
Alsford, S; Horn, D (2011) Elongator Protein 3b Negatively Regulates
Ribosomal DNA Transcription in African Trypanosomes. Molecular
and cellular biology, 31 (9). pp. 1822-32. ISSN 0270-7306 DOI:
10.1128/MCB.01026-10
Downloaded from:
http://researchonline.lshtm.ac.uk/1261/
DOI: 10.1128/MCB.01026-10
Usage Guidelines
Please refer to usage guidelines at http://researchonline.lshtm.ac.uk/policies.html or alterna-
tively contact researchonline@lshtm.ac.uk.
Available under li cense: http://creativecommons.org/licenses/by-nc-nd/2.5/

MOLECULAR AND CELLULAR BIOLOGY, May 2011, p. 1822–1832 Vol. 31, No. 9
0270-7306/11/$12.00 doi:10.1128/MCB.01026-10
Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Elongator Protein 3b Negatively Regulates Ribosomal DNA
Transcription in African Trypanosomes
䌤
†
Sam Alsford and David Horn*
London School of Hygiene and Tropical Medicine, Keppel Street, London WC1E 7HT, United Kingdom
Received 1 September 2010/Returned for modification 28 September 2010/Accepted 16 February 2011
Eukaryotic cells limit ribosomal DNA (rDNA) transcription by RNA polymerase I (RNAP-I) to maintain
genome integrity. African trypanosomes present an excellent model for studies on RNAP-I regulation because
they possess a bifunctional RNAP-I and because RNAP-II transcription appears unregulated. Since Elp3, the
catalytic component of Elongator, controls RNAP-II transcription in yeast and human cells, we predicted a role
for a trypanosome Elp3-related protein, ELP3a or ELP3b, in RNAP-I regulation. elp3b null and conditional
strains specifically exhibited resistance to a transcription elongation inhibitor, suggesting that ELP3b nega-
tively impacts elongation. Nascent RNA analysis and expression of integrated reporter cassettes supported this
interpretation and revealed negative control of rDNA transcription. ELP3b specifically localized to the nucle-
olus, and ELP3b loss rendered cells hypersensitive to DNA damage and to translation inhibition, suggesting
that anti-Elongator function was important to maintain genome integrity rather than to modulate ribosome
production. Finally, ELP3b displayed discrimination between RNAP-I compartments in the same cell. Our
results establish ELP3b as a major negative regulator of rDNA transcription and extend the roles of the
Elp3-related proteins to RNAP-I transcription units. ELP3b is also the first trypanosome protein shown
to distinguish between rDNA and variant surface glycoprotein transcription within different RNAP-I
compartments.
In eukaryotic cells, RNA polymerase II (RNAP-II) tran-
scription regulation appears to operate predominantly at the
level of elongation (18), and several factors that influence this
process have been described in human cells and in model
organisms (43). Much less is known about RNAP-I control, but
cells do limit ribosomal DNA (rDNA) transcription, and this is
important for the maintenance of genome integrity (22); extra
copies of rDNA allow for reduced transcription, which facili-
tates repair.
Trypanosomatids are protozoa that branched early from the
eukaryotic lineage and are important human and animal patho-
gens which are emerging as model organisms for the study of
epigenetic regulation (13). In trypanosomatids, RNAP-II
transcription of protein-coding genes is polycistronic and
apparently constitutive (10). In the bloodstream-form Afri-
can trypanosome Trypanosoma brucei, RNAP-I is bifunc-
tional, directing transcription of rDNA in the nucleolus and
monotelomeric transcription of a variant surface glycoprotein
(VSG) gene in an expression site body (ESB) that is distinct
from the nucleolus (32). RNAP-I is readily able to contribute
to mRNA production in trypanosomatids (45) because all ma-
ture mRNAs are fused to an RNAP-II-transcribed, trans-
spliced leader sequence (19). The high rate of transcription
achieved by RNAP-I may be important for VSG expression
since T. brucei bloodstream-form cells derive ⬎10% of total
cellular protein from a single VSG gene. These features lead us
to predict that conserved factors involved in RNAP-II tran-
scription elongation control in other eukaryotes, such as Elon-
gator, might function in RNAP-I control in trypanosomes.
Saccharomyces cerevisiae Elongator (49) associates with
elongating RNA polymerase II (RNAP-II) with a hyperphos-
phorylated carboxyl-terminal domain (37). The catalytic sub-
unit of the six-subunit Elongator complex (26), Elp3 (Elonga-
tor protein 3, also called KAT9), appears to provide a direct
link between histone acetylation and transcription by facilitat-
ing RNAP-II elongation in a chromatin- and acetyl coenzyme
A (acetyl-CoA)-dependent manner (56, 57). Human Elongator
also facilitates RNAP-II elongation through chromatin and
displays histone acetyltransferase activity with specificity for
lysine residues in the N-terminal tail of histone H3 (25). More
recently, S. cerevisiae Elp3 was shown to modulate transcrip-
tional silencing at telomeres and to modulate DNA repair
through an interaction with proliferating cell nuclear antigen
(28). As well as a GNAT-type acetyltransferase domain, Elp3
contains a radical S-adenosylmethionine (SAM) domain with
an iron-sulfur (FeS) cluster. Recent evidence points to a role
for the mouse Elp3 radical SAM domain in DNA demethyla-
tion (36). Surprisingly, S. cerevisiae Elongator (40, 41) and
human Elongator (25) localize predominantly to the cyto-
plasm, and roles have also been reported in exocytosis (41) and
tRNA metabolism in S. cerevisiae (21) and in tubulin acetyla-
tion in mouse neurons (11).
There are two Elp3-related proteins in trypanosomatids,
and we predicted a role for one or both of these proteins in
RNAP-I regulation. Here, we demonstrate that T. brucei
ELP3b negatively regulates rDNA transcription. This conclu-
sion is supported by elongation inhibitor resistance, increased
nascent rDNA transcription, and increased rDNA-integrated
* Corresponding author. Mailing address: London School of Hy-
giene and Tropical Medicine, Keppel Street, London WC1E 7HT,
United Kingdom. Phone: (44) 20 7927 2352. Fax: (44) 20 7636 8739.
E-mail: [email protected].
䌤
Published ahead of print on 28 February 2011.
† The authors have paid a fee to allow immediate free access to
this article.
1822
reporter expression in elp3b-depleted strains and ELP3b nu-
cleolar localization. Remarkably, ELP3b selectively controls
rDNA transcription, indicating an ability to distinguish be-
tween different RNAP-I transcription units.
MATERIALS AND METHODS
Trypanosomes. Bloodstream forms of T. brucei, MiTat 1.2 clone 221a, were
maintained, transfected, and differentiated as previously described (3). At least
5 h after transfection, transformants were exposed to puromycin (2 gml
⫺1
;
phosphonoacetic acid [PAA]), blasticidin (10 gml
⫺1
), hygromycin (2.5 g
ml
⫺1
), or phleomycin (2 gml
⫺1
) selection, as appropriate. ELP3b disruption
was confirmed by PCR and Southern blotting, using standard protocols (5). For
conditional strains, pHD1313 (Tet-R) (1), was integrated at the -tubulin locus
and pRP
iGFP
ELP3b was integrated at an rDNA locus in an ELP3b heterozygous
strain prior to disruption of the second native allele of ELP3b. Transformants
were screened by immunoblotting and immunofluorescence to confirm robust
regulated expression, and two strains were transformed with the final disruption
construct in the presence of tetracycline (Tet; 1 gml
⫺1
) to generate four
conditional strains. For growth assays, cultures were seeded at ⬃10
5
ml
⫺1
and
diluted back every 24 h. Cell counts were made using a hemocytometer and
carried out over at least 72 h. Cumulative counts were used to calculate popu-
lation doubling times, assuming exponential growth. For half-maximal effective
concentration (EC
50
) determinations, cells were seeded at 2 ⫻ 10
3
ml
⫺1
in
96-well plates in a 2-fold dilution series of drug. After ⬃3 days of growth, 20 l
of Alamar blue (AbD Serotec) was added to each well, and the plates were
incubated for a further 7 h. Fluorescence was determined using a fluorescence
plate reader (Molecular Devices) at an excitation wavelength of 530 nm, an
emission wavelength of 585 nm, and a filter cutoff of 570 nm (42). All assays were
carried out in the absence of any additional antibiotics.
