
I:\\16645dftR1.doc
4/20/16
Assay Development and
Validation for
Immunogenicity Testing of
Therapeutic Protein Products
Guidance for Industry
DRAFT GUIDANCE
This guidance document is being distributed for comment purposes only.
Comments and suggestions regarding this draft document should be submitted within 60 days of
publication in the Federal Register of the notice announcing the availability of the draft
guidance. Submit electronic comments to http://www.regulations.gov. Submit written
comments to the Division of Dockets Management (HFA-305), Food and Drug Administration,
5630 Fishers Lane, rm. 1061, Rockville, MD 20852. All comments should be identified with
the docket number listed in the notice of availability that publishes in the Federal Register.
For questions regarding this draft document, contact (CDER) Susan Kirshner at 301-827-1731;
(CBER) Office of Communication, Outreach and Development, 800-835-4709 or 240-402-8010;
or (CDRH) Office of Communication and Education, 800-638-2041 or 301-796-7100.
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Drug Evaluation and Research (CDER)
Center for Biologics Evaluation and Research (CBER)
Center for Devices and Radiological Health (CDRH)
April 2016
Pharmaceutical Quality/CMC
Revision 1

Assay Development and Validation
for Immunogenicity Testing of
Therapeutic Protein Products
Guidance for Industry
Additional copies are available from:
Office of Communications,
Division of Drug Information
Center for Drug Evaluation and Research
Food and Drug Administration
10001 New Hampshire Ave., Hillandale Bldg., 4
th
Floor
Silver Spring, MD 20993-0002
Phone: 855-543-3784 or 301-796-3400; Fax: 301-431-6353
Email: druginfo@fda.hhs.gov
http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
and/or
Office of Communication, Outreach and Development
Center for Biologics Evaluation and Research
Food and Drug Administration
10903 New Hampshire Ave., Bldg. 71, Room 3128
Silver Spring, MD 20993-0002
Phone: 800-835-4709 or 240-402-8010
Email: [email protected].gov
http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
and/or
Office of Communication and Education
CDRH-Division of Industry and Consumer Education
Center for Devices and Radiological Health
Food and Drug Administration
10903 New Hampshire Ave., Bldg. 66, Room 4621
Silver Spring, MD 20993-0002
Phone: 800-638-2041 or 301-796-7100; Fax: 301-847-8149
Email: DICE@fda.hhs.gov
http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/default.htm
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Drug Evaluation and Research (CDER)
Center for Biologics Evaluation and Research (CBER)
Center for Devices and Radiological Health (CDRH)
April 2016
Pharmaceutical Quality/CMC
Revision 1
Contains Nonbinding Recommendations
Draft — Not for Implementation
TABLE OF CONTENTS
I. INTRODUCTION............................................................................................................. 1
II. BACKGROUND ............................................................................................................... 2
III. GENERAL PRINCIPLES................................................................................................ 2
IV. ASSAY DESIGN ELEMENTS ........................................................................................ 4
A. Testing Strategy ............................................................................................................................. 4
1. Multi-Tiered Testing Approach ........................................................................................................ 4
2. Immunoglobulin Isotypes ................................................................................................................. 5
3. Epitope Specificity ........................................................................................................................... 5
B. Assay Cut Point .............................................................................................................................. 6
C. Sensitivity ........................................................................................................................................ 7
1. Assay Sensitivity ............................................................................................................................... 7
2. Drug Tolerance ................................................................................................................................ 8
D. Specificity and Selectivity .............................................................................................................. 8
1. Matrix Interference .......................................................................................................................... 9
2. Minimal Required Dilution ............................................................................................................ 10
E. Precision ........................................................................................................................................ 10
F. Reproducibility ............................................................................................................................. 11
G. Robustness and Sample Stability ................................................................................................ 11
H. Selection of Format ...................................................................................................................... 11
I. Selection of Reagents ................................................................................................................... 12
1. Development of Positive Control Antibodies ................................................................................. 12
2. Development of Negative Controls ................................................................................................ 13
3. Detection Reagent Consideration ..................................................................................................
13
4. Controlling Non-Specific Binding .................................................................................................. 14
J. Reporting Results for Qualitative and Semi-Quantitative Assays .......................................... 14
K. Other Considerations for Assay Development .......................................................................... 15
1. Pre-Existing Antibodies ................................................................................................................. 15
2. Rheumatoid Factor ........................................................................................................................ 15
3. Monoclonal Antibodies .................................................................................................................. 15
4. Conjugated Proteins ...................................................................................................................... 16
5. Products With Multiple Functional Domains ................................................................................ 16
V. ASSAY DEVELOPMENT ............................................................................................. 16
A. Development of Screening Assay ................................................................................................ 16
B. Development of Confirmatory Assay ......................................................................................... 17
1. Selection of Format for Confirmatory Assay ................................................................................. 17
2. Cut Point of Confirmatory Assay ................................................................................................... 17
C. Development of Titering Assay ................................................................................................... 17
Contains Nonbinding Recommendations
Draft — Not for Implementation
1. Titer Determination ....................................................................................................................... 17
2. Cut Point of Titering Assay ............................................................................................................ 18
D. Development of Neutralization Assay ........................................................................................ 18
1. Selection of Format for Neutralization Assay ................................................................................ 18
2. Activity Curve of Neutralization Assay .......................................................................................... 19
3. Considerations for Matrix Interference for Neutralization Assay ................................................. 20
4. Cut Point of Neutralization Assay .................................................................................................. 21
5. Additional Considerations for Neutralization Assay ..................................................................... 21
VI. ASSAY VALIDATION .................................................................................................. 22
A. General Considerations for Assay Validation ........................................................................... 22
B. Validation of Screening Assay .................................................................................................... 24
1. Sensitivity of Screening Assay ........................................................................................................ 24
2. Cut Point of Screening Assay ......................................................................................................... 24
C. Validation of Confirmatory Assay ............................................................................................. 24
D. Validation of Titering Assay ....................................................................................................... 25
E. Validation of Neutralization Assay ............................................................................................. 25
VII. IMPLEMENTATION OF ASSAY TESTING ............................................................. 26
A. Obtaining Patient Samples .......................................................................................................... 26
B. Concurrent Positive and Negative Quality Controls ................................................................ 27
C. Confirmation of Cut Point in the Target Population ................................................................ 27
VIII. DOCUMENTATION ...................................................................................................... 28
REFERENCES ............................................................................................................................ 29
Contains Nonbinding Recommendations
Draft — Not for Implementation
1
Assay Development and Validation for Immunogenicity Testing of 1
Therapeutic Protein Products 2
Guidance for Industry
1
3
4
5
This draft guidance, when finalized, will represent the current thinking of the Food and Drug 6
Administration (FDA or Agency) on this topic. It does not establish any rights for any person and is not 7
binding on FDA or the public. You can use an alternative approach if it satisfies the requirements of the 8
applicable statutes and regulations. To discuss an alternative approach, contact the FDA office 9
responsible for this guidance as listed on the title page. 10
11
12
13
14
I. INTRODUCTION 15
16
This guidance provides recommendations to facilitate industry’s development and validation of 17
immune assays for assessment of the immunogenicity of therapeutic protein products during 18
clinical trials. Specifically, this document includes guidance regarding the development and 19
validation of screening assays, confirmatory assays, titering assays, and neutralization assays.
2,3
20
For the purposes of this guidance, immunogenicity is defined as the propensity of the therapeutic 21
protein product to generate immune responses to itself and to related proteins or to induce 22
immunologically related adverse clinical events. The recommendations for assay development 23
and validation provided in this document apply to assays for detection of anti-drug antibody(ies) 24
(ADA).
4
This guidance may also apply to some combination products on a case-by-case basis.
5
25
1
This guidance has been prepared by the Office of Medical Policy in the Center for Drug Evaluation and Research
in cooperation with the Center for Biologics Evaluation and Research and the Center for Devices and Radiological
Health at the Food and Drug Administration.
2
This document specifically does not discuss the development or validation of anti-drug antibody(ies) (ADA)
assays for animal studies; however, some concepts discussed are relevant to the design of ADA studies for
nonclinical testing. Refer to the International Conference on Harmonisation (ICH) guidance for industry S6(R1)
Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals for more information regarding
immunogenicity assessments in animal toxicology studies. Also see the guidance for industry Immunogenicity
Assessment for Therapeutic Protein Products, where the topic “Utility of Animal Studies” is covered in more detail.
We update guidances periodically. To make sure you have the most recent version of a guidance, check the FDA
guidance Web page at http://www.fda.gov/RegulatoryInformation/Guidances/default.htm
.
3
For information on clinical immunogenicity assessment of proposed biosimilar biological products, see the
guidance for industry Scientific Considerations in Demonstrating Biosimilarity to a Reference Product.
4
This guidance does not pertain to immunogenicity assays for assessment of immune response to preventative and
therapeutic vaccines for infectious disease indications.
5
General information on combination products is available at
http://www.fda.gov/CombinationProducts/default.htm
.

Contains Nonbinding Recommendations
Draft — Not for Implementation
2
This document does not discuss the product and patient risk factors that may contribute to 26
immunogenicity.
6
This guidance, including any discussions of terminology used in this 27
guidance, does not apply to in vitro diagnostic products.
7
This guidance revises the draft 28
guidance for industry Assay Development for Immunogenicity Testing of Therapeutic Proteins 29
issued in December 2009. The information in this guidance has been reorganized for clarity and 30
includes new information on titering and confirmatory assays. 31
32
In general, FDA’s guidance documents do not establish legally enforceable responsibilities. 33
Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only 34
as recommendations, unless specific regulatory or statutory requirements are cited. The use of 35
the word should in Agency guidances means that something is suggested or recommended, but 36
not required. 37
38
39
II. BACKGROUND 40
41
Patient immune responses to therapeutic protein products have the potential to affect product 42
safety and efficacy.
8
The clinical effects of patient immune responses are highly variable, 43
ranging from no effect at all to extremely harmful effects to patient health. Detection and 44
analysis of ADA formation is a helpful tool in understanding potential patient immune responses. 45
Information on immune responses observed during clinical trials, particularly the incidence of 46
ADA induction and the implications of ADA responses for therapeutic protein product safety 47
and efficacy, is crucial for any therapeutic protein product development program. Accordingly, 48
such information, if applicable, should be included in the prescribing information as a subsection 49
of the ADVERSE REACTIONS section entitled “Immunogenicity.” Therefore, the development 50
of valid, sensitive, specific, and selective assays to measure ADA responses is a key aspect of 51
therapeutic protein product development. 52
53
54
III. GENERAL PRINCIPLES 55
56
The risk to patients of mounting an immune response to a therapeutic protein product will vary 57
with the product. FDA recommends adoption of a risk-based approach to evaluating and 58
mitigating immune responses to or immunologically related adverse clinical events associated 59
6
See the guidance for industry Immunogenicity Assessment for Therapeutic Protein Products, where these topics are
covered in more detail.
7
Per 21 CFR 809.3(a), “in vitro diagnostic products are those reagents, instruments, and systems intended for use in
the diagnosis of disease or other conditions, including a determination of the state of health, in order to cure,
mitigate, treat, or prevent disease or its sequelae. Such products are intended for use in the collection, preparation,
and examination of specimens taken from the human body. These products are devices as defined in section 201(h)
of the Federal Food, Drug, and Cosmetic Act (the act), and may also be biological products subject to section 351 of
the Public Health Service Act.”
8
See the guidance for industry Immunogenicity Assessment for Therapeutic Protein Products.

