
HAL Id: hal-04192479
https://hal.science/hal-04192479
Submitted on 31 Aug 2023
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-
entic research documents, whether they are pub-
lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diusion de documents
scientiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Distributed under a Creative Commons Attribution 4.0 International License
An Ecient Protocol for CUT&RUN Analysis of
FACS-Isolated Mouse Satellite Cells
Kamar Ghaibour, Joe Rizk, Claudine Ebel, Tao Ye, Muriel Philipps, Valérie
Schreiber, Daniel Metzger, Delphine Duteil
To cite this version:
Kamar Ghaibour, Joe Rizk, Claudine Ebel, Tao Ye, Muriel Philipps, et al.. An Ecient Protocol
for CUT&RUN Analysis of FACS-Isolated Mouse Satellite Cells. Journal of visualized experiments :
JoVE, 2023, 197 (e65215), pp.1-21. �10.3791/65215�. �hal-04192479�

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 1 of 21
An Efficient Protocol for CUT&RUN Analysis of FACS-
Isolated Mouse Satellite Cells
Kamar Ghaibour
*,1
, Joe Rizk
*,1
, Claudine Ebel
1
, Tao Ye
1
, Muriel Philipps
1
, Valérie Schreiber
1
, Daniel Metzger
1
, Delphine
Duteil
1
1
CNRS, Inserm, IGBMC UMR 7104- UMR-S 1258, Université de Strasbourg
*
These authors contributed equally
Corresponding Author
Delphine Duteil
Citation
Ghaibour, K., Rizk, J., Ebel, C., Ye, T.,
Philipps, M., Schreiber, V., Metzger, D.,
Duteil, D. An Efficient Protocol for
CUT&RUN Analysis of FACS-Isolated
Mouse Satellite Cells. J. Vis. Exp. (197),
e65215, doi:10.3791/65215 (2023).
Date Published
July 7, 2023
DOI
10.3791/65215
URL
jove.com/video/65215
Abstract
Genome-wide analyses with small cell populations are a major constraint for studies,
particularly in the stem cell field. This work describes an efficient protocol for the
fluorescence-activated cell sorting (FACS) isolation of satellite cells from the limb
muscle, a tissue with a high content of structural proteins. Dissected limb muscles
from adult mice were mechanically disrupted by mincing in medium supplemented with
dispase and type I collagenase. Upon digestion, the homogenate was filtered through
cell strainers, and cells were suspended in FACS buffer. Viability was determined
with fixable viability stain, and immunostained satellite cells were isolated by FACS.
Cells were lysed with Triton X-100 and released nuclei were bound to concanavalin
A magnetic beads. Nucleus/bead complexes were incubated with antibodies against
the transcription factor or histone modifications of interest. After washes, nucleus/
bead complexes were incubated with protein A-micrococcal nuclease, and chromatin
cleavage was initiated with CaCl
2
. After DNA extraction, libraries were generated and
sequenced, and the profiles for genome-wide transcription factor binding and covalent
histone modifications were obtained by bioinformatic analysis. The peaks obtained for
the various histone marks showed that the binding events were specific for satellite
cells. Moreover, known motif analysis unveiled that the transcription factor was bound
to chromatin via its cognate response element. This protocol is therefore adapted to
study gene regulation in adult mice limb muscle satellite cells.
Introduction
Skeletal striated muscles represent on average 40% of the
weight of the total human body
1
. Muscle fibers exhibit a
remarkable capacity for regeneration upon injury, which is
described by the fusion of newly formed myocytes and
the generation of new myofibers that replace the damaged
ones
2
. In 1961, Alexander Mauro reported a population of
mononuclear cells that he termed as satellite cells
3
. These
stem cells express the transcription factor paired box 7

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 2 of 21
(PAX7), and are located between the basal lamina and the
sarcolemma of muscle fibers
4
. They were reported to express
the cluster of differentiation 34 (CD34; a hematopoietic,
endothelial progenitor and mesenchymal stem cell marker),
integrin alpha 7 (ITGA7; a smooth, cardiac and skeletal
muscle marker), as well as the C-X-C chemokine receptor
type 4 (CXCR4; a lymphocyte, hematopoietic, and satellite
cell marker)
5
. In basal conditions, satellite cells reside in a
particular microenvironment that keeps them in a quiescent
state
6
. Upon muscle damage, they become activated,
proliferate, and undergo myogenesis
7
. However, contributing
only to a minor fraction of the total number of muscle cells,
their genome-wide analyses are particularly challenging,
especially under physiological settings (<1% of total cells).
Various methods for chromatin isolation from satellite
cells have been described, which involve chromatin
immunoprecipitation followed by massive parallel sequencing
(ChIP-seq) or cleavage under targets and tagmentation
(CUT&Tag) experiments. Nevertheless, these two techniques
present some significant limitations that remain unchallenged.
Indeed, ChIP-seq requires a high amount of starting
material to generate enough chromatin, a large proportion
of which is lost during the sonication step. CUT&Tag is
more appropriate for low cell number, but generates more
off-target cleavage sites than ChIP-seq due to the Tn5
transposase activity. In addition, since this enzyme has
a high affinity for open-chromatin regions, the CUT&Tag
approach might be preferentially used for analyzing histone
modifications or transcription factors associated with actively
transcribed regions of the genome, instead of silenced
heterochromatin
8 , 9
.
Presented here is a detailed protocol that allows the
isolation of mouse limb muscle satellite cells by FACS
for cleavage under targets and release using nuclease
(CUT&RUN)
10 , 11
analysis. The various steps involve the
mechanical disruption of tissue, cell sorting, and nuclei
isolation. The method's efficiency, regarding the preparation
of a viable cell suspension, was demonstrated by performing
CUT&RUN analysis for covalent histone modifications and
transcription factors. The quality of isolated cells makes
the described method particularly attractive for preparing
chromatin that captures the native genomic occupancy
state faithfully, and is likely to be suitable for capturing
the chromosome conformation in combination with high-
throughput sequencing at specific loci (4C-seq) or at genome-
wide levels (Hi-C).
Protocol
Mice were kept in an accredited animal house, in compliance
with National Animal Care Guidelines (European Commission
directive 86/609/CEE; French decree no.87-848) on the use
of laboratory animals for research. Intended manipulations
were submitted to the Ethical committee (Com'Eth,
Strasbourg, France) and to the French Research Ministry
(MESR) for ethical evaluation and authorization according to
the 2010/63/EU directive under the APAFIS number #22281.
1. Preparation of cell suspension for isolation
of satellite cells by fluorescence-activated cell
sorting (FACS) (Figure 1 )
1. Isolation of muscle tissue
1. Decontaminate the tools for muscle dissection,
including forceps, scalpels, and scissors, using a
cleaning agent (Table 1), and rinse thoroughly with
distilled water.

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 3 of 21
2. Prepare two 2 mL tubes (Table 1), each containing
1 mL of muscle isolation buffer, and place them on
ice for collecting harvested muscles.
3. Sacrifice two 10-week-old C57/Bl6J male mice by
CO
2
asphyxiation followed by cervical dislocation.
Spray 70% ethanol over each entire mouse. Peel off
the skin from the hind limb using forceps. Dissect out
all the limb muscles surrounding the femur, tibia, and
fibula (approximately 1 mg of muscles per mouse).
NOTE: The satellite cells number decreases after 15
weeks of age.
4. Place the harvested limb muscles into 2 mL tubes
containing 1 mL of muscle isolation buffer prepared
in step 1.1.2. Collect the muscles from the second
mouse following the same procedure. Mince the
harvested muscles with scissors on ice until smaller
than 1 mm
3
fragments are obtained.
NOTE: Muscles were collected and minced mainly
as described
12
. Perform tissue digestion either
following step 1.2 or 1.3.
2. Tissue digestion with collagenase enzyme
1. Transfer the minced muscle suspension from the
two mice by pouring them into a 50 mL tube
(Table 1) containing 18 mL of muscle isolation buffer
(supplemented with 5 mL [5 U/mL] of dispase and 5
mg of type I collagenase) (Table 1).
2. Close the tube tightly and seal it with laboratory film
(Table 1). Place it horizontally in a shaking water
bath (Table 1) at 37 °C at 100 rpm for 30 min.
3. After 30 min, add 5 mg of type I collagenase. Keep
tubes for another 30 min under agitation in a shaking
water bath at 37 °C at 100 rpm.
4. Upon digestion, pipette the muscle suspension up
and down 10 times with a 10 mL pipette to improve
the efficiency of dissociation. Centrifuge at 4 °C at
400 x g for 5 min. A clear pellet will be visible at the
bottom of the tube. Discard the supernatant using a
10 mL pipette, leaving 5 mL of medium in the tube.
NOTE: Leaving the medium helps avoid stressing
the cells. Add 10 mL of fresh muscle isolation buffer
and resuspend the pellet by pipetting up and down
with a 10 mL pipette.
5. Place 100 µm, 70 µm, and 40 µm cell strainers (one
of each kind) (Table 1) on open 50 mL tubes. Pipette
the suspension onto the successive cell strainers
(100 µm, 70 µm, 40 µm) and collect the flowthrough
into the 50 mL tubes, containing cells below 40 µm.
6. Centrifuge the suspension at 4 °C at 400 x g for 5
min. Discard the supernatant using a 10 mL pipette
until 2 mL remain, and then use a 0.2-1 mL pipette
until 100-200 µL remain. Resuspend the pellet in 2
mL of red blood cell lysis buffer (Table 2). Incubate
on ice for 3 min.
7. Centrifuge at 4 °C at 400 x g for 5 min and discard the
supernatant using a 20-200 µL pipette. Resuspend
the cells in 100 µL of cold FACS buffer (Table 2).
Place on ice.
3. Alternative method for tissue digestion with Liberase
thermolysin low (TL) enzyme
1. Follow the descriptions in step 1.1. for tissue
isolation.
2. For Liberase-mediated tissue disaggregation,
harvest muscles in 2 mL of Roswell Park Memorial
Institute (RPMI) isolation buffer (Table 2), instead of
muscle isolation buffer described in step 1.1.2.

