
Immunogenicity Testing
of Therapeutic Protein
Products — Developing
and Validating Assays for
Anti-Drug Antibody
Detection
Guidance for Industry
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Drug Evaluation and Research (CDER)
Center for Biologics Evaluation and Research (CBER)
January 2019
Pharmaceutical Quality/CMC

Immunogenicity Testing of Therapeutic
Protein Products — Developing and
Validating Assays for
Anti-Drug Antibody Detection
Guidance for Industry
Additional copies are available from:
Office of Communications,
Division of Drug Information
Center for Drug Evaluation and Research
Food and Drug Administration
10001 New Hampshire Ave., Hillandale Bldg., 4
th
Floor
Silver Spring, MD 20993-0002
Phone: 855-543-3784 or 301-796-3400; Fax: 301-431-6353
Email: druginfo@fda.hhs.gov
https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
and/or
Office of Communication, Outreach and Development
Center for Biologics Evaluation and Research
Food and Drug Administration
10903 New Hampshire Ave., Bldg. 71, Room 3128
Silver Spring, MD 20993-0002
Phone: 800-835-4709 or 240-402-8010
Email: [email protected].gov
https://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
U.S. Department of Health and Human Services
Food and Drug Administration
Center for Drug Evaluation and Research (CDER)
Center for Biologics Evaluation and Research (CBER)
January 2019
Pharmaceutical Quality/CMC
Contains Nonbinding Recommendations
i
TABLE OF CONTENTS
I. INTRODUCTION............................................................................................................. 1
II. BACKGROUND ............................................................................................................... 2
III. GENERAL PRINCIPLES................................................................................................ 3
A. Assays for ADA Detection ............................................................................................................. 3
B. Limitations in Comparing ADA Incidence Across Products ..................................................... 4
IV. ASSAY DESIGN ELEMENTS ........................................................................................ 5
A. Testing Strategy ............................................................................................................................. 5
1. Multi-Tiered Testing Approach ........................................................................................................ 5
2. Immunoglobulin Isotypes or Subtypes ............................................................................................. 6
3. Domain Specificity ........................................................................................................................... 6
B. Assay Cut-Point .............................................................................................................................. 7
C. Sensitivity ........................................................................................................................................ 8
1. Assay Sensitivity ............................................................................................................................... 8
2. Drug Tolerance, Sensitivity, and Assay Suitability .......................................................................... 9
D. Specificity ........................................................................................................................................ 9
E. Selectivity ...................................................................................................................................... 10
1. Matrix Interference ........................................................................................................................ 10
2. Minimal Required Dilution ............................................................................................................ 11
F. Precision ........................................................................................................................................ 11
G. Reproducibility ............................................................................................................................. 12
H. Robustness and Sample Stability ................................................................................................ 12
I. Selection of Format ...................................................................................................................... 12
J. Selection of Reagents ...................................................................................................................
13
1. Development of Positive Control Antibodies ................................................................................. 13
2. Development of Negative Controls ................................................................................................ 14
3. Controlling Non-Specific Binding .................................................................................................. 14
K. Reporting Results for Qualitative and Quasi-Quantitative Assays ......................................... 15
L. Other Considerations for Assay Development .......................................................................... 15
1. Pre-Existing Antibodies ................................................................................................................. 16
2. Rheumatoid Factor ........................................................................................................................ 16
3. Monoclonal Antibodies .................................................................................................................. 16
4. Conjugated Proteins ...................................................................................................................... 16
V. ASSAY DEVELOPMENT ............................................................................................. 17
A. Development of Screening Assay ................................................................................................ 17
B. Development of Confirmatory Assay ......................................................................................... 17
Contains Nonbinding Recommendations
ii
1. Selection of Format for Confirmatory Assay ................................................................................. 17
2. Cut-Point of Confirmatory Assay ................................................................................................... 18
C. Development of Titration Assay ................................................................................................. 18
D. Development of Neutralization Assay ........................................................................................ 18
1. Selection of Format for Neutralization Assay ................................................................................ 18
2. Activity Curve of Neutralization Assay .......................................................................................... 19
3. Considerations for Matrix Interference for Neutralization Assay ................................................. 20
4. Cut-Point of Neutralization Assay ................................................................................................. 21
5. Additional Considerations for Neutralization Assay ..................................................................... 21
VI. ASSAY VALIDATION .................................................................................................. 22
A. General Considerations for Assay Validation ........................................................................... 22
B. Validation of Screening Assay .................................................................................................... 24
1. Sensitivity of Screening Assay ........................................................................................................ 24
2. Cut-Point of Screening Assay ........................................................................................................ 24
C. Validation of Confirmatory Assay ............................................................................................. 24
D. Validation of Titration Assay ...................................................................................................... 25
E. Validation of Neutralization Assay ............................................................................................. 25
VII. IMPLEMENTATION OF ASSAY TESTING ............................................................. 26
A. Obtaining Subject Samples ......................................................................................................... 26
B. Concurrent Positive and Negative Quality Controls ................................................................ 27
C. Confirmation of Cut-Point in the Target Population ............................................................... 28
VIII. DOCUMENTATION ...................................................................................................... 28
REFERENCES ............................................................................................................................ 30
APPENDIX: MULTI-TIERED APPROACH TO ANTI-DRUG ANTIBODY TESTING 33