Plasmid construction. Enhanced green fluorescent protein (eGFP) and cMYC
fusions were generated using the pRP (ELP3a, ELP3b, RPB6z, and RPB6) and
pNAT (SNAP42 and RPC160) vectors (2). Protein-coding sequences were am-
plified from T. brucei genomic DNA using Phusion polymerase (New England
BioLabs). Primers were designed using the publicly available T. brucei genome
sequence (www.genedb.org/genedb/tryp/). For the generation of ELP3b disrup-
tion constructs, targeting fragments were amplified from genomic DNA and
cloned into pBLA (blasticidin S deaminase) and pPAC (puromycin N-acetyl-
transferase). ELP3a targeting fragments were cloned into pPAC; the PAC gene
was subsequently replaced with BLE and HYG to generate additional disruption
constructs. All transcription run-on probes were cloned in pBluescript (Strata-
gene) or pGEM-T Easy (Promega) with the exception of R3, which proved
refractory to cloning. All primer sequences are available upon request.
Protein analysis. Immunoblotting was carried out following SDS-PAGE of
whole-cell lysates and electroblotting using standard protocols (5) and an en-
hanced chemiluminescence kit (Amersham), according to the manufacturer’s
instructions. Immunofluorescence analysis was carried out on fixed cells settled
onto slides pretreated with 3-aminopropyl triethoxysilane (Sigma) and pro-
cessed as previously described (4). eGFP and cMYC fusions were detected
with rabbit polyclonal anti-GFP (Molecular Probes) or mouse monoclonal
anti-GFP (AbCam) and mouse anti-cMYC (9E10; Santa Cruz Biotechnology),
respectively. NOG1 and NUP1 were detected with rabbit anti-NOG1 (38) and
mouse anti-NUP1 (35), respectively. Images were captured using a Nikon Eclipse
E600 epifluorescence microscope in conjunction with a Coolsnap FX (Photo-
metrics) charge-coupled device (CCD) camera and processed in Metamorph 5.0
(Photometrics) and Adobe Photoshop elements 2.0 (Adobe).
Transcript analysis. Total RNA was isolated using an RNeasy kit (Qiagen),
fractionated on 5% polyacrylamide-7 M urea or 1.5% agarose-formaldehyde
gels, and analyzed by Northern blotting according to standard protocols (5).
Transcription run-on analysis was carried out using an adapted lysolecithin
permeabilization protocol (51). Briefly, ⬃2 ⫻ 10
8
cells were washed in transcrip
-
tion buffer (TB; 20 mM
L-glutamic acid monopotassium salt, 3 mM MgCl
2
, 150
mM sucrose, 1 mM dithiothreitol [DTT], 10 g ml 1-leupeptin [Sigma]) and
incubated for1min0.4mlTBcontaining 0.2 mg lysolecithin (
L-␣-lysophos-
phatidylcholine palmitoyl [Sigma]) on ice. Permeabilized cells were washed in TB
and then placed in labeling mix (20 mM
L-glutamic acid [monopotassium salt], 3
mM MgCl
2
, 1 mM DTT, 10 gml
⫺1
leupeptin, 25 mM creatine phosphate, 0.6
gml
⫺1
creatine phosphokinase [type I, rabbit muscle; Sigma], 2 mM ATP, 1
mM CTP, 1 mM GTP [Fermentas], 100 Ci [␣-
32
P]UTP [Perkin-Elmer]) in a
final volume of 200 l for 15 m at 37°C. Total RNA was isolated using an RNeasy
kit (Qiagen). Slot blots were generated in sets of three and included seven rDNA
probes: R1 (rDNA promoter; ⫺244 to ⫹255), R2 (including the rDNA promoter
and the first 400 bp of small-subunit [SSU] rDNA; RH6), R3 (⫹1 to 3310,
including SSU rDNA), R4 (⫹3339 to 6389, including 5.8S and large-subunit ␣
[LSU␣] rDNA), R5 (⫹6444 to 9455, including LSU rDNA), R6 (nontran-
scribed rDNA spacer), and 5S rDNA; four mRNA-associated probes, VSG2,
-tubulin, Procyclin, and spliced-leader RNA (SL-RNA); and a pBluescript neg-
ative control. Three micrograms of each plasmid (or 1.5 g of purified R3 PCR
product) was denatured in 0.4 M NaOH, transferred to nylon membrane under
vacuum, and UV cross-linked. Total labeled RNA was hybridized to the slot blots
overnight at 65°C and subsequently washed and processed according to standard
protocols (5). Hybridization signals were detected using a Typhoon phosphor-
imager (Amersham), quantified using ImageQuant (Amersham), and analyzed in
MS Excel.
RESULTS
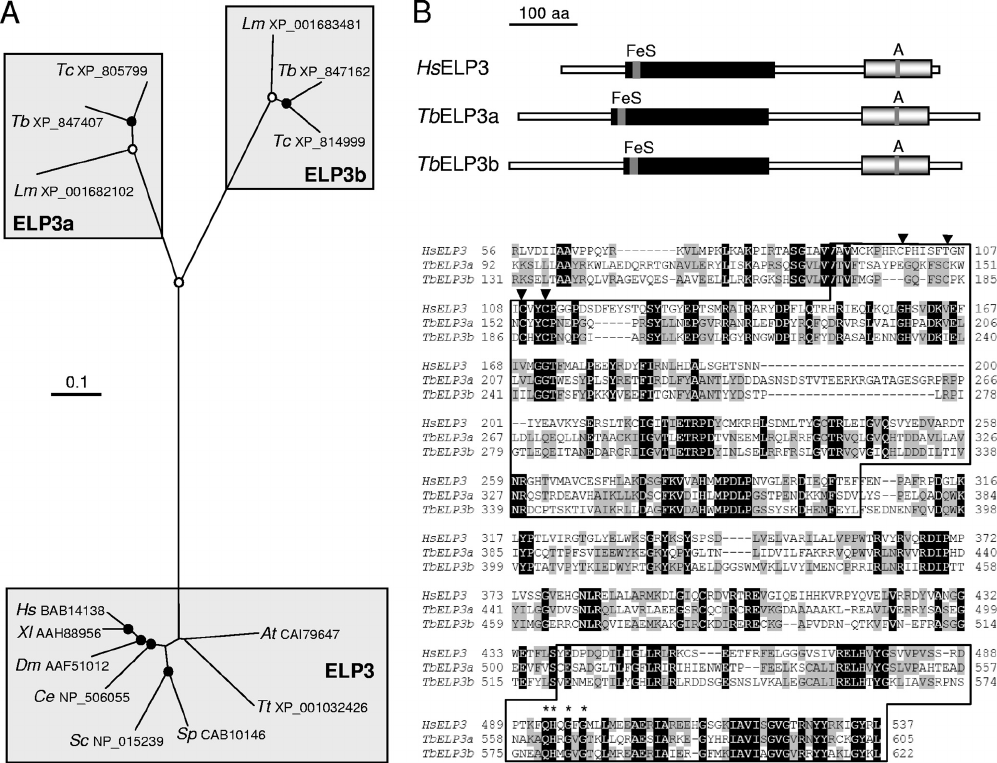
Trypanosomatid genomes encode two Elp3-related proteins.
Elp3 orthologues are conserved from archaea to humans. Pairs
of Elp3 orthologues were identified in T. brucei, Trypano-
soma cruzi, and Leishmania major (23), but we found no
organism beyond the trypanosomatids with more than one
Elp3 orthologue. As expected from the position in the Ex-
cavata, the trypanosomatid Elp3 orthologues, designated
ELP3a and ELP3b, are diverged relative to Elp3 orthologues
from the Opisthokonta, i.e., metazoa, plants, fungi, and alveo-
lates (Fig. 1A). In addition, the ELP3a and ELP3b paralogue
groups are monophyletic, suggesting a single gene duplication
that predates divergence of the trypanosomatids. Despite the
divergence, the residues required for substrate binding within
both the radical SAM and acetyltransferase domains are con-
served in both T. brucei ELP3a and ELP3b (Fig. 1B). Our
phylogenetic and sequence analysis indicated that ELP3a and
ELP3b have similar features regardless of the trypanosomatid
under consideration; we have focused on the T. brucei proteins.