Contains Nonbinding Recommendations
Draft — Not for Implementation
3
with therapeutic protein products that affect their safety and efficacy.
9
Immune responses may 60
have multiple effects, including neutralizing activity and the ability to induce hypersensitivity 61
responses. Immunogenicity tests should be designed to detect ADA that could mediate 62
unwanted biological or physiological consequences. 63
64
Screening assays, also known as binding antibody (BAb) assays, are used to detect all antibodies 65
that bind to the therapeutic protein product. The specificity of BAb for the therapeutic protein 66
product is established using confirmatory assays. ADA are further characterized using titering 67
and neutralization assays. Titering assays are used to characterize the magnitude of the ADA 68
response. It is important to characterize this magnitude with titering assays because the impact 69
of ADA on safety and efficacy may correlate with ADA titer and persistence rather than 70
incidence (Cohen and Rivera 2010). Neutralization assays assess the ability of ADA to interfere 71
with the therapeutic protein product-target interactions. Therefore, neutralizing antibodies 72
(NAb) are a subset of BAb. It is important to characterize neutralizing activity of ADA with 73
neutralization assays because the impact of ADA on safety and efficacy may correlate with NAb 74
activity rather than ADA incidence (Calabresi, Giovannoni, et al. 2007; Goodin, Frohman, et al. 75
2007; Cohen and Rivera 2010). Similarly, it may be important in some cases to establish NAb 76
titers. Additional characterization assays, such as isotyping, epitope mapping, and assessing 77
cross-reactivity, e.g., to endogenous counterparts or to other products, may be useful. 78
79
The optimal time to design, develop, and validate ADA assays during therapeutic protein product 80
development depends on the risk assessment of the product (Mire-Sluis, Barrett, et al. 2004; 81
Gupta, Indelicato, et al. 2007; Shankar, Devanarayan, et al. 2008; Gupta, Devanarayan, et al. 82
2011). The sponsor should provide a rationale for the immunogenicity testing paradigm, 83
preferably at the investigational new drug application (IND) stage, during phase 1. Because 84
ADA assays are critical when immunogenicity poses a high clinical risk (e.g., assessment of a 85
therapeutic protein product with a non-redundant endogenous counterpart) and real-time data 86
concerning patient responses are needed, the sponsor should implement preliminary validated 87
assays early, before and during phase 1, and obtain data in real time. Real-time assessments 88
entail analyses of the samples as soon as possible after sampling, before banking of the samples, 89
and prior to additional dosing when the dosing regimen allows. In lower risk situations, the 90
sponsor may bank patient samples so they can be tested when suitable assays are available. FDA 91
encourages sponsors to test samples during phase 1 and phase 2 studies using suitable assays. 92
Samples derived from pivotal studies should be tested with fully validated assays. At the time of 93
license application, the sponsor should provide data supporting full validation of the assays. 94
Recommendations regarding the timing of ADA sample collection can be found in section 95
VII.A.
10
96
97
9
See the guidance for industry Immunogenicity Assessment for Therapeutic Protein Products.
10
See the guidance for industry Immunogenicity Assessment for Therapeutic Protein Products, where
immunogenicity risk assessment and mitigation considerations are covered in more detail. Guidance on appropriate
assay development and validation for immunogenicity testing is also available in the ICH guidances for industry
Q2A Text on Validation of Analytical Procedures and Q2B Validation of Analytical Procedures: Methodology.

Contains Nonbinding Recommendations
Draft — Not for Implementation
4
Assays for detection of ADA facilitate understanding of the immunogenicity, safety, and efficacy 98
of therapeutic protein products. However, the detection of ADA is dependent on key operating 99
parameters of the assays (e.g., sensitivity, specificity), which vary between assays.
11
Although 100
information on ADA incidence is typically included in the prescribing information under an 101
“Immunogenicity” subsection of the ADVERSE REACTIONS section, FDA cautions that 102
comparison of ADA incidence among products, even for products that share sequence or 103
structural homology, can be misleading. This is because detection of ADA formation is highly 104
dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of 105
ADA (including NAb) positivity in an assay may be influenced by factors such as method, 106
sample handling, timing of sample collection, concomitant medications, and disease condition. 107
Therefore, comparing immunogenicity rates among therapeutic protein products with structural 108
homology for the same indication is unsound, even though fully validated assays are employed. 109
When a true comparison of immunogenicity across different therapeutic protein products that 110
have homology is needed, it should be obtained by conducting a head-to-head clinical study 111
using a standardized assay under the same conditions that has equivalent sensitivity and 112
specificity for both therapeutic protein products.
12
113
114
The recommendations on assay development and validation provided in this guidance are based 115
on common issues encountered by the Agency upon review of immunogenicity submissions. 116
Sponsors should contact FDA for any product-specific guidance. Isotyping and cross-reactivity 117
assay designs should be discussed with FDA. Other publications may also be consulted for 118
additional insight (see Mire-Sluis, Barrett, et al. 2004; Gupta, Indelicato, et al. 2007; Shankar, 119
Devanarayan, et al. 2008; Gupta, Devanarayan, et al. 2011). In general, FDA recommends that 120
sponsors develop assays that are optimized for sensitivity, specificity, selectivity, precision, 121
reproducibility, and robustness (see sections IV.C through G). 122
123
124
IV. ASSAY DESIGN ELEMENTS 125
126
This section applies to all types of assays for detection of ADA, unless specified otherwise. 127
128
A. Testing Strategy 129
130
1. Multi-Tiered Testing Approach 131
132
FDA recommends a multi-tiered ADA testing approach because of the size of some clinical trials 133
and the necessity of testing patient samples at several time points. In this paradigm, a rapid, 134
sensitive screening assay is initially used to assess clinical samples. The initial screening assay 135
should be sensitive to low levels of low- and high-affinity ADA (see section V.A). Samples 136
testing positive in the screening assay are then subjected to a confirmatory assay to demonstrate 137
11
See the United States Pharmacopeia (USP) General Chapter 1106 Immunogenicity Assays – Design and
Validation of Immunoassays to Detect Anti-Drug Antibodies for a broader discussion of various assay types.
12
For information on proposed biosimilar products, see the guidance for industry Scientific Considerations in
Demonstrating Biosimilarity to a Reference Product.
Contains Nonbinding Recommendations
Draft — Not for Implementation
5
that ADA are specific for the therapeutic protein product. For example, a competition assay 138
could confirm that antibody is specifically binding to the therapeutic protein product and that the 139
positive finding in the screening assay is not a result of non-specific interactions of the test serum 140
or detection reagent with other materials in the assay milieu such as plastic or other proteins. 141
142
Samples identified as positive in the confirmatory assay should be further characterized in other 143
assays, such as titering and neutralization assays. In some cases, assays to detect cross-reactivity 144
to other proteins with homology, such as the corresponding endogenous protein, may be needed. 145
Further, tests to assess the isotype of the antibodies and their epitope specificity may also be 146
recommended once samples containing antibodies are confirmed as positive. 147
148
2. Immunoglobulin Isotypes 149
150
The initial screening assay should be able to detect all relevant immunoglobulin (Ig) isotypes. 151
For non-mucosal routes of administration, and in the absence of anaphylaxis, the expected ADA 152
isotypes are IgM and IgG. For mucosal routes of administration, IgA isotype ADA are also 153
expected. Although FDA expects that all relevant isotypes be detected in screening assays, it is 154
not necessary that the screening assay establish which isotypes are being detected. For example, 155
assays using the bridging format may provide no information on which isotypes are being 156
detected. Bridging assay format can theoretically detect antibodies of most isotypes, but may not 157
detect IgG4 isotypes. In some circumstances the sponsor should develop assays that discriminate 158
between antibody isotypes. For example, for therapeutic protein products where the risk for 159
anaphylaxis is a concern, antigen-specific IgE assays should be developed. In addition, the 160
generation of IgG4 antibodies has been associated with immune responses generated under 161
conditions of chronic antigen exposure, such as with factor VIII treatment, and in erythropoietin-162
treated patients with pure red cell aplasia (Matsumoto, Shima, et al. 2001; Aalberse and 163
Schuurman 2002). Consequently, depending on the clinical concern, assessing for specific 164
isotypes may be needed. 165
166
3. Epitope Specificity 167
168
FDA recommends that the sponsor direct initial screening tests against the whole therapeutic 169
protein product and, when relevant, its endogenous counterpart. For some therapeutic protein 170
products, the sponsor may need to investigate the ADA to specific epitopes to which immune 171
responses are specifically generated. For example, determination of epitope specificity is 172
recommended for some fusion molecules because the region where the two molecules join may 173
form a neoantigen, and immune responses to this region may arise. Because of epitope 174
spreading, immune responses to other parts of the molecule may ensue, leading to the generation 175
of antibodies to the therapeutic protein product or its endogenous counterpart (Prummer 1997; 176
Miller, Korn, et al. 1999; Disis, Goodell, et al. 2004; Thrasyvoulides, Liakata, et al. 2007; van 177
der Woude, Rantapaa-Dahlqvist, et al. 2010; Hintermann, Holdener, et al. 2011). For these 178
therapeutic protein products, FDA encourages sponsors to investigate the initiating event in the 179
immune cascade. This knowledge may allow for modification to the protein to reduce its 180
potential immunogenicity. Similarly, for therapeutic protein products with modifications, such 181
as PEGylation, sponsors should develop assays to determine the specificity of ADA for the 182

Contains Nonbinding Recommendations
Draft — Not for Implementation
6
protein component as well as the modification to the therapeutic protein product. Also see 183
sections IV.K.4 and 5. 184
185
B. Assay Cut Point 186
187
The cut point of the assay is the level of response of the assay that defines the sample response as 188
positive or negative. Information specific to establishing the cut point for the respective assay 189
types is provided in sections V and VI. Establishing the appropriate cut point is critical to 190
ensuring acceptable assay sensitivity. 191
192
The cut point of the assay can be influenced by a myriad of interfering factors, such as pre-193
existing antibodies, rheumatoid factor (RF), human anti-mouse antibodies, and the levels of 194
product-related material or homologous proteins in the matrix. These factors should be 195
considered early on in assay development when defining the cut point. Because samples from 196
different target populations and disease states may have components that can cause the 197
background signal from the assay to vary, different cut points may be needed for discrete 198
populations being studied. 199
200
The cut point should be statistically determined using samples from treatment-naïve subjects.
13
201
By performing replicate assay runs with these samples, the variability of the assay can be 202
estimated. During assay development, a small number of samples may be used to estimate the 203
cut point. This may be done with as few as 5–10 samples from treatment-naïve subjects. 204
205
The specific approach employed to determine the cut point will depend on various factors. 206
Specifically, because the cut point should identify any samples that produce a signal beyond that 207
of the variability of the assay, the sponsor should consider the impact of statistically determined 208
outlier values as well as true-positive samples when establishing the cut point. The sponsor 209
should provide justification for the removal of any data points, along with the respective method 210
used to determine their status as outliers. Positive values and samples may derive from non-211
specific serum factors or the presence of pre-existing antibodies in patient samples (Ross, 212
Hansen, et al. 1990; Turano, Balsari, et al. 1992; Coutinho, Kazatchkine, et al. 1995; Caruso and 213
Turano 1997; van der Meide and Schellekens 1997; Boes 2000). Although pre-existing 214
antibodies to a variety of endogenous proteins are present in healthy individuals, these can be 215
much higher in some disease states. The sponsor should identify those samples with pre-existing 216
antibodies, for example, through immunodepletion approaches, and remove them from the cut 217
point analysis. If the presence of pre-existing antibodies is a confounding factor, it may be 218
necessary to assign positive responses or a cut point based on the difference between individual 219
patient results before and after exposure. It is possible to arrive at a reasonable value to define 220
assay cut point through careful design consideration, such as utilizing the minimal required 221
dilution (MRD) of the sample, removing statistical outliers from analyses, minimizing the impact 222
13
Treatment-naïve subjects could be healthy individuals or a patient population not exposed to therapeutic protein
product, depending on the stage of assay development or validation and on the availability of samples. Sponsors
should provide justification for the appropriateness of the samples used.

