns to which antigen
epitopes had been immobilized (Fig 4.5). Elution was achieved by lowering the pH to 2.5. The
required high throughput was obtained by using 12 modules of an ÄKTA system to purify 48
polyclonal antisera per day.
AC
Columns: 4 × HiTrap NHS-activated HP 1 ml, with immobilized antigens
Sample: 12 ml antiserum applied to each column
Binding buffer: 2 mM NaH
2
PO
4
, 8 mM Na
2
HPO
4
, 150 mM NaCl, 0.05% Tween,
pH 7.0 to 7.5
Elution buffer: 200 mM glycine, 1 mM EDTA, pH 2.5
Flow rate: 0.7 ml/min (sample loading); 1ml/min (elution)
System: ÄKTA system
DS
Columns: 2 × HiTrap Desalting, 5 ml
Flow rate: 5 ml/min
Buffer: 2 mM NaH
2
PO
4
, 8 mM Na
2
HPO
4
, 150 mM NaCl
System: ÄKTA system
mAU
Volume (ml)
400
300
200
100
0
-10 -5 50
AC
DS
Fig 4.5. Elution of purified antibody from the antigen-specific column and desalting on HiTrap Desalting, 5 ml
(courtesy of the Human Protein Atlas project).
Performing a purification
HiTrap NHS-activated HP
The protocol below describes the preparation of a prepacked HiTrap NHS-activated HP column
and is generally applicable to NHS-activated Sepharose chromatography media. A general
column packing procedure is described in Appendix 3.
The activated matrix is supplied in 100% isopropanol to preserve the stability before
coupling. Do not replace the isopropanol until it is time to couple the ligand.
Buffer preparation
Acidification solution: 1 mM HCl (kept on ice)
Coupling buffer: 200 mM NaHCO
3
, 500 mM NaCl, pH 8.3
Blocking buffer: 500 mM ethanolamine, 500 mM NaCl, pH 8.3
Wash buffer: 100 mM acetate, 500 mM NaCl, pH 4.0
Coupling within pH range 6.5 to 9.0, maximum yield is achieved at around pH 8.0.
Ligand and column preparation
1. Dissolve the ligand in the coupling buffer to a final concentration of 0.5 to 10 mg/ml
(for protein ligands) or perform a buffer exchange using a desalting column (see Buffer
exchange and desalting in Appendix 1). The optimal concentration depends on the
ligand. Dissolve the ligand in one column volume of buffer.
2. Remove the top cap from the column and apply a drop of ice-cold 1 mM HCl to the top
of the column to avoid air bubbles.
3. Connect the top of the column to the syringe or pump.
4. Remove the snap-off end.

18102229 AF 83
Ligand coupling
1. Wash out the isopropanol with 3 × 2 CV of ice-cold 1 mM HCl.
2. Inject 1 CV of ligand solution onto the column.
3. Seal the column. Leave for 15 to 30 min at 25°C (or 4 h at 4°C).
Recirculate the solution if larger volumes of ligand solution are used. For example, when
using a syringe, connect a second syringe to the outlet of the column and gently pump
the solution back and forth for 15 to 30 min or, if using a peristaltic pump, circulate the
ligand solution through the column.
Do not use excessive flow rates. Maximum recommended flow rates are 1 ml/min
(equivalent to approximately 30 drops/min when using a syringe) with HiTrap 1 ml
columns. For HiTrap 5 ml columns, the recommended flow rate is 5 ml/min (equivalent
to approximately 120 drops/min when using a syringe). The column contents can be
irreversibly compressed.
Measure the efficiency of protein ligand by comparing the A
280
values of the ligand
solution before and after coupling. Note that the N-hydroxysuccinimide, released during
the coupling procedure, absorbs strongly at 280 nm and should be removed from the
used coupling solution before measuring the concentration of the remaining ligand.
Use a small desalting column (see Buffer exchange and desalting, Appendix 1) to remove
N-hydroxysuccinimide from protein ligands. Alternative methods for the measurement of
coupling efficiency are described in Binding capacity, ligand density, and coupling efficiency
earlier in this chapter and in the HiTrap NHS-activated HP instructions, 71700600.
Washing and deactivation
This procedure deactivates any excess active groups that have not coupled to the ligand and
washes out nonspecifically bound ligands.
1. Inject 3 × 2 CV of blocking buffer.
2. Inject 3 × 2 CV of wash buffer.
3. Inject 3 × 2 CV of blocking buffer.
4. Let the column stand for 15 to 30 min.
5. Inject 3 × 2 CV of wash buffer.
6. Inject 3 × 2 CV of blocking buffer.
7. Inject 3 × 2 CV of wash buffer.
8. Inject 2 to 5 CV of a buffer with neutral pH.
The column is now ready for use.
Storage
Store the column in a solution that maintains the stability of the ligand and contains a
bacteriostatic agent, see Appendix 8.
pH stability of the chromatography medium when coupled to the chosen ligand will
depend upon the stability of the ligand itself.
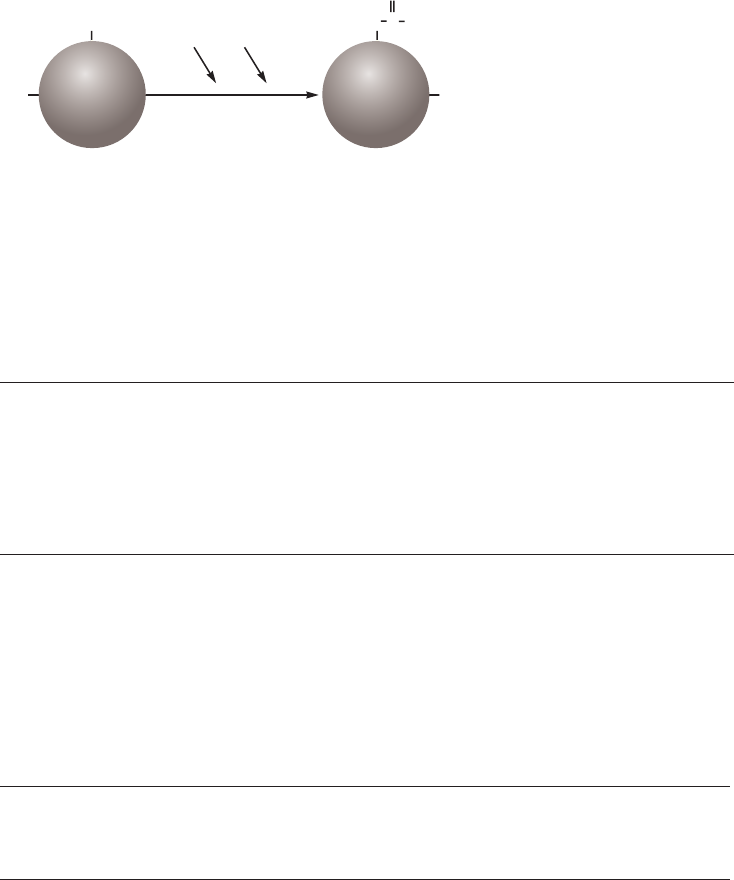
84 18102229 AF
CNBr-activated Sepharose
CNBr-activated Sepharose offers a well-established option for the attachment of larger ligands
and is an alternative to NHS-activated Sepharose.
Cyanogen bromide reacts with hydroxyl groups on Sepharose to form reactive cyanate ester
groups. Proteins, peptides, amino acids, or nucleic acids can be coupled to CNBr-activated
Sepharose, under mild conditions, via primary amino groups or similar nucleophilic groups. The
activated groups react with primary amino groups on the ligand to form isourea linkages (Fig 4.6).
The coupling reaction is spontaneous and requires no special chemicals or equipment. The
resulting multipoint attachment ensures that the ligand does not hydrolyze from the matrix.
The activation procedure also cross-links Sepharose and thus enhances its chemical stability,
offering considerable flexibility in the choice of elution conditions.
C
NH
isourea
NHR
CNBr
RNH
2
O
OH
OH
HO
Sepharose Sepharose
Fig 4.6 Activation by cyanogen bromide and coupling to the activated matrix.
Chromatography media characteristics
Characteristics of CNBr-activated Sepharose chromatography media are shown in Table 4.5.
Table 4.5. Characteristics of CNBr-activated Sepharose chromatography media
Product Composition
Binding capacity/ml
medium pH stability
1
Average
particle size
(µm)
CNBr-activated
Sepharose 4 Fast Flow
CNBr-activated
Sepharose 4B
Cyanogen bromide
reacts with hydroxyl
groups on Sepharose
to give a reactive
product for coupling
ligands via primary
amino groups or
similar nucleophilic
groups.
a-chymotrypsinogen,
13 to 26 mg
a-chymotrypsinogen,
25 to 60 mg
Short term: 3 to 11
Long term: 3 to 11
Short term: 3 to 11
Long term: 3 to 11
90
90
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Stability data refers
to the coupled medium provided that the ligand can withstand the pH. Long term refers to the pH interval over which the
matrix is stable over a long period of time without adverse effects on its subsequent chromatographic performance.
Purification options
Available CNBr-activated Sepharose chromatography media are shown in Table 4.6.
Table 4.6. Purification options for CNBr-activated Sepharose chromatography media
Product Spacer arm Coupling conditions
Maximum operating
flow velocity (cm/h)
1
Comments
CNBr-activated
Sepharose 4 Fast Flow
None pH 7 to 9; 2 to 16 h;
4°C to room temp.
400 Supplied as a freeze-
dried powder.
CNBr-activated
Sepharose 4B
None pH 8 to 10; 2 to 16 h;
4°C to room temp.
75 Supplied as a freeze-
dried powder.
1
See Appendix 4 to convert flow velocity (cm/h) to volumetric flow rate (ml/min). Maximum operating flow is calculated
from measurement in a packed column with a bed height of 10 cm and i.d. of 5 cm.
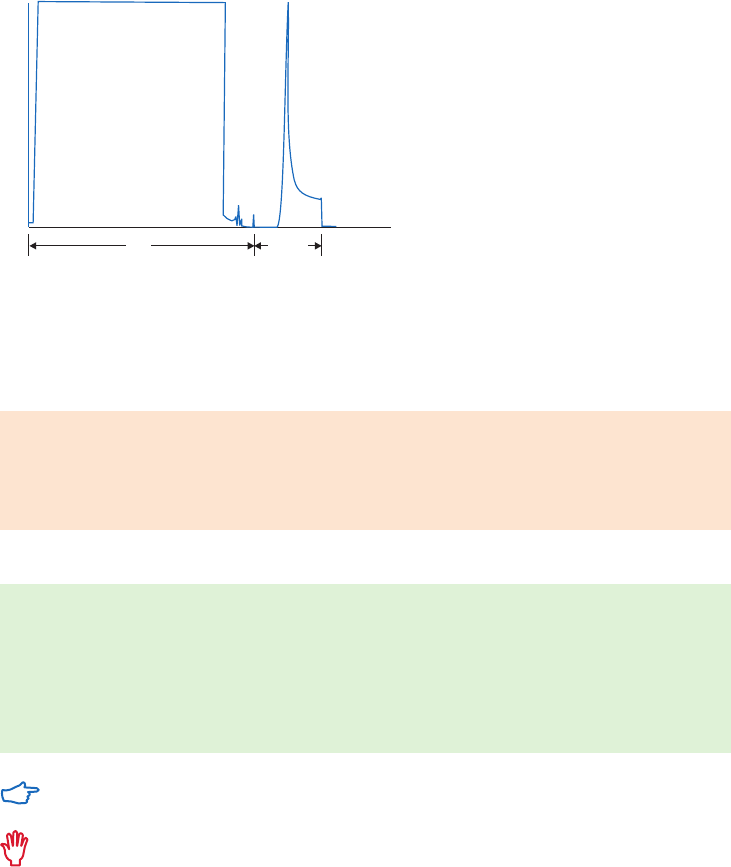
18102229 AF 85
Purification example
There are many examples in the literature of the use of CNBr-activated Sepharose.
Figure 4.7 shows the separation of a native outer envelope glycoprotein, gp120, from
HIV-1 infected T-cells. Galanthus nivalis agglutinin (GNA), a lectin from the snowdrop bulb,
was coupled to CNBr-activated Sepharose 4 Fast Flow to create a suitable AC medium.
A
280 nm
0.5
0
24 h 40 min Time
native gp120
Fig 4.7. Separation of native gp120 protein on GNA coupled to CNBr-activated Sepharose 4 Fast Flow.
From Gilljam, G. et al., Purification of native gp120 from HIV-1 infected T-cells; poster presented at
Recovery of Biological Products VII, San Diego, CA, USA (1994).
Performing a separation
Buffer preparation
Acidification solution: 1 mM HCl (kept on ice)
Coupling buffer: 200 mM NaHCO
3
, 500 mM NaCl, pH 8.3
Blocking buffer: 1 M ethanolamine or 200 mM glycine, pH 8.0
Wash buffer: 100 mM acetate, 500 mM NaCl, pH 4.0
Preparation of CNBr-activated Sepharose 4 Fast Flow and CNBr-activated Sepharose 4B
1. Suspend the required amount of freeze-dried powder in ice-cold 1 mM HCl (HCl
preserves the activity of the reactive groups that hydrolyze at high pH).
2. Wash for 15 min on a sintered glass filter (porosity G3), using a total of 200 ml of 1 mM
HCl per gram dry powder, added and removed by suction in several aliquots. The final
aliquot of 1 mM HCl is removed by suction until cracks appear in the cake.
3. Transfer the matrix immediately to the ligand solution.
Preparation of the matrix should be completed without delay since reactive groups on
the matrix hydrolyze at the coupling pH.
Do not use buffers containing amino groups at this stage since they will couple to the matrix.

86 18102229 AF
Ligand preparation
Dissolve the ligand in the coupling buffer to a final concentration of 0.5 to 10 mg/ml (for
protein ligands) or perform a buffer exchange using a desalting column (see page 133).
The optimal concentration depends on the ligand. Use a matrix:buffer ratio of 1:0.5 to 1:1.
Ligand coupling
1. Mix the ligand solution with suspension in an end-over-end or similar mixer for 2 h
at room temperature or overnight at 4°C. A matrix: buffer ratio of 1:0.5 to 1:1 gives a
suitable suspension for coupling.
2. Transfer the medium to blocking buffer for 16 h at 4°C or 2 h at room temperature to
block any remaining active groups. Alternatively, leave the medium for 2 h in Tris-HCl
buffer, pH 8.0.
3. Remove excess ligand and blocking agent by alternately washing with coupling
buffer followed by wash buffer. Repeat four or five times. A general column packing
procedure is described in Appendix 3.
Do not use magnetic stirrers as they can disrupt the Sepharose matrix.
The coupling reaction proceeds most efficiently when the amino groups on the ligand are
predominantly in the unprotonated form. A buffer at pH 8.3 is most frequently used for
coupling proteins. The high salt content of the coupling buffer minimizes protein-protein
adsorption caused by the polyelectrolyte nature of proteins.
Coupling of a-chymotrypsinogen by the method described here typically yields about 90%
coupled protein. It might be necessary to reduce the number of coupling groups on the matrix
to preserve the structure of binding sites in a labile molecule, or to facilitate elution when
steric effects reduce the binding efficiency of a large ligand. Reduced coupling activity may
be achieved by controlled hydrolysis of the activated matrix before coupling, or by coupling
at a lower pH. Prehydrolysis reduces the number of active groups available for coupling and
reduces the number of points of attachment between the protein and matrix as well as the amount
of protein coupled. In this way a higher binding activity of the product can be obtained. At pH 3.0,
coupling activity is lost only slowly, whereas at pH 8.3 activity is lost fairly rapidly. A large
molecule is coupled at about half as many points after 4 h of prehydrolysis at pH 8.3 (Fig 4.8).
a-chymotrypsinogen
Glycyl-leucine
Time of prehydrolysis (h)
0 21
3
4
20
40
60
Coupled substance (%)
0
100
24
A
B
A
B
80
Fig 4.8. Variation of coupling activity with time of pre-hydrolysis at pH 8.3. CNBr-activated Sepharose 4B
was washed at pH 3.0 and transferred to 100 mM NaHCO
3
, pH 8.3 for prehydrolysis. Samples were removed
after different times and tested for coupling activity towards a-chymotrypsinogen (A) and glycyl-leucine (B).