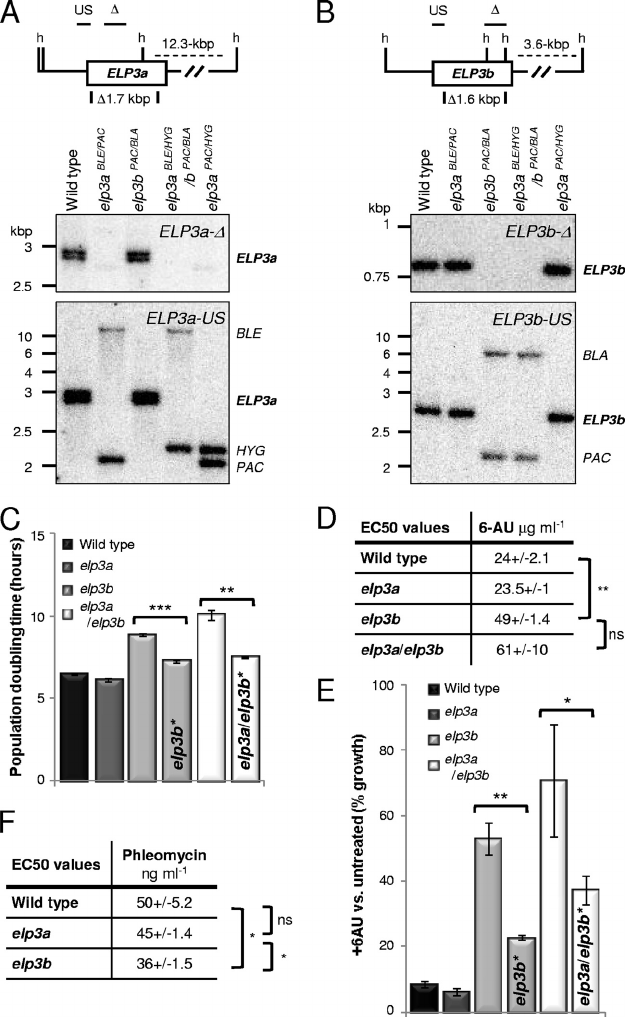
elp3b mutants are resistant to transcription elongation in-
hibition and hypersensitive to DNA-damaging agents. To ex-
plore the function of the trypanosomatid Elp3-related pro-
teins, we generated elp3a (Fig. 2A) and elp3b (Fig. 2B) null
strains. Double-null elp3a/elp3b strains were also generated
(Fig. 2A and B). These strains were indistinguishable from wild
type in relation to cell cycle phase distribution and differenti-
ation to the insect stage (data not shown), but the elp3b strains
displayed a growth defect relative to elp3a and wild-type try-
panosomes (Fig. 2C). Interestingly, repeating the analysis after
further growth suggested that the cells were adapting to the
defect, with population doubling time approaching that of
wild-type cells (Fig. 2C). Double-null elp3a/elp3b strains also
displayed a growth defect and partial reversal of this pheno-
type after prolonged growth. Transcription elongation defects
increase sensitivity to depleted nucleoside triphosphate (NTP)
substrate pools. Indeed, S. cerevisiae elp3 null strains were
hypersensitive to 6-azauracil (6AU) (57), which inhibits IMP
dehydrogenase (IMPDH) and depletes pools of GTP and UTP
(46). Surprisingly, elp3b cells displayed specific and significant
resistance to 6AU relative to that of elp3a and wild-type try-
panosomes (Fig. 2D), suggesting that ELP3b negatively con-
trols transcription elongation. This phenotype, like the growth
phenotype, was diminished after further growth (Fig. 2E).
Double-null elp3a/elp3b strains also displayed 6AU resistance
and partial reversal of this phenotype after prolonged growth.
Thus, elp3b null cells display an adaptation phenomenon char-
acterized by partial reversal of the growth and 6AU resistance
phenotypes, and ELP3a is not required for this adaptation.
VOL. 31, 2011 ELONGATOR REGULATES rDNA IN T. BRUCEI 1823
The data above are consistent with the idea that ELP3b
negatively controls transcription, and preliminary analysis of
nascent RNA did indeed reveal increased rDNA transcription
in elp3b cells (data not shown). Consistent with a link between
the growth and 6AU resistance phenotypes described above,
the rDNA transcription derepression phenotype was also un-
stable (data not shown but see below). Saccharomyces cerevi-
siae strains lacking Elp3 (28) or with increased rDNA tran-
scription (22) are hypersensitive to DNA-damaging agents,
and notably, elp3b null cells were also hypersensitive to phleo-
mycin (Fig. 2F), an agent that damages DNA via a mechanism
involving chelation of metal ions and the generation of free
radicals.
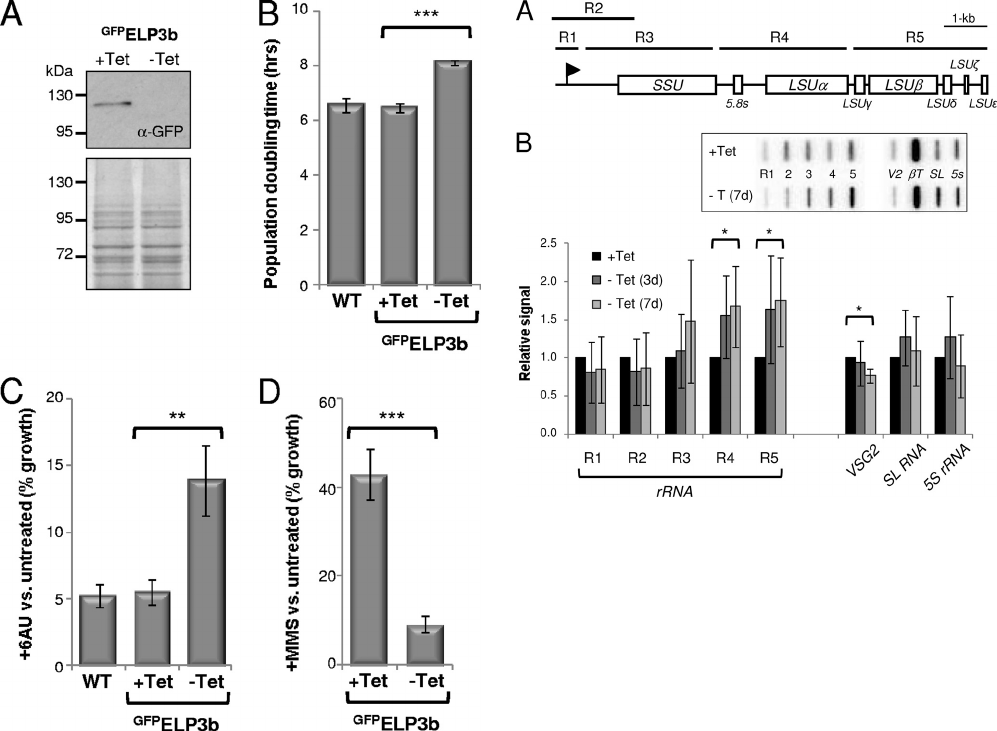
To facilitate studies of ELP3b function in a controlled en-
vironment, we generated strains with a conditional (Tet-on)
copy of
GFP
ELP3b in an elp3b null background. Cells in which
GFP
ELP3b expression was inactivated (Fig. 3
A) displayed de-
creased growth rate (Fig. 3B), reduced 6AU sensitivity (Fig.
3C), and increased sensitivity to methyl methanesulfonate
(MMS) (Fig. 3D), an alkylating agent that damages DNA and
is thought to stall replication forks. These results recapitulate
the elp3b null phenotypes and demonstrate that
GFP
ELP3b
functionally complements the elp3b defect.