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 4 of 21
3. Transfer the minced muscle suspension from the
two mice by pouring them into a 50 mL tube
(Table 1) containing 18 mL of RPMI isolation buffer
supplemented with 300 or 600 µL of Liberase TL at
5 mg/mL (Table 1) (i.e., 0.083 mg/mL and 0.167 mg/
mL final concentrations, respectively)
13
.
4. Close the tube tightly and seal it with laboratory film
(Table 1). Place it horizontally in a shaking water
bath (Table 1) at 37 °C at 100 rpm for 30 min.
5. Upon digestion, pipette the muscle suspension up
and down 10 times with a 10 mL pipette to dissociate
and improve the efficiency of dissociation.
6. Centrifuge at 4 °C at 400 x g for 5 min. A clear pellet
will be visible at the bottom of the tube. Discard the
supernatant using a 10 mL pipette, leaving 5 mL
of medium in the tube. Leaving the medium helps
avoid stressing the cells. Add 10 mL of fresh RPMI
isolation buffer, and resuspend the pellet by pipetting
up and down with a 10 mL pipette.
7. Place 100 µm, 70 µm, and 40 µm cell strainers (one
of each kind) (Table 1) on open 50 mL tubes.
8. Pipette the suspension onto successive cell
strainers (100 µm, 70 µm, 40 µm) and collect the
flowthrough into the 50 mL tubes, containing cells
below 40 µm.
9. Centrifuge the suspension at 4 °C at 400 x g for 5
min. Discard the supernatant using a 10 mL pipette
until 2 mL remain, and then use a 0.2-1 mL pipette
until 100-200 µL remain.
10. Resuspend the pellet in 2 mL of red blood cell lysis
buffer (Table 2). Incubate on ice for 3 min.
11. Centrifuge at 4 °C at 400 x g for 5 min and discard
the supernatant using a 20-200 µL pipette.
12. Resuspend the cells in 100 µL of cold FACS buffer
(Table 2). Place on ice.
4. Preparation of cell suspension for FACS isolation
1. Transfer 10 µL of the cell suspension obtained in
step 1.2.12 into a fresh 1.5 mL tube. This sample
will constitute the unstained control or the negative
control (Figure 2). Add 190 µL of FACS buffer,
transfer to a 5 mL tube (Table 1), and store on ice.
2. Centrifuge the remaining 90 µL of the cell
suspension obtained in step 1.2.12 at 4 °C at 400 x g
for 5 min and discard the supernatant using a pipette
(20-200 µL tip volume). Incubate the cells with 400
µL of fixable viability stain (Table 3) diluted in serum-
free Dulbecco's modified Eagle medium (DMEM) for
15 min at room temperature (RT).
3. Wash the cells by centrifugation at 4 °C at 400 x g for
5 min and add 100 µL of FACS buffer. Gently invert
the tubes three times and centrifuge again at 4 °C at
400 x g for 5 min.
4. During the centrifugation time, prepare 100 µL of
a master mix of primary antibodies coupled to
fluorophores and directed against CD11b, CD31,
CD45, TER119, CD34, ITGA7, and CXCR4 (Table
3), diluted in FACS buffer.
5. Centrifuge at 400 x g for 5 min at 4 °C, discard
the cell supernatant using a pipette (20-200 µL tip
volume), and add the 100 µL antibody mix. Gently
invert the tube three times. Do not vortex. Incubate
in the dark on ice for 30 min.

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 5 of 21
6. Centrifuge at 400 x g for 5 min at 4 °C. Discard the
supernatant using a 20-200 µL pipette and add 500
µL of 1x phosphate-buffered saline (PBS) to wash
the cells. Gently invert the tube three times. Re-
centrifuge at 400 x g for 5 min at 4 °C and discard
the supernatant using a 20-200 µL pipette.
7. Resuspend the cell pellet in 500 µL of FACS buffer
and transfer the suspension to a 5 mL tube.
NOTE: the cell suspension obtained from Liberase
digestion is processed in the same way.
5. Satellite cells selection by FACS
1. Briefly vortex the cell suspension (2-5 sec) and
process the cells on a flow cytometer equipped with
a 100 µm nozzle (Table 1).
2. Determine the various gate sizes based on the
unstained sample stored at step 1.4.1 (Figure 2).
3. Coat a 5 mL tube with 1 mL of pure fetal calf serum
(FCS) to improve cell collection and add 500 µL of
FACS buffer.
4. Exchange the unstained sample with the antibody-
labelled sample.
5. Select the population of interest according to the
forward scatter area (FSC-A) and side scatter area
(SSC-A) (Figure 3A), and remove doublet cells
with the FSC-A and forward scatter height (FSC-H)
(Figure 3B)
14
.
6. Identify living cells with fixable viability stain negative
staining (Figure 3C).
7. Select negative cells for CD31, CD45, TER119, and
CD11b (Figure 3D).
8. To identify satellite cells, select first the cells that are
positive for CD34 and ITGA7 (Figure 3E), and then
select the CXCR4-positive cells on the CD34- and
ITGA7-selected population (Figure 3F).
9. Collect the selected cells (between 40,000 and
80,000 cells, according to the quality of the
preparation) in the 5 mL coated tube containing 500
µL of FACS buffer.
2. Validation of the isolated population in tissue
culture
1. Slide coating with hydrogel
1. Dilute 280 µL of a pure hydrogel human embryonic
stem cell (hESC) qualified matrix (Table 1) in 12 mL
of serum-free DMEM/F12 medium.
2. Coat a chamber slide (Table 1) with the hydrogel
solution, and incubate it overnight at 4 °C.
3. The next day, incubate the chamber slide at 37 °C
and 5% CO
2
for 1 h before cell seeding.
2. Cell growth and differentiation
1. Plate out approximately 20,000 cells, obtained from
step 1.5.9, per well, and grow them in growth
medium (Table 2) for 5 days. Take phase-contrast
images using a brightfield microscope (Figure 4A),
before processing them for immunofluorescence
analysis to ensure the quality of the preparation
(Figure 4B).
2. To induce myogenesis, grow amplified satellite cells
from step 2.2.1 in myogenic medium (Table 2) for
an additional 7 days. Take phase-contrast images
using brightfield microscope (Figure 4C) before
processing them for immunofluorescence analysis
to ensure the quality of the preparation (Figure 4D).
3. Immunocytofluorescence analysis

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 6 of 21
1. Gently remove the medium, wash the cells that
were cultured on chamber slide with 100 µL of
1x PBS twice, and fix them with 100 µL of 4 %
paraformaldehyde (PFA) at RT for 1 h.
NOTE: This step should be performed with care.
A small volume of medium should always be kept
in the chamber to prevent stressing the cells, and
the PBS should be poured through the walls of the
chamber.
2. Wash the cells three times with 100 µL of 1x
PBS, supplemented with 0.1% Tween 20 (PBST) to
permeabilize the cell membranes.
3. Block unspecific signals by incubation in 100 µL of
1x PBST supplemented with 5% FCS (PBST-FCS)
at RT for 1 h.
4. Incubate the cells with 100 µL of a master mix of anti-
PAX7 and anti-dystrophin (DMD) antibodies (diluted
in 1x PBST-FCS) at 4 °C overnight, to detect satellite
cells and myofibers, respectively.
5. Wash the cells three times with 100 µL of 1x
PBST, and incubate them with 100 µL of goat anti-
mouse Cy3 or goat anti-rabbit Alexa 488 secondary
antibodies (Table 3) diluted in 1x PBST-FCS at RT
for 1 h.
6. Dissociate the chamber wells from the slide using
the equipment provided by the supplier, add 20 µL
of aqueous mounting medium with 4′,6-diamidino-2-
phenylindole (DAPI), and cover the slide with a
coverslip (Table 1).
7. Observe and capture the image of the stained cells
with a confocal microscope.
8. Process the images using image analysis software
(Figures 4B,D).
3. CUT&RUN analysis
1. Sample preparation for CUT&RUN analysis on FACS-
isolated satellite cells
NOTE: CUT&RUN was performed essentially as
described
10 , 15
. The buffer composition is presented in
Table 2.
1. For the CUT&RUN assay, use approximately 40,000
of the cells obtained in method 1, step 1.5.9, per
sample/antibody that must be tested.
2. Centrifuge the FACS-isolated satellite cells at RT at
500 x g for 10 min, then discard the supernatant
using a pipette (20-200 µL tip volume).
3. Wash the cells with 1 mL of 1x PBS, centrifuge at RT
at 500 x g for 5 min, discard the supernatant using a
pipette (0.2-1 mL tip volume), and resuspend them
in 1 mL of cold nuclear extraction buffer (Table 2).
Incubate on ice for 20 min.
4. During incubation, prepare one 1.5 mL tube
containing 850 µL of cold binding buffer (Table 2)
and add 20 µL of concanavalin A-coated magnetic
beads per sample (Table 1).
5. Wash the beads twice with 1 mL of cold binding
buffer using a magnetic rack (Table 1). For each
wash or buffer change throughout the procedure, let
the beads accumulate at the side of the tube on the
magnetic rack for 5 min before removing the cleared
supernatant with a pipette (0.2-1 mL tip volume).
Then, resuspend gently in 300 µL of cold binding
buffer.