18102229 AF 87
Coupling at low pH is less efficient, but can be advantageous if the ligand loses
biological activity when it is fixed too firmly, for example, by multipoint attachment, or
because of steric hindrance between binding sites which occurs when a large amount
of high molecular weight ligand is coupled. Use a buffer of approximately pH 6.0.
IgG is often coupled at a slightly higher pH, for example in 200 to 250 mM NaHCO
3
,
500 mM NaCl, pH 8.5 to 9.0.
Storage
Store the freeze-dried powder below 8°C in dry conditions.
Store the column in a solution that maintains the stability of the ligand and contains a
bacteriostatic agent, see Appendix 8 or 20% ethanol in a suitable buffer.
The pH stability of the chromatography medium when coupled to the chosen ligand will
depend upon the stability of the ligand itself.
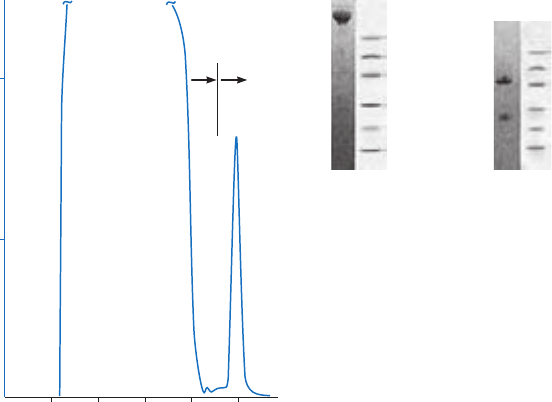
88 18102229 AF
Immunoaffinity chromatography
Immunoaffinity chromatography utilizes antigens or antibodies as ligands (sometimes
referred to as adsorbents, immunoadsorbents, or immunosorbents) to create highly selective
chromatography media for affinity purification.
Antibodies are extremely useful as ligands for antigen purification, especially when the
substance to be purified has no other apparent complementary ligand.
Similarly, highly purified antigens or anti-antibodies can provide highly specific ligands for
antibody purification. The handbook Affinity Chromatography Vol. 1: Antibodies, 18103746 from
GE covers the purification and application of antibodies in greater detail.
Immunoaffinity media are created by coupling the ligand (a pure antigen, an antibody, or an
antiantibody) to a suitable matrix. The simplest coupling is via the primary amine group of the
ligand, using NHS-activated Sepharose or CNBr-activated Sepharose. Figure 4.9 illustrates a
typical immunoaffinity purification.
1 2
M
r
14 400
20 100
30 000
45 000
66 000
97 000
M
r
14 400
20 100
30 000
45 000
66 000
97 000
1 2
2.0
1.0
20 40 60 80 100
Volume (ml)
Binding
buffer
Elution
buffer
A
280 nm
Flowthrough
material
Column: HiTrap NHS-activated HP 1 ml. Mouse IgG, (10 mg, 3.2 ml) was
coupled in 200 mM NaHCO
3
, 500 mM NaCl, pH 8.3, room temp.,
recycled with a peristaltic pump for 1 h.
The coupling yield was 95% (9.5 mg).
Sample: 50 ml sheep antimouse Fc serum, filtered 0.45 µm
Binding buffer: 75 mM Tris-HCl, pH 8.0
Elution buffer: 100 mM glycine-HCl, 500 mM NaCl, pH 2.7
Flow rate: 1 ml/min
Electrophoresis: SDS-PAGE, ExcelGel SDS Gradient 8–25, 1 µl sample, Coomassie stained
Lanes
1. Eluted material
2. LMW-SDS Marker Kit
Nonreducing
conditions
Reducing
conditions
Fig 4.9. Purification of antimouse Fc-IgG from sheep antiserum.
If there is no primary amine available (this group might be required for the specific interaction),
then preactivated medium for ligand attachment via carboxyl, thiol, or hydroxyl groups can
be considered.
Optimal binding and elution conditions will be different for each immunospecific reaction
according to the strength of interaction and the stability of the target proteins.
18102229 AF 89
Coupling small ligands through carboxyl groups via a spacer arm
EAH Sepharose 4B
The partial structure of EAH Sepharose 4B is shown in Figure 4.10.
OH
NH
6
2 2
2 2
CH
(CH )
NHO CH CH
Sepharose
Fig 4.10. Partial structure of EAH Sepharose 4B.
Ligands are coupled in a simple one-step procedure in the presence of a coupling reagent,
carbodiimide. The carbodiimides may be regarded as anhydrides of urea. The N,N’ di-substituted
carbodiimides promote condensation between a free amino and a free carboxyl group to form
a peptide link by acid-catalyzed removal of water. Thus EAH Sepharose 4B can be coupled with
carboxyl-containing ligands. The carbodiimide yields an isourea upon hydration. The coupling
reaction is shown in Figure 4.11.
1
NH
2
NH
O C
O
R
R
C N
R
Carbodiimide Active ester
COOH
R
N
+
2
R
1
R
NH
NH
O C
O
O
R
R
Active ester Peptide bond Urea derivate
(by-product)
NH
O
C
2
3
1
NH
NH
+
+
3
2
NH R
R
R
R
2
R
1
R
R and R = matrix or ligand
3
Fig 4.11. Carbodiimide coupling reaction.
Chromatography medium characteristics
Characteristics of EAH Sepharose 4B chromatography medium are shown in Table 4.7.
Table 4.7. Characteristics of EAH 4B chromatography medium
Product Composition pH stability
1
Average
particle size
(µm)
EAH Sepharose 4B Covalent linkage of 1,6-diamino-hexane by
epoxy coupling creates a stable, uncharged
ether link between a 10-atom spacer arm
and Sepharose 4B.
Short term: 3 to 14
Long term: 3 to 14
90
1
Stability data refers to the coupled medium provided that the ligand can withstand the pH. Short term refers to the pH
interval for regeneration, cleaning-in-place, and sanitization procedures. Long term refers to the pH interval over which
the matrix is stable over a long period of time without adverse effects on its subsequent chromatographic performance.
90 18102229 AF
Purification options
The purification options for EAH Sepharose 4B are shown in Table 4.8
Table 4.8. Purification options for EAH Sepharose 4B
Product
Spacer
arm
Substitution
(µmol/ml of
medium)
Coupling
conditions
Maximum
operating flow
velocity (cm/h)
1
Comments
EAH Sepharose 4B 11-atom 7 to 11
amino
groups
pH 4.5, 1.5 to
24 h, 4°C to
room temp.
75 Couple ligands containing
free carboxyl groups.
Supplied as a suspension
ready for use.
1
See Appendix 4 to convert flow velocity (cm/h) to volumetric flow rate (ml/min). Maximum operating flow is calculated
from measurement in a packed column with a bed height of 10 cm and i.d. of 5 cm.
Preparation of coupling reagent
Use a water-soluble carbodiimide such as N-ethyl-N’-(3-dimethylaminopropyl) carbodiimide
hydrochloride (EDC) or N-cyclohexyl-N’-2-(4’-methyl-morpholinium) ethyl carbodiimide
p-toluene sulfonate (CMC). These two carbodiimides have been used in a variety of
experimental conditions and at a wide range of concentrations (Table 4.9). EDC often gives
better coupling yields than CMC.
Table 4.9. Examples of conditions used during coupling via carbodiimides
Coupled ligand Carbodiimide
Conc. of carbodiimide
(mg/ml) pH
Reaction
time (h)
Methotrexate EDC 18 6.4 1.5
UDP-glucuronic acid EDC 32 4.8 24
p-amino-benzamidine CMC 2 4.75 5
Folic acid EDC 5 6.0 2
Mannosylamine EDC 19 4.5 to 6.0 24
Use a concentration of carbodiimide greater than the stoichiometric concentration, usually
10- to 100-fold greater than the concentration of spacer groups.
The coupling reaction is normally performed in distilled water adjusted to pH 4.5 to 6.0 to
promote the acid-catalyzed condensation reaction. Blocking agents are not usually required
after the coupling reaction if excess ligand has been used.
Always use freshly prepared carbodiimides.
Coupling buffer: Dissolve the carbodiimide in water and adjust to pH 4.5
Wash buffer: 100 mM acetate, 500 mM NaCl, pH 4.0
Avoid the presence of amino, phosphate, or carboxyl groups as these will compete with
the coupling reaction.
Preparation of EAH Sepharose 4B
Wash the required amount of matrix on a sintered glass filter (porosity G3) with distilled water
adjusted to pH 4.5 with HCl, followed by 500 mM NaCl (80 ml in aliquots/ml sedimented matrix).

18102229 AF 91
Ligand preparation
Dissolve the ligand and adjust to pH 4.5. The optimal concentration depends on the ligand.
Organic solvents can be used to dissolve the ligand, if necessary. If using a mixture of organic
solvent and water, adjust the pH of the water to pH 4.5 before mixing it with the organic
solvent. Solvents such as dioxane (up to 50%), ethylene glycol (up to 50%), ethanol, methanol,
and acetone have been used.
If organic solvents have been used, use pH paper to measure pH since solvents can
damage pH electrodes.
Ligand coupling
1. Add the ligand solution followed by the carbodiimide solution to the matrix suspension
and leave on an end-over-end or similar mixer. Use a matrix: ligand solution ratio of 1:2
to produce a suspension that is suitable for coupling. Typically the reaction takes place
overnight either at 4°C or room temperature.
2. Adjust the pH of the reaction mixture during the first hour (pH will decrease) by adding
100 mM sodium hydroxide.
3. Wash at pH 8.0 and pH 4.0 to remove excess reagents and reaction by-products.
If a mixture of aqueous solution and organic solvent has been used, use this mixture to
wash the final product as in Step 3. After Step 3 wash in distilled water, followed by the
binding buffer to be used for the affinity purification.
Do not use magnetic stirrers as they can disrupt the Sepharose matrix.
Storage
Store preactivated matrices 4°C to 8°C in 20% ethanol.
Store the column in a solution that maintains the stability of the ligand and contains a
bacteriostatic agent, see Appendix 8, or 20% ethanol in a suitable buffer.
The pH stability of the chromatography medium when coupled to a ligand will depend
upon the stability of the ligand.
Performing a separation
See Binding and elution conditions earlier in this chapter for a preliminary separation protocol
and Chapter 2 for general guidelines.

92 18102229 AF
Coupling through hydroxy, amino, or thiol groups
via a 12-carbon spacer arm
Epoxy-activated Sepharose 6B
Epoxy-activated Sepharose 6B is used for coupling ligands that contain hydroxyl, amino, or
thiol groups. Because of the long hydrophilic spacer arm, it is particularly useful for coupling
small ligands such as choline, ethanolamine, and sugars. The preactivated matrix is formed
by reacting Sepharose 6B with the bis-oxirane, 1,4 bis-(2,3-epoxypropoxy-)butane. The partial
structure is shown in Figure 4.12.
OH
4 22 22 2
CH CH
(CH )
O
O
O CH CHCH CH
Sepharose
O
Fig 4.12. Partial structure of Epoxy-activated Sepharose 6B.
A stable ether linkage is formed between the hydrophilic spacer and the matrix. Free oxirane
groups couple via stable ether bonds with hydroxyl-containing molecules such as sugars, via
alkylamine linkages with ligands containing amino groups, and via thioether linkages with
ligands containing thiol groups.
Chromatography medium characteristics
Characteristics of Epoxy-activated Sepharose 6B are shown in Table 4.11.
Table 4.11. Characteristics of Epoxy-activated Sepharose 6B medium
Product Composition pH stability
1
Average
particle size
(µm)
Epoxy-activated Sepharose 6B Sepharose 6B reacts with 1,4
bis- (2,3 epoxypropoxy-) butane
to form a stable ether linkage.
Short term: 2 to 14
Long term: 2 to 14
90
1
Short term refers to the pH interval for regeneration, cleaning-in-place, and sanitization procedures. Stability data refers
to the coupled medium provided that the ligand can withstand the pH. Long term refers to the pH interval over which the
matrix is stable over a long period of time without adverse effects on its subsequent chromatographic performance.
Purification options
Purification options for Epoxy-activated Sepharse 6B are shown in Table 4.12.
Table 4.12. Purification options for Epoxy-activated Sepharose 6B medium
Product
Spacer
arm
Substitution
(µmol/ml medium)
Coupling
conditions
Maximum
operating flow
velocity (cm/h)
1
Comments
Epoxy-activated
Sepharose 6B
12-atom 19 to 40 epoxy
groups
pH 9 to 13, 1 h
to several days,
20°C to 40°C
75 Supplied as a freeze-
dried powder.
1
See Appendix 4 to convert flow velocity (cm/h) to volumetric flow rate (ml/min). Maximum operating flow is calculated
from measurement in a packed column with a bed height of 10 cm and i.d. of 5 cm.

18102229 AF 93
Purification example
Capture and purification of fucose-specific lectin from a crude plant extract using Epoxy-
activated Sepharose 6B is shown in Figure 4.13.
Elution volume
Fucose-specific lectin
A
280 nm
Fig 4.13. Chromatography of a crude extract of Ulex europaeus on fucose coupled to Epoxy-activated
Sepharose 6B, column volume 11 ml. Extract was applied in 0.9% NaCl. Fucose-specific lectin was eluted with
5 ml fucose (50 mg/ml).
Alternative coupling solutions
Distilled water or aqueous buffers with sugars and carbohydrates are preferable.
Carbonate, borate, or phosphate buffers can be used.
Sodium hydroxide may be used for solutions of high pH.
Organic solvents such as dimethylformamide (up to 50%) and dioxane (up to 50%) may be
used to dissolve the ligand. The same concentration of organic solvent should be included
in the coupling solution.
Coupling procedure
1. Suspend the required amount of freeze-dried powder in distilled water (1 g freeze-dried
powder gives about 3.0 ml of final medium volume).
2. Wash immediately for 1 h on a sintered glass filter (porosity G3), using approximately
200 ml distilled water per gram freeze-dried powder, added in several aliquots.
3. Dissolve the ligand in the coupling buffer to a final concentration of 0.5 to 10 mg/
ml (for protein ligands) or transfer solubilized ligands into the coupling buffer using a
desalting column (see Buffer exchange and desalting in Appendix 1). Adjust the pH of
the aqueous phase.
4. Use a medium: buffer ratio of 1:0.5 to 1:1, mix the medium suspension with the ligand
solution for 16 h at 20°C to 40°C in a shaking water bath.
5. Block remaining excess groups with 1 M ethanolamine for at least 4 h or overnight, at
40°C to 50°C.
6. Wash away excess ligand with coupling solution followed by distilled water, 100 mM
NaHCO
3
, 500 mM NaCl, pH 8.0, and 100 mM NaCl, 100 mM acetate, pH 4.0.
If organic solvents have been used, use pH paper to measure pH since solvents can
damage pH electrodes.
Using the higher temperatures can decrease coupling times.
Do not use Tris, glycine, or other nucleophilic compounds as these will couple to the
oxirane groups.
Do not use magnetic, stirrers as they can disrupt the Sepharose medium.
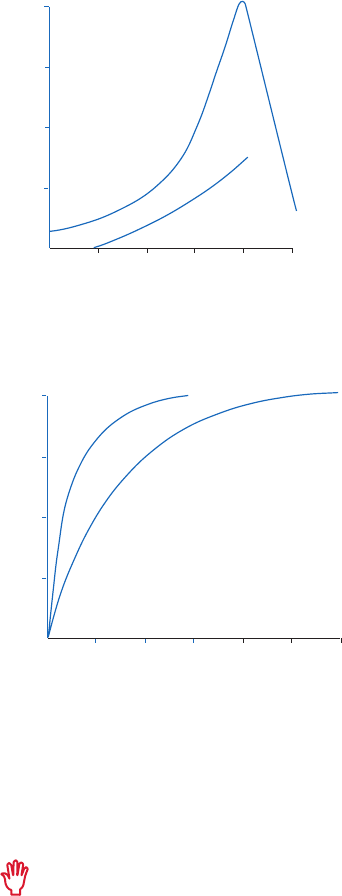
94 18102229 AF
When a ligand contains more than one kind of group (thiol, amino and hydroxyl), the coupling
pH will determine which of these groups is coupled preferentially. As a general rule, the order of
coupling is e-amino > thiol > a-amino > hydroxyl although the exact result will depend on the
detailed structure of the ligand.
The time of reaction depends greatly on the pH of the coupling solution, properties of the ligand,
and the coupling temperature. The stability of the ligand and the carbohydrate chains of the matrix
limit the maximum pH that can be used. Coupling is performed in the pH range of 9.0 to 13.0 as
shown in Figure 4.14 and the efficiency of coupling is pH- and temperature-dependent (Fig 4.15).
20 mg/ml
5 mg/ml
pH
9
10
11
12 13
25
50
75
)g/lomµ( etagujnoc dnagil delpuoC
0
100
14
Fig 4.14. pH dependence of coupling N-acetyl-D-galactosamine to Epoxy-activated Sepharose 6B.
Carbonate/bicarbonate buffers were used in the pH range of 9.0 to 11.0, sodium hydroxide solution in the
pH range of 12.0 to 14.0. Ligand concentrations: 5 mg/ml and 20 mg/ml.
pH 10.5, 40°CpH 11.0, 45°C
0
8
16 24 32
25
50
75
enicuellycylg delpuoC (%)
0
100
40
48
Time (h)
Fig 4.15. Efficiency of coupling glycyl-leucine to Epoxy-activated Sepharose 6B.
Storage
Store the freeze-dried powder dry below 8°C.
Store the column in a solution that maintains the stability of the ligand and contains a
bacteriostatic agent, see Appendix 8, or 20% ethanol in a suitable buffer.
The pH stability of the chromatography media when coupled to a ligand will depend
upon the stability of the ligand.
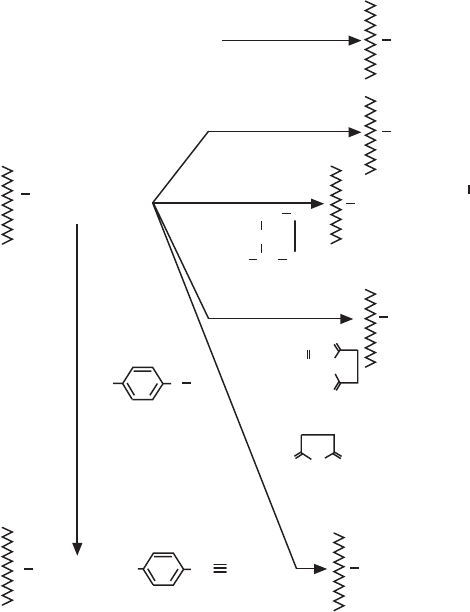
18102229 AF 95
Coupling other functional groups
EAH Sepharose 4B may be used as a starting material for coupling via alternative functional
groups (Fig 4.16). Phenolic groups may be attached via diazonium derivatives (VII) or via
the bromoacetamidoalkyl derivative (V) prepared by treating EAH Sepharose 4B with
O-bromoacetyl-N-hydroxysuccinimide. This derivative also couples via primary amino groups.
The spacer arm of EAH Sepharose 4B may be extended by reaction with succinic anhydride at
pH 6.0 (VI) to form a derivative to which amino groups can be coupled by carbodiimide reaction.
Carboxyl groups are coupled to EAH Sepharose 4B by the carbodiimide reaction (III). Thiol
derivatives, prepared by reaction (IV), couple carboxyl groups in the presence of carbodiimide
and the thiol ester bond may be cleaved specifically using hydroxylamine, thus providing a
simple and gentle method for eluting the intact ligand-protein complex.
CNBr -Activated Sepharose
I
NHR
RNH
2
carbodiimideIII
RCOOH
NH(CH ) NH
EAH Sepharose 4B
2
6
2
NH(CH )
2
6
O
O
O
O
BrCH CON
O
O
2
(O-bromoacetyl-N-hydroxysuccinimide)
CH S
CH
NH CH CO
(homocysteine thiolactone)
IV
V
2
2
2
NH(CH )
2
6
N
N
+
O N
C N
3
=
O
2
(1)
(p-nitrobenzoylazide)
(sodium dithionite)
(2)
HNO
(nitrous acid)
2
(3)
VI
VII
(succinic anhydride)
NHCOR
NH(CH )
2 2 2
6
NH
2
NHCOCH(CH )
SH
NH(CH )
2
6
2
NHCOCH
Br
NH(CH )
2 2 2
6
NHCO(CH ) COOH
NHCO
Na
2 2 4
S O
Fig 4.16. Reactions used to couple ligands to Sepharose.
96 18102229 AF

18102229 AF 97
Chapter 5
Magnetic beads for affinity chromatography
The magnetic bead format facilitates fast and efficient small-scale experiments without the
need for a chromatography system. The purification can simply be performed at the lab bench
in combination with a magnetic device. Magnetic beads also provide flexibility, allowing a wide
range of sample volumes and easy scaling up by varying the bead quantity.
Magnetic beads for AC from GE include Streptavidin beads for biotinylated biomolecules,
TiO
2
beads for phosphorylated biomolecules, and NHS- and carboxy-preactivated beads
for covalent coupling to obtain highly specific chromatography media. The beads are
superparamagnetic, which means that they do not retain magnetism once removed from a
magnetic field.
Magnetic beads for purification of antibodies and histidine-tagged proteins are also available
from GE. These products are described in Affinity Chromatography Handbook Vol. 1: Antibodies,
18103746 and Affinity Chromatography Handbook Vol. 2: Tagged Proteins, 18114275.
The magnetic beads are based on two different matrices: Mag Sepharose and Sera-Mag. The
major application area for Mag Sepharose beads is purification of proteins. The beads have a
particle size of 37 to 100 µm and consist of highly cross-linked spherical agarose (Sepharose)
containing magnetite. Since the beads share properties such as high porosity and high
capacity with Sepharose chromatography media, Mag Sepharose beads can also be used for
fast screening before scaling up to nonmagnetic Sepharose media with the same ligand.
Sera-Mag beads are small polymer based beads with a particle size of 1 µm. The
beads possess colloidal stability, resisting flocculation and aggregation, and have high
monodispersity with a narrow particle size range. Sera-Mag beads contains a single layer of
magnetite, while Sera-Mag SpeedBeads have two layers of magnetite which increases the
speed in response to the magnetic field (Fig 5.1). Sera-Mag and Sera-Mag SpeedBeads are
designed for diagnostic kits and for purification of proteins, nucleic acids, and peptides.
Encapsulation
Magnetite
CoreEncapsulation
Magnetite
CoreEncapsulation
Magnetite
Core
(A) (B)
Fig 5.1. Sera-Mag SpeedBeads (A) have a second layer of magnetite within the bead, resulting in twice the
speed in a magnetic field compared with Sera-Mag beads (B).
98 18102229 AF
The handling of magnetic beads is simple, and efficient purifications are quickly performed in
combination with a magnetic device such as MagRack 6 and MagRack Maxi (Fig 5.2). MagRack
6 enables preparation of up to six samples in 1.5 ml microcentrifuge tubes. The larger MagRack
Maxi is designed for sample volumes up to 50 ml. When the tubes are placed in the rack, the
magnetic beads are attracted to the magnet within a few seconds, allowing easy removal of
the supernatant from the magnetic beads, which remain in the tube (see description below).
Except for MagRack 6 and MagRack Maxi, a robotic device can be used for a large number of
samples for high throughput purification.
Although using a magnet is usually the most convenient and straightforward way to achieve
the separation, magnetic beads have a high density and can alternatively be separated from
liquid using centrifugation.
Fig 5.2. MagRack 6 (lower) and MagRack Maxi (upper) are designed for efficient small-scale purification
using magnetic beads.