ELP3b negatively controls ribosomal DNA transcription
elongation. We next measured nascent transcripts emanating
from different loci in cells expressing
GFP
ELP3b or in cells
FIG. 1. Phylogenetic and sequence analysis of Elp3 orthologues. (A) ELP3a and ELP3b were identified in T. brucei (Tb), T. cruzi (Tc), and
Leishmania major (Lm). The unrooted neighbor-joining tree was generated using CLUSTAL 1.8X and TreeView. Where excellent (ⱖ99.9%, open
circles) or very good (ⱖ90%, closed circles), branching confidence is indicated. Hs, Homo sapiens; Xl, Xenopus laevis; Dm, Drosophila melanogaster;
Ce, Caenorhabditis elegans; Sc, Saccharomyces cerevisiae; Sp, Schizosaccharomyces pombe; Tt, Tetrahymena thermophila; At, Arabidopsis thaliana.
All accession numbers are indicated. The GeneIDs for the T. brucei proteins are as follows: TbELP3a, Tb927.8.5770; TcELP3a,
Tc00.1047053503851.10; LmELP3a, LmjF16.0240; TbELP3b, Tb927.8.3310; TcELP3b, Tc00.1047053509769.110; LmELP3b, LmjF23.1350.
(B) Schematic representation of the predicted structures of T. brucei ELP3a and ELP3b compared with human Elp3. The radical SAM domains
(black boxes) and GNAT-type acetyltransferase domains (gray boxes) are indicated with the FeS cluster and motif A, respectively. The sequences
were aligned using ClustalW, and the conserved domains (boxed) are indicated. Residues that are shared between all three proteins are white on
a black background, and residues shared among any pair of proteins are on a gray background. Arrowheads indicate the Cys residues that form
part of the FeS cluster. Asterisks indicate the conserved residues of motif A, QHXGXG, in all Elp3 orthologues.
1824 ALSFORD AND HORN M
OL.CELL.BIOL.
where
GFP
ELP3b expression had been inactivated for 3 or 7
days (Fig. 4). In this case, we used a series of five probes (R1
to R5) covering the length of the rDNA transcription unit (Fig.
4A). Slot blots were also loaded with probes for VSG2, the
active VSG in all clones analyzed (RNAP-I), -tubulin genes
(RNAP-II), spliced-leader (SL-RNA) genes (RNAP-II), 5S
rDNA genes (RNAP-III), and several negative controls:
nontranscribed rDNA intergenic spacer, an insect-stage-
FIG. 2. ELP3b loss is associated with unstable resistance to transcription inhibition and hypersensitivity to a DNA-damaging agent. (A) South-
ern blot indicating ELP3a disruption. Genomic DNA was digested with HindIII. The map details the HindIII sites (h), deleted regions, and probes
used (horizontal bars). The deleted region was replaced with the selectable marker BLE, HYG,orPAC; the HYG and PAC cassettes each contain
a HindIII site. The elp3aPAC/HYG strain was used for the assay shown in panel F. (B) Southern blot indicating ELP3b disruption. The deleted
region was replaced with the selectable marker PAC or BLA. Other details are as in panel A. (C) Population doubling time in wild-type strain and
elp3a, elp3b, and elp3a/elp3b strains. elp3b* and elp3a/elp3b* strains were maintained in culture for more than 8 weeks. Standard deviations are
indicated, and P values were derived using an unpaired Student t test.
***
, P ⬍ 0.001;
**
, P ⬍ 0.01;
*
, P ⬍ 0.05; ns, not significant.
(D) Half-maximal effective concentrations (EC
50
) for 6AU. All data were derived from two wild-type samples and two independent null strains.
Other details are as in panel C above. (E) 6AU sensitivity (50 gml
⫺1
). Other details are as in panel C above. (F) EC
50
for phleomycin. Other
details are as in panel C above.
V
OL. 31, 2011 ELONGATOR REGULATES rDNA IN T. BRUCEI 1825
specific transcript that is not expressed in the bloodstream-
form cells used for this analysis (procyclin), and plasmid
vector DNA. The transcription run-on analysis indicated
significantly increased transcription through the rDNA unit
following
GFP
ELP3b inactivation (Fig. 4B). In the 7-day
samples, transcription was significantly increased (by ⬎60%)
across a region encompassed by probes in the middle and at
the distal end of the rDNA unit (Fig. 4B). The equivalent 3-day
samples also displayed increased transcription across this re-
gion but without achieving statistical significance. Probes en-
compassing the proximal end of the rDNA unit failed to show
an increase in transcription, suggesting little attenuation in this
region in wild-type cells. Thus, consistent with increased 6AU
resistance, cells depleted for ELP3b displayed a relative in-
crease in nascent rDNA promoter-distal transcripts; the large
increase is remarkable given the major contribution that rDNA
genes are thought to make to total transcription in unper-
turbed cells. Interestingly, VSG2 transcription was significantly
reduced (by ⬃20%) in these cells (Fig. 4B), which could reflect
depletion of the extranucleolar pool of RNAP-I. No negative-
control transcript, including nontranscribed rDNA intergenic
spacer, was significantly above background (data not shown),
and neither the RNAP-II (SL-RNA) or RNAP-III (5S rRNA)
transcripts displayed significant change (Fig. 4B).
ELP3b suppresses transcription of a reporter integrated
within ribosomal DNA. We next sought an independent ap-
proach to confirm negative control of transcription by ELP3b.
Trypanosomes are unusual in that all mature mRNAs are fused
to an RNAP-II-transcribed, trans-spliced leader sequence (19).
This allows RNAP-I to transcribe the protein-coding segment
of mRNA (45), as is the case for VSG genes. We took advan-
FIG. 3. ELP3 downregulation phenocopies ELP3b knockout.
(A) Western blotting with anti-GFP confirmed Tet-on (1 gml
⫺1
)
regulation of
GFP
ELP3b in an elp3b background. The lower panel
shows an equivalent Coomassie blue-stained gel as a loading con-
trol. Data from one representative cell line are shown. (B and C)
Population doubling times (B) and 6AU (50 gml
⫺1
) sensitivities
(C) in wild-type (WT) and
GFP
ELP3b strains. (D) Methyl methane
-
sulfonate (MMS; 0.0004%) sensitivity in
GFP
ELP3b strains (EC
50
:
⫹Tet, 0.00036% ⫾ 0.000015%; ⫺Tet, 0.00024% ⫾ 0.000016%).
The ⫺Tet,
GFP
ELP3b-depleted population was assessed 4 to 7 days
after Tet removal. Data in panels B to D were derived from four
independent clones. Standard deviations are indicated, and P values
were derived using a paired Student t test.
***
, P ⬍ 0.001;
**
, P ⬍
0.01.
FIG. 4. ELP3b negatively regulates transcription elongation at
rDNA loci. (A) Schematic of a T. brucei rDNA transcription unit and
the location of the R1 to R5 probes (horizontal bars) for nascent
transcript analysis. Each rDNA unit is approximately 10 kbp in length
(55). The promoter (flag) and the rDNA subunit coding regions are in-
dicated. (B) Transcription run-on analysis during depletion of
GFP
ELP3b. Phosphorimager signals were corrected against -tubulin
transcript abundance and expressed relative to the ⫹Tet value (set to
1). The inset shows a sample slot blot. V2, VSG2; T, -tubulin. VSG2
is a single-copy gene ⬃60 kbp from its promoter that is transcribed by
RNAP-I. The spliced-leader RNA (SL-RNA) is derived from a tan-
dem gene array and contributes a fragment that is trans-spliced to the
5⬘ end of every mRNA in trypanosomes. This RNA and -tubulin, used
as a loading control and also derived from a tandem gene array, are
transcribed by RNAP-II. 5S rRNA is also derived from a tandem gene
array and is transcribed by RNAP-III. Data were derived from four
independent
GFP
ELP3b strains. Error bars represent one standard
deviation, and P values were derived using a paired Student t test.