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 7 of 21
6. Centrifuge the nuclei at 4 °C at 600 x g for 5 min
and resuspend them gently in 600 µL of nuclear
extraction buffer. Gently mix the 600 µL of extracted
nuclei with the 300 µL of concanavalin A bead slurry,
and incubate at 4 °C for 10 min.
7. Remove the supernatant using a magnetic rack, as
described in step 3.1.4, and resuspend the bead-
bound nuclei gently with 1 mL of cold blocking buffer
(Table 2). Incubate at RT for 5 min.
8. Remove the supernatant using a magnetic rack
and wash the bead-bound nuclei twice with 1 mL
of cold wash buffer (Table 2). During the second
wash, equally split the bead-bound nuclei into 1.5
mL tubes. Each tube will be treated with a specific
antibody in the following step.
NOTE: In this example, 250 µL of bead-bound nuclei
were split in four 1.5 mL tubes.
9. Separate the supernatant using a magnetic rack, as
described in step 3.1.4, and aspirate with a pipette.
Gently resuspend the nucleus/bead complexes with
a specific primary antibody (Table 3), or an IgG of
another species (here rabbit) diluted in 250 µL of
cold wash buffer. Incubate at 4 °C overnight with
gentle agitation.
NOTE: The antibodies used here are directed
against AR, H3K4me2, and H3K27ac.
10. Remove the supernatant with a magnetic rack, as
described in step 3.1.4, wash the bead-bound nuclei
twice with 1 mL of cold wash buffer, and resuspend
in 100 µL of cold wash buffer.
11. Dilute protein A-micrococcal nuclease at 1.4 ng/µL
in 100 µL per sample of cold wash buffer.
12. Add 100 µL of protein A-micrococcal nuclease to the
100 µL of sample obtained in 3.1.11 and incubate at
4 °C for 1 h with agitation.
13. Remove the supernatant with a magnetic rack, as
described in step 3.1.4, wash twice with 1 mL of cold
wash buffer, and resuspend the bead-bound nuclei
in 150 µL of cold wash buffer.
14. To initiate DNA cleavage, add 3 µL of 100 mM
of CaCl
2
to the 150 µL of sample, mix quickly
by flicking, and incubate on ice for 30 min. Stop
the reaction by adding 150 µL of stop buffer and
incubate at 37 °C for 20 min to digest the RNA and
release the DNA fragments.
15. For DNA extraction, centrifuge the samples at
16,000 x g at 4 °C for 5 min.
16. Transfer the supernatant to a new microfuge tube
and discard the pellet and beads.
17. Add 3 µL of 10% sodium dodecyl sulfate (SDS) and
2.5 µL of 20 mg/mL proteinase K. Mix by inversion.
Incubate for 10 min at 70 °C (no shaking).
18. Add 300 µL of phenol/chloroform/isoamyl alcohol,
vortex, transfer to 2 mL phase-lock tubes (pre-
spinned for 5 min at 16,000 x g), and centrifuge for
5 min at 16,000 x g at 4 °C.
19. Add 300 µL of chloroform to the same tube and
centrifuge for 5 min at 16,000 x g at 4 °C. Collect the
supernatant (~300 µL) with a pipette (0.2-1 mL tip
volume) and transfer into a new 1.5 mL tube.
20. Add 1 µL of glycogen (20 mg/mL concentration).
21. Add 750 µL of 100% ethanol and precipitate
overnight at -20 °C.

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 8 of 21
22. Pellet the DNA by centrifugation for 15 min at 16,000
x g at 4 °C. Wash the pellet with 1 mL of 100%
ethanol, centrifuge for 5 min at 16,000 x g, discard
the supernatant, centrifuge for 30 s at 16,000 x g,
and remove the liquid with a pipette (20-200 µL tip
volume).
23. Air-dry the pellet for ~5 min. Resuspend in 25
µL of 1 mM Tris-HCl (pH 8) and 0.1 mM
ethylenediaminetetraacetic acid (EDTA; pH 8).
2. Bioinformatics analysis
1. Prepare libraries from immunocleaved DNA and
sequence them as paired-end 100 bp reads with the
help of the genomic platform as described
16
.
2. Remove reads overlapping with the ENCODE
blacklist region (V2) and separate the remaining
reads into two groups: fragment size <120 bp
(without nucleosome, in general for transcription
factors) and fragment size >150 bp (with
nucleosomes, normally for histone marks). Map
to the mm10 reference genome using Bowtie 2
(v2.3.4.3)
17
.
3. Generate bigwig files with bamCoverage (deeptools
3.3.0: bamCoverage --normalizeUsing RPKM --
binSize 20).
4. Retain uniquely mapped reads for further analysis.
5. Generate raw bedgraph files with
genomeCoverageBed (bedtools v2.26.0).
6. Use the SEACR 1.3 algorithm (stringent option) for
the peak calling. Load the target data bedgraph
file in UCSC bedgraph format that omits regions
containing zero signal, and control (IgG) data
bedgraph file to generate an empirical threshold for
peak calling
18
.
7. Perform a Pearson correlation analysis with
deeptools to determine the similarity between
the samples
19
. Use the command line
multiBamSummary bins --bamfiles file1.bam
file2.bam -o results.npz, followed by
plotCorrelation -in results.npz --corMethod
pearson --skipZeros --plotTitle "Pearson
Correlation of Read Counts" --whatToPlot
heatmap --colorMap RdYlBu --plotNumbers
-o heatmap_PearsonCorr_readCounts.png --
outFileCorMatrix PearsonCorr_readCounts.tab.
8. Visualize the genome-wide intensity profiles with
IGV
20
using bedgraph files and the bed file peaks
obtained from SEACR.
9. Use HOMER for peak annotation and motif
search
21
.
10. Finally, compare the datasets with previously
published ones with ChIP-Atlas Peak browser to
visualize them on IGV, and/or enrichment analysis
using the SEACR-generated bed files as an input
dataset
22
.
Representative Results
Satellite cells from mouse skeletal muscles were isolated by
combining the protocols of Gunther et al. (hereafter Protocol
1)
12
and of Liu et al.
23
(hereafter Protocol 2). Since non-
digested muscle fibers were observed after digestion when
using the concentration of collagenase and dispase proposed
in Protocol 1, the quantity of enzymes was increased to
improve muscle fiber dissociation, as described in steps 1.2.1
and 1.2.3. As indicated in Protocol 2, the samples were