18102229 AF 99
General magnetic bead separation steps
When performing magnetic bead separation, it is recommended to use MagRack 6 for test
tubes up to 1.5 ml and MagRack Maxi for test tubes up to 50 ml.
1. Remove the magnet before adding liquid.
2. Insert the magnet before removing liquid.
When using volumes above 50 ml, the beads can be spun down using a swing-out
centrifuge.
Dispensing the medium slurry
1. Prior to dispensing the medium slurry, make sure it is homogeneous by vortexing the
vial thoroughly.
2. When the medium slurry is resuspended, immediately pipette the required amount of
medium slurry into the desired tube.
3. Due to the fast sedimentation of the beads, it is important to repeat the resuspension
between each pipetting.
Handling liquids
1. Before application of liquid, remove the magnet from the magnetic rack.
2. After addition of liquid, resuspend the beads by vortexing or manual inversion of the tube.
When processing multiple samples, manual inversion of the magnetic rack is
recommended.
3. Use the magnetic rack with the magnet in place for each liquid removal step. Pipette or
pour off the liquid. If needed, a pipette can be used to remove liquid from the lid of the
test tube.
Incubation
During incubation, make sure the magnetic beads are well resuspended and kept in
solution by end-over-end mixing or by using a benchtop shaker.
Incubation generally takes place at room temperature. However, incubation can take
place at 4°C if this is the recommended condition for the specific sample.
When purifying samples of large volumes, an increase of the incubation time may
be necessary.

100 18102229 AF
Purification or removal of biotin and biotinylated biomolecules with
magnetic beads
Streptavidin Mag Sepharose, Sera-Mag Streptavidin coated,
Sera-Mag SpeedBeads Streptavidin-Coated, Sera-Mag SpeedBeads
Streptavidin-Blocked, Sera-Mag SpeedBeads Neutravidin™-Coated
The magnetic beads for separation of biotinylated biomolecules have a Streptavidin ligand.
Streptavidin is an M
r
60 000 protein from Streptomyces avidinii, and is a tetramer containing
four biotin binding sites. Since the affinity between streptavidin and biotin is exceptionally
high, harsh conditions are required for disruption, such as the use of SDS in the sample buffer.
Therefore, it is also possible to use the beads for immunoprecipitation and elute binding
partners in an interaction complex without coeluting the biotinylated component. The principle
of immunoprecipitation is shown in Figure 5.3.
Streptavidin Mag Sepharose is used for efficient enrichment of biotinylated proteins, such
as antibodies and immunoprecipitation. Streptavidin-coated Sera-Mag and Sera-Mag
SpeedBeads are designed for isolation of biotinylated targets such as PCR products, oligos,
and antibodies. The beads are generally used for increased throughput and precision in
immunoassays. Neutravidin-coated and Streptavidin-blocked beads have reduced nonspecific
binding, which can be beneficial in some applications.
Bead characteristics
Characteristics of Mag Sepharose, Sera-Mag, and Sera-Mag SpeedBeads for the capture of
biotin and biotinylated substances are shown in Table 5.1.
Table 5.1. Characteristics of Mag Sepharose, Sera-Mag, and Sera-Mag SpeedBeads for capture of biotin
and biotinylated molecules
Product Ligand Matrix Binding capacity
Average
particle size
(µm)
Streptavidin
Mag Sepharose
Streptavidin Highly cross-linked
agarose with magnetite
300 µg biotinylated
BSA/ml medium slurry
37 to 100
Sera-Mag
Streptavidin-Coated
Streptavidin Polymer beads with single
layer of magnetite
Biotin (pmol/mg):
Low 2500 to 3500
Medium 3500 to 4500
High 4500 to 5500
1
Sera-Mag SpeedBeads
Streptavidin-Coated
Streptavidin Polymer beads with
double layer of magnetite
Biotin (pmol/mg):
Low 2500 to 3500
Medium 3500 to 4500
High 4500 to 5500
1
Sera-Mag SpeedBeads
Streptavidin-Blocked
Streptavidin Polymer beads with
double layer of magnetite
Fluorescein (pmol/mg):
Medium ~ 3500
1
Sera-Mag SpeedBeads
Neutravidin-Coated
Neutravidin Polymer beads with
double layer of magnetite
Biotin (pmol/mg):
Medium 3500 to 4500
1
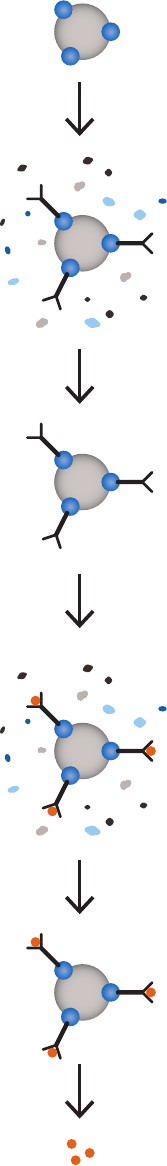
18102229 AF 101
Equilibration
Binding of biotinylated biomolecule
(often antibody)
Washing
Binding of the target protein
Washing
Elution of target protein
Fig 5.3. Principle of immunoprecipitation.
102 18102229 AF
Purification examples
Immunoprecipitation of low concentration transferrin from large sample volumes
The ability to use different volumes of sample and medium slurry is one of the key advantages
of the magnetic beads separation method. Streptavidin Mag Sepharose was used to purify
human transferrin spiked in 5 mg/ml of E. coli lysate. Purification was scaled up 10-fold
from 50 to 500 µl of Streptavidin Mag Sepharose and 3 to 30 ml of sample, respectively. The
experiment was performed in duplicate using MagRack Maxi. The transferrin concentration
was 0.75 µg/ml, which corresponds to ~ 0.015% of the total E. coli protein content. Transferrin
was captured by immunoprecipitation using a biotinylated polyclonal rabbit antihuman
transferrin immobilized on the medium and the yield of transferrin was estimated by SDS-PAGE
of the eluted fractions.
The results (Fig 5.4) show the inherent flexibility of Streptavidin Mag Sepharose. A corresponding
increase in the purity and recovery of transferrin was observed when the immunoprecipitation
was scaled up 10-fold. The yields of transferrin were 1.2 and 13 µg using 50 and 500 µl of
Streptavidin Mag Sepharose slurry, respectively. The average purity was 78% (5200-fold
enrichment) and 75% (5000-fold enrichment).
Magnetic beads: Strepatvidin Mag Sepharose
Medium slurry volume: 50 or 500 µl
Sample: 0.75 µg/ml human transferrin in 5 mg/ml E. coli lysate
Sample volume: 3 or 30 ml
Antibody: Polyclonal rabbit antihuman transferrin (biotinylated)
Binding buffer: Tris-buffered saline (TBS: 50 mM Tris, 150 mM NaCl), pH 7.5
Wash buffer: TBS, 2 M urea, pH 7.5
Elution buffer: 100 mM glycine-HCl, pH 2.9
14.4
20.1
30.0
45.0
66.0
97.
Transferrin
0
LMW markers
Start material (diluted 1:30)
Elution pool, 3 ml sample, replicate 1
Elution pool, 3 ml sample, replicate 2
Elution pool, 30 ml sample, replicate 1
Elution pool, 30 ml sample, replicate 2
M
r
× 10
3
Fig 5.4. SDS-PAGE (reducing conditions) stained with Deep Purple Total Protein Stain. The purity and
recovery obtained were equally high when the scale of purification was increased 10-fold. Quantitation
of the eluted transferrin was performed using standard curves with known amounts of transferrin
(data not shown).
Performing a separation: Streptavidin Mag Sepharose
The protocols recommended below are suitable as starting points for most purifications
involving biotinylated biomolecules. However, the optimal parameters depend on the specific
biomolecules used and optimization may be required for best results.

18102229 AF 103
Examples of parameters that might require optimization are:
• Amount of beads
• Amount of biotinylated biomolecules
• Amount of biomolecules to be enriched in immunoprecipitation
• Incubation time
• Number of washes
• Buffer compositions and pH.
Recommended buffers for capture and elution of biotinylated proteins:
Binding buffer: Tris-buffered saline (TBS; 50 mM Tris-HCl, 150 mM NaCl), pH 7.5
Washing buffer: TBS, 2 M urea, pH 7.5
Elution buffer: 2% SDS
Recommended buffers for immunoprecipitation:
Binding buffer: TBS, pH 7.5
Washing buffer: TBS, 2 M urea, pH 7.5
Elution buffer: 100 mM glycine-HCl, 2 M urea, pH 2.9
Alternative buffers for optimization:
Wash buffer: 100 mM triethanolamine, 500 mM NaCl, pH 9.0
Elution buffer: - 100 mM glycine, pH 2.5 to pH 3.1
- 100 mM citric acid, pH 2.5 to pH 3.1
- 100 mM ammonium hydroxide, pH 10.0 to pH 11.0
Sample preparation
Adjust the sample to the composition and pH of the binding buffer. pH can be adjusted by either
diluting the sample with binding buffer or by buffer exchange using PD-10 MiniTrap™ G-25 or
HiTrap Desalting columns (see Appendix 1). Clarify the sample before applying it to the beads, if
needed. Inhibiting protease activity in the sample prevents degradation of the target protein.
Purification of biotinylated proteins
Use the magnetic rack with the magnet in place to attract the beads before each liquid
removal step.
1. Prepare the Mag Sepharose beads
A. Mix the medium slurry thoroughly by vortexing. Dispense 100 µl of the
homogenous medium slurry into an Eppendorf™ tube.
B. Place the Eppendorf tube in the magnetic rack to attract the beads.
C. Remove the storage solution.
2. Equilibration
A. Add 500 µl binding buffer and resuspend the medium.
B. Remove the liquid.
3. Apply the sample
A. Add 300 µl of sample. If the sample volume is less than 300 µl, dilute to 300 µl with
binding buffer.
B. Resuspend the medium and incubate for 30 min with slow end-over-end mixing or
by using a benchtop shaker.
C. Remove the sample.

104 18102229 AF
4. Wash (perform this step three times)
A. Add 500 µl washing buffer and resuspend the medium.
B. Remove the liquid.
5. Elute biotinylated proteins
A. Add 100 µl elution buffer.
B. Resuspend the medium and incubate at 95°C to 100°C for 5 min.
C. Remove and collect the eluted fraction. The collected fraction contains the main
part of the protein. If needed, repeat the elution.
The streptavidin-biotin bond can be broken efficiently only by harsh denaturing
conditions. Hence, dissociation of biotin from streptavidin will denature both—the
biotinylated protein and streptavidin, causing a leakage of the streptavidin monomer.
Immunoprecipitation
Use the magnetic rack with the magnet in place to attract the beads before each
liquid removal step.
1. Prepare the Mag Sepharose beads
A. Mix the medium slurry thoroughly by vortexing. Dispense 50 µl of the homogenous
medium slurry into an Eppendorf tube.
B. Place the Eppendorf tube in the magnetic rack to attract the beads.
C. Remove the storage solution.
2. Equilibration
A. Add 500 µl binding buffer and resuspend the medium.
B. Remove the liquid.
3. Binding of biotinylated antibody
A. Add 300 µl of biotinylated antibody solution (~ 0.2 to 0.4 mg/ml). If the sample
volume is less than 300 µl, dilute to 300 µl with the binding buffer.
B. Resuspend the medium and incubate for 30 min with slow end-over-end mixing or
by using a benchtop shaker.
C. Remove the liquid.
4. Wash (perform this step twice)
A. Add 500 µl washing buffer and resuspend the medium.
B. Remove the liquid.
5. Binding of the target protein
A. Add 300 µl of sample. If the sample volume is less than 300 µl, dilute to 300 µl with
binding buffer.
B. Resuspend the medium and incubate for 60 min with slow end-over-end mixing or
by using a benchtop shaker.
C. Remove the liquid.
6. Wash (perform this step three times)
A. Add 500 µl washing buffer and resuspend the medium.
B. Remove the liquid.
7. Elution
A. Add 50 µl of elution buffer.
B. Resuspend the medium and incubate for 2 min.
C. Remove and collect the elution fraction. The collected elution fraction contains the
main part of the protein. If needed, repeat the elution.