*
,
P ⬍ 0.05.
1826 ALSFORD AND HORN M
OL.CELL.BIOL.
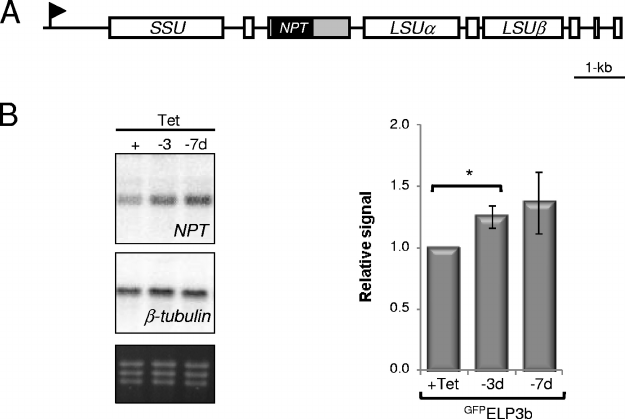
tage of this feature and used a selectable marker as a reporter
of transcription through rDNA (Fig. 5A). A neomycin phos-
photransferase (NPT) gene was inserted between the 5.8S and
LSU␣ genes in cells engineered for conditional expression of
GFP
ELP3b in an elp3b null background, and three independent
clones were analyzed (Fig. 5B); correct integration was con-
firmed using PCR assays (data not shown). NPT expression
was increased 3 and 7 days after inactivation of
GFP
ELP3b
expression, achieving statistical significance in the 3-day sam-
ples (Fig. 5B). The results confirm negative control of tran-
scription through rDNA by ELP3b and suggest suppression of
transcription through individual rDNA units rather than com-
plete transcription blockade in a subset.
Having established that ELP3b negatively controls transcrip-
tion at rDNA loci, we now revisited the adaptation phenome-
non in the elp3b strains described above. We predicted that
adaptation would involve reduced rDNA transcription, either
through change in rDNA gene copy number or through the
action of a second negative regulator. There are nine complete
rDNA units annotated in the haploid T. brucei genome se-
quence, with one unit each on chromosomes 1 and 7, three on
chromosome 2, and four on chromosome 3 (6). Using elp3b
null cells grown in culture for several weeks (elp3b*), we saw
no evidence for altered rDNA gene dosage (data not shown),
but as predicted, transcription run-on analysis revealed re-
duced rDNA transcription (Fig. 6B). In elp3b* cells, transcrip-
tion was reduced by ⬎50% compared to the wild type in a
region encompassed by three probes at the proximal end of the
rDNA unit. This does not reflect more transcription in the
distal region but is readily explained by a two-stage process
involving increased elongation after ELP3b loss which is com-
pensated for by less initiation (compare Fig. 6B and Fig. 4B).
Consistent with adaptation in the absence of ELP3a (in elp3a/
elp3b double-null strains [Fig. 2C and E]), nascent transcript
analysis in elp3a strains did not reveal changes in rDNA tran-
scription that were statistically significant (Fig. 6C), implicating
another, unknown factor in reduced initiation and adaptation
in elp3b null cells.
ELP3b localizes to the nucleolus but not to the VSG expres-
sion site body. We proceeded to explore the subcellular
location of
GFP
ELP3a and
GFP
ELP3b in bloodstream-form
trypanosomes engineered for tetracycline (Tet)-inducible
expression (3). Microscopic analysis of these strains did not
reveal any substantial GFP signal in uninduced cultures,
while fluorescence or immunofluorescence analysis of induced
cells indicated specific accumulation in distinct subnuclear
compartments (data not shown). To directly compare these
compartments, we established trypanosomes constitutively
expressing both
GFP
ELP3a and
MYC
ELP3b (Fig. 7
A). Im-
munofluorescence analysis revealed little overlap in the lo-
cation of the two proteins; the subnuclear compartment
occupied by
GFP
ELP3a is punctate and typically at the nu
-
clear periphery, while
MYC
ELP3b occupies a more central
compartment (Fig. 7B). We obtained similar results using
insect-stage, procyclic trypanosomes (data not shown). Inter-
estingly, the accumulation of
MYC
ELP3b was specifically dis
-
rupted after transcription inhibition (Fig. 7B), which could
indicate engagement with active transcription factors.
Diploid T. brucei nuclei contain a single nucleolus, the site of
rDNA transcription driven by RNAP-I, and apparent ELP3b
accumulation at this site is consistent with the phenotypes
described above. However, bloodstream-form T. brucei try-
panosomes are unusual in that they also use RNAP-I to tran-
scribe VSG mRNA at an extranucleolar site known as the
expression site body (ESB) (32). To examine ELP3b localiza-
tion with respect to both RNAP-I compartments, we estab-
FIG. 5. ELP3b suppresses transcription of an mRNA reporter integrated at an rDNA locus. (A) Schematic of a T. brucei rDNA transcription
unit showing the location of the neomycin phosphotransferase (NPT) reporter. The flanking 5⬘-procyclin and 3⬘-aldolase untranscribed regions are
represented by gray boxes. (B) Northern analysis of NPT mRNA expression following
GFP
ELP3b depletion. One representative Northern blot is
shown. An ethidium bromide-stained gel is included to show loading. Phosphorimager signals were processed as described for Fig. 4B. Data were
derived from three independent
GFP
ELP3b (rDNA::NPT) strains. Error bars represent 1 standard deviation, and P values were derived using a
paired Student t test.
*
, P ⬍ 0.05.
V
OL. 31, 2011 ELONGATOR REGULATES rDNA IN T. BRUCEI 1827
lished trypanosomes expressing
MYC
ELP3b and
GFP
RPB6z
(Fig. 7A), the latter being a well-characterized RNAP-I sub-
unit found in both RNAP-I compartments (12, 33); N-termi-
nally tagged RPB6z was previously shown to be fully functional
and does not interfere with RNAP-I activity in vitro (33). Im-
munofluorescence staining of
GFP
RPB6z revealed the nucleo
-
lus and the ESB as expected (Fig. 7C). Dual detection of
GFP
RPB6z and
MYC
ELP3b revealed a strong
MYC
ELP3b signal
at the nucleolus, but no detectable
MYC
ELP3b signal that co
-
incided with the smaller ESB, the compartment involved in
VSG transcription (Fig. 7C).
The data above indicated nucleolar sequestration of ELP3b
and little or no association with the ESB. However, we de-
tected some nuclei with a second compartment of
MYC
ELP3b
staining, and we speculated that these represented nascent
nucleoli. This was confirmed using cells expressing
GFP
ELP3b
(Fig. 3A). Dual immunofluorescence detection of
GFP
ELP3b
and NOG1, a nucleolar G protein (38) that is not detected in
the ESB, indicated that the second focus of
GFP
ELP3b staining
always colocalized with NOG1 (Fig. 7D). In addition, nuclear
and mitochondrial (kinetoplast) DNAs, stained with DAPI
(4⬘,6-diamidino-2-phenylindole), provide excellent cytological
markers that define the position in the cell cycle (58), and as
expected, two nucleoli and two
GFP
ELP3b foci in a single nucleus
were observed in trypanosomes only in the G
2
/M phases. Thus,
ELP3b was sequestered in the nucleolus and showed little or no
association with the ESB regardless of whether the protein was
fused to GFP or a MYC epitope. The results show that nucle-
olus-enriched
GFP
ELP3b complemented the growth, 6AU re
-
sistance, and transcription phenotypes seen in elp3b cells (see
above). We also demonstrated that elp3b cells were indistin-
guishable from the wild type in relation to ELP3a, NOG1, and
RPB6z localization (data not shown).
We used a similar approach to further examine the compart-
ment(s) occupied by the other Elp3-related protein, ELP3a.