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 9 of 21
subjected to a gentle agitation in a water bath to maintain
cell viability. We performed filtration through cell strainers,
as mentioned in Protocol 1, and incubation with red blood
cell lysis buffer (see steps 1.2.7 to 1.2.10). In Protocol 1,
cells were loaded on a Percoll density gradient of 30%/70%
to isolate mononucleated cells at the interphase, which may
have led to the loss of cells of interest. Thus, this step was
omitted, as proposed in Protocol 2.
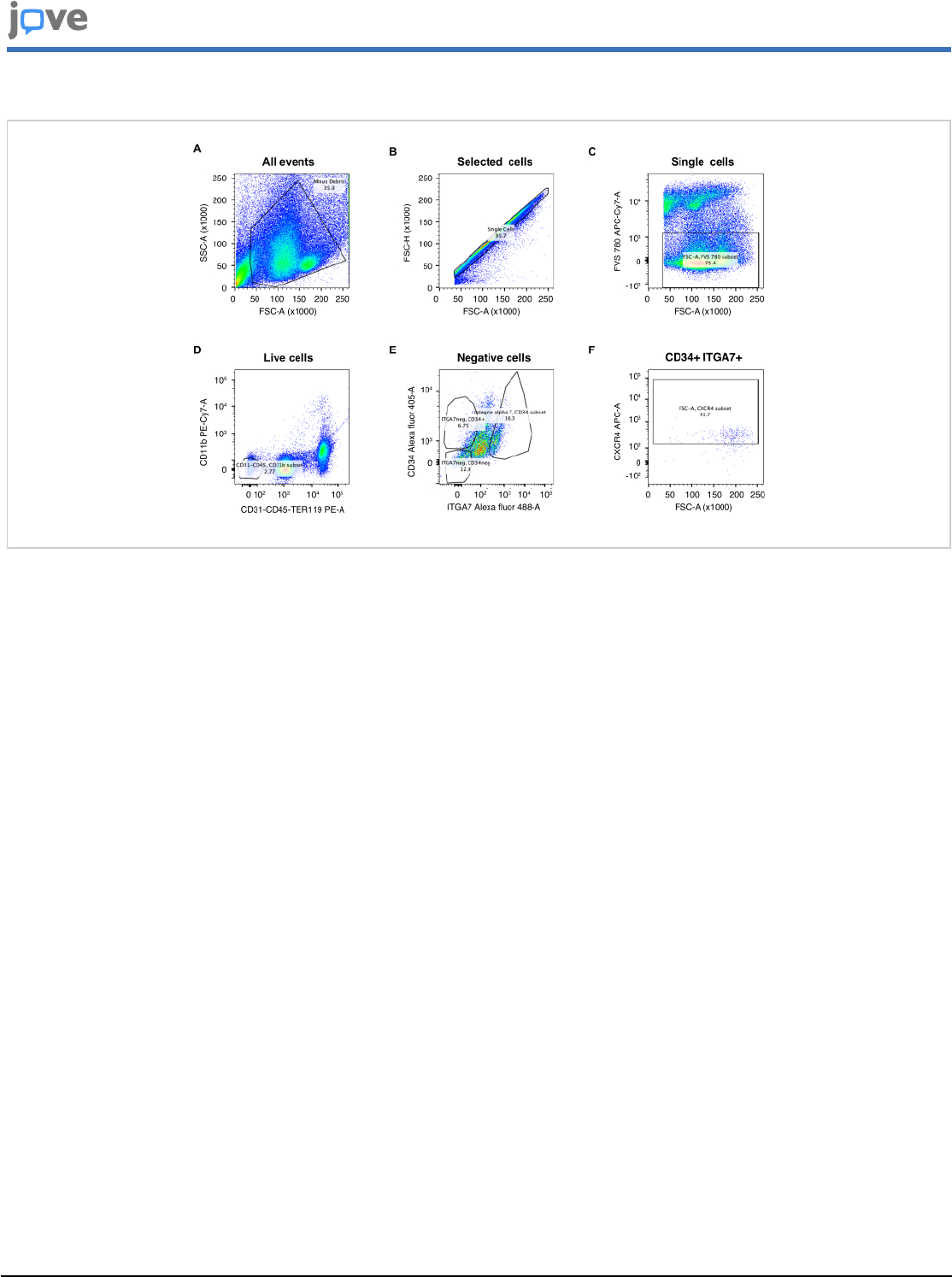
FSC-A versus SSC-A gating was used to identify
mononucleated cells based on size (FSC) and granularity
(SSC) (Figure 3A). Debris were excluded by ignoring events
below 40 K on the FSC-A axis, and 38.8% ± 3.6% of cells
were selected. FSC-A versus forward scatter height (FSC-H)
gating density plots were used for doublet exclusion (Figure
3B). After selecting the cells negative for the fixable viability
stain marker, an average of 34.3% ± 7.7% of live cells was
obtained (Figure 3C).
The percentage shown in the dot plot in Figure 3C, 75.4%,
corresponds to the percentage of single living cells, calculated
from the parent cell population, which is in this case the
singles. 95.7% of single cells are obtained from the live
events which make up 35% of the total events. Thus, the
percentage 34.3% obtained on average, is calculated from
the total events and not the parent populations.
About 3% of single living cells were negative for leukocyte-
(CD45), monocyte- (CD11b), endothelial- (CD31), and
erythroid-specific (TER119) markers (Figure 3D). CD11b/
CD45/CD31/TER119 negative cells were then selected
according to their expression of CD34 (hematopoietic,
endothelial progenitors, and mesenchymal stem cells) and
ITGA7 (cardiac, smooth, and skeletal muscle cells) markers
(Figure 3E). A final gating for CXCR4 (lymphocytes,
hematopoietic, and satellite cells) was performed to select
putative satellite cells (Figure 3F). From the CD34+/ITGA7+
cells, ~80% were found to be positive for CXCR4, which
represents an average of 1% ± 0.15% of total single living
cells, and an absolute number of 60,000 ± 14,000 putative
satellite cells per mouse limb muscles among 14 independent
experiments. An additional resorting assessment on the
CXCR4+ cell fraction revealed that about 80% of living cells
were obtained after post-sorting, of which almost 70% were
CXCR4+ (Figure S1), showing the high viability and purity of
this FACS-isolated cell population.
Since various studies compared the efficiency of collagenase
and Liberase TL enzymes for cell isolation
24 , 25
, these two
digestion methods have been processed in parallel. With 300
µL of Liberase TL, the digestion was less efficient than with
collagenase, as undigested fibers remained. In addition, more
cell debris and large events were observed (Figures S2A,B),
and only 17.3% of single living cells on average were obtained
after FVS 780 selection (Figure S2C). An additional concern
with Liberase digestion was the low number of CD34+/
ITGA7+ cells compared to collagenase (Figures S2D-S2E),
even though CXCR4 gating was similar (Figure S2F). With
600 µL of Liberase TL, the digestion was more efficient.
However, the amount of cell debris remained elevated and
the cell viability substandard (16.3%) (Figure S3). Thus,
digestion with Liberase TL was less efficient for satellite cell
isolation.
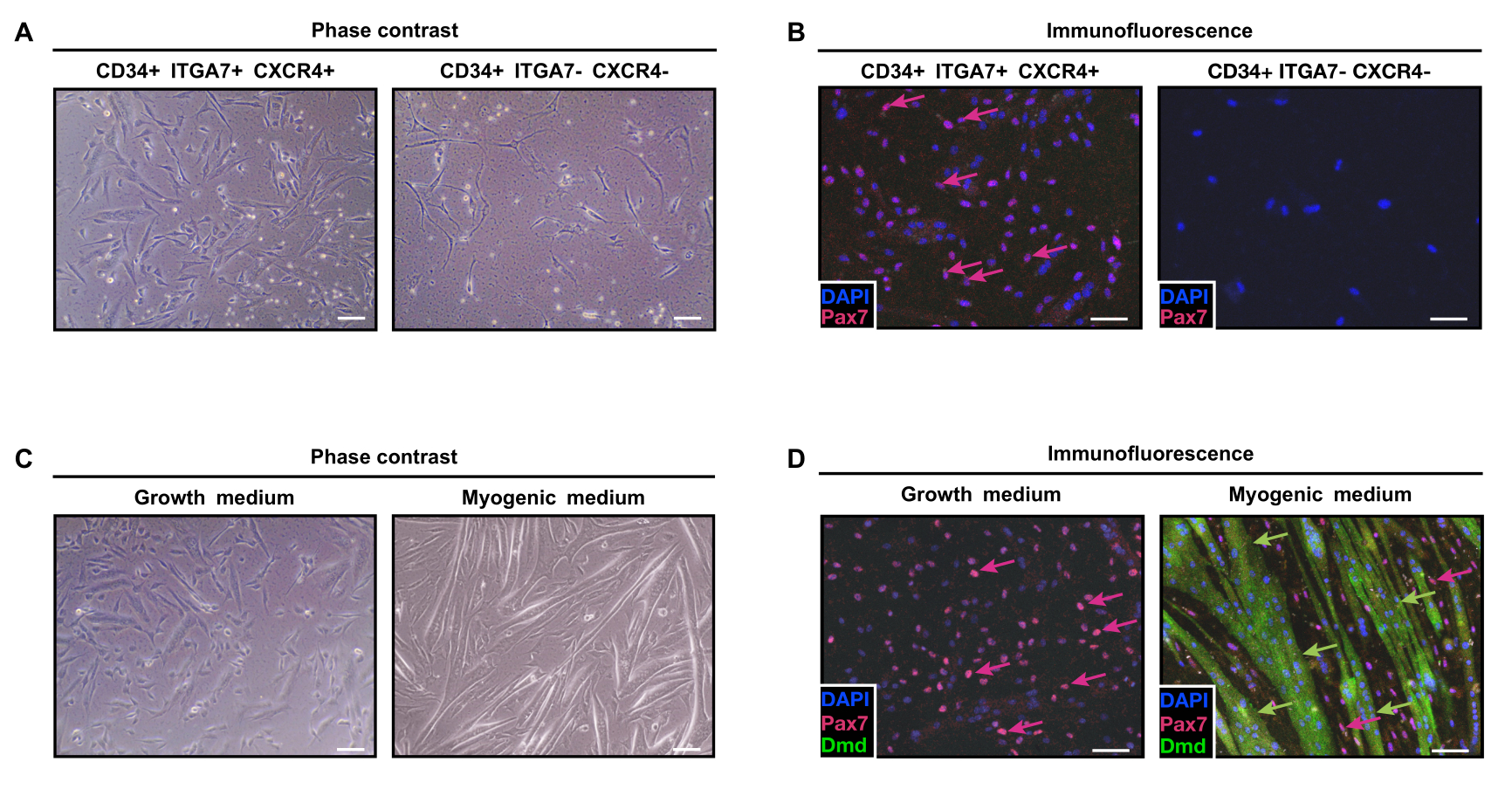
When seeded, more than 70% of the CD34+/ITGA7+/
CXCR4+ cells (Figure 4A) expressed PAX7 (Figure 4B),
contrary to CD34+/ITGA7-/CXCR4- cells that were PAX7-
negative (Figure 4C). CD34+/ITGA7+/CXCR4+ cells were
able to differentiate into myofibers when grown in a
myogenic medium for 7 additional days, as shown by
dystrophin (DMD) staining (Figure 4D), confirming their