18102229 AF 105
Performing a separation: Sera-Mag SpeedBeads Streptavidin-Blocked
Sample preparation
Combine antigen sample with 10 µg of biotinylated antibody (or biomolecule). Incubate 1 to 2 h
at room temperature or overnight at 4°C with mixing.
Dilute each sample to a minimum volume of 300 µl with cell lysis buffer or
binding/wash buffer.
Immunoprecipitation
Use the magnetic rack with the magnet in place to attract the beads before each liquid
removal step.
See recommended buffers for Streptavidin Mag Sepharose.
1. Prepare the Sera-Mag beads
A. Mix the medium slurry thoroughly by vortexing. Dispense 50 µl (0.5 mg) of the
homogenous medium slurry into an Eppendorf tube.
B. Place the Eppendorf tube in the magnetic rack to attract the beads.
C. Remove the storage solution.
2. Equilibration
A. Add 1 ml of binding buffer and resuspend the medium.
B. Remove the liquid.
3. Binding of biotinylated antibody/antigen sample
A. Add 300 µl of biotinylated antibody/antigen mixture.
B. Resuspend the medium and incubate at room temperature for 1 h with slow
end-over-end mixing or by using a bench-top shaker.
C. Remove the liquid.
4. Washing (perform this step twice)
A. Add 300 µl of wash buffer and resuspend the medium.
B. Remove the liquid.
5. Elution
A. Add 100 µl of elution buffer.
B. Resuspend the medium and incubate at room temperature with mixing for 5 min.
C. Remove and collect the elution fraction. The collected fraction contains the main
part of the protein. If needed, repeat the elution.
If a low pH elution buffer is selected for elution, streptavidin may leach from the
particles. Low pH elution buffers are effective for most antibody-antigen interactions.
However, to ensure efficient release of target antigen from the antibody, prerinse the
beads with 300 µl of 0.1% Tween 20 in water (no buffering capacity) before adding
low pH elution buffer.
Alternative elution for recovery of antigen: Add 100 µl of SDS reducing sample buffer to the
tube and heat the samples at 96°C to 100°C in a heating block for 5 min. Magnetically
separate the particles and save the supernatant containing the target antigen.
106 18102229 AF
Purification or removal of phosphorylated biomolecules
TiO
2
Mag Sepharose
Phosphorylation is a common reversible post-translational modification involved in the regulation
of many essential biological processes. Phosphoproteins and phosphopeptides are usually
present at very low concentrations and ionize poorly, making their detection by MS difficult.
TiO
2
Mag Sepharose magnetic beads simplify capture and enrichment of phosphopeptides
by titanium dioxide (TiO
2
)-based chromatography (Fig 5.5). TiO
2
has high affinity for
phosphopeptides and provides efficient enrichment of phosphopeptides from complex samples.
Fig 5.5. TiO
2
Mag Sepharose is designed for efficient small-scale enrichment of phosphopeptides.
Bead characteristics
Characteristics of TiO
2
Mag Sepharose beads are shown in Table 5.2.
Table 5.2. Characteristics of Mag Sepharose beads
Product Ligand Matrix Binding capacity
Average
particle size
(µm)
TiO
2
Mag Sepharose TiO
2
Highly cross-linked
agarose with magnetite
~ 35 µg phosphopeptide/ml
medium slurry
37 to 100
Purification examples
Two phosphorylated proteins (a-casein and b-casein) and one nonphosphorylated protein
(bovine serum albumin) were reduced and alkylated with Tris(2-carboxyethyl) phosphine (TCEP)
and iodoacetamide, respectively, followed by trypsin-digestion. A total of 50 pmol of each
protein digest was mixed and applied to the magnetic beads. After enrichment, the eluates
were lyophilized and dissolved in 20% acetonitrile with 0.1% trifluoroacetic acid (TFA, 20 µl) and
analyzed by MALDI-ToF MS.
TiO
2
Mag Sepharose detected five peptides, with a ratio of 2.5 between phosphopeptides
and nonphosphorylated peptides. Also, after a 100-fold dilution of the eluate, two
phophopeptides could still be detected. The experimental conditions and mass spectrograms
are shown in Figure 5.6.
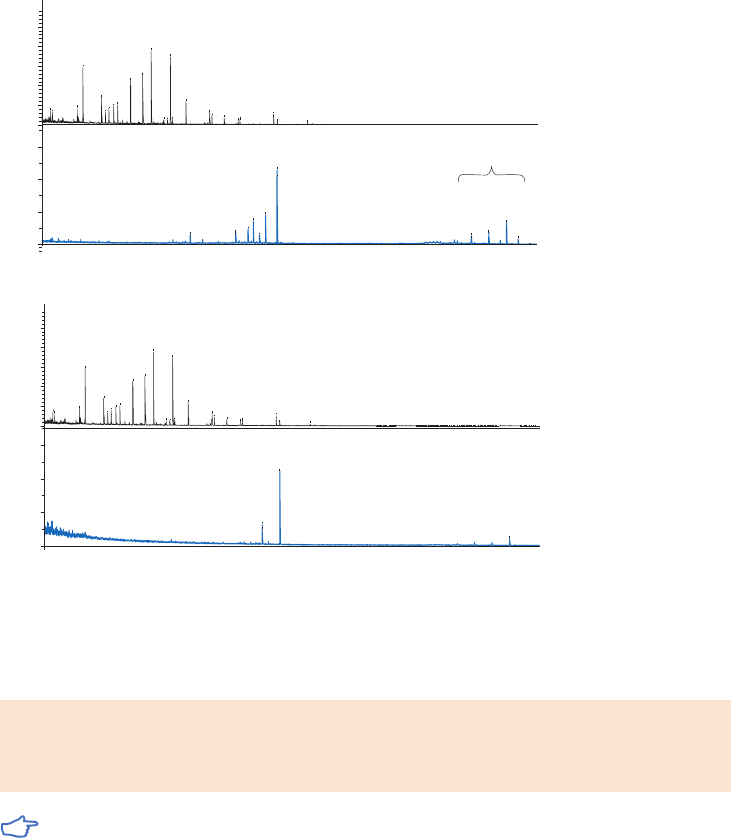
18102229 AF 107
Magnetic beads: TiO
2
Mag Sepharose
Sample: 100 µl mix of tryptic fragments from BSA, a-casein, and b-casein (50 pmol each)
Conditioning/equilibration: 1 × 500 µl binding buffer (1 M glycolic acid, 5% TFA, 80% acetonitrile)
Binding: 1 × 30 min
Washing: 1 × 500 µl binding buffer 2 × 500 µl washing buffer (1% TFA, 80% acetonitrile)
Elution: 2 × 50 µl elution buffer (5% ammonia)
Analysis: MALDI-ToF MS
14 79 .990
15 67 .938
11 63 .778
13 83 .977
12 49 .769
16 40 .142
13 24 .896
11 38 .657
10 14 .714
20 45 .229
17 49 .885
18 17 .182
18 89 .125
15 38 .000
22 02 .364
0.50
1.00
×10
4
Intens. [a.u.]
20 61 .319
20 08 .500
19 51 .488
31 21 .648
18 70 .350
16 60 .389
30 39 .496
29 59 .277
31 75 .616
0
0
2000
4000
6000
Intens. [a.u.]
I
II
*
*
*
*
*
•
14 79.99 0
15 67.93 8
11 63.77 8
13 83.97 7
12 49.76 9
16 40 .142
13 24.89 6
11 38.65 7
10 14.71 4
20 45.22 9
17 49.88 5
18 17.18 2
18 89.12 5
15 38.00 0
22 02.36 4
0.00
0.25
0.50
0.75
1.00
1.25
×10
4
Intens. [a.u.]
20 61.91 4
19 80.77 6
31 22.30 8
0
2000
4000
6000
Intens. [a.u.]
*
*
I
II
•
14 79 .990
15 67 .938
11 63 .778
13 83 .977
12 49 .769
16 40 .142
13 24 .896
11 38 .657
10 14 .714
20 45 .229
17 49 .885
18 17 .182
18 89 .125
15 38 .000
22 02 .364
0.50
1.00
×10
4
Intens. [a.u.]
20 61 .319
20 08 .500
19 51 .488
31 21 .648
18 70 .350
16 60 .389
30 39 .496
29 59 .277
31 75 .616
0
0
2000
4000
6000
Intens. [a.u.]
I
II
*
*
*
*
*
•
14 79.99 0
15 67.93 8
11 63.77 8
13 83.97 7
12 49.76 9
16 40 .142
13 24.89 6
11 38.65 7
10 14.71 4
20 45.22 9
17 49.88 5
18 17.18 2
18 89.12 5
15 38.00 0
22 02.36 4
0.00
0.25
0.50
0.75
1.00
1.25
×10
4
Intens. [a.u.]
20 61.91 4
19 80.77 6
31 22.30 8
0
2000
4000
6000
Intens. [a.u.]
*
*
I
II
•
(A)
(A)
Fig 5.6. MALDI-ToF MS analysis of trypsin-digested protein mix (50 pmol each of BSA, a-casein, and b-casein)
enriched using three different chromatographic media. (A) Spotting from lyophilized eluates dissolved in
20 µl and (B) eluates diluted 100-fold before spotting. The spectra show start material (Panel I) and eluates
from TiO
2
Mag Sepharose (Panel II). Identified phophorylated peptides are marked with asterisks*.
Performing a separation
Binding buffer: 1 M glycolic acid in 80% acetonitrile, 5% trifluoroacetic acid
Wash buffer: 80% acetonitrile, 1% trifluoroacetic acid
Elution buffer: 5% ammonium hydroxide, pH ~ 12.0
Use high-purity water and chemicals for buffer preparation.
Sample preparation
For complex samples, such as cell lysate digests, it is recommended to perform a desalting step
by use of for example an RPC/C18 cartridge or similar for efficient phosphopeptide enrichment.
Dilute the sample with a minimum of four volumes of binding buffer or dissolve lyophilized
sample in binding buffer. Keep sample volumes small, preferably max 100 µl, however up to
250 µl may be used.

108 18102229 AF
Enrichment of phosphorylated proteins
Use the magnetic rack with the magnet in place to attract the beads before each liquid
removal step.
1. Prepare the Mag Sepharose beads
A. Mix the medium slurry throughly by vortexing. Dispense 50 µl of the homogenous
medium slurry into an Eppendorf tube.
B. Place the Eppendorf tube in the magnetic rack to attract the beads.
C. Remove the storage solution.
2. Equilibration
A. Add 500 µl binding buffer and resuspend the medium.
B. Remove the liquid.
3. Apply the sample
A. Add 50 µl to 250 µl sample.
B. Resuspend the medium and incubate for 30 min with slow end-over-end mixing or
by using a benchtop shaker.
C. Remove the liquid.
4. Wash 1
A. Add 500 µl binding buffer and resuspend the medium.
B. Remove the liquid.
5. Wash 2 and 3 (perform this step twice)
A. Add 500 µl wash buffer and resuspend the medium.
B. Remove the liquid.
6. Elution
A. Add 50 µl elution buffer.
B. Resuspend the medium and incubate for 5 min.
C. Remove and collect the eluted fraction. The collected fraction contains the main part
of the protein. If needed repeat the elution.
MS analysis
Eluates must be evaporated or neutralized with formic acid or trifluoroacetic acid before analysis
with MALDI-ToF. Suitable solvent for evaporated samples is 20% acetonitrile acidified with 0.1%
trifluoroacetic acid. For LC-MS analysis using reversed phase chromatography (RPC) the eluates
must firstly be evaporated and resuspended in formic acid to a final concentration of 0.1%.
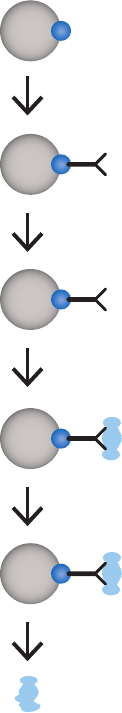
18102229 AF 109
Preactivated magnetic beads
NHS Mag Sepharose, Sera-Mag Carboxylate-Modified,
Sera-Mag SpeedBeads Carboxylate-Modified
Preactivated magnetic beads are designed for covalent coupling of antibodies, aptamers, and
proteins. After coupling is performed, proteins of interest can be affinity captured and enriched
using immunoprecipitation (see example in Fig 5.7). The preactivated magnetic beads include
NHS Mag Sepharose, Sera-Mag Carboxylate-Modified, and Sera-Mag SpeedBeads Carboxylate-
Modified Magnetic Particles.
Preactivated Mag Sepharose or Sera-Mag bead
Binding of antibodies
Blocking of residual active groups
Binding of target protein
Wash
Elution
Fig 5.7. Principle for coupling of biomolecules.
NHS Mag Sepharose has an N-hydroxysuccinimide ligand to which molecules with primary
amino groups bind covalently. This enables enrichment of target protein for further
downstream analyses such as LC-MS and electrophoresis techniques.
Sera-Mag and Sera-Mag SpeedBeads Carboxylate-Modified Magnetic Particles have carboxylic
groups on the surface that permit easy covalent coupling using simple carbodiimide chemistry.
Proteins bind to carboxylate-modified particles by adsorption. Adsorption is mediated by
hydrophobic and ionic interactions between the protein and the surface of the particles.
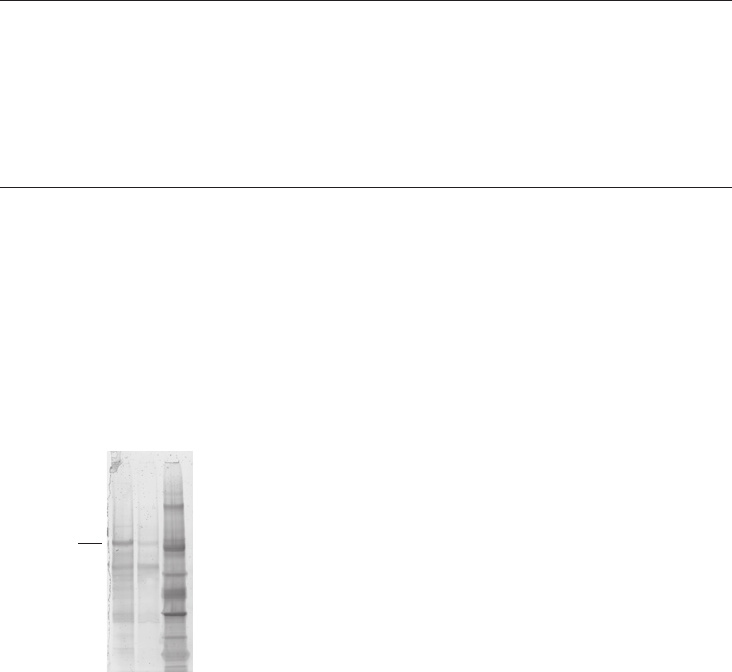
110 18102229 AF
In addition to being adsorbed, proteins can be covalently attached to the surface of
carboxylate-modified particles. Carboxyl groups on the particles, activated by the water-
soluble carbodiimide 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDAC), react with free
amino groups of the adsorbed protein to form amide bonds.
Bead characteristics
Characteristics of Mag Sepharose, Sera-Mag, and Sera-Mag SpeedBeads preactivated
magnetic beads are shown in Table 5.3.
Table 5.3. Characteristics of Mag Sepharose, Sera-Mag, and Sera-Mag SpeedBeads preactivated
magnetic beads
Product Ligand Matrix
Binding
capacity
Average
particle size
(µm)
NHS Mag Sepharose N-hydroxysuccinimide Highly cross-linked
agarose with
magnetite
8 to 14 µmol/ml
medium
37 to 100
Sera-Mag
Carboxylate-Modified
Carboxylate Polymer beads
with single layer
of magnetite
Sample dependent,
no data available
1
Sera-Mag SpeedBeads
Carboxylate-Modified
Carboxylate Polymer beads
with double layers
of magnetite
Sample dependent,
no data available
1
Purification example
Enrichment of plasminogen from human plasma
Human plasma contains a vast number of proteins and can be difficult to work with due to
the great range of protein concentrations. In this experiment, plasminogen was enriched
from human plasma by immunoprecipitation. Monoclonal antiplasminogen mouse IgG1 was
covalently coupled to NHS Mag Sepharose for capture of plasminogen. The eluted fractions
were analyzed by SDS-PAGE (Fig 5.8) and showed efficient enrichment of the target protein.
Enrichment and identification of plasminogen were confirmed by LC-MS/MS analysis.
M
r
× 10
3
97 000
66 000
45 000
30 000
20 100
14 400
1 2 3
Plasminogen
Lanes
1. First elution
2. Second elution
3. LMW markers
Magnetic beads: 25 µl of NHS Mag Sepharose
Sample: Human plasma, 6 ml
Antibodies: Antiplasminogen mouse monoclonal IgG
1
Coupling buffer: 150 mM triethanolamine, 500 mM NaCl, pH 8.3
Binding buffer: Tris buffered saline (TBS: 50 mM Tris, 150 mM NaCl, pH 7.5)
Wash buffer: TBS, 2 M urea, pH 7.5
Elution buffer: 2 × 50 µl of 100 mM glycine/HCl, 2 M urea, pH 2.9
Fig 5.8. SDS-PAGE results of the enrichment of plasminogen from human plasma using NHS Mag Sepharose
magnetic beads. The gel was post-stained with Deep Purple Total Protein Stain and scanned.

18102229 AF 111
Performing a separation
NHS Mag Sepharose
The optimal parameters for protein enrichment are dependent on the specific combination of
biomolecules used. Optimization may be required for each specific combination to obtain the
best result. Examples of parameters which may require optimization are:
• Amount of beads
• Amount of antibodies
• Amount of protein (antigen) to be enriched
• Incubation times
• Choice of buffers
• Number of washes
Equilibration buffer: 1 mM HCl (ice cold)
Coupling buffers: 150 mM triethanolamine, 500 mM NaCl, pH 8.3
200 mM NaHCO
3
, 500 mM NaCl, pH 8.3
Blocking buffer A: 500 mM ethanolamine, 500 mM NaCl, pH 8.3
Blocking buffer B: 100 mM Na-acetate, 500 mM NaCl, pH 4.0
Binding buffer: TBS (50 mM Tris, 150 mM NaCl, pH 7.5)
Wash buffer: TBS with 2 M urea, pH 7.5
Elution buffer: 100 mM glycine-HCl, 2 M urea, pH 2.9
Alternative buffers:
Blocking buffers: 50 mM Tris-HCl, 1 M NaCl, pH 8.0
50 mM glycine-HCl, 1 M NaCl, pH 3.0
Elution buffers: 100 mM glycine-HCl, pH 2.5 to 3.1
100 mM citric acid, pH 2.5 to 3.1
2.5% acetic acid
2% SDS
100 mM ammonium hydroxide, pH 10.0 to 11.0

112 18102229 AF
Preparation of antibody solution
Prepare the antibody solution by dilution in coupling buffer and keep it on ice.
Coupling and purification of target protein
Use the magnetic rack with the magnet in place to attract the beads before each liquid
removal step.
1. Prepare the Mag Sepharose beads
A. Mix the medium slurry throughly by vortexing. Dispense 25 µl of medium slurry into
an Eppendorf tube.
B. Place the Eppendorf tube in the magnetic rack to attract the beads.
C. Remove the storage solution.
2. Equilibration
A. Add 500 µl ice cold equilibration buffer and resuspend the medium.
B. Remove the liquid.
3. Binding of antibody
A. Immediately after equilibration, add the antibody solution (at least 50 µl).
B. Resuspend the medium and incubate at least 15 min with slow end-over-end mixing
or by using a benchtop shaker.
C. Remove the liquid.
4. Blocking of residual active groups
A. Add 500 µl blocking buffer A and remove the liquid.
B. Add 500 µl blocking buffer B and remove the liquid.
C. Add 500 µl blocking buffer A.
D. Incubate for 15 min with slow end-over-end mixing.
E. Remove the liquid.
F. Add 500 µl blocking buffer B and remove the liquid.
G. Add 500 µl blocking buffer A and remove the liquid.
H. Add 500 µl blocking buffer B and remove the liquid.
5. Equilibration for binding
A. Add 500 µl binding buffer and resuspend the medium.
B. Remove the liquid.
6. Binding of target protein
A. Add sample, diluted in, for example, binding buffer.
B. Resuspend the medium and incubate for 10 to 60 min with slow end-over-end mixing.
C. Remove the liquid.
7. Wash (perform this step three times)
A. Add 500 µl wash buffer.
B. Remove the liquid.
8. Elution (perform this step twice)
A. Add 50 µl elution buffer.
B. Resuspend the medium and incubate for at least 2 min.
C. Remove and collect the elution fraction. The collected elution fraction contains the
main part of the protein. If needed, repeat the elution.
Do not use an amine-containing buffer (e.g. Tris or glycine) for the antibody/protein that
is to be coupled since the amines in the buffer will compete for the coupling sites.
Remove potential amines before coupling with antibody solution, for example by
dialysis or buffer exchange with a desalting column.
18102229 AF 113
Sera-Mag and Sera-Mag SpeedBeads Carboxylate-Modified
Sera-Mag and Sera-Mag SpeedBeads Carboxylate-Modified Magnetic Particles feature
carboxylic groups on the surface that permit easy covalent coupling using simple carbodiimide
chemistry (Fig 5.9).
Second layer of magnetite with
carboxylated polymer surface
Layer of magnetite locked
in by polymer encapsulation
First Layer of magnetite
Polystyrene Core
H
O
C
O
PS PS PS PS
Fig 5.9. Sera-Mag SpeedBead Carboxylate-Modified Magnetic Particles feature carboxylic groups on the
surface that permit easy covalent coupling using simple carbodiimide chemistry.
Two procedures exist for coupling of proteins and there is also a method for coupling of
oligonucleotides. The one-step covalent coupling procedure with EDAC is recommended for
covalent coupling of most proteins. However, if EDAC is found to damage the protein of interest,
the two-step procedure, which includes a preaactivation (active ester) step prior to introducing
the protein, can be used for covalent coupling.
Coupling buffer: 2-(N-morpholino)-ethanesulfonic acid (MES) buffer.
Prepare a 10× stock buffer of 500 mM MES,
pH 6.1. Store the 10× stock at 4°C and discard if
yellowed or contaminated.
Activation solution: 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
hydrochloride (EDAC)
Just before use, weigh approximately 10 mg
of EDAC on an analytical balance. Add 1 ml of
deionized water for each 10.0 mg to obtain a final
concentration of approximately 52 µmol/ml.
For two-step coupling procedure only: N-hydroxysuccinimide (NHS), 50 mg/ml in water.
Additional buffers for coupling 100 mM imidazole at pH 6.0 and 100 mM sodium
of oligonucleotides: bicarbonate buffer (pH-adjustment not needed).
EDAC is very sensitive to moisture and undergoes rapid hydrolysis in aqueous solutions.
Therefore, EDAC should be stored in a desiccator at -20°C and brought to room
temperature just before weighing.
Sample preparation
The protein used to coat the particles should be completely dissolved and not too
concentrated. A concentration of 1 to 10 mg/ml in water is recommended for most proteins.