GFP
ELP3a was coexpressed with other tagged transcription
factors, and expression of proteins of the predicted size was
confirmed by Western blotting (Fig. 8A). ELP3a appeared to
occupy a compartment that was distinct from all three major
RNA polymerases (Fig. 8B). A peripheral nuclear localization
appeared more pronounced during mitosis (Fig. 8C), and
partial colocalization with NUP-1, a putative nuclear lamina
component (44), supported an association with the nuclear
envelope (Fig. 8D). We also examined spindle microtubule
acetylation, telomere position-effect repression (16), and VSG
expression site silencing (54) in elp3a cells, as well as cytosine
methylation (31) in elp3a and elp3b cells (data not shown), but
detected no significant differences from the wild type.
ELP3b-depleted cells are hypersensitive to translation inhi-
bition. We considered the possible benefits of limiting tran-
scription through rDNA. This could facilitate DNA replication
and DNA damage tolerance (Fig. 2F and 3D) (22) or could
contribute to modulating rRNA synthesis to satisfy cellular
demands for translation capacity. Indeed, rDNA transcription
elongation is the rate-limiting step for rRNA synthesis in hu-
man cells (48). To explore a role for ELP3b in regulating
rRNA synthesis, we examined the downstream consequences
of increased rDNA transcription in ELP3b-deficient cells (Fig.
9). The trypanosomatid rDNA transcription unit is unusual in
that it encodes several small rRNAs (55), but no major change
in the relative steady-state abundance of any of these tran-
scripts was seen in elp3a, elp3b*,orelp3a/elp3b null cells
(Fig. 9A). Furthermore, Northern blot analysis revealed no
major change in relative SSU or LSU transcript abundance
in ELP3b-depleted cells (Fig. 9B). To ask whether increased
rDNA transcription is reflected at the level of sensitivity to
translation inhibition, we assessed growth in G418. Surpris-
ingly, ELP3b-depleted cells were hypersensitive to G418 (Fig.
9C). This was also the case in elp3b* cells, while elp3a cells
were indistinguishable from the wild type (data not shown).
This result could reflect disruption of the ribosome assembly
process, causing limiting factors to be channeled into a non-
productive pathway, or ELP3b could play an additional role in
tRNA modification (21). Taken together, the results are con-
sistent with a role for rDNA transcription control by ELP3b in
maintaining genome stability rather than in modulating trans-
lation capacity.
FIG. 6. Adaptation to the elp3b defect involves downregulation of
rRNA transcription. (A) Schematic of a T. brucei rDNA transcription
unit reproduced from Fig. 4A. (B) Transcription run-on analysis of
adapted elp3b* null strains. Data were derived from two independent
clones. Other details are as in Fig. 4B, except that corrected values are
expressed relative to the wild type (WT). (C) Transcription run-on
analysis of elp3a null strains. Data were derived from four independent
clones. Other details are as in panel B above.
1828 ALSFORD AND HORN M
OL.CELL.BIOL.

DISCUSSION
rDNA genes encode the core components of the ribosomes,
the molecular machines that drive mRNA translation into pro-
tein. Sixty to 80 percent of transcription in rapidly growing
yeast cells is at rDNA loci mediated by RNAP-I. The elonga-
tion rate has been estimated to be ⬃60 nucleotides/s with a
reinitiation rate of ⬍1 s (14). Despite the important link to
genome stability (22), the balance between silencing and acti-
vating complexes and their contributions to the formation of
alternative chromatin states at these loci remain poorly under-
stood (30). A number of factors that typically control multiple
classes of RNA polymerase have been shown to exert positive
(7, 59) and negative (30) control on RNAP-I elongation at
rDNA genes. In addition, structural analysis reveals that S.
cerevisiae RNAP-I contains a built-in elongation factor related
to the RNAP-II-associated factor TFIIF (27), and a mutated
phosphorylation site on S. cerevisiae RNAP-I increases resis-
tance to 6AU, consistent with a role in negative control of
elongation (15). We have demonstrated negative control by
ELP3b that is specific to nucleolar rDNA genes in trypano-
somes, and our reporter analysis suggests suppression of indi-
vidual rDNA transcription units rather than complete block-
ade of a subset.
Nucleosomes are depleted or disordered at actively tran-
scribed rDNA loci (30). Thus, ELP3b may generate a more
stable or “closed” chromatin state that promotes premature
termination. How might this be achieved? rDNA gene regula-
tion involves histone modification and DNA methylation (50),
and ELP3b, like other Elp3 orthologues, has two major do-
mains, an acetyltransferase domain and a radical SAM do-
main. Histone acetylation is important for transcription initi-
ation and elongation, and a specific enrichment of histone
H4K10 acetylation, H2AZ and H2BV histone variants, and the
BDF3 bromodomain factor is seen at probable RNAP-II tran-
scription start sites in trypanosomes (47). These same variants
are depleted within the nontranscribed rDNA spacer, and
FIG. 7. ELP3b localizes to the nucleolus but is not detected in the ESB in bloodstream-form T. brucei. (A) Western analysis of strains used for
ELP3 localization studies. Blots were incubated with anti-MYC or anti-GFP. A Coomassie blue-stained gel is included to show loading. Predicted
molecular masses of the fusions:
GFP
ELP3a, 104 kDa;
MYC
ELP3b, 77 kDa;
GFP
RPB6z, 42 kDa. The lower anti-GFP panel is a shorter exposure.
(B) Dual localization of
GFP
ELP3a and
MYC
ELP3b. The effect of actinomycin D treatment is also shown. The regions outlined in the phase images
indicate the regions shown in the immunofluorescence panels. The nucleus (n) and kinetoplast (k) were stained with the DNA intercalating dye
4⬘,6-diamidino-2-phenylindole (DAPI), and these images were merged with the immunofluorescence images. Bar, 5 m. (C)
MYC
ELP3b localizes
specifically to the nucleolus but not to the smaller ESB (arrowhead), as revealed by
GFP
RPB6z. (D) Colocalization of
GFP
ELP3b and the nucleolar
protein, NOG1, through the cell cycle. G
1
/S cells have a single nucleolus, while a second nucleolus can be seen during G
2
/M (identified by two
kinetoplasts). Postmitotic (Post-M) cells have a single nucleolus in each nucleus. Other details are as in panel B above.
V
OL. 31, 2011 ELONGATOR REGULATES rDNA IN T. BRUCEI 1829
BDF3 is depleted in the transcribed rDNA region, revealing a
distinct chromatin architecture at these loci (see the region
encompassing Tb927.3.3421 to Tb927.3.3455 at http://tritrypdb
.org/). Importantly, histone acetylation has also been associated
with negative control of transcription (9, 24, 29, 53). Thus, acet-
ylation by ELP3b may negatively control rDNA transcription
elongation. The radical SAM domain could equally be responsi-
ble for negative control, possibly via DNA demethylation (36).
The nucleus is highly heterogeneous, containing euchro-
matic and heterochromatic compartments thought to be per-
missive and repressive for transcription, respectively. RNAP-I
transcribes rDNA genes in the nucleolar compartment, and, in
T. brucei, RNAP-I also synthesizes a subset of abundant pre-
mRNAs. In fact, T. brucei is the only known eukaryote with a
multifunctional RNAP-I and presents a unique opportunity to
study RNAP-I regulators. The trypanosomatid RNAP-I com-
plex (34, 52) contains three “specialized” subunits, RPB5z,
RPB6z, and RPB10z (12), as well as RPA31 (33) and the RPB7
subunit, typically associated with RNAP-II (39). Trypanosome
RNAP-I transcription also depends upon a novel complex
known as class I transcription factor A (8). ELP3b has not been
identified in fractions containing T. brucei RNAP-I (8, 33, 34,
52), possibly due to weak or little direct interaction or because
the majority of RNAP-I is engaged in processive transcrip-
tion. Remarkably though, ELP3b specifically impacts rela-
tively short RNAP-I transcription units (rDNA, 10 kbp) and
appears to be excluded from a much longer RNAP-I transcrip-
tion unit at the ESB (VSG, 60 kbp). Several additional nucle-
olar factors are undetectable in the ESB, but ELP3b is the first
of these factors shown to distinguish between rDNA and VSG
mRNA synthesis. The distinct promoters that operate at these
loci could determine this differential association.