Contains Nonbinding Recommendations
Draft — Not for Implementation
7
of interfering factors, improving assay drug tolerance, and using an approach to account for pre-223
existing antibodies. 224
225
C. Sensitivity 226
227
1. Assay Sensitivity 228
229
The sponsor should determine the sensitivity of the assay to have confidence when reporting 230
immunogenicity rates. Assay sensitivity represents the lowest concentration at which the 231
antibody preparation consistently produces either a positive result or readout equal to the cut 232
point determined for that particular assay.
14
FDA recommends that screening and confirmatory 233
ADA assays achieve a sensitivity of at least 100 nanograms per milliliter (ng/mL). Although 234
traditionally FDA has recommended sensitivity of at least 250–500 ng/mL, recent data suggest 235
that concentrations as low as 100 ng/mL may be associated with clinical events (Plotkin 2010; 236
Zhou, Hoofring, et al. 2013). However, it is understood that neutralization assays may not 237
always achieve that level of sensitivity. 238
239
The assays should have sufficient sensitivity to enable detection of low levels of ADA before the 240
amount of ADA reaches levels that can be associated with altered pharmacokinetic, 241
pharmacodynamic, safety, or efficacy profiles. Because assessment of patient antibody levels 242
will occur in the presence of biological matrix, testing of assay sensitivity should be performed 243
with the relevant dilution of the same biological matrix (e.g., serum or plasma, with the same 244
anticoagulant as the diluent, from the target population). The final sensitivity should be 245
expressed as mass of antibody detectable/mL of undiluted matrix. Therefore, assay sensitivity 246
should be reported after factoring in the MRD. Assay sensitivity should not be reported as titer. 247
During development, sensitivity should be assessed using both individual as well as pooled 248
samples from treatment-naïve subjects so that the suitability of the negative control can be 249
established. 250
251
Assay sensitivity should be determined by testing serial dilutions of a positive control antibody 252
of known concentration in pooled negative control matrix. The dilution series should be no 253
greater than two- or threefold, and a minimum of five dilutions should be tested. Alternatively, 254
sensitivity can be calculated by interpolating the linear portion of the dilution curve to the assay 255
cut point. As noted previously, assay sensitivity should be reported in mass units per volume of 256
undiluted matrix. 257
258
A purified preparation of antibodies specific to the therapeutic protein product should be used to 259
determine the sensitivity of the assay so that assay sensitivity can be reported in mass units/mL 260
of matrix. Antibodies used to assess sensitivity can take the form of affinity purified polyclonal 261
preparations or monoclonal antibodies (mAb). 262
263
A low positive system suitability control containing a concentration of ADA slightly above the 264
sensitivity of the assay should be used to ensure that the sensitivity of the assay is consistent 265
14
See the USP General Chapter 1106 Immunogenicity Assays – Design and Validation of Immunoassays to Detect
Anti-Drug Antibodies for a discussion on Relative Sensitivity.

Contains Nonbinding Recommendations
Draft — Not for Implementation
8
across assay runs. The low positive system suitability control should be designed to fail in 1% of 266
the runs (see section IV.I.1). 267
268
2. Drug Tolerance 269
270
Therapeutic protein product or the endogenous counterpart present in the serum may interfere 271
with the sensitivity of the assay. Specifically, complexes formed between ADA and the 272
therapeutic protein product, also called ADA-drug complexes, that prevent detection of ADA in 273
the test format can form if product-related materials are present in the test sample. This is 274
because ADA assays are generally designed to detect uncomplexed ADA. The assessment of 275
assay sensitivity in the presence of the expected levels of interfering therapeutic protein product, 276
also known as the assay’s drug tolerance, is critical to understanding the suitability of the method 277
for detecting ADA in dosed patients.
15
FDA recommends that the sponsor examine assay drug 278
tolerance early in assay development. The sponsor may examine drug tolerance by deliberately 279
adding different known amounts of purified ADA into individual ADA-negative control samples 280
in the absence or presence of different quantities of the therapeutic protein product under 281
consideration and determining quantitatively whether the therapeutic protein product interferes 282
with ADA detection. Results obtained in the absence and presence of different quantities of the 283
therapeutic protein product under consideration should be compared. There should be a 284
relationship between the quantity of antibody and the amount of therapeutic protein product 285
required for a specified degree of inhibition. Data from pharmacokinetic studies may be useful 286
in establishing optimal sample collection times. Acid dissociation pretreatment or other 287
approaches may be used to disrupt circulating ADA-drug complexes, which may lead to 288
increased assay drug tolerance. Interference from the therapeutic protein product can be 289
minimized if the sponsor collects patient samples at a time when the therapeutic protein product 290
has decayed to a level where it does not interfere with assay results. 291
292
D. Specificity and Selectivity 293
294
Demonstrating assay specificity and selectivity is critical to the interpretation of immunogenicity 295
assay results. Specificity refers to the ability of a method to detect ADA that bind the therapeutic 296
protein product but not assay components such as surfaces or reagents. The assays should 297
exclusively detect the target analyte, in this case the ADA.
16
The selectivity of an ADA assay is 298
its ability to identify therapeutic protein product-specific ADA in a matrix such as serum or 299
plasma that may contain potential interfering substances. Assay results may be affected by 300
interference from the matrix or from on-board therapeutic protein product.
17
Lack of assay 301
specificity or selectivity can lead to false-positive results, which could obscure relationships 302
between ADA response and clinical safety and efficacy measures. Demonstrating the specificity 303
15
See the USP General Chapter 1106 Immunogenicity Assays – Design and Validation of Immunoassays to Detect
Anti-Drug Antibodies.
16
See the USP General Chapter 1106 Immunogenicity Assays – Design and Validation of Immunoassays to Detect
Anti-Drug Antibodies.
17
See the USP General Chapter 1106 Immunogenicity Assays – Design and Validation of Immunoassays to Detect
Anti-Drug Antibodies.
Contains Nonbinding Recommendations
Draft — Not for Implementation
9
and selectivity of antibody responses to mAb, Fc-fusion protein, and Ig-fusion proteins poses 304
particular challenges because of the high concentration of Ig in human serum. The sponsor 305
should clearly demonstrate that the assay method specifically detects anti-mAb and not the mAb 306
product itself, non-specific endogenous antibodies, or antibody reagents used in the assay. 307
Similarly, for patient populations with a high incidence of RF, the sponsor should demonstrate 308
that RF does not interfere with the detection method. Host cell proteins and other product-309
related impurities may interfere with demonstrating the assay specificity and selectivity as well. 310
311
A straightforward approach to addressing specificity and selectivity is to demonstrate that 312
binding can be blocked by soluble or unlabeled purified therapeutic protein product. One 313
approach is to incubate positive and negative control antibody samples with the purified 314
therapeutic protein product or its components under consideration. Inhibition of signal in the 315
presence of the relevant therapeutic protein product or its components demonstrates that the 316
response is specific and selective. For responses to mAb products, inclusion of another mAb 317
with the same Fc but different variable region can be critical. For responses to other proteins, an 318
unrelated protein of similar size and charge can be used. If the assay is specific and selective for 319
the protein in question, generally the addition of that protein in solution should reduce the 320
response to background or the cut point, whereas the addition of an unrelated protein of similar 321
size and charge should have no effect. Conversely, addition of the protein in question should 322
have little effect on antibodies specific to an unrelated protein. Selectivity should further be 323
evaluated by performing recovery studies, in which positive control antibodies are spiked into 324
matrix at defined concentrations, and the positive control antibody signal is compared to that 325
obtained from antibody spiked into assay buffer alone. 326
327
1. Matrix Interference 328
329
An important consideration is how interference from the assay matrix, which is composed of the 330
sample and the diluent, can affect assay performance. Components in the matrix other than 331
therapeutic protein product can interfere with assay results. For example, different 332
anticoagulants used during sample collection may have different effects in the assay, potentially 333
affecting the assay sensitivity and linearity. Sponsors should evaluate different salt anticoagulant 334
sample collection solutions for their effect on assay results. 335
336
Endogenous and exogenous components in serum or plasma may influence assay results, and it 337
is usually necessary to dilute patient samples for testing to minimize such effects. The sponsor 338
should examine the effect of such interferents by performing spike-and-recovery studies. The 339
sponsor should define the dilution factor that will be used for preparation of patient samples 340
before performing validation studies assessing potential interference of this matrix on assay 341
results (see section IV.D.2 on MRD). 342
343
Buffer components that are chemically related to the therapeutic protein product may also 344
interfere in the assay. For example, polysorbate is chemically similar to polyethylene glycol 345
(PEG) and therefore may interfere in the detection of anti-PEG antibodies. The chemical 346
composition of the buffer should be carefully considered during assay development. 347
348

Contains Nonbinding Recommendations
Draft — Not for Implementation
10
The sponsor may examine matrix interference by spiking different known amounts of purified 349
ADA into the assay buffer in the absence or presence of different matrix components. 350
Comparing the recovery of ADA in buffer alone with that in the matrix can provide input on the 351
degree of interference from matrix components. Furthermore, such analysis may guide decisions 352
on the MRD recommended for sample testing. In addition, the sponsor should examine other 353
parameters affecting patient samples, such as hemolysis, lipemia, presence of bilirubin, and 354
presence of concomitant medications that a patient population may be using. Samples that have 355
very high antibody titers may need additional testing, such as with different dilutions of the 356
competing product in the confirmatory assay, to ensure their identification. 357
358
2. Minimal Required Dilution 359
360
Matrix components can contribute to non-specific signal if undiluted, thereby obscuring positive 361
results. Therefore, there is frequently a need to dilute patient samples to maintain a reasonable 362
ability to detect ADA (sensitivity). Ideally, the MRD is the sample dilution that yields a signal 363
close to that of the assay diluent and allows for the highest signal-to-noise ratio. MRD typically 364
ranges from 1:5 to 1:100. 365
366
FDA recommends that the sponsor determine the MRD from a panel of appropriate number of 367
samples from treatment-naïve subjects. Determination of MRD usually involves serially diluting 368
treatment-naïve ADA-negative samples, as well as testing known amounts of purified antibody 369
(at high, medium, and low concentrations) in serially diluted matrix in comparison to the same 370
amount of antibody in buffer. This ensures a reasonable signal-to-noise ratio throughout the 371
range of the assay. The MRD should be calculated using at least 10 individual serum samples; 372
the appropriate number of samples will depend on various factors, including the variability of the 373
individual samples. 374
375
Although the MRD ultimately selected by the sponsor will depend on the assay design and 376
patient population, FDA recommends that dilutions not exceed 1:100. Higher dilution may 377
result in the spurious identification of a negative response when patients may actually possess 378
low levels of therapeutic protein product-specific antibodies, the occurrence of which can be 379
related to significantly altered pharmacokinetics, pharmacodynamics, safety, or efficacy profiles. 380
However, in some instances greater initial dilutions may be required, and the overall effect of 381
such dilutions on assay sensitivity and immunogenicity risk assessment should be considered. 382
383
E. Precision 384
385
Precision is a measure of the variability in a series of measurements for the same material run in 386
a method. Results should be reproducible within and between assay runs to assure adequate 387
precision.
18
Demonstrating assay precision is critical to the assessment of ADA because assay 388
variability is the basis for determining the cut points and ensuring that low positive samples are 389
18
For more information on precision, see the guidance for industry Bioanalytical Method Validation. Also see the
USP General Chapter 1106 Immunogenicity Assays – Design and Validation of Immunoassays to Detect Anti-Drug
Antibodies.