114 18102229 AF
Optimizing the amount of EDAC and protein
1. Determine the optimal molar ratio of EDAC:COOH.
For one-step coupling: Perform an EDAC titration while holding the amount of protein
constant. We recommend EDAC:COOH ratios of 0, 0.5, 1, 2.5, 5 and 10:1 for optimization.
For two-step coupling: Optimize the EDAC:COOH ratio, starting with a recommended
2.5:1 ratio. A molar ratio of 20:1 NHS:COOH is recommended for all reactions.
2. Determine the amount of protein to add.
Perform a protein titration, holding the determined EDAC concentration fixed. The
optimal amount of protein to use depends on several factors:
• Surface area available: surface area/mg of particles increases linearly with
decreasing particle diameter.
• Colloidal stability: proteins can have stabilizing or destabilizing effects on the particles.
• Immunoreactivity: the optimal amount of bound sensitizing protein must ultimately
be determined by functional assay. Performing a protein titration or binding isotherm
is a good first experiment. It is recommended to start with protein concentrations of
0, 25, 50, 75, 100, and 150 or 200 µg/mg of particle.
Calculation of required amount of EDAC
Use the following information to calculate the amount of EDAC required. See Optimizing
the amount of EDAC and protein above for details. Reactions of 1 ml are recommended for
optimization.
The magnetic bead acid content, provided in mEq/g, is equivalent to µmol/mg.
Note! 1 ml of 1% medium slurry contains 10 mg of magnetic beads.
The µmol EDAC required = (acid content, in µmol/mg) × 10 mg of magnetic beads × desired ratio.
Use this value in the following equation to determine how much of the EDAC stock to add:
µmol EDAC required
= ml of EDAC stock for a 1 ml reaction
52 µmol/ml
One-step coupling procedure for proteins
Use the magnetic rack with the magnet in place to attract the beads before each liquid
removal step.
1. Prepare Sera-Mag beads
A. Mix the 10% medium slurry throughly by vortexing. Dispense 100 µl of the
homogenous medium slurry into an Eppendorf tube.
B. Add 50 µl of 10× MES buffer (to 25 mM final concentration) and water to bring
reaction up to 1.0 ml final volume.
C. Add protein stock solution (see Optimizing the amount of EDAC and protein for details)
D. Resuspend the medium and incubate for approximately 15 min by end-over-end
mixing or a benchtop device.
2. Activation and coupling
A. Prepare the EDAC solution immediately before use.
B. Mix the calculated volume of EDAC solution rapidly into the reaction by pipetting up
and down repeatedly.
C. Incubate for 1 h by end-over-end mixing. The beads may flocculate during this time,
but this is not unusual or harmful.
D. Perform centrifugation (10 to 30 min in a standard microcentrifuge)
E. Decant the supernatant.

18102229 AF 115
3. Wash (perform this step twice)
A. Add 50 µl of 10× MES buffer (to 25 mM final concentration) and water to bring reaction up
to 1.0 ml, or use a higher pH buffer of choice. Use ultrasonication for resuspension.
B. Perform centrifugation as in previous step.
C. Decant the supernatant.
4. Resuspension
A. Add buffer that does not contain blocking proteins (use MES buffer or a higher pH
buffer of choice)
B. Resuspend the coupled magnetic beads to the desired medium slurry concentration.
For example, if the target of solid concentration is 1.0%, add 0.97 ml of the buffer,
which accounts for a small amount of liquid that will remain after pellet formation.
The amount of protein bound on the magnetic beads is determined by a BCA protein assay.
For long-term colloidal stability, a stabilizing storage buffer will be needed. After performing
the protein analysis, coated beads can be centrifuged and resuspended in a variety of storage
buffers, and the colloidal stability and reactivity can be optimized.
Covalently bound protein will not elute when subjected to detergent washes or buffer changes.
Thus, covalently coupled reagents are compatible with a wide variety of buffer additives.
Active ester two-step coupling procedure for proteins
Use the magnetic rack with the magnet in place to attract the beads before each liquid
removal step.
1. Prepare Sera-Mag beads
A. Mix the 10% medium slurry throughly by vortexing. Dispense 100 µl of the
homogenous medium slurry into an Eppendorf tube.
B. Add 100 µl of 10× MES buffer (to 50 mM final concentration) and water to bring
reaction up to 1.0 ml final volume.
C. Add 230 µl NHS solution: 100 mM final concentration and resuspend the medium.
2. Activation
A. Prepare the EDAC solution immediately before use.
B. Mix the calculated volume of EDAC solution rapidly into the reaction by pipetting up
and down repeatedly.
C. Incubate for 30 min by end-over-end mixing. The beads may flocculate during this
time, but this is not unusual or harmful.
D. Perform centrifugation (10 to 30 min in a standard microcentrifuge).
E. Decant the supernatant.
3. Wash (perform this step twice)
A. Add 1 ml of 50 mM MES buffer, pH 6.1. Use ultrasonication for resuspension.
B. Perform centrifugation as in previous step.
C. Decant the supernatant.
4. Coupling
A. Add the protein stock solution (see Optimizing the amount of EDAC and protein for details)
B. Resuspend the medium and incubate for 1 h by end-over-end mixing.
C. Perform centrifugation as in previous step.
D. Decant the supernatant.
5. Wash (perform this step twice)
A. Add 100 µl of 500 mM MES buffer and fill up to 1 ml with water. Use ultrasonication to
resuspend pellet.
B. Perform centrifugation as in previous step.
C. Decant the supernatant.

116 18102229 AF
6. Resuspension
A. Add buffer that does not contain blocking proteins (use MES buffer or a higher pH
buffer of choice)
B. Resuspend the coupled magnetic beads to the desired medium slurry concentration.
For example, if the target concentration for solids is 1.0%, add 0.97 ml of the buffer,
which accounts for a small amount of liquid that will remain after pellet formation.
The amount of protein bound on the magnetic beads is determined by a BCA protein assay.
For long-term colloidal stability, a stabilizing storage buffer will be needed. After performing
the protein analysis, coated beads can be centrifuged and resuspended in a variety of storage
buffers, and the colloidal stability and reactivity can be optimized.
Note that covalently bound protein will not elute when subjected to detergent washes
or buffer changes. Thus, covalently coupled reagents are compatible with a wide
variety of buffer additives.
Covalent coupling of oligonucleotides
Use the magnetic rack with the magnet in place to attract the beads before each liquid
removal step.
1. Prepare the Sera-Mag beads
A. Mix the 10% medium slurry thoroughly by vortexing. Dispense 200 µl of the
homogenous medium slurry into an Eppendorf tube.
B. Add 100 µl of 10× MES buffer (to 50 mM final concentration)
C. Add amine-modified oligonucleotide modified in water
D. Add DNase/RNase-free water to 1 ml final volume
2. Activation and coupling
A. Prepare the EDAC solution immediately before use.
B. Add 100 µl EDAC solution rapidly into the reaction by pipetting up and down
repeatedly.
C. Incubate for overnight by end-over-end mixing at 37°C. The beads might flocculate
during this time, but this is not unusual or harmful.
D. Remove the liquid.
3. Wash 1 (perform this step twice)
A. Add 1 ml of DNase/RNase-free water and resuspend the medium.
B. Remove the liquid.
4. Wash 2 (perform this step twice)
A. Add 1 ml of 100 mM imidazole (pH 6.0).
B. Resuspend the medium and incubate for 5 min at 37°C by end-over-end mixing.
C. Remove the liquid.
5. Wash 3 (perform this step twice)
A. Add 1 ml of 100 mM sodium bicarbonate.
B. Resuspend the medium and incubate for 5 min at 37°C by end-over-end mixing.
C. Remove the liquid.
6. Wash 4 (perform this step twice)
A. Add 1 ml of 100 mM sodium bicarbonate.
B. Resuspend the medium and incubate for 30 min at 65°C by end-over-end mixing.
C. Remove the liquid.
7. Resuspension
A. Add DNase/RNase-free water or buffer of choice for storage.
18102229 AF 117
Chapter 6
Affinity chromatography in a purification
strategy (CIPP )
AC separates proteins on the basis of a reversible interaction between a protein (or group of
proteins) and a specific ligand coupled to a chromatography matrix. With such high selectivity
and hence high resolution for the protein(s) of interest, purification levels in the order of several
thousand-fold with high recovery of active material are achievable. Samples are concentrated
during binding and the target protein(s) is collected in a purified, concentrated form. AC can
therefore offer immense time-saving over less selective multistep procedures.
In many cases, the high level of purity achieved in affinity purification requires, at most,
only a second step on a SEC column to remove unwanted small molecules, such as salts or
aggregates.
For an even higher degree of purity, or when there is no suitable ligand for affinity purification,
an efficient multistep process can be developed using the purification strategy of capture,
intermediate purification, and polishing (
C
IPP), shown in Figure 6.1.
C
IPP is used in both the pharmaceutical industry and in the research laboratory to ensure faster
method development, a shorter time to pure product and good economy. AC can be used, in
combination with other chromatography techniques, as an effective capture or intermediate
step in a
CIPP strategy.
This chapter gives a brief overview of the approach recommended for any multistep protein
purification. The Strategies for Protein Purification Handbook, 28983331 from GE is highly
recommended as a guide to planning efficient and effective protein purification strategies and
for the selection of the correct chromatography medium for each step and scale of purification.
Purity
Step
Preparation, extraction, clarification
Capture
Isolate, concentrate, and stabilize
Intermediate purification
Remove bulk impurities
Polishing
Achieve final high-level purity
Fig 6.1. Preparation and CIPP.

118 18102229 AF
Applying CIPP
Imagine the purification has three phases: Capture, Intermediate Purification, and Polishing.
Assign a specific objective to each step within the purification process.
The issues associated with a particular purification step will depend greatly upon the
properties of the starting material. Thus, the objective of a purification step will vary according
to its position in the process.
In the capture phase, the objectives are to isolate, concentrate, and stabilize the target product.
The product should be concentrated and transferred to an environment that will conserve
potency/activity.
During the intermediate purification phase, the objectives are to remove most of the bulk
impurities, such as other proteins and nucleic acids, endotoxins, and viruses.
In the polishing phase, most impurities have already been removed. The objective is to achieve
final purity by removing any remaining trace impurities or closely related substances.
The optimal selection and combination of purification techniques for Capture, Intermediate
Purification, and Polishing is crucial for an efficient purification.
CIPP does not mean that there must always be three purification steps. For example,
capture and intermediate purification might be achievable in a single step, as might
intermediate purification and polishing. Similarly, purity demands can be so low that
a rapid capture step is sufficient to achieve the desired result. For purification of
therapeutic proteins, a fourth or fifth purification step might be required to fulfill the
highest purity and safety demands. The number of steps used will always depend upon
the purity requirements and intended use of the protein.
Selection and combination of purification techniques
Proteins are purified using purification techniques that separate according to differences in
specific properties, as shown in Table 6.1.
Table 6.1. Protein properties used during purification
Protein property Chromatography technique
Size Size exclusion chromatography (SEC)
Charge Ion exchange chromatography (IEX)
Hydrophobicity Hydrophobic interaction chromatography (HIC)
Reversed phase chromatography (RPC)
Biorecognition (ligand specificity) Affinity chromatography (AC)
There are four important performance parameters to consider when planning each purification
step: resolution, capacity, speed, and recovery. Optimization of any one of these four
parameters can be achieved only at the expense of the others, and each purification step will
be a compromise (Fig 6.2). The importance of each parameter will vary depending on whether
a purification step is used for capture, intermediate purification, or polishing. Purification
methods should be selected and optimized to meet the objectives for each purification step.

18102229 AF 119
Resolution
Capacity
RecoverySpeed
Fig 6.2. Key performance parameters for protein purification. Each purification step should be optimized
for one or two of the parameters.
Capacity, in the simple model shown, refers to the amount of target protein loaded during
purification. In some cases the amount of sample that can be loaded will be limited by volume (as
in SEC) or by large amounts of contaminants rather than the amount of the target protein.
Speed is most important at the beginning of purification where contaminant such as proteases
must be removed as quickly as possible.
Recovery becomes increasingly important as the purification proceeds because of the
increased value of the purified product. Recovery is influenced by destructive processes in
the sample and by unfavourable conditions on the column.
Resolution is achieved by the selectivity of the technique and the efficiency and selectivity of
the chromatography matrix in producing narrow peaks. In general, resolution is most difficult
to achieve in the final stages of purification when impurities and target protein are likely to
have very similar properties.
Select a technique to meet the objectives for the purification step.
Choose logical combinations of purification techniques based on the main benefits of
the technique and the condition of the sample at the beginning or end of each step.
A guide to the suitability of each purification technique for the stages in
CIPP is shown in Table 6.2.

120 18102229 AF
Table 6.2. Suitability of purification techniques for CIPP
Method
Typical
characteristics
Purification
phase
Sample start
conditions
Sample end
conditions
Resolution
Capacity
Capture
Intermediate
Polishing
AC +++
or
++
+++
or
++
+++ ++ + Various binding conditions Specific elution conditions
SEC ++ + + +++ Most conditions accept-
able, limited sample
volume
Buffer exchange possible,
diluted sample
IEX +++ +++ +++ +++ +++ Low ionic strength.
pH depends on protein
and IEX type
High ionic strength or pH
changed
HIC +++ ++ ++ +++ +++ High ionic strength,
addition of salt required
Low ionic strength
RPC +++ ++ + ++ Ion-pair reagents and
organic modifiers might
be required
Organic solvents (risk for
loss of biological activity)
Minimize sample handling between purification steps by combining techniques to avoid
the need for sample conditioning. The product should be eluted from the first column in
conditions suitable for the start conditions of the next column (see Table 6.2).
Ammonium sulfate, often used for sample clarification and concentration (see Appendix
1), leaves the sample in a high salt environment. Consequently HIC, which requires
high salt to enhance binding to the chromatography media, becomes the excellent
choice as the capture step. The salt concentration and the total sample volume will be
significantly reduced after elution from the HIC column. Dilution of the fractionated
sample or rapid buffer exchange using a desalting column will prepare it for the next
IEX or AC step.
SEC is a nonbinding technique unaffected by buffer conditions, but with limited volume
capacity. SEC is well-suited for use after any of the concentrating techniques (IEX, HIC,
AC) since the target protein will be eluted in a reduced volume and the components
from the buffer will not affect the size exclusion process.
Selection of the final strategy will always depend upon specific sample properties and the
required level of purification. Logical combinations of techniques are shown in Figure 6.3.

18102229 AF 121
Capture
Polishing
IEX
HIC
GF
HIC
GF
IEX
HIC
HIC
IEX
Intermediate
SEC or
IEX
SEC SEC SEC
AC
IEX
AC
AC
(NH
4
)
2
SO
4
precipitation
Fig 6.3. Examples of logical combinations of chromatography steps.
For any capture step, select the technique showing the most effective binding to
the target protein while binding as few of the contaminants as possible, that is, the
technique with the highest selectivity and/or capacity for the target protein.
A sample is purified using a combination of techniques and alternative selectivities. For
example, in an IEX-HIC-SEC strategy, the capture step selects according to differences in
charge (IEX), the intermediate purification step according to differences in hydrophobicity (HIC),
and the final polishing step according to differences in size (SEC).
If nothing is known about the target protein, use IEX-HIC-SEC. This combination of
techniques can be regarded as a standard protocol.
Consider the use of both AIEX and CIEX to give different selectivities within the same
purification strategy.
122 18102229 AF

18102229 AF 123
Appendix 1
Sample preparation
Samples for chromatographic purification should be clear and free from particulate matter.
Simple steps to clarify a sample before beginning purification will avoid clogging the column, reduce
the need for stringent washing procedures, and extend the life of the chromatographic medium.
Sample extraction procedures and the selection of buffers, additives, and detergents are
determined largely by the source of the material, the stability of the target molecule, the
chromatographic techniques that will be employed and the intended use of the product.
These subjects are dealt with in general terms in the Strategies for Protein Purification
Handbook and more specifically according to target molecule in the handbooks Affinity
Chromatography, Vol. 1: Antibodies, 18103746 and Vol. 2: Tagged Proteins, 18114275 available
from GE.
Sample stability
In the majority of cases, biological activity needs to be retained after purification. Retaining
the activity of the target molecule is also an advantage when following the progress of
the purification, since detection of the target molecule often relies on its biological activity.
Denaturation of sample components often leads to precipitation or enhanced nonspecific
adsorption, both of which will impair column function. Hence there are many advantages to
checking the stability limits of the sample and working within these limits during purification.
Proteins generally contain a high degree of tertiary structure, kept together by van der Waals’
forces, ionic and hydrophobic interactions, and hydrogen bonding. Any conditions capable of
destabilizing these forces can cause denaturation and/or precipitation. By contrast, peptides
contain a low degree of tertiary structure. Their native state is dominated by secondary
structures, stabilized mainly by hydrogen bonding. For this reason, peptides tolerate a much
wider range of conditions than proteins. This basic difference in native structures is also
reflected in that proteins are not easily renatured, while peptides often renature spontaneously.
It is advisable to perform some stability tests before beginning to develop a purification
protocol. The list below shows examples of such testing:
• Test pH stability in steps of one pH unit between pH 2.0 and pH 9.0.
• Test salt stability with 0 to 2 M NaCl and 0 to 2 M (NH
4
)
2
SO
4
in steps of 500 mM.
• Test the stability towards acetonitrile and methanol in 10% steps between 0% and 50%.
• Test the temperature stability in 10°C steps from 4°C to 40°C.
• Test the stability and occurrence of proteolytic activity by leaving an aliquot of the sample
at room temperature overnight. Centrifuge each sample and measure activity and UV
absorbance at 280 nm in the supernatant.
Sample clarification
Centrifugation and filtration are standard laboratory techniques for sample clarification and
are used routinely when handling small samples.
It is highly recommended to centrifuge and filter samples immediately before
chromatographic purification.