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 10 of 21
myogenic potential. Thus, combined tissue culture and
immunofluorescence analyses showed that FACS-isolated
CD34+/ITGA7+/CXCR4+ cells are satellite cells.
To determine whether isolated satellite cells are suitable
for CUT&RUN analysis, the genomic profile of acetylated
lysine 27 (H3K27ac) and dimethylated lysine 4 (H3K4me2)
of histone H3, two histone modifications found at active
promoter and enhancer regions, was determined. Our data
uncovered 68,694 and 13,514 peaks for H3K4me2 and
H3K27ac, respectively, with a similar genomic repartition
between the two histone marks (Figure S4A). In detail, we
found that one-quarter of the peaks were located ± 2 kb
from the transcription start site (TSS) of the nearest gene,
the majority being located either -100 kb to -10 kb, or 10
kb to 100 kb from the TSS (Figure S4B). Of note, Pearson
analysis showed an 80% correlation between H3K27ac and
H3K4me2 read profiles (Figure S4C). To assess that the
chromatin prepared from the abovementioned protocol was
isolated from satellite cells, the presence of H3K27ac and
H3K4me2 at the promoter of satellite cell-specific genes was
determined. H3K4me2 was enriched around the TSS of Pax7,
Itga7, Lamb2, Cxcr4, and Vcam1 (Figure 5A), but not at those
of Itgam (CD11b) and Ptprc (CD45) (immune cells), Pecam
(CD31; endothelial cells) (Figure 5B), Ckm (developing
muscle fibers), or Myh3 (myosin heavy chain fast embryonic,
myofibers) (Figure 5C). Similar results were obtained with
H3K27ac (Figure 5). Almost no signal was obtained with
the IgG sample (Figure 5). Moreover, comparison of the
H3K27ac peaks obtained from SEACR with those resulting
from MACS2 peak calling from published ChIP-seq datasets
showed a high correlation, as noted below each panel (ChIP-
Atlas track; Figure 5). Together, these results indicate that the
reads obtained after the bioinformatics analysis originate from
the chromatin of FACS-isolated satellite cells and correlate
with those previously identified by ChIP-seq analyses.
While single-cell RNA sequencing studies in mouse satellite
cells showed a striking stress response, caused by the
isolation procedure
26
, the presence of H3K4me2 or H3K27ac
was determined at stress response genes, as exemplified
with Atf3, Azin1, Gls, and Elf2. The results provide evidence
that the H3K27ac mark is not deposited at the promoter
of such genes, and that levels of H3K4me2 remain low
compared to those obtained for satellite cell-specific genes
(Figure S5). Thus, these data highlight how mild the isolation
procedure is.
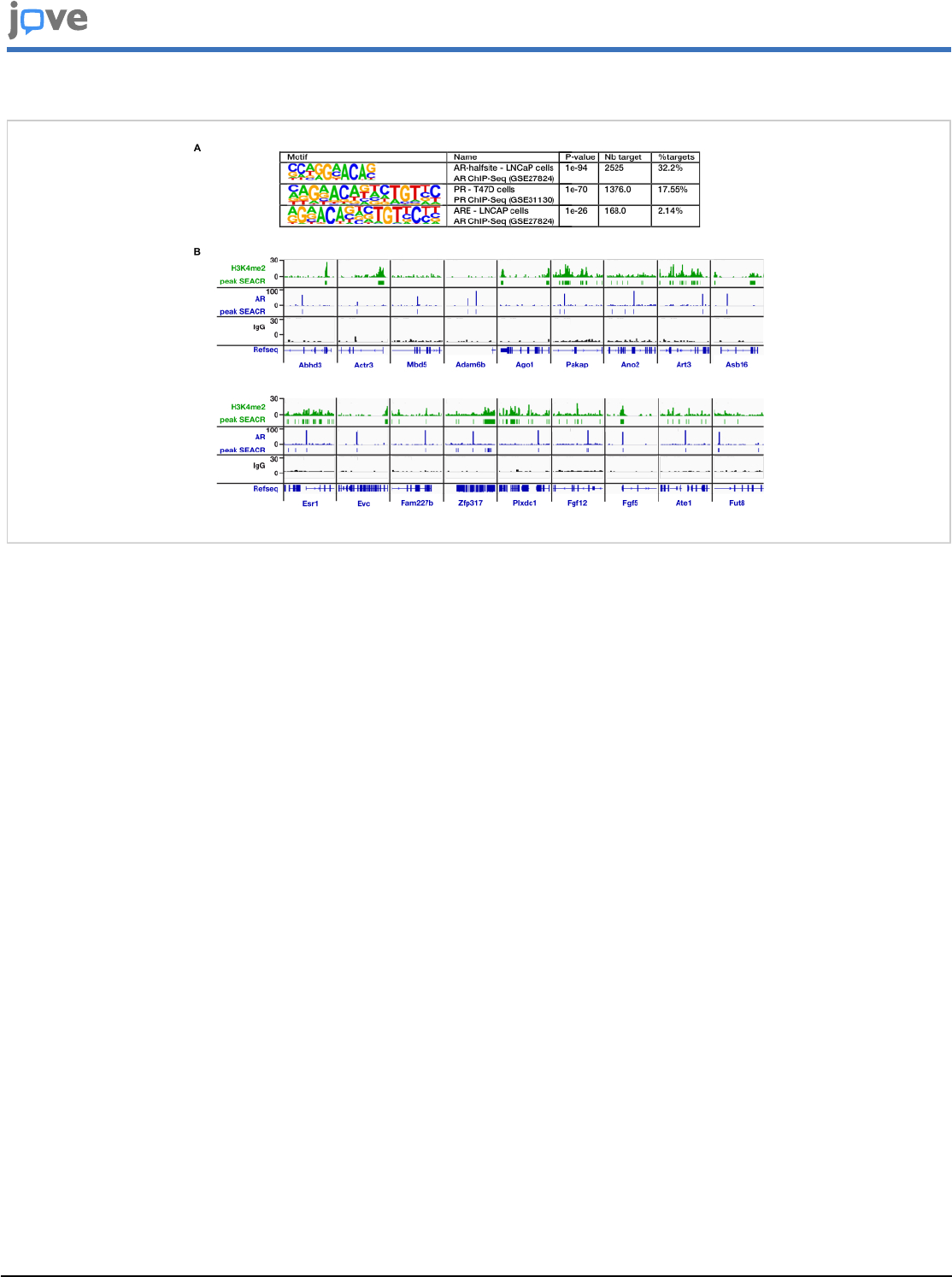
Next, the suitability of our isolation method for transcription
factors was examined. CUT&RUN analysis for the androgen
receptor (AR), a transcription factor that belongs to the
nuclear receptors superfamily and that plays an important
role in myogenic differentiation
27
, unraveled 7,840 peaks.
These peaks were mainly located at intron and intergenic
regions (Figure S4A), either -100 kb to -10 kb, or 10
kb to 100 kb from the TSS (Figure S4B). Phylogenetic
analyses revealed that AR, alongside the glucocorticoid
(GR/Nr3c1), the mineralocorticoid (MR/Nr3c2), and the
progesterone (PR/Nr3c3) receptors, is a member of the
oxosteroid nuclear receptors subfamily
28
, that bind as
homodimers to DNA segments which are made up of
two 5′-RGAACA-3′ palindromic half-sites separated by
three base pairs
29
. Searching for a known motif by
means of hypergeometric optimization of motif enrichment
(HOMER, http://homer.ucsd.edu/homer/) revealed that AR is
bound to the 5′-RGRNCA-3′ AR half-site motif, to the typical
5′-RGNACAnnnTGTNC-3′ oxosteroid motif (referred in the
figure as PR), and to the 5′-RGNACAnnnTGTNCY-3′ AR
consensus motif in >32%, 17%, and 2% of the targeted
Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 11 of 21
regions, respectively (Figure 6A). It should be noted
that similar proportions were previously obtained for the
glucocorticoid receptor in skeletal muscle
16
.
SEACR analysis unveiled almost 500 peaks with a peak
score >50, with more than 200 of them with a score >100;
some of which are exemplified in Figure 6B. Our analysis
also uncovered a modest enrichment of AR at the previously
described binding sites of genes involved in polyamine
biosynthesis (Amd1, Oat, and Smox) and in prostate cancer
(Acox1, Fkbp5, and Tmprss2) (Figure S6). Interestingly, the
AR was found at the loci of genes involved in satellite cell
stemness (Pax7, Cxcr4, and Cd34) (Figure S7). Of note, the
oxosteroid response element was found at each of these AR-
enriched regions (Figure S6 and Figure S7), demonstrating
that the AR signal seen in the CUT&RUN data is highly
specific.
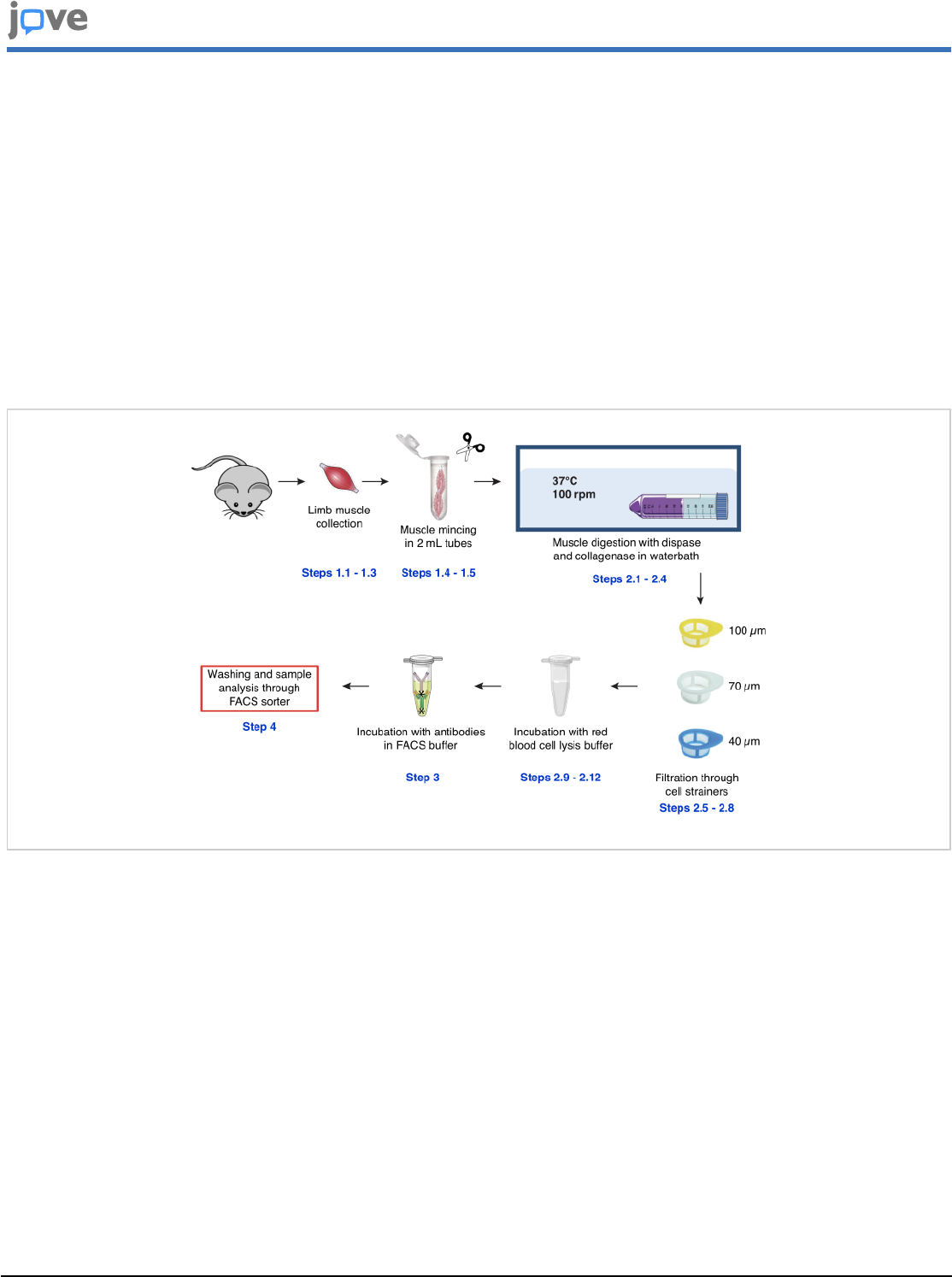
Figure 1: Schematic representation of the protocol used for satellite cell isolation from mouse limb muscles. The
protocol comprises four mains steps. Briefly, mice are sacrificed, and the hind limb muscles are harvested (steps 1.1.1-1.1.3)
and mechanically minced using scissors (steps 1.4-1.5). This is followed by steps 1.2.1-1.2.4, during which muscles are
dissociated in a water-bath using collagenase and dispase. In steps 1.2.5-1.2.8, the cell suspension is filtered through
100, 70, and 40 µm cell strainers, consecutively, and erythrocytes are eliminated using red blood cell lysis buffer (steps
1.2.9-1.2.12). The remaining cell populations are labelled in step 1.3 with antibodies coupled to fluorophores, which are
directed against specific cellular phenotypical markers. Step 1.4 includes samples passing through FACS to collect satellite
cells for further application. Please click here to view a larger version of this figure.