Contains Nonbinding Recommendations
Draft — Not for Implementation
11
detected as positive. To provide reliable estimates, the sponsor should evaluate both intra-assay 390
(repeatability) and inter-assay (intermediate precision) variability of assay responses. 391
392
F. Reproducibility 393
394
Reproducibility is an important consideration if an assay will be run by two or more independent 395
laboratories during a study, and a sponsor should establish the comparability of the data 396
produced by each laboratory.
19
In addition, the assays should have the same precision between 397
different laboratories under the established assay operating conditions (for example, using the 398
same instrument platform). 399
400
G. Robustness and Sample Stability 401
402
Assay robustness is an indication of the assay’s reliability during normal usage
20
and is assessed 403
by the capacity of the assay to remain unaffected by small but deliberate variations in method 404
and instrument performance that would be expected under relevant, real-life circumstances in 405
routine laboratory practice. For example, changes in temperature, incubation times, or buffer 406
characteristics, such as pH and salt concentration, can all impact assay results. The complexity 407
of bioassays makes them particularly susceptible to variations in assay conditions, and it is 408
essential to evaluate and optimize parameters such as cell passage number, incubation times, and 409
culture media components. The sponsor should examine robustness during the development 410
phase, and if small changes in specific steps in the assay affect results, specific precautions 411
should be taken to control their variability. FDA recommends storing patient samples in a 412
manner that preserves antibody reactivity at the time of testing. FDA recommends that the 413
sponsor avoid freeze-thaw cycles because freezing and thawing patient samples may also affect 414
assay results. However, studies evaluating long-term stability of positive control antibodies may 415
be useful.
21
416
417
H. Selection of Format 418
419
A number of different assay formats and instrumentation are available that can be employed for 420
detection of ADA. These include, but are not limited to, direct binding assays, bridging assays, 421
and equilibrium binding assays. Each assay format has advantages and disadvantages, including 422
rapidity of throughput, sensitivity, selectivity, dynamic range, ability to detect various Ig 423
isotypes, ability to detect rapidly dissociating antibodies, and availability of reagents. One of the 424
major differences between each of these assay formats is the number and vigor of washes, which 425
19
For more information on reproducibility, see the guidance for industry Bioanalytical Method Validation. Also see
the USP General Chapter 1106 Immunogenicity Assays – Design and Validation of Immunoassays to Detect Anti-
Drug Antibodies, the USP General Chapter 1225 Validation of Compendial Procedures, and the ICH guidance for
industry Q2B Validation of Analytical Procedures: Methodology.
20
For more information on robustness, see the ICH guidance for industry Q2B Validation of Analytical Procedures:
Methodology. Also see the USP General Chapter 1106 Immunogenicity Assays – Design and Validation of
Immunoassays to Detect Anti-Drug Antibodies.
21
For more information on stability studies, see the guidance for industry Bioanalytical Method Validation.