124 18102229 AF
Centrifugation
Centrifugation removes lipids and particulate matter, such as cell debris. If the sample is still
not clear after centrifugation, use filter paper or a 5 µm filter as a first step and one of the filters
below as a second-step filter.
• For small sample volumes or proteins that adsorb to filters, centrifuge at 10 000 × g for 15 min.
• For cell lysates, centrifuge at 40 000 to 50 000 × g for 30 min.
• Serum samples can be filtered through glass wool after centrifugation to remove
remaining lipids.
Filtration
Filtration removes particulate matter. Whatman™ syringe filters, which give the least amount
of nonspecific binding of proteins, are composed of cellulose acetate (CA), regenerated
cellulose (RA), or polyvinylidene fluoride (PVDF) (Table A1.1).
Table A1.1. Whatman syringe filters for filtration of samples
Filter pore size (µm) Up to sample volume (ml) Whatman syringe filter
1
Membrane
0.8 100 Puradisc FP 30 CA
0.45 1 Puradisc 4 PVDF
0.45 10 Puradisc 13 PVDF
0.45 100 Puradisc 25 PVDF
0.45 10 SPARTAN™ 13 RC
0.45 100 SPARTAN 30 RC
0.45 100 Puradisc FP 30 CA
0.2 1 Puradisc 4 PVDF
0.2 10 Puradisc 13 PVDF
0.2 100 Puradisc 25 PVDF
0.2 10 SPARTAN 13 RC
0.2 100 SPARTAN 30 RC
0.2 100 Puradisc FP 30 CA
1
The number indicates the diameter (mm) of the syringe filter.
For sample preparation before chromatography, select a filter pore size in relation to the bead
size of the chromatographic medium (Table A1.2).
Table A1.2. Selecting a sample filter based on the bead size of the chomatographic medium used
Nominal pore size of filter (µm) Particle size of chromatographic medium (µm)
1.0 90 and upwards
0.45 30 or 34
0.22 3, 10, 15 or when extra clean samples or sterile filtration is required
Check the recovery of the target protein in a test run. Some proteins adsorb
nonspecifically to filter surfaces.
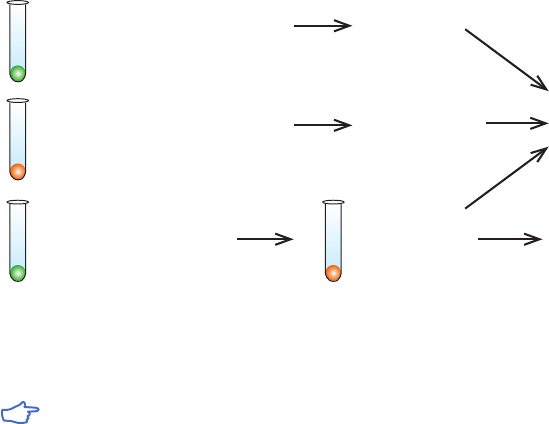
18102229 AF 125
Desalting
Desalting columns are suitable for any sample volume and will rapidly remove LMW contaminants
in a single step at the same time as transferring the sample into the correct buffer conditions.
Centrifugation and/or filtration of the sample before desalting is still recommended. Detailed
procedures for buffer exchange and desalting are given in the section Buffer exchange and
desalting later in this appendix.
At laboratory scale, when samples are reasonably clean after filtration or centrifugation, the
buffer exchange and desalting step can be avoided. For AC or HIC, it might be sufficient to
adjust the pH of the sample. For IEX, it might be sufficient to dilute the sample to reduce the
ionic strength.
Specific sample preparation steps
Specific sample preparation steps might be required if the crude sample is known to contain
contamininants such as lipids, lipoproteins, or phenol red that might build up on a column.
Gross impurities, such as bulk protein, should be removed before any chromatographic step.
Fractional precipitation
Fractional precipitation is occasionally used at laboratory scale to remove gross impurities
from the sample. Precipitation techniques separate fractions by the principle of differential
solubility. Because proteins differ in their degree of hydrophobicity, increased salt
concentrations can enhance hydrophobic interactions between the proteins and cause
precipitation. Fractional precipitation can be applied to remove gross impurities in three
different ways, as shown in Figure A1.1.
Clarification
Bulk proteins and particulate
matter precipitated
Extraction, Clarification, Concentration
Target protein precipitated
with proteins of similar solubility
Chromatography
Note: if precipitating
agent is incompatible
with next purification step,
use Sephadex™ G-25 for
desalting and buffer
exchange, for example,
HiTrap Desalting or
PD-10 columns
Redissolve pellet
1
Extraction, Clarification
Bulk proteins and particulate
matter precipitated
1
Remember: not all proteins are easy to redissolve, yield can be reduced
Supernatant
Redissolve pellet
1
Concentration
Target protein
precipitated
with proteins of
similar solubility
Fig A1.1. Three ways to use precipitation.
Precipitation techniques can be affected by temperature, pH, and sample
concentration. These parameters should be controlled to ensure reproducible results.
Examples of precipitation agents are reviewed in Table A1.3. The most common precipitation
method using ammonium sulfate is described in more detail.
126 18102229 AF
Table A1.3. Examples of precipitation techniques
Precipitation agent Typical conditions for use Sample type Comment
Ammonium sulfate As described below. > 1 mg/ml proteins
especially immuno-
globulins.
Stabilizes proteins, no
denaturation, supernatant
can go directly to HIC.
Helps to reduce lipid content.
Dextran sulfate Add 0.04 ml 10% dextran
sulfate and 1 ml of
1 M CaCl
2
per ml sample,
mix 15 min, centrifuge
10 000 × g, discard pellet.
Samples with high
levels of lipoprotein,
e.g., ascites.
Precipitates lipoprotein.
Polyvinylpyrrolidine Add 3% (w/v), stir 4 h,
centrifuge 17 000 × g,
discard pellet.
Samples with high
levels of lipoprotein,
e.g., ascites.
Alternative to dextran sulfate.
Polyethylene glycol
(PEG, M
r
> 4000)
Up to 20% w/v. Plasma proteins. No denaturation,
supernatant goes directly
to IEX or AC, complete
removal might be difficult.
Stabilizes proteins.
Acetone (cold) Up to 80% v/v at
± 0°C. Collect pellet
after centrifugation
at full speed in a
microcentrifuge.
Can denature protein
irreversibly.
Useful for peptide
precipitation or
concentration of sample
for electrophoresis.
Polyethyleneimine 0.1% w/v. Precipitates aggregated
nucleoproteins.
Protamine sulfate 1% w/v. Precipitates aggregated
nucleoproteins.
Streptomycin sulfate 1% w/v. Precipitation of nucleic acids.
Caprylic acid (X/15) g where X =
volume of sample.
Antibody concentration
should be > 1 mg/ml.
Precipitates bulk of proteins
from sera or ascites, leaving
immunoglobulins in solution.
Details taken from: Scopes R. K., Protein Purification, Principles and Practice, Springer, (1994), J. C. Janson and L. Rydén, Protein
Purification, Principles, High Resolution Methods and Applications, second ed. Wiley Inc, (1998). Personal communications.
Ammonium sulfate precipitation
Some proteins can be damaged by ammonium sulfate. Take care when adding
crystalline ammonium sulfate; high local concentrations can cause contamination of
the precipitate with unwanted proteins.
For routine, reproducible purification, precipitation with ammonium sulfate should be
avoided in favor of chromatography.
In general, precipitation is rarely effective for protein concentrations below 1 mg/ml.
Solutions needed for precipitation:
Saturated ammonium sulfate solution (add 100 g ammonium sulfate to 100 ml distilled
water, stir to dissolve).
1 M Tris-HCl, pH 8.0.
Buffer for first purification step.
18102229 AF 127
1. Filter (0.45 µm) or centrifuge the sample (10 000 × g at 4°C).
2. Add 1 part 1 M Tris-HCl, pH 8.0 to 10 parts sample volume to maintain pH.
3. Stir gently. Add ammonium sulfate solution, drop by drop. Add up to 50% saturation
1
.
Stir for 1 h.
4. Centrifuge 20 min at 10 000 × g.
5. Remove supernatant. Wash the pellet twice by resuspension in an equal volume of
ammonium sulfate solution of the same concentration (i.e. a solution that will not
redissolve the precipitated protein or cause further precipitation). Centrifuge again.
6. Dissolve pellet in a small volume of the buffer to be used for the next step.
7. Ammonium sulfate is removed during clarification/buffer exchange steps with
Sephadex G-25, using desalting columns (see Buffer exchange and desalting later
in this appendix).
1
The percent saturation can be adjusted either to precipitate a target molecule or to precipitate contaminants.
The quantity of ammonium sulfate required to reach a given degree of saturation varies
according to temperature. Table A1.4 shows the quantities required at 20°C.
Table A1.4. Quantities of ammonium sulfate required to reach given degrees of saturation at 20°C
Final percent saturation to be obtained
20 25 30 35 40 45 50 55 60 65 70 75 80 85 90 95 100
Starting
percent
saturation
Amount of ammonium sulfate to add (gram) per liter of solution at 20°C
0 113 144 176 208 242 277 314 351 390 430 472 516 561 608 657 708 761
5 85 115 146 179 212 246 282 319 358 397 439 481 526 572 621 671 723
10 57 86 117 149 182 216 251 287 325 364 405 447 491 537 584 634 685
15 28 58 88 119 151 185 219 255 293 331 371 413 456 501 548 596 647
20 0 29 59 89 121 154 188 223 260 298 337 378 421 465 511 559 609
25 0 29 60 91 123 157 191 228 265 304 344 386 429 475 522 571
30 0 30 61 92 125 160 195 232 270 309 351 393 438 485 533
35 0 30 62 94 128 163 199 236 275 316 358 402 447 495
40 0 31 63 96 130 166 202 241 281 322 365 410 457
45 0 31 64 98 132 169 206 245 286 329 373 419
50 0 32 65 99 135 172 210 250 292 335 381
55 0 33 66 101 138 175 215 256 298 343
60 0 33 67 103 140 179 219 261 305
65 0 34 69 105 143 183 224 267
70 0 34 70 107 146 186 228
75 0 35 72 110 149 190
80 0 36 73 112 152
85 0 37 75 114
90 0 37 76
95 0 38
128 18102229 AF
Resolubilization of protein precipitates
Many proteins are easily resolubilized in a small amount of the buffer to be used in the next
chromatographic step. However, a denaturing agent may be required for less soluble proteins.
Specific conditions will depend upon the specific protein. These agents must always be
removed to allow complete refolding of the protein and to maximize recovery of mass and
activity. A chromatographic step often removes a denaturant during purification. Table A1.5
gives examples of common denaturing agents.
Table A1.5. Denaturing agents used for resolubilization of relatively insoluble proteins
Denaturing agent
Typical conditions
for use (molar, M) Removal/comment
Urea 2 to 8 Remove using Sephadex G-25
Guanidine hydrochloride 3 to 6 Remove using Sephadex G-25
Details taken from: Scopes R. K., Protein Purification, Principles and Practice, Springer, (1994), J. C. Janson and L. Rydén,
Protein Purification, Principles, High Resolution Methods and Applications, second ed. Wiley Inc, (1998) and other sources.
Buffer exchange and desalting
Dialysis is frequently mentioned in the literature as a technique to remove salt or other small
molecules and to exchange the buffer composition of a sample. However, dialysis is generally
a very slow technique, requiring large volumes of buffer. There is also a risk of losing material
during handling or as a result of proteolytic breakdown or nonspecific binding to the dialysis
membranes. A simpler and much faster technique is to use a desalting column, packed with
Sephadex G-25, to perform a group separation between HMW and LMW substances. Proteins are
separated from salts and other small molecules.
In a fast, single step, the sample is desalted, transferred into a new buffer and LMW materials
are removed.
Desalting columns are used not only to remove LMW contaminants, such as salt, but also for
buffer exchange before or after different chromatographic steps and for the rapid removal of
reagents to terminate a reaction.
Sample volumes up to 30% of the total volume of the desalting column can be processed.
Sample concentration does not influence the separation as long as the concentration of
proteins does not exceed 70 mg/ml when using normal aqueous buffers. The sample should be
fully dissolved. Centrifuge or filter to remove particulate material.
For small sample volumes, it is possible to dilute the sample with the start buffer that is
to be used for chromatographic purification, but cell debris and particulate matter must
still be removed.
Volatile buffers such as 100 mM ammonium acetate or 100 mM ammonium
hydrogen carbonate can be used if it is necessary to avoid the presence of NaCl.
Figure A1.2 shows a typical buffer exchange and desalting separation. The process can be
monitored by following changes in UV absorption and conductivity.
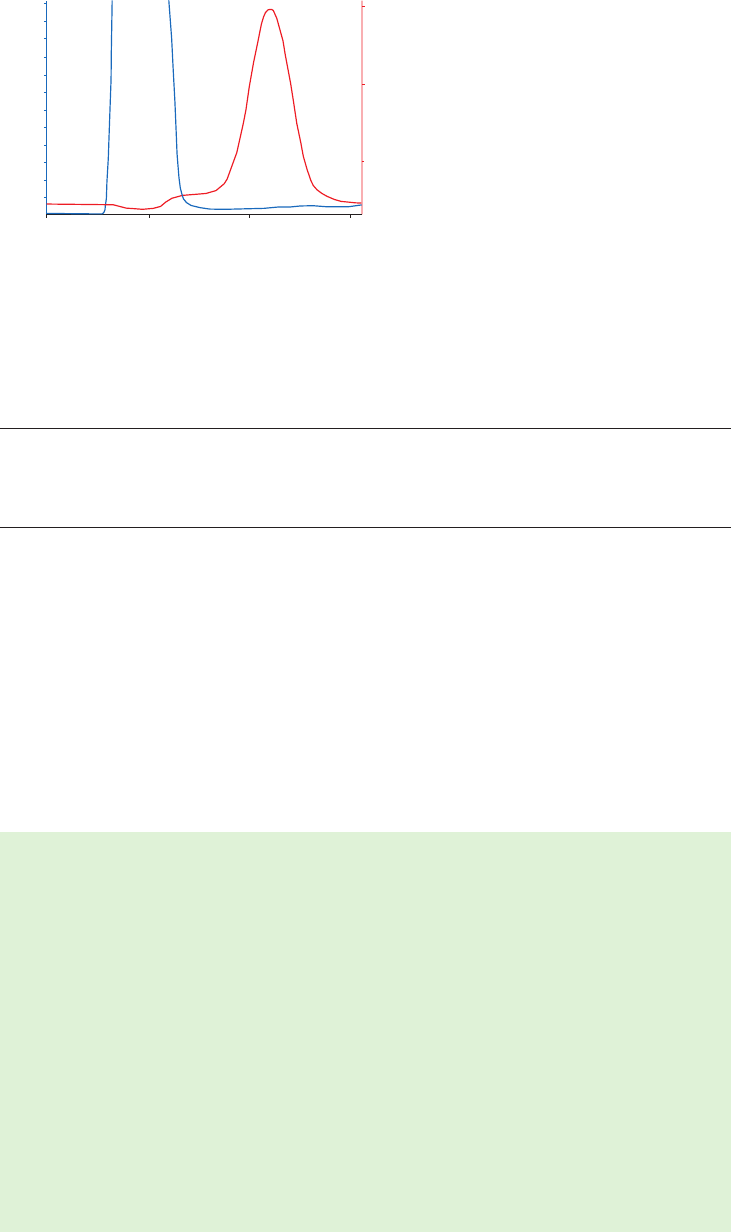
18102229 AF 129
01
23
0.00
0.05
0.10
0.15
0.20
0.25
0.30
A
280
5
10
15
Conductivity (mS/cm)
protein salt
Time (min)
Fig A1.2. Buffer exchange of mouse plasma (10 ml) on HiPrep 26/10 Desalting.
For laboratory-scale operations, Table A1.6 shows examples of prepacked, ready-to-use
desalting and buffer exchange columns (see Size Exclusion Chromatography Handbook,
18102218, for additional formats).
Table A1.6. Examples of desalting and buffer exchange columns
Column Sample volume (ml) Sample elution volume (ml)
PD MiniTrap G-25 0.2 to 0.5 0.1 to 0.5
PD-10 (gravity feed column) 1.0 to 2.5 3.5
HiTrap Desalting, 5 ml 0.25 to 1.5 1.0 to 2.0
HiPrep 26/10 Desalting 2.5 to 15 7.5 to 20
To desalt larger sample volumes:
• Connect up to five HiTrap Desalting 5 ml columns in series to increase the sample volume
capacity, for example, two columns: sample volume 3 ml, five columns: sample volume 7.5 ml.
• Connect up to four HiPrep 26/10 Desalting columns in series to increase the sample volume
capacity, for example two columns: sample volume 30 ml, four columns: sample volume 60 ml.
Even with four columns in series, the sample can be processed in 20 to 30 min, at room
temperature, in aqueous buffers.
Instructions are supplied with each column. Desalting and buffer exchange can take less than
5 min per sample with greater than 95% recovery for most proteins.
Manual desalting with HiTrap Desalting 5 ml using a syringe
1. Fill the syringe with buffer. Remove the stop plug. To avoid introducing air into the
column, connect the column “drop to drop” to the syringe (via the adapter provided).
2. Remove the snap-off end.
3. Wash the column with 25 ml buffer at 5 ml/min to remove completely the 20% ethanol
(supplied as storage buffer). If air is trapped in the column, wash with degassed buffer
until the air disappears. Air bubbles introduced onto the column by accident during
sample application do not influence the separation.
4. Apply the sample (0.25 to 1.5 ml) using a 2 to 5 ml syringe at a flow rate between 1 to
10 ml/min. Discard the liquid eluted from the column.
5. If the sample volume is less than 1.5 ml, change to buffer and proceed with the
injection until a total of 1.5 ml has been eluted. Discard the eluted liquid.
6. Elute the protein with the appropriate volume selected from Table A1.7 and collect the
desalted protein.
Note: 5 ml/min corresponds to approximately 120 drops/min when using a HiTrap 5 ml
column. A simple peristaltic pump or a chromatography system can also be used
for the desalting procedure.
130 18102229 AF
The maximum recommended sample volume is 1.5 ml. See Table A1.7 for the effect of
reducing the sample volume applied to the column.
Table A1.7. Recommended sample and elution volumes using a syringe
Sample load
(ml)
Add buffer
(ml)
Elute and collect
(ml)
Yield
(%)
Remaining salt
(%)
Dilution
factor
0.25 1.25 1.0 > 95 0.0 4.0
0.50 1.0 1.5 > 95 < 0.1 3.0
1.00 0.5 2.0 > 95 < 0.2 2.0
1.50 0 2.0 > 95 < 0.2 1.3
Removal of lipoproteins
Lipoproteins and other lipid material can rapidly clog chromatography columns and it is advisable
to remove them before beginning purification. Precipitation agents such as dextran sulfate and
polyvinylpyrrolidine, described under Fractional precipitation above, are recommended to remove
high levels of lipoproteins from samples such as ascitic fluid.
Centrifuge samples when performing precipitation to avoid the risk of nonspecific binding
of the target molecule to a filter.
Samples such as serum can be filtered through glass wool to remove remaining lipids.
Removal of phenol red
Phenol red is frequently used at laboratory scale as a pH indicator in cell culture. Although not
directly interfering with purification, phenol red binds to certain purification media and should
be removed as early as possible to avoid the risk of contamination. It is known to bind to AIEX
media at pH > 7.0.
Use a desalting column to simultaneously remove phenol red (a low molecular weight
molecule) and transfer sample to the correct buffer conditions for further purification,
as described under Buffer exchange and desalting earlier in this appendix.
Removal of LMW contaminants
If samples contain a high level of LMW contaminants, use a desalting column before
the first chromatographic purification step, as described under Buffer exchange and
desalting earlier in this appendix.
18102229 AF 131
Appendix 2
Selection of purification equipment
Simple AC purification with step elution can be performed using a syringe or peristaltic
pump with prepacked HiTrap columns. A chromatography system is required when reproducible
results are important and when manual purification becomes too time-consuming and
inefficient. This can be the case when large sample volumes are handled, or when there are
many different samples to be purified. The progress of the purification can be monitored
automatically and high-resolution separations with accurately controlled linear-gradient
elution can be performed.
Table A2.1 lists the standard ÄKTA system configurations for currently available systems, see
also ÄKTA Laboratory-scale Systems: Instrument Management Handbook, 29010831.
Table A2.1. Ways of working with standard ÄKTA chromatography systems
Way of working ÄKTA start ÄKTAprime plus ÄKTAxpress ÄKTA pure ÄKTA avant
Simple, one-step desalting,
buffer exchange
Research 4
• • • •
Process development 4
•
Automated and reproducible
protein purification using all
common techniques including
support for gradient elution
• • • • •
Software compatible with
regulatory requirements, e.g.,
good laboratory practice (GLP)
• • •
Method development and
optimization using design
of experiments (DoE)
o
• •
Automatic buffer preparation
including pH scouting
•
o
•
Automatic chromatography
medium or column scouting
o
•
o
•
Automatic, multistep purification
o
•
o o
Scale-up, process development
o o o
•
Flow rate (ml/min)
Max. operating pressure (MPa)
0.5 to 5.0
0.5
0.1 to 50.0
1
0.1 to 65.0
3
0.001 to 25.0
(ÄKTA pure 25)/
0.01 to 150
(ÄKTA pure 150)
20/5
0.001 to 25/0.01
to 150
20/5
Software
1
for system control
and data handling
UNICORN™
start
PrimeView™
2
UNICORN 5 UNICORN 6
or later
UNICORN 6
or later
1
A specific software version might be needed for the chosen system. See the web page for each respective system at
www.gelifesciences.com/AKTA.
2
With PrimeView, you can monitor results and evaluate data but not create methods nor control the system.
• = included
o
= optional