FIG. 8. ELP3a occupies nuclear territories distinct from those occupied by the transcription machinery. (A) Western analysis of strains used
for ELP3 localization studies. Predicted molecular masses of the fusions:
6MYC
RPB6z, 24 kDa;
6MYC
RPB6, 25 kDa;
GFP
RPB6, 43 kDa;
SNAP42
12MYC
, 60 kDa; RPC160
12MYC
, 188 kDa. (B)
GFP
ELP3a and the tagged polymerase subunits occupy distinct nuclear territories.
6MYC
RPB6z, RNAP-I;
6MYC
RPB6 and SNAP42
12MYC
, RNAP-II (colocalization of these factors is associated with sites of SL-RNA transcription
[30, 52]); RPC160
12MYC
, RNAP-III. (C) Localization of
GFP
ELP3a and SNAP42
12MYC
through the cell cycle. (D)
GFP
ELP3a shows partial
colocalization with NUP1. Other details are as in Fig. 7B.
1830 ALSFORD AND HORN M
OL.CELL.BIOL.
Microarray analysis of steady-state transcripts in Elongator-
defective S. cerevisiae revealed 52 genes that were downregu-
lated and 44 genes that were upregulated (26). Under the
conditions examined here, ELP3b negatively controls rDNA
transcription and can be considered an “anti-Elongator.” We
suggest that this may also be the case at the upregulated loci in
S. cerevisiae. Indeed, Elp3 was recently shown to have a role in
maintaining silencing at telomeres and at mating-type loci in S.
cerevisiae (28). Our results indicate a negative role for ELP3b
in processivity, while, at this stage, the role of ELP3a remains
unknown.
The purpose of limited transcription through rDNA appears
to be to improve genome stability (22). Our results are consis-
tent with the idea that unregulated transcription of rDNA
genes is indeed toxic and compromises the capacity for DNA
repair at these loci. Our findings could reflect a conserved role
for Elp3 proteins in this process. Indeed, Elp3 is concentrated
in nucleoli in HeLa cells (20). We favor a model whereby
ELP3b, like Elp3 in S. cerevisiae, associates with elongating
RNAP and modifies chromatin structure, but we cannot rule
out other possible scenarios to explain negative control of
RNAP-I processivity at this stage. Targeted meganuclease
cleavage (17) could now be used to further explore the impact
of ELP3b transcription control on double-strand break repair
within rDNA transcription units. It will also be important to
determine what contributions the ELP3b acetyltransferase and
radical SAM domains make to the novel anti-Elongator func-
tion described here.
ACKNOWLEDGMENTS
This work was supported by The Wellcome Trust (project grants
079457 and 083648).
We also thank John Kelly (LSHTM) for critical reading of the
manuscript, Marilyn Parsons (Seattle Biomedical Research Institute)
for NOG1 antisera, Klaus Ersfeld (University of Hull, United King-
dom) for NUP1 antisera, and Elisabetta Ullu (Yale University) for the
transcription run-on protocol.
REFERENCES
1. Alibu, V. P., L. Storm, S. Haile, C. Clayton, and D. Horn. 2005. A doubly
inducible system for RNA interference and rapid RNAi plasmid construc-
tion in Trypanosoma brucei. Mol. Biochem. Parasitol. 139:75–82.
2. Alsford, S., and D. Horn. 2008. Single-locus targeting constructs for reliable
regulated RNAi and transgene expression in Trypanosoma brucei. Mol.
Biochem. Parasitol. 161:76–79.
3. Alsford, S., T. Kawahara, L. Glover, and D. Horn. 2005. Tagging a T. brucei
RRNA locus improves stable transfection efficiency and circumvents induc-
ible expression position effects. Mol. Biochem. Parasitol. 144:142–148.
4. Alsford, S., T. Kawahara, C. Isamah, and D. Horn. 2007. A sirtuin in the
African trypanosome is involved in both DNA repair and telomeric gene
silencing but is not required for antigenic variation. Mol. Microbiol. 63:724–
736.
5. Ausubel, F. M., et al. 1998. Current protocols in molecular biology. John
Wiley and Sons, Inc., New York, NY.
6. Berriman, M., et al. 2005. The genome of the African trypanosome Trypano-
soma brucei. Science 309:416–422.
7. Birch, J. L., et al. 2009. FACT facilitates chromatin transcription by RNA
polymerases I and III. EMBO J. 28:854–865.
8. Brandenburg, J., et al. 2007. Multifunctional class I transcription in Trypano-
soma brucei depends on a novel protein complex. EMBO J. 26:4856–4866.
9. Braunstein, M., R. E. Sobel, C. D. Allis, B. M. Turner, and J. R. Broach.
1996. Efficient transcriptional silencing in Saccharomyces cerevisiae requires
a heterochromatin histone acetylation pattern. Mol. Cell. Biol. 16:4349–
4356.
10. Clayton, C. E. 2002. Life without transcriptional control? From fly to man
and back again. EMBO J. 21:1881–1888.
11. Creppe, C., et al. 2009. Elongator controls the migration and differentiation
of cortical neurons through acetylation of ␣-tubulin. Cell 136:551–564.
12. Devaux, S., et al. 2007. Diversification of function by different isoforms of
conventionally shared RNA polymerase subunits. Mol. Biol. Cell 18:1293–
1301.
13. Figueiredo, L. M., G. A. Cross, and C. J. Janzen. 2009. Epigenetic regulation
in African trypanosomes: a new kid on the block. Nat. Rev. Microbiol.
7:504–513.
14. French, S. L., Y. N. Osheim, F. Cioci, M. Nomura, and A. L. Beyer. 2003. In
exponentially growing Saccharomyces cerevisiae cells, rRNA synthesis is de-
termined by the summed RNA polymerase I loading rate rather than by the
number of active genes. Mol. Cell. Biol. 23:1558–1568.
15. Gerber, J., et al. 2008. Site specific phosphorylation of yeast RNA polymer-
ase I. Nucleic Acids Res. 36:793–802.
16. Glover, L., and D. Horn. 2006. Repression of polymerase I-mediated gene
expression at Trypanosoma brucei telomeres. EMBO Rep. 7:93–99.
FIG. 9. ELP3b loss is associated with hypersensitivity to translation
inhibition. (A) Total RNA from the wild type and pairs of elp3a, elp3b,
and elp3a/elp3b null strains was fractionated by agarose-formaldehyde
(upper panel) or polyacrylamide-urea (lower panel) gel electropho-
resis. (B) Northern analysis of steady-state rRNA transcripts fol-
lowing depletion of
GFP
ELP3b. One representative blot is shown.
An ethidium bromide-stained gel is included to show loading. Phos-
phorimager signals were processed as described for Fig. 4B. Data were
derived from four independent
GFP
ELP3b strains. (C) EC
50
values for
G418 and
GFP
ELP3b strains, ⫹ and ⫺Tet. ⫺Tet cultures were main
-
tained in Tet-free medium for 3 days prior to G418 treatment. Data
were derived from four independent
GFP
ELP3b strains. Standard de
-
viations are indicated, and P values were derived using a paired Stu-
dent t test.
***
, P ⬍ 0.001.
V
OL. 31, 2011 ELONGATOR REGULATES rDNA IN T. BRUCEI 1831
17. Glover, L., R. McCulloch, and D. Horn. 2008. Sequence homology and
microhomology dominate chromosomal double-strand break repair in Afri-
can trypanosomes. Nucleic Acids Res. 36:2608–2618.
18. Guenther, M. G., S. S. Levine, L. A. Boyer, R. Jaenisch, and R. A. Young.
2007. A chromatin landmark and transcription initiation at most promoters
in human cells. Cell 130:77–88.
19. Gunzl, A. 2010. The pre-mRNA splicing machinery of trypanosomes: com-
plex or simplified? Eukaryot. Cell 9:1159–1170.
20. Hawkes, N. A., et al. 2002. Purification and characterization of the human
elongator complex. J. Biol. Chem. 277:3047–3052.
21. Huang, B., M. J. Johansson, and A. S. Bystrom. 2005. An early step in
wobble uridine tRNA modification requires the Elongator complex. RNA
11:424–436.