Contains Nonbinding Recommendations
Draft — Not for Implementation
12
can have an effect on assay sensitivity. All assays should be evaluated for their ability to detect 426
rapidly dissociating antibodies such as IgM, which are common in early immune responses. 427
Failure to detect such antibodies in early immune responses to therapeutic protein products may 428
result in under-detection of true-positive antibody samples. Epitope exposure is also important 429
to consider because binding to plastic or coupling to other agents, such as reporters (i.e., 430
fluorochromes, enzymes, or biotin), can result in conformational changes of the antigen that can 431
obscure, expose, modify, or destroy relevant antibody binding sites on the therapeutic protein 432
product in question. 433
434
I. Selection of Reagents 435
436
Many components of the assays for ADA detection may be standard or obtained from 437
commercial sources, for example, commercially available reagents such as Protein A/G coated 438
resins used in the depletion approach for confirmatory assays. Other components, however, 439
including positive control antibodies, negative controls, and system suitability controls, may 440
need to be generated specifically for the particular assay. 441
442
1. Development of Positive Control Antibodies 443
444
Sponsors may use different or the same positive control antibodies to establish and monitor 445
system suitability during routine assessment of assay performance, as well as to determine that 446
the assay employed is fit for purpose. For system suitability controls, a positive control 447
antibody, either mono- or polyclonal, used at concentrations adjusted to control the cut point and 448
dynamic range levels, may be suitable. 449
450
Positive control antibodies frequently are generated by immunizing animals in the absence or 451
presence of adjuvants. FDA recommends that positive control antibodies generated by 452
immunizing animals be affinity purified using the therapeutic protein product. This approach 453
enriches the polyclonal antibody preparation for ADA, which enables a more accurate 454
interpretation of sensitivity assessment results. The selection of animal species when generating 455
positive control antibodies should be carefully considered. For example, if an anti-human Ig 456
reagent will be used as a secondary reagent to detect patient antibodies, the positive control 457
antibodies and quality control (QC) samples should be detectable by that same reagent. When 458
the positive control antibody is not detectable by that same reagent, an additional secondary 459
reagent to detect the positive control antibody may be needed. In those cases, an additional 460
positive control antibody for the secondary reagent used to detect human antibodies should be 461
implemented to ensure that the reagent performs as expected. In some instances, the sponsor 462
may be able to generate a positive control antibody from patient samples.
22
Although such 463
antibodies can be very valuable, such samples are generally not available in early trials. 464
Alternatively, individual mAb or panels of mAb may be used for positive control antibodies. 465
Sponsors should discuss with FDA alternative approaches to assay development and validation 466
in the rare event that a sponsor is not able to generate a positive control antibody. 467
468
22
Proper informed consent from patients is needed and should be planned ahead of time.
Contains Nonbinding Recommendations
Draft — Not for Implementation
13
Ideally, the positive control antibody used to determine assay applicability for the purpose of the 469
respective assay should reflect the anticipated immune response that will occur in humans. For 470
therapeutic mAb, the sponsor should give special consideration to the selection of a positive 471
control antibody for the assay. When animals are immunized with a chimeric, humanized, or 472
human mAb to develop a positive control antibody, the humoral response may be against the 473
human Fc and not the variable region of the molecule. Such positive control antibodies may not 474
be relevant for the anticipated immune response in patients where the response is primarily 475
directed to the antigen-binding regions. 476
477
Once a source of a positive control antibody has been identified, the sponsor should use that 478
source to assess assay performance characteristics such as sensitivity, selectivity, specificity, and 479
reproducibility. FDA recommends that sponsors generate and reserve positive control antibody 480
solution for use as a quality or system suitability control. For assay development and validation, 481
dilutions should be representative of a high, medium, and low value in the assay. This is needed 482
even for qualitative assays to understand whether assay performance is acceptable across a broad 483
range of antibody concentrations. Although high- and low-value QC samples should be used, 484
medium-value QC samples for detection of ADA are generally not needed for monitoring system 485
suitability during routine assessment of assay performance. 486
487
2. Development of Negative Controls 488
489
For negative control samples, it is recommended that when possible, the control population 490
should have the same disease condition. The control samples should represent a similar gender, 491
age, and concomitant medications so that the sample matrix is representative of the study 492
population. Similarly, control samples should be collected and handled in the same manner as 493
study samples with respect to, for example, type of anticoagulant used, sample volume, and 494
sample preparation and storage, because these pre-analytical variables can impact the 495
performance of control samples in the assay. It is frequently the case that such control samples 496
are not available for use during development or pre-study validation exercises. In those 497
situations, it is acceptable to use purchased samples or samples from healthy donors, but 498
important parameters of assay performance such as cut point, sensitivity, and selectivity should 499
be confirmed when samples from treatment-naïve subjects from the appropriate target population 500
become available. 501
502
FDA recommends that the sponsor establish a negative control for validation studies and patient 503
sample testing. In this regard, a pool of sera from an appropriate number of treatment-naïve 504
subjects can serve as a useful negative control. Importantly, the value obtained for the negative 505
control should be below but close to the cut point determined for the assay in the patient 506
population being tested. Negative controls that yield values far below the mean value derived 507
from individual serum samples used to establish the cut point may not be useful in ensuring 508
proper assay performance. 509
510
3. Detection Reagent Consideration 511
512
The selection of a suitable detection reagent (i.e., reporter) depends on the assay format chosen. 513
It is critical to minimize the non-specific signal from the detection reagent. The detection 514
Contains Nonbinding Recommendations
Draft — Not for Implementation
14
reagent chosen should have the adequate sensitivity required for the particular assay. These 515
factors should be taken into consideration when deciding on the detection reagent. 516
517
4. Controlling Non-Specific Binding 518
519
Every reagent, from the plastic of the microtiter plates to the developing agent, can affect assay 520
sensitivity and non-specific binding. One of the most critical elements is the selection of the 521
proper assay buffer and blocking reagents used to prevent non-specific binding to the solid 522
surface. The sponsor should carefully consider the number and timing of wash steps as well as 523
the detergents added to the assay buffer (i.e., blocking or wash buffer) to reduce background 524
noise, but still maintain sensitivity. A variety of proteins can be used as blocking reagents to 525
provide acceptable signal-to-noise ratio. However, these proteins may not all perform 526
equivalently in specific immunoassays. For example, they may not bind well to all types of solid 527
phases or may show unexpected cross-reactivity with the detecting reagent. Therefore, the 528
sponsor may need to test several blocking agents to optimize assay performance. Moreover, 529
including uncoated wells is insufficient to assess non-specific binding. Rather, determining the 530
capacity of ADA to bind to an unrelated protein of similar size and charge that may be present in 531
the sample may prove to be a better test of binding specificity. 532
533
J. Reporting Results for Qualitative and Semi-Quantitative Assays 534
535
Several approaches may be used to report positive antibody responses, and the appropriateness of 536
the approach used should be evaluated on a case-by-case basis. The most common approach is 537
qualitative, with patients reported as having a positive or negative antibody response. 538
539
For patients who are confirmed to be ADA positive, determining antibody levels can be 540
informative because it allows for the stratified assessment of ADA levels and their impact on 541
safety and efficacy. These relationships may not be elucidated unless ADA levels are 542
determined. Positive antibody responses may be reported as a titer (e.g., the reciprocal of the 543
highest dilution that gives a readout at or just above the cut point of the assay), when appropriate. 544
The MRD should be factored in the calculations of titers and provided when reporting titers. 545
Reporting levels of antibodies in terms of titers is appropriate and generally understood by the 546
medical community. Values may also be reported as amount of mass units of therapeutic protein 547
product neutralized per volume serum with the caveat that these are arbitrary in vitro assay units 548
and cannot be used to directly assess therapeutic protein product availability in vivo. 549
550
Unless the assay method used allows for independent determination of mass, antibody levels 551
reported in mass units are generally not acceptable because they are based on interpolation of 552
data from standard curves generated with a positive control antibody, and parallelism between 553
the reference standard and test article cannot be assumed. Thus, FDA does not consider it 554
necessary nor desirable for the sponsor to report patient antibody results in terms of mass units 555
unless (1) the results are determined by quantitative means or (2) a universally accepted and 556
accessible source of validated antibody is available as a control and parallelism between the 557
dilution curves of the control antibody and patient samples has been demonstrated. Furthermore, 558
even if parallelism is demonstrated, because the reference standard and test articles are likely to 559
Contains Nonbinding Recommendations
Draft — Not for Implementation
15
contain different populations of antibodies, the absolute mass units cannot be calculated. 560
Therefore, FDA understands that the mass units reported are relative rather than absolute values. 561
562
K. Other Considerations for Assay Development 563
564
A myriad of factors can affect the assessment of antibody levels, such as patient sample 565
variability, therapeutic protein product-dose response of the cells used to generate the standard 566
curve in a cell-based neutralization bioassay, affinity and avidity of the ADA, and concentration 567
of competing product in confirmatory assays. Accounting for such factors is important to 568
understand and analyze assay variability and avoid errors. Common factors that should be 569
considered include the following: 570
571
1. Pre-Existing Antibodies 572
573
A growing body of evidence in the medical literature suggests that B-cells and T-cells with 574
specificity for a number of self-proteins exist naturally and may even be heightened in some 575
disease states, such as in patients subjected to cytokine therapy or suffering from a variety of 576
immunological or immunoinflammatory diseases (Coutinho, Kazatchkine, et al. 1995; van der 577
Meide and Schellekens 1997; Boes 2000). For example, antibodies to interferon can be found in 578
normal individuals (Ross, Hansen, et al. 1990; Turano, Balsari, et al. 1992; Caruso and Turano 579
1997). Less surprisingly, subjects may have pre-existing antibodies to foreign antigens, such as 580
bacterial products, most likely as a result of exposure to the organism or cross-reactivity. Pre-581
existing antibodies may have clinical effects and may affect the efficacy of the therapeutic 582
protein product being tested. An alternative to the qualitative screening assay approach may be 583
needed to assess the quantity and quality of ADA when pre-existing antibodies are present. For 584
example, testing samples for an increase in ADA using a semi-quantitative assay type such as a 585
titering assay (see sections V.C and VI.D) can provide information on the impact of a therapeutic 586
protein product on product immunogenicity that is not provided by a qualitative assay. 587
588
2. Rheumatoid Factor 589
590
Measuring immune responses to therapeutic protein products that possess Ig tails, such as mAb 591
and Fc-fusion proteins, may be particularly difficult when RF is present in serum or plasma. RF 592
is generally an IgM antibody that recognizes IgG, although other Ig specificities have been 593
noted. Consequently, RF will bind Fc regions, making it appear that specific antibody to the 594
therapeutic protein product exists. Several approaches for minimizing interference from RF have 595
proven useful, including treatment with aspartame (Ramsland, Movafagh, et al. 1999) and 596
careful optimization of reagent concentrations so as to reduce background binding. When 597
examining immune responses to Fc-fusion proteins in clinical settings where RF is present, FDA 598
recommends developing an assay specific for the non-Fc region of the proteins. 599
600
3. Monoclonal Antibodies 601
602
Some special considerations pertain to the detection of antibodies against mAb. Animal-derived 603
mAb, particularly those of rodent origin, are expected to be immunogenic with the immune 604
response directed against the whole mAb molecule. In the early days of the therapeutic mAb 605
Contains Nonbinding Recommendations
Draft — Not for Implementation
16
industry, this was a key reason for the failure of clinical trials (Kuus-Reichel, Grauer, et al. 606
1994). 607
608
Technologies reducing the presence of non-human sequences in mAb, such as chimerization and 609
humanization, have led to a dramatic reduction but not elimination of immunogenicity. In these 610
cases, the immune responses are directed largely against the variable regions of the mAb 611
(Harding, Stickler, et al. 2010; van Schouwenburg, Kruithof, et al. 2014). As immune responses 612
against the variable regions of human mAb are anticipated, FDA does not expect that the use of 613
human mAb will further reduce immunogenicity by a significant margin. The assays that can 614
detect the reactivity against variable regions are considered more appropriate to evaluate the 615
potential impact of antibodies against mAb-based therapeutics in patients. However, engineering 616
of Fc portion (e.g., modification of the levels of afucosylation) in human antibodies may affect 617
immunogenicity. Many of these concerns also pertain to Fc-fusion proteins containing a human 618
Fc region. 619
620
4. Conjugated Proteins 621
622
Because antibody-drug conjugates (ADCs) are antibodies conjugated with small molecule drugs, 623
they represent a classic hapten-carrier molecule. Therefore, the immunogenicity assays should 624
be able to measure the responses to all components of the ADC therapeutic protein product, 625
including the antibody, linker-drug, and new epitopes that may result from conjugation. When 626
ADCs need to be labeled for immunogenicity assays, the conjugation should be performed 627
carefully because ADCs are already modified. The potential for increased hydrophobicity of the 628
labeled molecules may cause aggregation, and therefore the stability and solubility of these 629
capture reagents should be adequately characterized. 630
631
5. Products With Multiple Functional Domains 632
633
Some proteins possess multiple domains that function in different ways to mediate clinical 634
efficacy. An immune response to one domain may inhibit a specific function while leaving 635
others intact. Examination of immune responses to therapeutic protein products with multiple 636
functional domains may require development of multiple assays to measure immune responses to 637
different domains of the molecules. 638
639
640
V. ASSAY DEVELOPMENT 641
642
Information specific to development of respective assay types is provided in sections A through 643
D below. These sections supplement information relevant to all assay types provided in 644
section IV. 645
646
A. Development of Screening Assay 647
648
Based on the multi-tiered approach discussed previously in section IV.A, the first assay to be 649
employed for detection of ADA should be a highly sensitive screening assay that detects low- 650
and high-affinity ADA. Approximately 10 individual samples may be used to estimate the cut 651
Contains Nonbinding Recommendations
Draft — Not for Implementation
17
point early in assay development; however, this may need to be adjusted when treatment-naïve 652
samples from the target population become available. A low but defined false-positive rate is 653
desirable for the initial screening assay because it maximizes detection of true positives. 654
Subsequent assays can be employed to exclude false-positive results when determining the true 655
incidence of immunogenicity. 656
657
B. Development of Confirmatory Assay 658
659
Because the screening assay is designed to broadly detect the presence of antibodies that bind 660
product in serum samples with a defined false-positive rate, FDA recommends that the sponsor 661
develop assays to confirm the binding of antibodies that are specific to the therapeutic protein 662
product. Implementation of a suitable confirmatory assay is important to prevent data on ADA 663
false-positive patients from confounding the analyses of the impact of ADA on safety and 664
efficacy. 665
666
1. Selection of Format for Confirmatory Assay 667
668
It is expected that the selected confirmatory assay will be at least as sensitive as the screening 669
assay but have higher specificity and at least as good selectivity in order to identify any false-670
positive samples. The method and instrument platform selected may be similar to or different 671
from those used for the screening assay. Frequently, both screening and confirmatory assays use 672
the same method and instrument platform. In such cases, the sensitivity of each assay will need 673
to be determined in mass units and confirmed using system suitability controls to ensure that the 674
assay is sensitive to the presence of binding antibody. When using a binding competition assay, 675
the concentration of competing product should be optimized to confirm the presence of 676
antibodies throughout and above the range of the assay. 677
678
2. Cut Point of Confirmatory Assay 679
680
If a competitive inhibition format is selected, a recommended approach to determining the cut 681
point uses the data from the binding of antibody-negative treatment-naïve patient samples in the 682
presence of the competitor, which is usually the therapeutic protein product. In this case, the 683
amount of therapeutic protein product used to establish the cut point should be the same as the 684
amount of therapeutic protein product that will be used as a competitive inhibitor in the assay. 685
However, this approach may not be appropriate when dealing with samples where pre-existing 686
antibodies are present in the treatment-naïve population. In those cases, the sponsor should 687
exclude true positives from the cut point assessment. In rare cases when baseline negative 688
samples are not available, sponsors may evaluate changes in titer or use an orthogonal method to 689
confirm samples that screen positive. 690
691
C. Development of Titering Assay 692
693
1. Titer Determination 694
695
Titers are defined as the maximal dilution where a sample gives a value above the screening cut 696
point. Titers are often informative and can be linked to clinical impact of the ADA. Titering 697
Contains Nonbinding Recommendations
Draft — Not for Implementation
18
assays can be particularly informative when patients have pre-existing antibodies. Titering 698
assays most often are performed using the same platform as the screening assay. Sera are tested 699
in sequential dilutions. Alternatively, titer may be determined by extrapolating the dilution to the 700
assay cut point using the linear portion of the dose response curve. 701
702
2. Cut Point of Titering Assay 703
704
When patients have pre-existing ADA, treatment-boosted ADA responses may be identified by 705
post-treatment increases in titer. A cut point for defining the treatment-emergent or boosted 706
responses is needed. Frequently this cut point is determined as a titer that is two dilution steps 707
greater than the pre-treatment titer, when twofold dilutions are used to determine the titer. If titer 708
is established by extrapolating the dilution curve to the assay cut point, treatment-emergent 709
responses may be determined using estimates of assay variability. 710
711
D. Development of Neutralization Assay 712
713
In vitro neutralization assays provide an indication of the potential of the ADA to inhibit the 714
biological activity of the product. Such NAb can interfere with the clinical activity of a 715
therapeutic protein product by preventing the product from reaching its target or by interfering 716
with receptor-ligand interactions. The testing method selected to assess neutralizing potential for 717
ADA-positive samples should be based on the mechanism of action of the therapeutic protein 718
product. 719
720
1. Selection of Format for Neutralization Assay 721
722
Two formats of assays have been used to measure NAb activity: cell-based bioassays and non-723
cell-based competitive ligand-binding assays. Selection of the appropriate assay format depends 724
on various factors. These factors include, but are not limited to, the mechanism of action of the 725
therapeutic protein product, its ability to reflect the in vivo situation most closely, and the 726
selectivity, sensitivity, precision, and robustness of the assay. FDA recommends that 727
neutralization assays use a cell-based bioassay format depending on the therapeutic protein 728
product’s mechanism of action because, frequently, cell-based bioassays more closely reflect the 729
in vivo situation and therefore provide more relevant information than ligand-binding assays. 730
Because the cell-based bioassays are often based on the product’s potency, historically the 731
format of these assays has been extremely variable. The choice and design of potency bioassays 732
are generally based on a cell line’s ability to respond to the product in question and the potency 733
bioassay’s relevance to the therapeutic protein product’s mechanism of action. 734
735
Contains Nonbinding Recommendations
Draft — Not for Implementation
19
736
The cellular responses measured in these bioassays are numerous and can include outcomes such 737
as phosphorylation of intracellular substrates, calcium mobilization, proliferation, and cell death. 738
In some cases, sponsors have developed cell lines to express relevant receptors or reporter 739
constructs. When therapeutic protein products directly stimulate a cellular response, the direct 740
effect of NAb on reducing bioactivity in the bioassay can be measured. When therapeutic 741
protein products indirectly impact cellular activity; for example, by blocking a receptor-ligand 742
interaction, the indirect effect of the NAb on restoring bioactivity in a bioassay can be measured. 743
Generally, bioassays have significant variability and a limited dynamic range for their activity 744
curves. Such problems can make development and validation of neutralization assays difficult. 745
746
There are cases when ligand-binding assay formats may be used. One such case is when 747
sufficiently sensitive or selective cell-based bioassays cannot be developed. Another case is 748
when the therapeutic protein product does not have a cell-based mechanism of action; for 749
example, enzyme therapeutic protein products that target serum proteins. Ligand-binding assays 750
may also be appropriate for therapeutic protein products that bind serum ligands, preventing 751
them from interacting with their receptor. However, cell-based bioassays may still be more 752
appropriate for such therapeutic protein products to demonstrate that ADA are inhibiting cellular 753
activity. Sponsors should discuss using ligand-binding assays with FDA in such cases. 754
755
2. Activity Curve of Neutralization Assay 756
757
The sponsor should carefully consider the dose response curve (product concentration versus 758
activity) before examining other elements of neutralization assay validation. Assays with a small 759
dynamic range may not prove useful for determination of neutralizing activity. Generally, the 760
neutralization assay will employ a single concentration of therapeutic protein product with a 761
single dilution of antibody. Consequently, the sponsor should choose a therapeutic protein 762
product concentration whose activity readout is sensitive to inhibition. If the assay is performed 763
at concentrations near the plateau of the dose-response curve (marked “No” in Figure 1, below), 764
it may not be possible to discern samples with low amounts of NAb. FDA recommends that the 765
neutralization assay be performed at therapeutic protein product concentrations that are on the 766
linear range of the curve (marked “Yes” in Figure 1). The assay should also give reproducible 767
results. 768
769
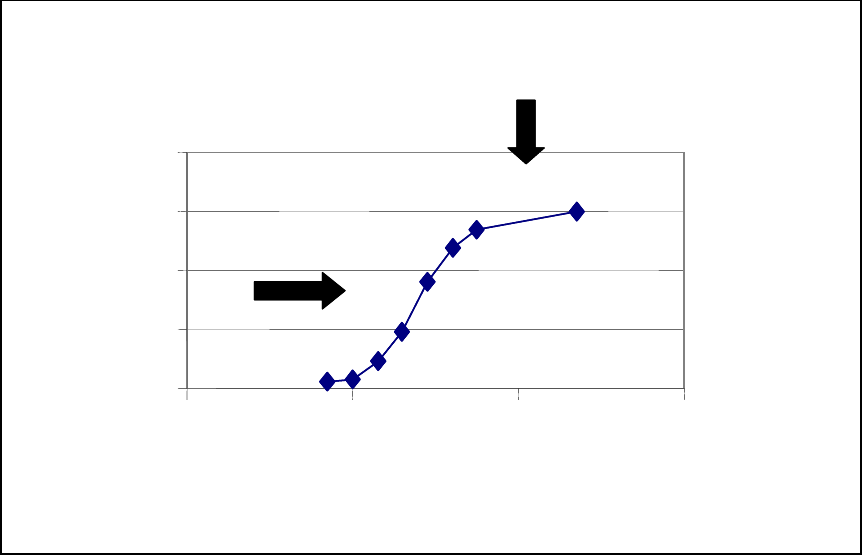
Contains Nonbinding Recommendations
Draft — Not for Implementation
20
770
Figure 1. Activity Curve for a Representative Therapeutic Protein Product 771
772
The x-axis (Concentration) indicates a concentration of the therapeutic protein product, and the 773
y-axis (Activity) indicates resultant activity; for example, the concentration of cytokine secretion 774
of a cell line upon stimulation with the therapeutic protein product. The curve demonstrates a 775
steep response to a therapeutic protein product that plateaus at approximately 300. The “No” 776
arrow indicates a concentration of a therapeutic protein product that would be inappropriate to 777
use in a single dose neutralization assay because it would represent a range of concentrations 778
where the activity induced by the therapeutic protein would be relatively insensitive to inhibition 779
by NAb. The “Yes” arrow represents a range of concentrations on the linear part of the curve 780
where the activity induced by the therapeutic protein product would be sensitive to neutralization 781
by antibody. 782
783
3. Considerations for Matrix Interference for Neutralization Assay 784
785
The matrix can cause interference with neutralization assays, particularly as serum or plasma 786
components may enhance or inhibit the activity of a therapeutic protein product in bioassays. 787
For example, sera from patients with particular diseases may contain elevated levels of one or 788
more cytokines that might serve to activate cells in the bioassay and obscure the presence of 789
NAb by increasing the response to the original stimulatory factor or therapeutic protein product. 790
Therefore, the sponsor should understand matrix effects in these assays. Approaches such as 791
enriching for ADA from serum or plasma samples may be appropriate for these types of 792
situations. However, this approach may result in the loss of NAb, and consequently will require 793
careful examination and validation by the sponsor. 794
100
200
300
400
100 10000 1000000
Concentration
{
{
Yes
No
Activity
100
200
300
400
100 10000 1000000
Concentration
{
{
Yes
No
Activity
Contains Nonbinding Recommendations
Draft — Not for Implementation
21
795
The concentration of therapeutic protein product employed in the neutralization assay has a 796
critical impact on assay sensitivity. FDA recognizes that although the use of low concentrations 797
of therapeutic protein product may lead to a neutralization assay that is more sensitive to 798
inhibition by antibodies, very low concentrations of therapeutic protein product may result in 799
poor precision of the assay. Also see section IV.D.1 for general information on matrix 800
interference. 801
802
4. Cut Point of Neutralization Assay 803
804
Determination of assay cut point has historically posed a great challenge for neutralization 805
assays. As with all assays, the cut point should be determined based on the assay variability 806
established using samples from treatment-naïve subjects. If neutralization assays are performed 807
on samples that tested positive in screening and confirmatory assays, a 1% false-positive rate is 808
acceptable. If neutralization assays are used for screening, a 5% false-positive rate should be 809
used (see section VI.B.2). If the degree of sample variation makes it difficult to assess NAb 810
activity, other approaches may be considered but should be discussed with FDA before 811
implementation. Alternatively, exploring other assay formats that lead to less variability and 812
provide a more accurate assignment of cut point may be necessary. Also see section IV.B for 813
general information on assay cut point. 814
815
5. Additional Considerations for Neutralization Assay 816
817
Because neutralization assays are most commonly performed only on samples that are confirmed 818
to have antigen-specific ADA, confirmatory approaches are not usually necessary. However, 819
because of the complexity of bioassays, confirmation of assay specificity may be useful in 820
determining whether patients have mounted a true NAb response. The sponsor should consider 821
the following approaches: 822
823
a. Unrelated inhibitory molecules may cause neutralizing activity, and sometimes it may 824
be unclear whether the observed neutralizing activity is caused by neutralizing 825
antibodies or by other inhibitory molecules. Test results from baseline pre-exposure 826
samples may be informative. When there is concern that there is non-specific 827
inhibition, antibody depletion assays should be performed to evaluate whether the 828
neutralizing activity is truly caused by ADA and not caused by other inhibitory 829
molecules. 830
831
b. Cell lines may be responsive to multiple stimuli other than the therapeutic protein 832
product under study. In such cases, the presence of NAb can be examined in the 833
presence of the therapeutic protein product, which should be blocked by a specific 834
NAb response, versus alternative stimuli, which should not be blocked by a specific 835
NAb response. 836
837
c. Serum may contain components such as soluble receptors or endogenous product 838
counterparts that may yield false results in the neutralization assay. In such instances, 839