132 18102229 AF
Appendix 3
Column packing and preparation
Prepacked columns from GE will ensure reproducible results and the highest performance.
Use small prepacked columns for chromatography media scouting and method
optimization and to increase efficiency in method development.
Efficient column packing is essential for AC separation, especially when using gradient elution.
A poorly packed column gives rise to poor and uneven flow, band broadening, and loss of
resolution. If column packing is required, the following guidelines will apply at all scales of
operation:
• With a high binding capacity medium, use short, wide columns (typically 5 to 15 cm bed
height) for rapid purification, even at low flow velocity.
• The amount of AC medium required will depend on the binding capacity of the medium and
the amount of sample. Binding capacities for each medium are given in this handbook and
supplied with the product instructions. Estimate the amount of medium required to bind
the sample of interest and use five times this amount to pack the column. The amount of
medium required can be reduced if resolution is satisfactory.
• Once separation parameters have been determined, scale up a purification by increasing
the diameter of the column to increase column volume. Avoid increasing the length of the
column, if possible, as this will alter separation conditions.
AC media can be packed in either Tricorn, XK, or HiScale columns available from GE (Fig A3.1).
Fig A3.1. Column packing in progress.

18102229 AF 133
1. Equilibrate all materials to the temperature at which the separation will be performed.
2. Eliminate air by flushing column end pieces with the recommended buffer. Ensure no
air is trapped under the column net. Close column outlet leaving 1 to 2 cm of buffer in
the column.
3. Gently resuspend the medium.
Note that AC media from GE are supplied ready to use. Decanting of fines that could clog the
column is unnecessary.
Avoid using magnetic stirrers since they can damage the chromatography matrix.
4. Estimate the amount of slurry (resuspended medium) required on the basis of the
recommendations supplied in the instruction manual.
5. Pour the required volume of slurry into the column. Pouring down a glass rod held
against the wall of the column will minimize the introduction of air bubbles.
6. Immediately fill the column with buffer.
7. Mount the column top piece/adapter and connect to a pump.
8. Open the column outlet and set the pump to the desired flow rate (for example,
15 ml/min in an XK 16/20 column).
When slurry volume is greater than the total volume of the column, connect a second
glass column to act as a reservoir (see Ordering information for details). This ensures
that the slurry has a constant diameter during packing, minimizing turbulence and
improving column packing conditions.
If the recommended flow rate cannot be obtained, use the maximum flow rate the
pump can deliver.
Do not exceed the maximum operating pressure of the medium or column.
9. Maintain the packing flow rate for at least 3 CV after a constant bed height is obtained.
Mark the bed height on the column.
Do not exceed 70% of the packing flow rate during any purification.
10. Stop the pump and close the column outlet. If a second column has been used:
Remove the top piece and carefully fill the rest of the column with buffer to form an
upward meniscus at the top. Insert the adapter into the column at an angle, ensuring
that no air is trapped under the net.
11. Slide the adapter slowly down the column (the outlet of the adapter should be open)
until the mark is reached. Lock the adapter in position.
12. Connect the column to the pump and begin equilibration. Reposition the adapter
if necessary.
The medium must be thoroughly washed to remove the storage solution, usually 20%
ethanol. Residual ethanol can interfere with subsequent procedures.
Many chromatography media equilibrated with sterile phosphate-buffered saline
containing an antimicrobial agent may be stored at 4°C for up to 1 mo, but always follow
the specific storage instructions supplied with the product.
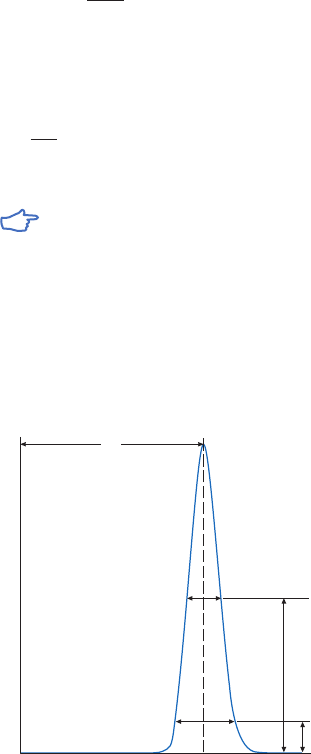
134 18102229 AF
Column packing and efficiency
Column efficiency is expressed as the number of theoretical plates per meter chromatography bed
(N) or as H (height equivalent to a theoretical plate, HETP), which is the bed length (L) divided
by the plate number. Column efficiency is related to the band broadening that can occur on a
column and can be calculated from the expression:
N = 5.54 ×
(
V
R
)
2
W
h
V
R
= volume eluted from the start of sample application to the peak maximum
w
h
= peak width measured as the width of the recorded peak at half of the peak height
H is calculated from the expression:
H =
L
N
L = height of packed bed.
Measurements of V
R
and w
h
can be made in distance (mm) or volume (ml) but both
parameters must be expressed in the same unit.
Column performance should be checked at regular intervals by injecting acetone to determine
column efficiency (N) and peak symmetry (asymmetry factor, A
s
). Since the observed value for
N depends on experimental factors such as flow rate and sample loading, comparisons must
be made under identical conditions. In AC, efficiency is measured under isocratic conditions by
injecting acetone (which does not interact with the medium) and measuring the eluted peak as
shown in Figure A3.2.
V
e
ab
50%
10%
w
h
Volume
Absorbance
Fig A3.2. Measurements taken to calculate column efficiency.
As a general rule, a good H value is about two to three times the average particle diameter of
the medium being packed. For a 90 µm particle, this means an H value of 0.018 to 0.027 cm.

18102229 AF 135
The asymmetry factor (A
s
) is expressed as:
A
s
=
b
a
where
a = First half peak width at 10% of peak height
b = Second half peak width at 10% of peak height
A
s
should be as close as possible to 1.0. A reasonable A
s
value for a short column as used in
AC is 0.80 to 1.80.
An extensive leading edge is usually a sign that the medium is packed too tightly and
extensive tailing is usually a sign that the medium is packed too loosely.
Run at least 2 CV of buffer through a newly packed column to ensure that the medium
is equilibrated with start buffer. Use pH monitoring to check the pH of the eluent.
Custom column packing
A service for packing of laboratory columns or filling of 96-well plates is supplied when columns
or plates with suitable chromatography media are not available from the standard portfolio.
The Custom Products group works in close collaboration with you to deliver packed columns
for specialized purification requirements. Visit www.gelifesciences.com/custom-column-packing
for more information.

136 18102229 AF
Appendix 4
Converting from flow velocity to
volumetric flow rates
It is convenient when comparing results for columns of different sizes to express flow as flow
velocity (cm/h). However, flow is usually measured in volumetric flow rate (ml/min). To convert
between flow velocity and volumetric flow rate use one of the formulae below.
From flow velocity (cm/h) to volumetric flow rate (ml/min)
Volumetric flow rate (ml/min) =
Flow velocity (cm/h)
× column cross sectional area (cm
2
)
60
=
Y
×
p × d
2
60 4
where
Y = flow velocity in cm/h
d = column inner diameter in cm
Example:
What is the volumetric flow rate in an XK 16/70 column (i.d. 1.6 cm) when the flow velocity
is 150 cm/h?
Y = flow velocity = 150 cm/h
d = inner diameter of the column = 1.6 cm
Volumetric flow rate =
150
× p × 1.6 × 1.6
ml/min
60 × 4
= 5.03 ml/min
From volumetric flow rate (ml/min) to flow velocity (cm/h)
Flow velocity (cm/h) =
Volumetric flow rate (ml/min) × 60
column cross sectional area (cm
2
)
= Z × 60 ×
4
p × d
2
where
Z = volumetric flow rate in ml/min
d = column inner diameter in cm
Example:
What is the linear flow in a Tricorn 5/50 column (i.d. 0.5 cm) when the volumetric flow rate is 1 ml/min?
Z = volumetric flow rate = 1 ml/min
d = column inner diameter = 0.5 cm
Flow velocity = 1 × 60 ×
4
cm/h
p × 0.5 × 0.5
From volumetric flow rate (ml/min) to using a syringe
1 ml/min = approximately 30 drops/min on a HiTrap 1 ml column
5 ml/min = approximately 120 drops/min on a HiTrap 5 ml column

18102229 AF 137
Appendix 5
Conversion data: proteins, column pressures
Proteins
Mass (g/mol) 1 µg 1 nmol Protein A
280
for 1 mg/ml
10 000 100 pmol; 6 × 10
13
molecules 10 µg IgG 1.35
50 000 20 pmol; 1.2 × 10
13
molecules 50 µg IgM 1.20
100 000 10 pmol; 6.0 × 10
12
molecules 100 µg IgA 1.30
150 000 6.7 pmol; 4.0 × 10
12
molecules 150 µg Protein A 0.17
Avidin 1.50
Streptavidin 3.40
Bovine serum albumin 0.70
1 kb of DNA = 333 amino acids of coding capacity
= 37 000 g/mol
270 bp DNA = 10 000 g/mol
1.35 kb DNA = 50 000 g/mol
2.70 kb DNA = 100 000 g/mol
Average molecular weight of an amino acid = 120 g/mol.
Column pressures
The maximum pressure drop over the packed bed refers to the pressure above which the column
contents might begin to compress.
Pressure units may be expressed in megaPascal (MPa), bar, or pounds per square inch (psi) and
can be converted as follows: 1 MPa = 10 bar = 145 psi

138 18102229 AF
Three-letter Single-letter
Amino acid code code Structure
Alanine Ala A
Arginine Arg R
Asparagine Asn N
Aspartic acid Asp D
Cysteine Cys C
Glutamic acid Glu E
Glutamine Gln Q
Glycine Gly G
Histidine His H
Isoleucine Ile I
Leucine Leu L
Lysine Lys K
Methionine Met M
Phenylalanine Phe F
Proline Pro P
Serine Ser S
Threonine Thr T
Tryptophan Trp W
Tyrosine Tyr Y
Valine Val V
CH
3
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
CH
2
CH
2
CH
2
NHC
NH
2
NH
CH
2
CONH
2
CH
2
COOH
CH
2
SH
CH
2
CH
2
COOH
CH
2
CH
2
CONH
2
H
CH
2
CH(CH
3
)CH
2
CH
3
CH
2
CH
CH
3
CH
3
CH
2
CH
2
CH
2
CH
2
NH
2
CH
2
CH
2
SCH
3
CH
2
HOOC
H
2
N
HOOC
H
2
N
HOOC
H
2
N
CH
2
OH
CHCH
3
OH
CH
2
CH(CH
3
)
2
HOOC
H
2
N
OH
CH
2
NH
NH
N
NH
Appendix 6
Table of amino acids

18102229 AF 139
Middle unit
residue (-H
2
0) Charge at Hydrophobic Uncharged Hydrophilic
Formula M
r
Formula M
r
pH 6.0 to 7.0 (nonpolar) (polar) (polar)
C
3
H
7
NO
2
89.1 C
3
H
5
NO 71.1 Neutral •
C
6
H
14
N
4
O
2
174.2 C
6
H
12
N
4
O 156.2 Basic (+ve) •
C
4
H
8
N
2
O
3
132.1 C
4
H
6
N
2
O
2
114.1 Neutral •
C
4
H
7
NO
4
133.1 C
4
H
5
NO
3
115.1 Acidic(-ve) •
C
3
H
7
NO
2
S 121.2 C
3
H
5
NOS 103.2 Neutral •
C
5
H
9
NO
4
147.1 C
5
H
7
NO
3
129.1 Acidic (-ve) •
C
5
H
10
N
2
O
3
146.1 C
5
H
8
N
2
O
2
128.1 Neutral •
C
2
H
5
NO
2
75.1 C
2
H
3
NO 57.1 Neutral •
C
6
H
9
N
3
O
2
155.2 C6H
7
N
3
O 137.2 Basic (+ve) •
C
6
H
13
NO
2
131.2 C
6
H
11
NO 113.2 Neutral •
C
6
H
13
NO
2
131.2 C
6
H
11
NO 113.2 Neutral •
C
6
H
14
N
2
O
2
146.2 C
6
H
12
N
2
O 128.2 Basic (+ve) •
C
5
H
11
NO
2
S 149.2 C
5
H
9
NOS 131.2 Neutral •
C
9
H
11
NO
2
165.2 C
9
H
9
NO 147.2 Neutral •
C
5
H
9
NO
2
115.1 C
5
H
7
NO 97.1 Neutral •
C
3
H
7
NO
3
105.1 C
3
H
5
NO
2
87.1 Neutral •
C
4
H
9
NO
3
119.1 C
4
H
7
NO
2
101.1 Neutral •
C
11
H
12
N
2
O
2
204.2 C
11
H
10
N
2
O 186.2 Neutral •
C
9
H
11
NO
3
181.2 C
9
H
9
NO
2
163.2 Neutral •
C
5
H
11
NO
2
117.1 C
5
H
9
NO 99.1 Neutral •
140 18102229 AF
Appendix 7
Analytical assays during purification
Analytical assays are essential to follow the progress of purification. They are used to assess the
effectiveness of each step in terms of yield, biological activity, and recovery as well as to help
during optimization of experimental conditions. The importance of a reliable assay for the
target molecule cannot be overemphasized.
When testing chromatographic fractions, ensure that the buffers used for purification
do not interfere with the assay.
Total protein determination
Lowry or Bradford assays are used most frequently to determine the total protein content.
The Bradford assay is particularly suited to samples where there is a high lipid content that
can interfere with the Lowry assay.
Purity determination
Purity is most often estimated by SDS-PAGE. Alternatively, IEF, capillary electrophoresis, RPC, or
MS may be used.
SDS-PAGE analysis
The general steps involved in SDS-PAGE analysis are summarized below.
1. Prepare samples by mixing with equal volume of 2x SDS loading buffer
2. Vortex briefly and heat for 5 min at 90°C to 100°C.
3. Load the samples and, optionally, a MW marker onto a SDS-polyacrylamide gel.
4. Run the gel.
5. Stain the gel with Coomassie Blue (Coomassie Blue Tablets, PhastGel Blue R-350) or
silver (PlusOne Silver Staining Kit, Protein).
The percentage of acrylamide in the SDS gel should be selected according to the
expected molecular weight of the protein of interest (see Table A8.1).
Table A8.1. Percentage of acrylamide used in SDS gels for proteins of different molecular weights
Acrylamide in resolving gel (%) Mol. weight range
Homogeneous: 5 36 000 to 200 000
7.5 24 000 to 200 000
10 14 000 to 200 000
12.5 14 000 to 100 000
15 14 000 to 60 000
1
Gradient: 5 to 15 14 000 to 200 000
5 to 20 10 000 to 200 000
10 to 20 10 000 to 150 000
1
The larger proteins fail to move significantly into the gel.

18102229 AF 141
The gel is usually stained after electrophoresis in order to make the protein bands visible by, for
example, Coomassie Blue or silver staining. A more recent way of making protein visible is by
prelabeling the proteins by fluorescent dye (Amersham™ WB Cy™5 dye reagent) before loading
the sample in the gel. By doing in this way the gel image can be acquired directly after finished
electrophoresis by laser scanner or CCD camera and the result is obtained much faster. This
workflow is outlined below.
Protein prelabeling with CyDye™
1. Prepare samples by prelabeling with Amersham WB Cy5 dye reagent.
2. Vortex briefly and heat for 5 min at 90°C to 100°C.
3. Load the samples and, optionally, a MW marker onto a SDS-polyacrylamide gel.
4. Run the gel and proceed directly to image capture.
For information and advice on electrophoresis techniques, refer to the handbook
2-D Electrophoresis, Principles and Methods, 80642960. For information on the
Amersham WB system and accessories including Amersham WB Cy5 prelabeling
reagents, visit www.gelifesciences.com/westernblotting.
Functional assays
Immunospecific interactions have enabled the development of many alternative assay systems
for the assessment of active concentration of target molecules.
• Western blot analysis is used to confirm protein identity and quantitate the level of
target molecule.
1. Separate the protein samples by SDS-PAGE.
2. Transfer the separated proteins from the gel to an appropriate membrane, depending
on the choice of detection reagents. Amersham Protran™ (NC) or Amersham Hybond™ P
(PVDF) membranes are recommended for chemiluminescent detection using
Amersham ECL™ start, Amersham ECL, Amersham ECL Prime, or Amersham ECL Select™
Western blotting detection reagents. Amersham Protran Premium (NC) or
Amersham Hybond LFP (PVDF) membranes are recommended for fluorescent detection
with Amersham ECL Plex™ Western blotting detection system.
3. Develop the membrane with the appropriate specified reagents.
Electrophoresis, protein transfer, and probing may be accomplished using a variety
of equipment and reagents. The Amersham WB system is an automated system that
can be used for all these steps including software evaluation. For more information,
visit www.gelifesciences.com/westernblotting. For further on the basic principles and
methods used in Western blotting, refer to the Western Blotting Handbook, 28999897
and the instruction manuals supplied with the detection kits.
• ELISAs are most commonly used as activity assays.
• Functional assays using the phenomenon of surface plasmon resonance (SPR) to detect
immunospecific interactions (e.g., using Biacore™ systems) enable the determination of
active concentration, epitope mapping, and studies of interaction kinetics.
The Biacore Assay Handbook, 29019400 gives a general overview of the different types
of SPR-based applications. The handbook also provides advice on sample preparation,
design, and optimization of different assays.
142 18102229 AF
Detection and assay of tagged proteins
SDS-PAGE, Western blotting, and ELISA can also be applied to the detection and assay of
genetically engineered molecules to which a specific tag has been attached. In some cases,
an assay based on the properties associated with the tag itself can be developed, for example,
the GST Detection Module for enzymatic detection and quantitation of GST-tagged proteins.
Further details on the detection and quantitation of GST and his-tagged proteins are available in
the Affinity Chromatography, Vol 2: Tagged Proteins, 18114275 and the GST Gene Fusion System
Handbook, 18115758 from GE.