22. Ide, S., T. Miyazaki, H. Maki, and T. Kobayashi. 2010. Abundance of
ribosomal RNA gene copies maintains genome integrity. Science 327:693–
696.
23. Ivens, A. C., et al. 2005. The genome of the kinetoplastid parasite, Leishma-
nia major. Science 309:436–442.
24. Kawahara, T., et al. 2008. Two essential MYST-family proteins display
distinct roles in histone H4K10 acetylation and telomeric silencing in try-
panosomes. Mol. Microbiol. 69:1054–1068.
25. Kim, J. H., W. S. Lane, and D. Reinberg. 2002. Human Elongator facilitates
RNA polymerase II transcription through chromatin. Proc. Natl. Acad. Sci.
U. S. A. 99:1241–1246.
26. Krogan, N. J., and J. F. Greenblatt. 2001. Characterization of a six-subunit
holo-elongator complex required for the regulated expression of a group of
genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 21:8203–8212.
27. Kuhn, C. D., et al. 2007. Functional architecture of RNA polymerase I. Cell
131:1260–1272.
28. Li, Q., et al. 2009. The elongator complex interacts with PCNA and modu-
lates transcriptional silencing and sensitivity to DNA damage agents. PLoS
Genet. 5:e1000684.
29. Liang, G., et al. 2004. Distinct localization of histone H3 acetylation and
H3-K4 methylation to the transcription start sites in the human genome.
Proc. Natl. Acad. Sci. U. S. A. 101:7357–7362.
30. McStay, B., and I. Grummt. 2008. The epigenetics of rRNA genes: from
molecular to chromosome biology. Annu. Rev. Cell Dev. Biol. 24:131–157.
31. Militello, K. T., et al. 2008. African trypanosomes contain 5-methylcytosine
in nuclear DNA. Eukaryot. Cell 7:2012–2016.
32. Navarro, M., and K. Gull. 2001. A pol I transcriptional body associated with
VSG mono-allelic expression in Trypanosoma brucei. Nature 414:759–763.
33. Nguyen, T. N., B. Schimanski, and A. Gunzl. 2007. Active RNA polymerase
IofTrypanosoma brucei harbors a novel subunit essential for transcription.
Mol. Cell. Biol. 27:6254–6263.
34. Nguyen, T. N., B. Schimanski, A. Zahn, B. Klumpp, and A. Gunzl. 2006.
Purification of an eight subunit RNA polymerase I complex in Trypanosoma
brucei. Mol. Biochem. Parasitol. 149:27–37.
35. Ogbadoyi, E., K. Ersfeld, D. Robinson, T. Sherwin, and K. Gull. 2000.
Architecture of the Trypanosoma brucei nucleus during interphase and
mitosis. Chromosoma 108:501–513.
36. Okada, Y., K. Yamagata, K. Hong, T. Wakayama, and Y. Zhang. 2010. A role
for the elongator complex in zygotic paternal genome demethylation. Nature
463:554–558.
37. Otero, G., et al. 1999. Elongator, a multisubunit component of a novel RNA
polymerase II holoenzyme for transcriptional elongation. Mol. Cell 3:109–
118.
38. Park, J. H., B. C. Jensen, C. T. Kifer, and M. Parsons. 2001. A novel
nucleolar G-protein conserved in eukaryotes. J. Cell Sci. 114:173–185.
39. Penate, X., et al. 2009. RNA pol II subunit RPB7 is required for RNA pol
I-mediated transcription in Trypanosoma brucei. EMBO Rep. 10:252–257.
40. Pokholok, D. K., N. M. Hannett, and R. A. Young. 2002. Exchange of RNA
polymerase II initiation and elongation factors during gene expression in
vivo. Mol. Cell 9:799–809.
41. Rahl, P. B., C. Z. Chen, and R. N. Collins. 2005. Elp1p, the yeast homolog
of the FD disease syndrome protein, negatively regulates exocytosis inde-
pendently of transcriptional elongation. Mol. Cell 17:841–853.
42. Raz, B., M. Iten, Y. Grether-Buhler, R. Kaminsky, and R. Brun. 1997. The
Alamar Blue assay to determine drug sensitivity of African trypanosomes
(T.b. rhodesiense and T.b. gambiense) in vitro. Acta Trop. 68:139–147.
43. Roberts, J. W., S. Shankar, and J. J. Filter. 2008. RNA polymerase elonga-
tion factors. Annu. Rev. Microbiol. 62:211–233.
44. Rout, M. P., and M. C. Field. 2001. Isolation and characterization of sub-
nuclear compartments from Trypanosoma brucei. Identification of a major
repetitive nuclear lamina component. J. Biol. Chem. 276:38261–38271.
45. Rudenko, G., H. M. Chung, V. P. Pham, and L. H. Van der Ploeg. 1991. RNA
polymerase I can mediate expression of CAT and neo protein-coding genes
in Trypanosoma brucei. EMBO J. 10:3387–3397.
46. Shaw, R. J., and D. Reines. 2000. Saccharomyces cerevisiae transcription
elongation mutants are defective in PUR5 induction in response to nucleo-
tide depletion. Mol. Cell. Biol. 20:7427–7437.
47. Siegel, T. N., et al. 2009. Four histone variants mark the boundaries of
polycistronic transcription units in Trypanosoma brucei. Genes Dev. 23:1063–
1076.
48. Stefanovsky, V., F. Langlois, T. Gagnon-Kugler, L. I. Rothblum, and T.
Moss. 2006. Growth factor signaling regulates elongation of RNA polymer-
ase I transcription in mammals via UBF phosphorylation and r-chromatin
remodeling. Mol. Cell 21:629–639.
49. Svejstrup, J. Q. 2007. Elongator complex: how many roles does it play? Curr.
Opin. Cell Biol. 19:331–336.
50. Tucker, S., A. Vitins, and C. S. Pikaard. 2010. Nucleolar dominance and
ribosomal RNA gene silencing. Curr. Opin. Cell Biol. 22:351–356.
51. Ullu, E., and C. Tschudi. 1990. Permeable trypanosome cells as a model
system for transcription and trans-splicing. Nucleic Acids Res. 18:3319–3326.
52. Walgraffe, D., et al. 2005. Characterization of subunits of the RNA polymer-
ase I complex in Trypanosoma brucei. Mol. Biochem. Parasitol. 139:249–260.
53. Wang, A., S. K. Kurdistani, and M. Grunstein. 2002. Requirement of Hos2
histone deacetylase for gene activity in yeast. Science 298:1412–1414.
54. Wang, Q. P., T. Kawahara, and D. Horn. 2010. Histone deacetylases play
distinct roles in telomeric VSG expression site silencing in African trypano-
somes. Mol. Microbiol. 77:1237–1245.
55. White, T. C., G. Rudenko, and P. Borst. 1986. Three small RNAs within the
10 kb trypanosome rRNA transcription unit are analogous to domain VII of
other eukaryotic 28S rRNAs. Nucleic Acids Res. 14:9471–9489.
56. Winkler, G. S., A. Kristjuhan, H. Erdjument-Bromage, P. Tempst, and J. Q.
Svejstrup. 2002. Elongator is a histone H3 and H4 acetyltransferase impor-
tant for normal histone acetylation levels in vivo. Proc. Natl. Acad. Sci.
U. S. A. 99:3517–3522.
57. Wittschieben, B. O., et al. 1999. A novel histone acetyltransferase is an
integral subunit of elongating RNA polymerase II holoenzyme. Mol. Cell
4:123–128.
58. Woodward, R., and K. Gull. 1990. Timing of nuclear and kinetoplast DNA
replication and early morphological events in the cell cycle of Trypanosoma
brucei. J. Cell Sci. 95:49–57.
59. Zhang, Y., M. L. Sikes, A. L. Beyer, and D. A. Schneider. 2009. The Paf1
complex is required for efficient transcription elongation by RNA polymer-
ase I. Proc. Natl. Acad. Sci. U. S. A. 106:2153–2158.
1832 ALSFORD AND HORN MOL.CELL.BIOL.