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 12 of 21
Figure 2: Flow cytometry analysis of an unstained cell preparation. (A) Selection of the population of interest based on
FSC-A and SSC-A parameters. (B) Single cell identification based on FSC-A and FSC-H. (C) Characterization of the auto-
fluorescence threshold of collected cells for APC-Cy7 conjugated to the fixable viability stain (FVS 780) marking dead cells.
(D-F). Auto-fluorescence measurement for PE-Cy7 conjugated to CD11b antibody and PE-conjugated to TER119/CD45/
CD31 markers (D), as well as for Alexa fluor 488 conjugated to ITGA7, Alexa fluor 405 conjugated to CD34 (E), and APC
fluorochrome conjugated to CXCR4 (F). Gates are represented as black boxes. Please click here to view a larger version of
this figure.
Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 13 of 21
Figure 3: Flow cytometer gating strategy for satellite cell sorting. (A) Selection of the population of interest based on
FSC-A and SSC-A parameters. (B) Single cell identification based on FSC-A and FSC-H. (C) Identification of living cells
with FVS 780. (D) Negative cell selection based on CD11b, CD31, CD45, and TER119 antigens. (E-F) Positive cell selection
based on CD34 and ITGA7 (E), as well as CXCR4 (F) antigens. Gates are represented as black boxes. Please click here to
view a larger version of this figure.

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 14 of 21
Figure 4: Analysis of FACS-isolated CD34+/ITGA7+/CXCR4+ and CD34+/ITGA7-/CXCR4- cells. (A) Phase contrast
images of CD34+/ITGA7+/CXCR4+ and CD34+/ITGA7-/CXCR4- cells cultured in growth medium. (B) Immunofluorescence
analysis of CD34+/ITGA7+/CXCR4+ and CD34+/ITGA7-/CXCR4- cells cultured in growth medium using antibodies directed
against PAX7 (red) and dystrophin (DMD, green). Nuclei were stained with DAPI (blue). Magenta arrows indicate PAX7-
positive cells. (C) Phase contrast images of CD34+/ITGA7+/CXCR4+ cells grown for 5 days in growth medium (left panel)
and for 7 additional days in myogenic medium (right panel). (D) Immunofluorescence analysis images of CD34+/ITGA7+/
CXCR4+ cells grown for 5 days in growth medium (left panel) and for 7 additional days in myogenic medium (right panel)
using antibodies directed against PAX7 (red) and dystrophin (DMD, green). Nuclei were stained with DAPI (blue). Magenta
arrows indicate PAX7-positive cells. Green arrows indicate DMD-positive cells. Scale bars: brightfield panels = 25 µm;
immunofluorescence panels = 50 µm. Please click here to view a larger version of this figure.