Contains Nonbinding Recommendations
Draft — Not for Implementation
22
adding test serum or plasma samples directly to the bioassay in the absence of 840
therapeutic protein product may be useful in understanding assay results. 841
842
843
VI. ASSAY VALIDATION 844
845
Assay validation is a process of demonstrating, by the use of specific laboratory investigations, 846
that the performance characteristics of the ADA assay employed are suitable for its intended 847
use.
23
The level of validation depends on the stage of product development and the risks of 848
consequences of immunogenicity to patients associated with the therapeutic protein product. A 849
partial validation involving assessments of assay sensitivity, specificity, and precision 850
requirements with less emphasis on robustness, reproducibility, and stability may be adequate for 851
the earlier stages of clinical development such as phase 1 and phase 2 studies. However, as a 852
scientific matter, as stated in section VI.A, fully validated assays should be used for pivotal and 853
postmarketing studies. 854
855
Information specific to validation of respective assay types is provided in sections VI.B 856
through E. These sections supplement information relevant to all assay types provided in 857
sections IV and VI.A. 858
859
A. General Considerations for Assay Validation 860
861
Samples derived from pivotal studies should be tested with fully validated assays. At the time of 862
license application, the sponsor should provide data supporting full validation of the assays. 863
Validation includes all of the procedures that demonstrate that a particular assay used for 864
quantitative measurement of ADA in a given sample is reliable and reproducible for the intended 865
use. The fundamental parameters for validation include (1) cut point, (2) sensitivity, 866
(3) specificity and selectivity, (4) precision, (5) reproducibility when relevant, and (6) robustness 867
of some assay features and stability of reagents and control samples. The acceptability of 868
clinical data generated by an assay corresponds directly to the criteria used to validate the assay. 869
870
Determination of cut point is a fundamental aspect of assay validation. If treatment-naïve 871
samples from the appropriate patient population are not available for the pre-study validation 872
exercise, alternative samples may be used. Frequently these are samples from commercial 873
sources. When alternative samples are used to determine the cut point in the validation exercise, 874
the cut point should be determined again once samples from the appropriate population (e.g. 875
treatment-naïve patients) are available. The cut point validated using the appropriate samples 876
should be used to determine whether samples are positive for ADA. 877
878
For validation of the fundamental assay parameters, FDA recommends, at the minimum, that 879
inter-assay precision be evaluated on at least 3 different days with two analysts each preparing a 880
23
See the USP General Chapter 1106 Immunogenicity Assays – Design and Validation of Immunoassays to Detect
Anti-Drug Antibodies. Also see the guidance for industry Bioanalytical Method Validation, the USP General
Chapter 1225 Validation of Compendial Procedures, and the ICH guidance for industry Q2(R1) Validation of
Analytical Procedures: Text and Methodology.