18102229 AF 143
Appendix 8
Storage of biological samples
The advice given here is of a general nature and cannot be applied to every biological
sample. Always consider the properties of the specific sample and its intended use
before following any of these recommendations.
General recommendations
• Add stabilizing agents, when necessary. Stabilizing agents are often required for storage of
purified proteins.
• Serum, culture supernatants, and ascitic fluid should be kept frozen at -20°C or -70°C, in
small aliquots.
• Avoid repeated freeze/thawing or freeze drying/redissolving that can reduce biological activity.
• Avoid conditions close to stability limits for example pH or salt concentrations, reducing or
chelating agents.
• Keep refrigerated at 4°C in a closed vessel to minimize bacterial growth and protease
activity. Above 24 h at 4°C, add a preserving agent if possible (e.g., merthiolate 0.01%).
Sodium azide can interfere with many coupling methods and some biological
assays and can be a health hazard. It can be removed by using a desalting column
(see Appendix 1, Sample preparation).
Specific recommendations for purified proteins
• Store as a precipitate in high concentration of ammonium sulfate, for example 4.0 M.
• Freeze in 50% glycerol, especially suitable for enzymes.
• Avoid the use of preserving agents if the product is to be used for a biological assay.
Preserving agents should not be added if in vivo experiments are to be performed.
Store samples in small aliquots and keep frozen.
• Sterile filter to prolong storage time.
• Add stabilizing agents such as glycerol (5% to 20%) or serum albumin (10 mg/ml) to help
maintain biological activity. Remember that any additive will reduce the purity of the protein
and might need to be removed at a later stage.
• Avoid repeated freeze/thawing or freeze drying/redissolving that can reduce biological activity.
Certain proteins, including some mouse antibodies of the IgG
3
subclass, should not be
stored at 4°C as they precipitate at this temperature. Keep at room temperature in the
presence of a preserving agent.

144 18102229 AF
Related literature
Code number
Purification handbooks
Affinity Chromatography, Vol. 1: Antibodies 18103746
Affinity Chromatography, Vol. 2: Tagged Proteins 18114275
Affinity Chromatography, Vol. 3: Specific Groups of Biomolecules 18102229
Ion Exchange Chromatography 11000421
Hydrophobic Interaction and Reversed Phase Chromatography 11001269
Multimodal Chromatography 29054808
Protein Sample Preparation 28988741
Purifying Challenging Proteins 28909531
Size Exclusion Chromatography 18102218
Strategies for Protein Purification 28983331
ÄKTA Laboratory-scale Chromatography Systems 29010831
Protein analysis handbooks
Biacore Assay 29019400
Biacore Sensor Surface BR100571
Western Blotting 28999897
Selection guides
Affinity chromatography columns and media, Selection guide 18112186
Prepacked chromatography columns for ÄKTA systems, Selection guide 28931778

18102229 AF 145
Ordering information
Product Quantity Code number
Prepacked AC columns
Albumin & IgG Depletion SpinTrap 10 columns 28948020
HiScreen Blue FF 1 × 4.7 ml 28978243
HiScreen Capto Blue 1 × 4.7 ml 28992474
HiTrap Benzamidine FF (high sub) 2 × 1 ml 17514302
5 × 1 ml 17514301
1 × 5 ml 17514401
HiScreen Capto Chelating 1 × 4.7 ml 17548510
HiScreen Capto Lentil Lectin 1 × 4.7 ml 29157958
HiScreen IXSelect 1 × 4.7 ml 17371410
HiTrap IXSelect 5 × 1 ml 17371411
1 × 5 ml 17371412
HiTrap Albumin & IgG Depletion 2 × 1 ml 28946603
HiTrap AVB Sepharose HP 5 × 1 ml 28411211
1 × 5 ml 28411212
HiTrap Blue HP 5 × 1 ml 17041201
1 × 5 ml 17041301
HiTrap Capto Chelating 5 × 1 ml 17548511
5 × 5 ml 17548512
HiTrap Capto Lentil Lectin 5 × 1 ml 17548911
5 × 5 ml 17548912
HiTrap Chelating HP 5 × 1 ml 17040801
1 × 5 ml 17040901
HiTrap GCSFSelect 5 × 1 ml 17548311
5 × 5 ml 17548312
HiTrap Heparin HP 5 × 1 ml 17040601
1 × 5 ml 17040701
HiTrap Streptavidin HP 5 × 1 ml 17511201
HiPrep Heparin FF 16/10 1 × 20 ml 28936549
Streptavidin HP SpinTrap 16 columns 28903130
Streptavidin HP SpinTrap Buffer Kit 1 28913568
Prepacked 96-well plates
Streptavidin HP MultiTrap 4 × 96-well plates 28903131

146 18102229 AF
Product Quantity Code number
Magnetic beads
Sera-Mag Streptavidin-Coated (low biotin binding) 1 ml 30152103011150
5 ml 30152103010150
100 ml 30152103010350
Sera-Mag Streptavidin-Coated (med. biotin binding) 1 ml 30152104011150
5 ml 30152104010150
100 ml 30152104010350
Sera-Mag Streptavidin-Coated (high biotin binding) 1 ml 30152105011150
5 ml 30152105010150
100 ml 30152105010350
Sera-Mag SpeedBead Streptavidin-Coated (med. biotin binding) 1 ml 66152104011150
5 ml 66152104010150
100 ml 66152104010350
Sera-Mag SpeedBeads Streptavidin-Blocked 1 ml 21152104011150
5 ml 21152104010150
100 ml 21152104010350
Sera-Mag SpeedBeads Neutravidin-Coated 1 ml 78152104011150
5 ml 78152104010150
100 ml 78152104010350
Sera-Mag Carboxylate-Modified (hydrophilic) 15 ml 24152105050250
100 ml 24152105050350
Sera-Mag Carboxylate-Modified (hydrophobic) 15 ml 44152105050250
100 ml 44152105050350
Sera-Mag SpeedBeads Carboxylate-Modified (hydrophilic) 15 ml 45152105050250
100 ml 45152105050350
Sera-Mag SpeedBeads Carboxylate-Modified (hydrophobic) 15 ml 65152105050250
100 ml 65152105050350
Streptavidin Mag Sepharose 2 × 1 ml, 10% slurry 28985738
5 × 1 ml, 10% slurry 28985799
TiO
2
Mag Sepharose 1 × 500 µl 28944010
4 × 500 µl 28951377
NHS Mag Sepharose 1 × 500 µl 28944009
4 × 500 µl 28951380
Chromatography media
2´5´ ADP Sepharose 4B 5 g 17070001
IXSelect 25 ml 17371401
200 ml 17371402
VIISelect 25 ml 17547701
100 ml 17547702
VIIISelect 25 ml 17545001

18102229 AF 147
Product Quantity Code number
AVB Sepharose High Performance 25 ml 28411210
75 ml 28411201
Benzamidine Sepharose 4 Fast Flow (high sub) 25 ml 17512310
100 ml 17512301
Benzamidine Sepharose 4 Fast Flow (low sub) 25 ml 28410899
100 ml 28410801
Blue Sepharose 6 Fast Flow 50 ml 17094801
Capto Blue 25 ml 17544801
Capto Blue (high sub) 25 ml 17545201
Capto DeVirS 25 ml 17546601
100 ml 17546602
Capto Heparin 25 ml 17546201
200 ml 17546202
Capto Lentil Lectin 25 ml 17548901
100 ml 17548902
Calmodulin Sepharose 4B 10 ml 17052901
Chelating Sepharose Fast Flow 50 ml 17057501
Con A Sepharose 4B 5 ml 17044003
100 ml 17044001
GCSFSelect 25 ml 17548301
200 ml 17548302
Gelatin Sepharose 4B 25 ml 17095601
Heparin Sepharose 6 Fast Flow 50 ml 17099801
250 ml 17099825
Heparin Sepharose High Performance 25 ml 90100019
100 ml 90100401
Lentil Lectin Sepharose 4B 25 ml 17044401
Streptavidin Sepharose High Performance 5 ml 17511301
Preactivated media and columns for ligand coupling
HiTrap NHS-activated HP 5 × 1 ml 17071601
1 × 5 ml 17071701
NHS-activated Sepharose 4 Fast Flow 25 ml 17090601
CNBr-activated Sepharose 4 Fast Flow 10 g 17098101
CNBr-activated Sepharose 4B 15 g 17043001
Epoxy-activated Sepharose 6B 15 g 17048001
EAH Sepharose 4B 50 ml 17056901
Activated Thiol Sepharose 4B 15 g 17064001
Prepacked desalting columns
HiTrap Desalting 1 × 5 ml 29048684
5 × 5 ml 17140801
HiPrep 26/10 Desalting 1 × 53 ml 17508701
4 × 53 ml 17508702
PD-10 Desalting Column 30 17085101

148 18102229 AF
Product index
IXSelect
41–43, 45
VIISelect
41–42, 44
VIIISelect
41–42, 45
A
Activated Thiol Sepharose 4B
76
2’5’ ADP Sepharose 4B
60–61
ÄKTA avant
131
ÄKTA pure
131
ÄKTA start
131
Albumin & IgG SpinTrap
31, 33
Albumin & IgG Sepharose
High Performance
31
ÄKTAxpress
141
Amersham ECL
141
Amersham ECL Plex
141
Amersham ECL Prime
141
Amersham ECL Select
141
Amersham Hybond LFP
141
Amersham Hybond P
141
Amersham Protran Premium
141
Amersham WB
(Western blotting) Cy5
141
Amersham WB system
141
AVB Sepharose
High Performance
71–74
B
Benzamidine Sepharose 4 Fast Flow
(high sub)
24, 67–68
Benzamidine Sepharose 4 Fast Flow
(low sub)
67–68
Blue Sepharose 6 Fast Flow
27–30, 59
C
Calmodulin Sepharose 4B
39
Capto Blue
27–28, 59
Capto Blue (high sub)
27–28, 59
Capto DeVirS
71–73, 74
Capto Heparin
41, 46–47, 50
Capto Lentil Lectin
52–53
Chelating Sepharose Fast Flow
63–64, 66
CNBr-activated Sepharose 4B
76–77, 84–85
CNBr-activated Sepharose 4
Fast Flow
76, 84–85
Con A Sepharose 4B
52–55
E
EAH Sepharose 4B
76, 89–90, 95
Epoxy-activated Sepharose 6B
76, 92–94
G
GCSFSelect
57–58
Gelatin Sepharose 4B
51
GSTrap FF
69
H
Heparin Sepharose 6 Fast Flow
41, 46–47, 49
Heparin Sepharose
High Performance
41, 46–47, 49
HiPrep 26/10 Desalting
131
HiScreen Capto Blue FF
28
HiScreen Capto Blue
28
HiScreen Capto Chelating
64
HiScreen Capto Lentil Lectin
53
HiScreen IXSelect
42
HiTrap Albumin & IgG Depletion
31–32
HiTrap AVB Sepharose HP
72
HiTrap Benzamidine FF (high sub)
67–69
HiTrap Blue HP
28–29
HiTrap Capto Chelating
64
HiTrap Capto Lentil Lectin
53
HiTrap Chelating HP
64, 65, 66
HiTrap Desalting
82, 103, 125, 129
HiTrap GCSFSelect
57
HiTrap Heparin HP
44, 47–48
HiTrap NHS-activated HP
79, 81–83, 88
HiTrap Phenyl FF (high sub)
40
HiTrap Q FF
40
HiTrap IXSelect
42
HiTrap Streptavidin HP
34, 35–36
L
Lentil Lectin Sepharose 4B
52–53, 55
LMW-SDS Marker Kit
44, 48, 49, 69, 88
N
NHS-activated
Sepharose 4 Fast Flow
76, 80–81
NHS Mag Sepharose
109–110, 111

18102229 AF 149
P
PD-10 desalting column
29, 48
PD MiniTrap G-25
129
PhastGel Gradient 8–25
44
PhastSystem
electrophoresis unit
44
PlusOne Coomassie Tablets,
PhastGel Blue R-350
140
PlusOne Silver Staining Kit,
Protein
140
PrimeView software
131
Protein G Sepharose High
Performance
31
Puradisc 4 Syringe Filter,
0.2 µm, PVDF
124
Puradisc 13 Syringe Filter,
0.2 µm, PVDF
124
Puradisc 25 Syringe Filter,
0.2 µm, PVDF
124
Puradisc 4 Syringe Filter,
0.45 µm, PVDF
124
Puradisc 13 Syringe Filter,
0.45 µm, PVDF
124
Puradisc 25 Syringe Filter,
0.45 µm, PVDF
124
Puradisc FP 30 Syringe Filter,
0.2 µm, CA
124
Puradisc FP 30 Syringe Filter,
0.45 µm, CA
124
S
Sephadex G-25
127, 128
SeraMag Carboxylate-Modified
109, 110
SeraMag Neutravidin-Coated
100
SeraMag SpeedBeads
Streptavidin-Blocked
100, 105
SeraMag SpeedBeads
Streptavidin-Coated
100
SeraMag SpeedBeads
Neutravidin-Coated
100
SeraMag Streptavidin-Blocked
100, 105
SeraMag Streptavidin-Coated
100
SPARTAN 13 mm HPLC-Certified
Syringe Filter, RC, 0.2 µm
124
SPARTAN 13 mm HPLC-Certified
Syringe Filter, RC, 0.45 µm
124
SPARTAN 30 mm HPLC-Certified
Syringe Filter, RA, 0.2 µm
124
SPARTAN 30 mm HPLC-Certified
Syringe Filter, RA, 0.45 µm
124
Streptavidin HP MultiTrap
34, 37
Streptavidin HP SpinTrap
34–35, 38
Streptavidin Mag Sepharose
34, 100, 102–103
Streptavidin Sepharose
High Performance
34
T
TiO
2
Mag Sepharose
106–108
U
UNICORN software
131
150 18102229 AF

Affinity Chromatography – Vol. 3: Specific Groups of Biomolecules
www.gelifesciences.com
GE, GE monogram, Amersham, ÄKTA, Biacore, BioProcess, Capto, Cy, CyDye, ECL, ECL Plex, ECL Select,
ExcelGel, GSTrap, HiPrep, HiScale, HiScreen, HiTrap, ImageQuant, Hybond, MiniTrap, MabSelect, MiniTrap,
MultiTrap, PhastGel, PhastSystem, PrimeView, Protran, Sephadex, Sepharose, SPARTAN, SpinTrap,
Superdex, Typhoon, Tricorn, UNICORN, and Whatman are trademarks of General Electric Company.
Coomassie is a trademark of Thermo Fisher Scientific LLC. Neutravidin is a trademark of Pierce
Biotechnology, Inc. Nonidet is a trademark of Air Products and Chemicals, Inc. Pefabloc is a trademark
of DSM IP Assets B.V. Tween is a trademark of Croda Group of Companies. All other third-party
trademarks are the property of their respective owners.
The purchase of CyDye products includes a limited license to use the CyDye products for internal
research and development but not for any commercial purposes. A license to use the Cy and CyDye
trademarks for commercial purposes is subject to a separate license agreement with GE Healthcare.
Commercial use shall include:
1. Sale, lease, license or other transfer of the material or any material derived or produced from it.
2. Sale, lease, license or other grant of rights to use this material or any material derived or produced from it.
3. Use of this material to perform services for a fee for third parties, including contract research and
drug screening.
If you require a commercial license to use the Cy and CyDye trademarks please contact: [email protected].
VIIISelect incorporates BAC BV’s proprietary ligand technology, which has been exclusively licensed to
GE Healthcare in the field of purification of beta domain depleted recombinant factor VIII. Other use of
this product may require a separate license from BAC BV, Huizerstraatweg 28, 1411 GP Naarden, The
Netherlands.
IXSelect incorporates BAC BV’s proprietary ligand technology, which has been exclusively licensed to
GE Healthcare for use in chromatography separation.
GCSFSelect incorporates BAC BV’s proprietary ligand technology, which has been exclusively licensed
to GE Healthcare for use in chromatography separation.
© 1988–2016 General Electric Company. First published 1988.
All goods and services are sold subject to the terms and conditions of sale of the company within
GE Healthcare that supplies them. A copy of these terms and conditions is available on request.
Contact your local GE Healthcare representative for the most current information.
GE Healthcare UK Ltd, Amersham Place, Little Chalfont, Buckinghamshire, HP7 9NA, UK
GE Healthcare Bio-Sciences Corp. 100 Results Way, Marlborough, MA 01752, USA
GE Healthcare Dharmacon, Inc., 2650 Crescent Dr., Lafayette, CO 80026, USA
HyClone Laboratories, Inc., 925 W 1800 S, Logan, UT 84321, USA
GE Healthcare Europe GmbH, Munzinger Strasse 5, D-79111 Freiburg, Germany
GE Healthcare Japan Corporation, Sanken Bldg. 3-25-1, Hyakunincho, Shinjuku-ku, Tokyo 169-0073, Japan
For local office contact information, visit www.gelifesciences.com/contact
18102229 AF 03/2016
GE Healthcare Bio-Sciences AB
Björkgatan 30
751 84 Uppsala
Sweden
Affinity Chromatography
Vol. 3: Specific Groups of Biomolecules
GE Healthcare
imagination at work
imagination at work
imagination at work