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 15 of 21
Figure 5: Genomic profiles of satellite cell chromatin. Localization of H3K4me2 and H3K27ac at satellite cell-specific
genes (A), immune and endothelial cell-specific genes (B), and myofiber-specific genes (C) on the chromatin of satellite cells
by CUT&RUN. Active promoters are boxed in green, whereas inactive promoters are boxed in brown. CUT&RUN performed
with IgG was used as a negative control. H3K27ac enrichment analysis for histone marks is presented below each track.
Enrichment score showing the levels of confidence of published datasets is depicted as a heatmap. Brown stars (*) refer
to H3K27ac ChIP-seq peaks performed in muscle satellite cells, blue stars to H3K4me3, and the pink one to H4K16me1.
Please click here to view a larger version of this figure.
Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 16 of 21
Figure 6: AR genomic distribution on satellite cell chromatin. (A) HOMER-known motif analysis of AR peaks in satellite
cells. PR: progesterone receptor. Nb target refers to the number of peaks presenting a particular motif. (B) Localization of
H3K4me2 and AR at indicated genes on the chromatin of satellite cells determined by CUT&RUN. CUT&RUN performed
with IgG was used as a negative control. Please click here to view a larger version of this figure.
Table 1: List of materials, reagents, and software. Please
click here to download this Table.
Table 2: Buffer compositions. Please click here to
download this Table.
Table 3: Antibody references and concentrations. Please
click here to download this Table.
Supplementary Figure 1: Flow cytometry analysis
of the cell preparation post-sorting. (A) Selection of
the population of interest based on FSC-A and SSC-A
parameters. (B) Single cell identification based on FSC-
A and FSC-H. (C) Identification of living cells with fixable
viability stain (FVS 780). (D) Negative cell selection based on
CD11b, CD31, CD45, and TER119 antigens. (E-F) Positive
cell selection based on CD34 and ITGA7 (E) as well as
CXCR4 (F) antigens. Gates are represented as black boxes.
Please click here to download this File.
Supplementary Figure 2: Flow cytometer gating strategy
for satellite cell sorting after digestion with 300 µL of
Liberase TL. (A) Selection of the population of interest
based on FSC-A and SSC-A parameters. (B) Single cell
identification based on FSC-A and FSC-H. (C) Identification of
living cells with FVS 780. (D) Negative cell selection based on
CD11b, CD31, CD45, and TER119 antigens. (E-F) Positive
cell selection based on CD34 and ITGA7 (E) as well as
CXCR4 (F) antigens. Gates are represented as black boxes.
Please click here to download this File.
Supplementary Figure 3: Flow cytometer gating strategy
for satellite cell sorting after digestion with 600 µL of

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 17 of 21
Liberase TL. (A) Selection of the population of interest
based on FSC-A and SSC-A parameters. (B) Single cell
identification based on FSC-A and FSC-H. (C) Identification of
living cells with FVS 780. (D) Negative cell selection based on
CD11b, CD31, CD45, and TER119 antigens. (E-F) Positive
cell selection based on CD34 and ITGA7 (E) as well as
CXCR4 (F) antigens. Gates are represented as black boxes.
Please click here to download this File.
Supplementary Figure 4: Characterization of H3K4me2,
H3K27ac, and AR genomic locations. (A) Pie charts
depicting the peak distribution of H3K4me2, H3K27ac, and
AR according to genome features in satellite cells. (B) Pie
charts depicting the peak distribution of H3K4me2, H3K27ac,
and AR according to their distance to the nearest TSS in
satellite cells. (C) Heatmap showing the Pearson correlation
between H3K4me2, H3K27ac, and IgG control. Please click
here to download this File.
Supplementary Figure 5: Genomic profiles of satellite cell
chromatin at stress-response induced genes. Localization
of H3K4me2 and H3K27ac at indicated genes on the
chromatin of satellite cells. Immunoprecipitation with IgG was
used as a negative control. Please click here to download this
File.
Supplementary Figure 6: Genomic profiles of satellite
cell chromatin at known AR target genes. Localization
of H3K4me2, H3K27ac, and AR at indicated genes on the
chromatin of satellite cells determined by CUT&RUN. AR
peaks are boxed in blue, and corresponding AR-responsive
elements are presented below. CUT&RUN performed with
IgG was used as a negative control. Please click here to
download this File.
Supplementary Figure 7: Genomic profiles of satellite
cell chromatin at their selectively expressed genes.
Localization of H3K4me2, H3K27ac, and AR at indicated
genes on the chromatin of satellite cells determined by
CUT&RUN. AR peaks are boxed in blue, and corresponding
AR-responsive elements are presented below. CUT&RUN
performed with IgG was used as a negative control. Please
click here to download this File.
Discussion
The present study reports a standardized, reliable, and easy-
to-perform method for the isolation and culture of mouse
satellite cells, as well as the assessment of transcriptional
regulation by the CUT&RUN method.
This protocol involves several critical steps. The first is
muscle disruption and fiber digestion to ensure a high
number of collected cells. Despite the increased enzyme
concentration, more living cells were obtained than using
Protocol 1. Satellite cells express a specific pattern of
various membrane proteins. To increase the stringency of
our sorting, we used a combination of previously described
negative (CD31, TER119, CD45, and CD11b) and positive
(CD34, ITGA7, and CXCR4) satellite cell markers
30 , 31
.
Using this strategy, an average of 1% of living putative
satellite cells was obtained. This outcome is in the range
of what was expected, since satellite cells represent 2%-7%
of muscle cell population at adulthood in mice
32
. As a
control for the satellite cell selection markers, CD34+/ITGA7-/
CXCR4- cells were isolated. Immunofluorescent staining
demonstrated that, whereas CD34+/ITGA7-/CXCR4- cells
did not express PAX7, 70% of CD34+/ITGA7+/CXCR4+
sorted cells were positive for this satellite cell-specific marker,
thereby demonstrating that the FACS isolated population
corresponds to satellite cells. In addition, 70% of satellite

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 18 of 21
cells grown in myogenic medium for 7 days were PAX7-/DMD
+, as shown by immunostaining, and formed an elongated
multinucleated myofiber, demonstrating that isolated satellite
cells conserved their stemness potential.
The major limitation in sample preparation might be the use
of a high concentration of enzymes, which results in a large
amount of dissociated biological material, but may contribute
to increased cell death. To resolve this issue, an assay
requiring lower enzyme quantity was tested. In this context,
Liberase TL, a purified form of the traditional collagenase
33
,
was tested. Although digestion was apparently more efficient
with this enzyme, the amount of cell debris remained
elevated, and the cell viability was slightly lowered. These
observations are in agreement with previous reports that
compared Liberase-mediated tissue digestion to this with
recombinant collagenase or custom collagenase
24 , 25
. In
addition, even though the proportion of endothelial and
immune cells was similar between collagenase and Liberase
digestion, the percentage of satellite cells was lower
in the Liberase-processed sample, overall showing that
Liberase is not recommended for satellite cell isolation. The
use of this enzyme is suitable for the phenotypic and
quantitative analysis of immune cells and could eventually be
recommended in the context of muscle and/or other tissues.
This is in concordance with what has been reported on
Liberase TL as the most suited to effectively isolate viable
immune cells
34 , 35 , 36
.
Another potential issue is the stress caused by both
dissociating and sorting satellite cells. However, the absence
of H3K27ac and the low H3K4me2 levels at the promoter
region of stress response genes identified by single-cell
RNA sequencing in mouse satellite cells
26
show that the
procedure is mild enough not to induce the stress response
transcriptional repertoire. Other limitations to be noted
are the selected antigens that remain empiric. However,
studies show a high overlap between unique surface marker
combinations that are enriched for skeletal muscle satellite
cells
30 , 31
.
An additional critical step is the purification of nuclei from
sorted cells. For this, the sorting speed is maintained low in
order not to damage the cells, but fast enough that they are
not stored for too long in FACS buffer. Indeed, CUT&RUN
nuclei are not fixed, and a longer incubation time might
influence the read obtained from the protocol. A typical
preparation from the two hind limbs of one mouse allows
CUT&RUN with one antibody and one IgG control, with an
amount of 2.5 ng of chromatin for each.
CUT&RUN experiments, designed to assess transcription
factor binding and epigenetic modifications, provided strong
and robust read signals for H3K4me2 and H3K27ac. The
high read levels of H3K4me2 and H3K27ac on genes that
are known to be expressed in satellite cells, compared to
genes expressed in immune or endothelial cells as well
as in myofibers, showed that the signal was amplified
predominantly from the satellite cells nuclei. In addition, this
protocol made the identification of the cistrome of the AR in
muscle stem cells possible, which to date remains a technical
issue inherent to the poor quality of mouse AR antibodies
and the low AR expression levels in cells that are not highly
androgen-sensitive, such as epithelial prostate cells. The
data presented here unveiled AR-bound regions with high
confidence peak scores. As only one replicate was originally
assessed per antibody, the robustness of our datasets will
be improved by adding at least two additional biological
replicates per condition. However, AR known target genes
and respective histone marks, originally described as highly