Contains Nonbinding Recommendations
Draft — Not for Implementation
23
minimum of six otherwise independent preparations of the same sample using the same 881
instrument platform and model. Intra-assay precision should be evaluated with a minimum of 882
six independent preparations of the same sample per plate independently prepared by the same 883
analyst. In cases where intra-assay or inter-assay precision has a coefficient of variance (%CV) 884
greater than 20%, sponsors should consider the need to refine the assay parameters to optimize 885
the assay precision to the extent possible or provide justification to explain why higher %CV 886
should be acceptable. Alternatively, in assays with low throughput (e.g., titer assay) when it may 887
not be possible to run six independent preparations of the same sample on a plate, intra-assay 888
precision should be evaluated with a minimum of three independent preparations of the same 889
sample per plate and at least nine total independent preparations of the same samples. Samples 890
should include negative controls and positive samples whose testing yields values in the low, 891
medium, and high levels of the assay dynamic range. The sponsor should evaluate inter-892
instrument and inter-operator precision when relevant. Assays should have comparable precision 893
between different operators under the same operating conditions. 894
895
When changes are made to a previously validated method, the sponsor should exercise judgment 896
as to how much additional validation is needed. During the course of a typical product 897
development program, a defined ADA assay may undergo modifications. Occasionally, samples 898
may need to be re-tested with the optimized validated assay; therefore, provisions should be 899
made to preserve sufficient sample volume under conditions that allow for re-testing until the 900
assays have been completely validated and evaluated by the Agency.
24
901
902
Critical method parameters, for example, incubation times and temperatures, should be validated 903
to demonstrate that the assay performs as expected within predetermined ranges for these 904
parameters. Generally, the low, middle, and high values of the allowed range are tested in the 905
validation exercise. 906
907
Additional parameters may need to be validated depending on the method (or technology) and 908
instrument platform used for the assay. For example, surface plasmon resonance assays should 909
be validated for surface stability upon regeneration, and criteria should be set for baseline 910
performance of the chip. The efficiency and stability of the labeled
25
reagents should be 911
established. The sponsor should examine robustness during the development phase, and if small 912
changes in specific steps in the assay affect results, specific precautions should be taken to 913
control their variability. 914
915
24
See the guidance for industry Bioanalytical Method Validation for different types and levels of validation. Also
see the USP General Chapter 1106 Immunogenicity Assays – Design and Validation of Immunoassays to Detect
Anti-Drug Antibodies.
25
A reagent is considered labeled if it is conjugated or fused to a moiety that will aid in its capture or visualization;
for example, conjugation to biotin, streptavidin, or a fluorochrome. Unlabeled reagent is a reagent (for example, a
drug) that is not labeled.

Contains Nonbinding Recommendations
Draft — Not for Implementation
24
B. Validation of Screening Assay 916
917
1. Sensitivity of Screening Assay 918
919
All the general considerations for assay validation discussed previously apply to validation of 920
screening assay. As noted earlier, the sensitivity is particularly important in the initial screening 921
assay because these results dictate the further analysis of the sample. 922
923
2. Cut Point of Screening Assay 924
925
The cut point should be determined statistically with a minimum of 50 samples tested on at least 926
3 different days by at least two analysts using suitable statistical methods. FDA recommends 927
that the cut point for screening assays be determined by a 90% one-sided lower confidence 928
interval for the 95
th
percentile of the negative control population (Shen, Dong, et al. 2015). This 929
will assure at least a 5% false-positive rate with a 90% confidence level. This approach 930
improves the probability of the assay identifying all patients who may develop antibodies. The 931
statistical method used to determine the cut point should be based on the statistical distribution of 932
the data. For example, the 95
th
percentile of the normal distribution is estimated by the mean 933
plus 1.645 standard deviation. Other approaches may be used for estimating 95
th
percentile, 934
including the use of median and median absolute deviation value instead of mean and standard 935
deviation. 936
937
The mean response of negative control samples may be constant or may vary between assays, 938
plates, or analysts. When the mean is constant, a cut point may be established during assay 939
validation that can be applied to the assay in-study. This is frequently called a fixed cut point. 940
When the mean varies between assays, plates, or analysts but the variance around the mean is 941
constant, a normalization factor can be statistically determined and applied in-study. This is also 942
known as a floating cut point. When both the mean and variance vary, a cut point must be 943
established for each assay, plate, or analyst. This is known as a dynamic cut point. One 944
drawback of the dynamic cut point is the need to have more replicates of the negative control in 945
the assay. Dynamic cut points should not be used to compensate for deficient assay 946
optimization. 947
948
C. Validation of Confirmatory Assay 949
950
Confirmatory assays should be fully validated in a manner similar to screening and neutralization 951
assays because these assays raise some specific issues. As a scientific matter, the studies to 952
validate the assay will depend on the assay format and instrumentation chosen. If these assays 953
are based on competition for antigen binding
26
by the antibodies in patient samples and the 954
measurement is loss of response, it is critical to identify the degree of inhibition or depletion that 955
will be used to ascribe positivity to a sample. In the past, fixed percentages of binding reduction 956
were used, but these numbers were often arbitrary and are unlikely to be relevant for all assays. 957
26
Competition for antigen binding refers to a competition assay where the ability of antigen-specific antibodies to
bind to either labeled or plate-bound antigen is inhibited by unlabeled or soluble antigen.

Contains Nonbinding Recommendations
Draft — Not for Implementation
25
FDA recommends establishing a cut point based on the assessment of the binding changes 958
observed in samples that are known to lack the antibodies when competing antigen is added. 959
FDA also recommends that the sensitivity of the confirmatory assay be confirmed using a low 960
concentration of the positive control antibody. 961
962
For the estimation of the confirmatory assay cut point, an 80% one-sided lower confidence 963
interval for the 99
th
percentile is recommended. Because the purpose of this assay is to eliminate 964
false-positive samples arising as a result of non-specific binding, it is adequate to use a 1% false-965
positive rate for the calculation of the confirmatory cut point. The use of tighter false-positive 966
rates such as 0.1% is not recommended because it will lead to an increased risk of false-negative 967
results. See section IV.B for general information on assay cut point. 968
969
If the confirmatory assay format is a competiton assay in which a competitor, usually unlabeled 970
therapeutic protein product,
27
will be added to the reaction mixture to inhibit ADA binding to the 971
capture reagent for the cut point assay, the same concentration of unlabeled therapeutic protein 972
product should be added to the samples when determining the confirmatory cut point. 973
974
D. Validation of Titering Assay 975
976
The principles of assay validation described in section VI.A apply in general to validation of 977
titering assays. The cut point of the titration assay may be the same as or different from that of 978
the screening assay. When the titering assay is not used for screening and the cut point is 979
different than that of the screening assay, the validation of the separate titration method cut point 980
can become necessary; for example, when the signal from the assay diluent or matrix causes 981
higher results than the screening assay cut point because of a blocking effect of serum or if 982
samples at a dilution higher than the MRD do not generate consistently negative results, i.e., 983
when the screening cut point falls on the lower plateau of the positive-control dilution curve.
28
984
985
E. Validation of Neutralization Assay 986
987
A minimum of 30 samples tested on at least 3 different days by at least two analysts should be 988
used to determine the cut point, using suitable statistical methods. 989
990
FDA recognizes that not all ADA are neutralizing, and it can be difficult to identify positive 991
control antibodies with neutralizing capacity. Further, if an affinity purified polyclonal positive 992
control antibody preparation is used, it is likely that only a portion of the antibodies are 993
neutralizing, which can make the assay appear less sensitive. Therefore, it is important to 994
validate assay sensitivity. 995
996
Sponsors should validate assay specificity for cell-based neutralization bioassays. As mentioned, 997
for cells that may be responsive to stimuli other than the specific therapeutic protein product, the 998
27
See footnote 25.
28
See the USP General Chapter 1106 Immunogenicity Assays – Design and Validation of Immunoassays to Detect
Anti-Drug Antibodies.
Contains Nonbinding Recommendations
Draft — Not for Implementation
26
ability to demonstrate that NAb only inhibit the response to therapeutic protein product and not 999
the response to other stimuli is a good indication of assay specificity. In such studies, FDA 1000
recommends that the other stimuli be employed at a concentration that yields an outcome similar 1001
to that of the therapeutic protein product. The sponsor should also confirm the absence of 1002
alternative stimuli in patient serum (see sections IV.C and D). 1003
1004
Cell-based neutralization bioassays frequently have reduced precision when compared to ligand-1005
binding assays because biologic responses can be inherently more variable than carefully 1006
controlled binding studies. Consequently, the sponsor should perform more replicates for 1007
assessment of precision and assessment of patient responses than for the screening assay (see 1008
section IV.E). 1009
1010
Additional parameters that should be validated are assay performance when cells at the low, 1011
middle, and high range of the allowed passage numbers, cell density, and cell viability are used 1012
(see section IV.G). 1013
1014
1015
VII. IMPLEMENTATION OF ASSAY TESTING 1016
1017
A. Obtaining Patient Samples 1018
1019
FDA recommends that the sponsor obtain pre-exposure samples from all patients. Because there 1020
is the potential for pre-existing antibodies or confounding components in the matrix, 1021
understanding the degree of reactivity before treatment is essential. The sponsor should obtain 1022
subsequent samples, with the timing depending on the frequency of dosing. Optimally, samples 1023
taken 7 to 14 days after the first exposure can help elucidate an early IgM response. Samples 1024
taken at 4 to 6 weeks after the first exposure are generally optimal for determining IgG 1025
responses. For individuals receiving a single dose of therapeutic protein product, the above time 1026
frame may be adequate. However, for patients receiving a therapeutic protein product at 1027
multiple times during the trial, the sponsor should obtain samples at appropriate intervals 1028
throughout the trial and also obtain a sample approximately 30 days after the last exposure. 1029
1030
Obtaining samples at a time when there will be minimal interference from the therapeutic protein 1031
product present in the serum is essential. A sponsor should consider the therapeutic protein 1032
product’s half-life to help determine appropriate times for sampling. This is especially important 1033
for mAb products because these products can have half-lives of several weeks or more; and 1034
depending on the dosing regimen, the therapeutic mAb itself could remain present in the serum 1035
for months. Under circumstances when testing for IgE is needed, the timing of sample collection 1036
should be discussed with FDA. 1037
1038
The level of therapeutic protein product that interferes with the assay, as determined by immune 1039
competition, may also help define meaningful time points for sampling. If therapeutic protein 1040
product-free samples cannot be obtained during the treatment phase of the trial, the sponsor 1041
should take additional samples after an appropriate washout period (e.g., five half-lives). 1042
Obtaining samples to test for meaningful antibody results can also be complicated if the 1043
therapeutic protein product in question is itself an immune suppressant. In such instances, the 1044