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 19 of 21
expressed and/or easily detectable in other tissue contexts
such as the prostate, present lower expression levels and are
very challenging to detect in satellite cells.
One limitation of the CUT&RUN approach is that it is
performed in unfixed cells. Thus, we might have missed
some AR targets because of the labile nature of the
interactions between transcription factors and their binding
site. Similarly, some post-translational modifications, such as
acetylation, can also be rather labile. Therefore, including a
light fixation step with formaldehyde or paraformaldehyde,
after FACS sorting, and before isolating the nuclei, could
be considered to stabilize the protein/DNA interactions and
histone modifications.
Although this paper shows that CUT&RUN assays can be
conducted on FACS-isolated satellite cells, other analyses
that use non-fixed chromatin such as ATAC-seq might also
be performed. In addition, even though here muscles were
harvested in basal physiological conditions, this protocol may
be applied to pathological contexts such as injury or aging.
All these protocol uses will allow a detailed understanding of
gene regulatory mechanisms in satellite cells, and may help
in understanding how these mechanisms might be altered by
pathophysiological conditions.
In summary, this low-cost and time-efficient protocol provides
an effective experimental setting to study transcription factor
recruitment and chromatin landscape in skeletal muscle
precursor cells. Chromatin prepared by this protocol has
provided the first genome-wide analysis of AR cistrome
in satellite cells and will facilitate future studies on gene
regulation.
Disclosures
The authors declare that they have no competing financial
interests.
Acknowledgments
We thank Anastasia Bannwarth for providing excellent
technical assistance. We thank the IGBMC animal house
facility, the cell culture, the Mouse Clinical Institute (ICS,
Illkirch, France), the imaging, the electron microscopy, the
flow cytometry, and the GenomEast platform, a member of
the 'France Génomique' consortium (ANR-10-INBS-0009).
This work of the Interdisciplinary Thematic Institute IMCBio,
as part of the ITI 2021-2028 program of the University
of Strasbourg, CNRS and Inserm, was supported by
IdEx Unistra (ANR-10-IDEX-0002) and by SFRI-STRAT'US
project (ANR 20-SFRI-0012) and EUR IMCBio (ANR-17-
EURE-0023) under the framework of the French Investments
for the Future Program. Additional funding was delivered
by INSERM, CNRS, Unistra, IGBMC, Agence Nationale
de la Recherche (ANR-16-CE11-0009, AR2GR), AFM-
Téléthon strategic program 24376 (to D.D.), INSERM young
researcher grant (to D.D.), ANR-10-LABX-0030-INRT, and a
French State fund managed by the ANR under the frame
program Investissements d'Avenir (ANR-10-IDEX-0002-02).
J.R. was supported by the Programme CDFA-07-22 from the
Université franco-allemande and Ministère de l'Enseignement
Supérieur de la Recherche et de l'Innovation, and K.G. by the
Association pour la Recherche à l'IGBMC (ARI).
References
1. Frontera, W. R., Ochala, J. Skeletal muscle: a brief
review of structure and function. Calcified Tissue
International. 96 (3), 183-195 (2015).

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 20 of 21
2. Tedesco, F. S., Dellavalle, A., Diaz-Manera, J., Messina,
G., Cossu, G. Repairing skeletal muscle: regenerative
potential of skeletal muscle stem cells. The Journal of
Clinical Investigation. 120 (1), 11-19 (2010).
3. Mauro, A. Satellite cell of skeletal muscle fibers. The
Journal of Biophysical and Biochemical Cytology. 9 (2),
493-495 (1961).
4. Buckingham, M. Skeletal muscle progenitor cells and the
role of Pax genes. Comptes Rendus Biologies. 330 (6-7),
530-533 (2007).
5. Tosic, M. et al. Lsd1 regulates skeletal muscle
regeneration and directs the fate of satellite cells. Nature
Communications. 9 (1), 366 (2018).
6. Kuang, S., Gillespie, M. A., Rudnicki, M. A. Niche
regulation of muscle satellite cell self-renewal and
differentiation. Cell Stem Cell. 2 (1), 22-31 (2008).
7. Collins, C. A. et al. Stem cell function, self-renewal, and
behavioral heterogeneity of cells from the adult muscle
satellite cell niche. Cell. 122 (2), 289-301 (2005).
8. Robinson, D. C. L. et al. Negative elongation factor
regulates muscle progenitor expansion for efficient
myofiber repair and stem cell pool repopulation.
Developmental Cell. 56 (7), 1014-1029, (2021).
9. Machado, L. et al. In situ fixation redefines quiescence
and early activation of skeletal muscle stem cells. Cell
Reports. 21 (7), 1982-1993 (2017).
10. Hainer, S. J., Fazzio, T. G. High-resolution chromatin
profiling using CUT&RUN. Current Protocols in
Molecular Biology. 126 (1), e85 (2019).
11. Meers, M. P., Bryson, T. D., Henikoff, J. G., Henikoff, S.
Improved CUT&RUN chromatin profiling tools. eLife. 8,
(2019).
12. Gunther, S. et al. Myf5-positive satellite cells contribute to
Pax7-dependent long-term maintenance of adult muscle
stem cells. Cell Stem Cell. 13 (5), 590-601 (2013).
13. Donlin, L. T. et al. Methods for high-dimensional analysis
of cells dissociated from cryopreserved synovial tissue.
Arthritis Research & Therapy. 20 (1), 139 (2018).
14. Rico, L. G. et al. Accurate identification of cell doublet
profiles: Comparison of light scattering with fluorescence
measurement techniques. Cytometry. Part A. 103 (3),
447-454 (2022).
15. Schreiber, V. et al. Extensive NEUROG3 occupancy
in the human pancreatic endocrine gene regulatory
network. Molecular Metabolism. 53, 101313 (2021).
16. Rovito, D. et al. Myod1 and GR coordinate myofiber-
specific transcriptional enhancers. Nucleic Acids
Research. 49 (8), 4472-4492 (2021).
17. Langmead, B., Salzberg, S. L. Fast gapped-read
alignment with Bowtie 2. Nature Methods. 9 (4), 357-359
(2012).
18. Meers, M. P., Tenenbaum, D., Henikoff, S. Peak calling
by Sparse Enrichment Analysis for CUT&RUN chromatin
profiling. Epigenetics Chromatin. 12 (1), 42 (2019).
19. Ramirez, F. et al. deepTools2: a next generation web
server for deep-sequencing data analysis. Nucleic Acids
Research. 44 (W1), W160-W165 (2016).
20. Thorvaldsdottir, H., Robinson, J. T., Mesirov, J. P.
Integrative Genomics Viewer (IGV): high-performance
genomics data visualization and exploration. Briefings in
Bioinformatics. 14 (2), 178-192 (2013).
21. Heinz, S. et al. Simple combinations of lineage-
determining transcription factors prime cis-regulatory

Copyright © 2023 JoVE Journal of Visualized Experiments
jove.com
July 2023
•
197
•
e65215
•
Page 21 of 21
elements required for macrophage and B cell identities.
Molecular Cell. 38 (4), 576-589 (2010).
22. Zou, Z., Ohta, T., Miura, F., Oki, S. ChIP-Atlas 2021
update: a data-mining suite for exploring epigenomic
landscapes by fully integrating ChIP-seq, ATAC-seq and
Bisulfite-seq data. Nucleic Acids Research. 50 (W1),
W175-W182 (2022).
23. Liu, L., Cheung, T. H., Charville, G. W., Rando,
T. A. Isolation of skeletal muscle stem cells by
fluorescence-activated cell sorting. Nature Protocols. 10
(10), 1612-1624 (2015).
24. Brandhorst, H. et al. Successful human islet isolation
utilizing recombinant collagenase. Diabetes. 52 (5),
1143-1146 (2003).
25. Nikolic, D. M. et al. Comparative analysis of collagenase
XI and liberase H1 for the isolation of human pancreatic
islets. Hepatogastroenterology. 57 (104), 1573-1578
(2010).
26. Machado, L. et al. Tissue damage induces a conserved
stress response that initiates quiescent muscle stem cell
activation. Cell Stem Cell. 28 (6), 1125-1135, (2021).
27. Diel, P., Baadners, D., Schlupmann, K., Velders, M.,
Schwarz, J. P. C2C12 myoblastoma cell differentiation
and proliferation is stimulated by androgens and
associated with a modulation of myostatin and Pax7
expression. Journal of Molecular Endocrinology. 40 (5),
231-241 (2008).
28. Gronemeyer, H., Gustafsson, J. A., Laudet, V. Principles
for modulation of the nuclear receptor superfamily.
Nature Reviews Drug Discovery. 3 (11), 950-964 (2004).
29. Billas, I., Moras, D. Allosteric controls of nuclear receptor
function in the regulation of transcription. Journal of
Molecular Biology. 425 (13), 2317-2329 (2013).
30. Garcia-Prat, L. et al. FoxO maintains a genuine muscle
stem-cell quiescent state until geriatric age. Nature Cell
Biology. 22 (11), 1307-1318 (2020).
31. Maesner, C. C., Almada, A. E., Wagers, A. J.
Established cell surface markers efficiently isolate highly
overlapping populations of skeletal muscle satellite cells
by fluorescence-activated cell sorting. Skeletal Muscle.
6, 35 (2016).
32. Schultz, E. A quantitative study of the satellite cell
population in postnatal mouse lumbrical muscle. The
Anatomical Record. 180 (4), 589-595 (1974).
33. Hyder, A. Effect of the pancreatic digestion with liberase
versus collagenase on the yield, function and viability of
neonatal rat pancreatic islets. Cell Biology International.
29 (9), 831-834 (2005).
34. Liang, F. et al. Dissociation of skeletal muscle for
flow cytometric characterization of immune cells in
macaques. Journal of Immunological Methods. 425,
69-78 (2015).
35. Park, J. Y., Chung, H., Choi, Y., Park, J. H. Phenotype
and tissue residency of lymphocytes in the murine oral
mucosa. Frontiers in Immunology. 8, 250 (2017).
36. Skulska, K., Wegrzyn, A. S., Chelmonska-Soyta, A.,
Chodaczek, G. Impact of tissue enzymatic digestion
on analysis of immune cells in mouse reproductive
mucosa with a focus on gammadelta T cells. Journal of
Immunological Methods. 474, 112665 (2019).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}