Contains Nonbinding Recommendations
Draft — Not for Implementation
27
sponsor should obtain samples from patients who have undergone a washout period either 1045
because the treatment phase has ended or because the patient has dropped out of the study. 1046
1047
Samples to determine serum concentrations of therapeutic protein product should be obtained at 1048
the same time as immunogenicity samples. Testing such samples can provide information on 1049
whether the therapeutic protein product in the samples may be interfering with ADA testing and 1050
whether ADA may be altering the therapeutic protein product’s pharmacokinetics. 1051
1052
B. Concurrent Positive and Negative Quality Controls 1053
1054
If the sponsor completes the proper validation work and makes the cut point determinations, the 1055
immunogenicity status of patients should be straightforward to determine. However, positive 1056
control or QC samples are critical and should be run concurrently with patient samples. We 1057
recommend that these samples span a level of positivity with QC samples having a known 1058
negative, low, and high reactivity in the assay. More important, the QC samples should be 1059
diluted in the matrix in which patient samples will be examined; for example, the same percent 1060
serum or plasma (specify salt anticoagulant used). In this way, the sponsor ensures that the assay 1061
is performing to its optimal degree of accuracy and that patient samples are correctly evaluated. 1062
For the low-positive QC sample, we recommend that a concentration be selected that, upon 1063
statistical analysis, would lead to the rejection of an assay run 1% of the time. Such an approach 1064
would ensure the appropriate sensitivity of the assay when performed on actual patient samples. 1065
The concentration of high-positive QC samples should be set to monitor prozone effects.
29
1066
1067
FDA also recommends that these QC samples be obtained from humans or animals possessing 1068
antibodies that are detected by the secondary detecting reagent, to ensure that negative results 1069
that might be observed are truly caused by lack of antigen reactivity and not caused by failure of 1070
the secondary reagent. This issue is not a problem for antigen bridging assays because labeled 1071
antigen is used for detection. 1072
1073
C. Confirmation of Cut Point in the Target Population 1074
1075
Samples from different populations can have different background activity in ADA assays. 1076
Therefore, it is necessary to confirm that the cut point determined during assay validation is 1077
suitable for the population being studied. Similarly, if samples used to determine the cut point 1078
during assay validation were not obtained and handled in a manner that represents how samples 1079
will be obtained and handled in-study, the cut point should also be confirmed with appropriate 1080
samples in-study. A sufficient number of samples from the target population should be used, and 1081
justification for the number used should be provided. If sufficient numbers of samples are not 1082
available, agreement with the Agency should be sought for the number of samples to be used. 1083
1084
1085
29
Prozone effects (also referred to as hook effects) are a reduction in signal that may occur as a result of the
presence of a high concentration of a particular analyte or antibody and may cause false-negative results.

Contains Nonbinding Recommendations
Draft — Not for Implementation
28
VIII. DOCUMENTATION 1086
1087
The rationale and information for the immunogenicity testing paradigm should be provided in 1088
module 5.3.1.4 of the electronic common technical document (eCTD) on Reports of 1089
Bioanalytical and Analytical Methods for Human Studies.
30
The standard operating procedure of 1090
the respective assay being used should be provided to the FDA, together with the results of the 1091
validation studies and relevant assay development information for parameters that were not 1092
validated, such as the MRD, the stimulatory concentration of therapeutic protein product used in 1093
the NAb assay, and some robustness parameters that are critical for assay performance (see 1094
section VII. Documentation in the draft guidance for industry Bioanalytical Method 1095
Validation.)
31
1096
1097
1098
30
See the FDA Web site for further information on eCTD submissions, available at
http://www.fda.gov/Drugs/DevelopmentApprovalProcess/FormsSubmissionRequirements/ElectronicSubmissions/uc
m153574.htm. For more information about the agreed-upon common format for the preparation of a well-structured
Efficacy section of the CTD for applications that will be submitted to regulatory authorities, see the ICH guidance
for industry M4E: The CTD — Efficacy. For more information on how sponsors and applicants must organize the
content they submit to the Agency electronically for all submission types under section 745A(a) of the FD&C Act,
see the guidance for industry (and the technical specification documents it incorporates by reference) Providing
Regulatory Submissions in Electronic Format — Certain Human Pharmaceutical Product Applications and Related
Submissions Using the eCTD Specifications.
31
When final, this guidance will represent the FDA’s current thinking on this topic. To make sure you have the most
recent version of a guidance, check the FDA guidance Web page at
http://www.fda.gov/RegulatoryInformation/Guidances/default.htm
.
Contains Nonbinding Recommendations
Draft — Not for Implementation
29
REFERENCES 1099
1100
Aalberse, R. C. and J. Schuurman (2002). "IgG4 breaking the rules." Immunology 105(1): 9-19. 1101
1102
Boes, M. (2000). "Role of natural and immune IgM antibodies in immune responses." Mol 1103
Immunol 37(18): 1141-1149. 1104
1105
Calabresi, P. A., G. Giovannoni, et al. (2007). "The incidence and significance of anti-1106
natalizumab antibodies: results from AFFIRM and SENTINEL." Neurology 69(14): 1391-1403. 1107
1108
Caruso, A. and A. Turano (1997). "Natural antibodies to interferon-gamma." Biotherapy 10(1): 1109
29-37. 1110
1111
Cohen, B. A. and V. M. Rivera (2010). "PRISMS: the story of a pivotal clinical trial series in 1112
multiple sclerosis." Curr Med Res Opin 26(4): 827-838. 1113
1114
Coutinho, A., M. D. Kazatchkine, et al. (1995). "Natural autoantibodies." Curr Opin Immunol 1115
7(6): 812-818. 1116
1117
Disis, M. L., V. Goodell, et al. (2004). "Humoral epitope-spreading following immunization with 1118
a HER-2/neu peptide based vaccine in cancer patients." J Clin Immunol 24(5): 571-578. 1119
1120
Goodin, D. S., E. M. Frohman, et al. (2007). "Neutralizing antibodies to interferon beta: 1121
assessment of their clinical and radiographic impact: an evidence report: report of the 1122
Therapeutics and Technology Assessment Subcommittee of the American Academy of 1123
Neurology." Neurology 68(13): 977-984. 1124
1125
Gupta, S., V. Devanarayan, et al. (2011). "Recommendations for the validation of cell-based 1126
assays used for the detection of neutralizing antibody immune responses elicited against 1127
biological therapeutics." J Pharm Biomed Anal 55(5): 878-888. 1128
1129
Gupta, S., S. R. Indelicato, et al. (2007). "Recommendations for the design, optimization, and 1130
qualification of cell-based assays used for the detection of neutralizing antibody responses 1131
elicited to biological therapeutics." J Immunol Methods 321(1-2): 1-18. 1132
1133
Harding, F. A., M. M. Stickler, et al. (2010). "The immunogenicity of humanized and fully 1134
human antibodies: residual immunogenicity resides in the CDR regions." MAbs 2(3): 256-265. 1135
1136
Hintermann, E., M. Holdener, et al. (2011). "Epitope spreading of the anti-CYP2D6 antibody 1137
response in patients with autoimmune hepatitis and in the CYP2D6 mouse model." J Autoimmun 1138
37(3): 242-253. 1139
1140
Kuus-Reichel, K., L. S. Grauer, et al. (1994). "Will immunogenicity limit the use, efficacy, and 1141
future development of therapeutic monoclonal antibodies?" Clin Diagn Lab Immunol 1(4): 365-1142
372. 1143
1144
Contains Nonbinding Recommendations
Draft — Not for Implementation
30
Matsumoto, T., M. Shima, et al. (2001). "Immunological characterization of factor VIII 1145
autoantibodies in patients with acquired hemophilia A in the presence or absence of underlying 1146
disease." Thromb Res 104(6): 381-388. 1147
1148
Miller, L. L., E. L. Korn, et al. (1999). "Abrogation of the hematological and biological activities 1149
of the interleukin-3/granulocyte-macrophage colony-stimulating factor fusion protein PIXY321 1150
by neutralizing anti-PIXY321 antibodies in cancer patients receiving high-dose carboplatin." 1151
Blood 93(10): 3250-3258. 1152
1153
Mire-Sluis, A. R., Y. C. Barrett, et al. (2004). "Recommendations for the design and 1154
optimization of immunoassays used in the detection of host antibodies against biotechnology 1155
products." J Immunol Methods 289(1-2): 1-16. 1156
1157
Plotkin, S. A. (2010). "Correlates of protection induced by vaccination." Clin Vaccine Immunol 1158
17(7): 1055-1065. 1159
1160
Prummer, O. (1997). "Treatment-induced antibodies to interleukin-2." Biotherapy 10(1): 15-24. 1161
1162
Ramsland, P. A., B. F. Movafagh, et al. (1999). "Interference of rheumatoid factor activity by 1163
aspartame, a dipeptide methyl ester." J Mol Recognit 12(5): 249-257. 1164
1165
Ross, C., M. B. Hansen, et al. (1990). "Autoantibodies to crude human leucocyte interferon 1166
(IFN), native human IFN, recombinant human IFN-alpha 2b and human IFN-gamma in healthy 1167
blood donors." Clin Exp Immunol 82(1): 57-62. 1168
1169
Shankar, G., V. Devanarayan, et al. (2008). "Recommendations for the validation of 1170
immunoassays used for detection of host antibodies against biotechnology products." J Pharm 1171
Biomed Anal 48(5): 1267-1281. 1172
1173
Shen, M., X. Dong, et al. (2015). "Statistical evaluation of several methods for cut-point 1174
determination of immunogenicity screening assay." J Biopharm Stat 25(2): 269-279. 1175
1176
Thrasyvoulides, A., E. Liakata, et al. (2007). "Spreading of antibody reactivity to non-thyroid 1177
antigens during experimental immunization with human thyroglobulin." Clin Exp Immunol 1178
147(1): 120-127. 1179
1180
Turano, A., A. Balsari, et al. (1992). "Natural human antibodies to gamma interferon interfere 1181
with the immunomodulating activity of the lymphokine." Proc Natl Acad Sci U S A 89(10): 1182
4447-4451. 1183
1184
van der Meide, P. H. and H. Schellekens (1997). "Anti-cytokine autoantibodies: epiphenomenon 1185
or critical modulators of cytokine action." Biotherapy 10(1): 39-48. 1186
1187
van der Woude, D., S. Rantapaa-Dahlqvist, et al. (2010). "Epitope spreading of the anti-1188
citrullinated protein antibody response occurs before disease onset and is associated with the 1189
disease course of early arthritis." Ann Rheum Dis 69(8): 1554-1561. 1190
Contains Nonbinding Recommendations
Draft — Not for Implementation
31
1191
van Schouwenburg, P. A., S. Kruithof, et al. (2014). "Functional analysis of the anti-adalimumab 1192
response using patient-derived monoclonal antibodies." J Biol Chem 289(50): 34482-34488. 1193
1194
Zhou, L., S. A. Hoofring, et al. (2013). "Stratification of antibody-positive subjects by antibody 1195
level reveals an impact of immunogenicity on pharmacokinetics." AAPS J 15(1): 30-40. 1196
1197