Contains Nonbinding Recommendations
1
Immunogenicity Testing of Therapeutic Protein Products —
Developing and Validating Assays for
Anti-Drug Antibody Detection
Guidance for Industry
1
This guidance represents the current thinking of the Food and Drug Administration (FDA or Agency) on
this topic. It does not establish any rights for any person and is not binding on FDA or the public. You
can use an alternative approach if it satisfies the requirements of the applicable statutes and regulations.
To discuss an alternative approach, contact the FDA office responsible for this guidance as listed on the
title page.
I. INTRODUCTION
This guidance provides recommendations to facilitate industry’s development and validation of
assays for assessment of the immunogenicity of therapeutic protein products during clinical
trials. Specifically, this document includes guidance regarding the development and validation
of screening assays, confirmatory assays, titration assays, and neutralization assays.
2,3
For the
purposes of this guidance, immunogenicity is defined as the propensity of a therapeutic protein
product to generate immune responses to itself and to related proteins or to induce
immunologically related adverse clinical events. The recommendations for assay development
and validation provided in this document apply to assays for the detection of one or more anti-
1
This guidance has been prepared by the Office of Medical Policy in the Center for Drug Evaluation and Research
in cooperation with the Center for Biologics Evaluation and Research at the Food and Drug Administration.
2
This document specifically does not discuss the development or validation of anti-drug antibody (ADA) assays for
animal studies; however, some concepts discussed are relevant to the design of ADA studies for nonclinical testing.
Refer to the International Conference on Harmonisation (ICH) guidance for industry S6(R1) Preclinical Safety
Evaluation of Biotechnology-Derived Pharmaceuticals for more information regarding immunogenicity assessments
in animal toxicology studies. Also see the guidance for industry Immunogenicity Assessment for Therapeutic
Protein Products, where the topic “Utility of Animal Studies” is covered in detail. We update guidances
periodically. For the most recent version of a guidance, check the FDA guidance web page at
https://www.fda.gov/RegulatoryInformation/Guidances/default.htm
.
3
In general, this guidance provides recommendations related to the development of therapeutic protein products
intended for submission in a “stand-alone” biologics license application (BLA) under section 351(a) of the Public
Health Service (PHS) Act or for submission as proposed biosimilar and interchangeable biological products under
section 351(k) of the PHS Act. For additional information on clinical immunogenicity assessment of proposed
biosimilar and interchangeable biological products, see the guidances for industry Scientific Considerations in
Demonstrating Biosimilarity to a Reference Product and Considerations in Demonstrating Interchangeability with a
Reference Product, respectively.

Contains Nonbinding Recommendations
2
drug antibodies (ADAs).
4
This guidance may also apply to some peptides, oligonucleotides, and
combination products on a case-by-case basis.
5
In general, this document does not discuss the rationale for ADA testing or the subject- and
product-specific risk factors that may contribute to immunogenicity.
6
Also, this guidance,
including any discussions of terminology used in this guidance, does not apply to in vitro
diagnostic products.
7
In general, FDA’s guidance documents do not establish legally enforceable responsibilities.
Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only
as recommendations, unless specific regulatory or statutory requirements are cited. The use of
the word should in Agency guidances means that something is suggested or recommended, but
not required.
II. BACKGROUND
Immune responses to therapeutic protein products have the potential to affect product
pharmacokinetics, pharmacodynamics, safety, and efficacy.
8
The clinical effects of immune
responses in subjects are highly variable, ranging from no measurable effect to extremely
harmful. Detection and analysis of ADA formation is a helpful tool in understanding potential
immune responses. Information on immune responses observed during clinical trials,
particularly the incidence of ADA induction or any implications of ADA responses affecting
pharmacokinetics, pharmacodynamics, safety, or efficacy, is crucial for any therapeutic protein
product development program. Accordingly, such information, if applicable, should be included
in the prescribing information as a subsection of the ADVERSE REACTIONS section entitled
4
This guidance does not pertain to immunogenicity assays for assessment of immune response to preventative and
therapeutic vaccines for infectious disease indications or to cell and gene therapy products.
5
General information on combination products is available at
https://www.fda.gov/CombinationProducts/default.htm
.
6
See the guidance for industry Immunogenicity Assessment for Therapeutic Protein Products, where some of these
topics are covered in detail.
7
Per 21 CFR 809.3(a), “In vitro diagnostic products are those reagents, instruments, and systems intended for use in
the diagnosis of disease or other conditions, including a determination of the state of health, in order to cure,
mitigate, treat, or prevent disease or its sequelae. Such products are intended for use in the collection, preparation,
and examination of specimens taken from the human body. These products are devices as defined in section 201(h)
of the Federal Food, Drug, and Cosmetic Act (the act), and may also be biological products subject to section 351 of
the Public Health Service Act.”
8
See the guidance for industry Immunogenicity Assessment for Therapeutic Protein Products.

Contains Nonbinding Recommendations
3
Immunogenicity.
9
Therefore, the development of valid, sensitive, specific, and selective assays
to measure ADA responses is a key aspect of therapeutic protein product development.
III. GENERAL PRINCIPLES
The risk to subjects from mounting an ADA-generating immune response to a therapeutic
protein product will vary with the product. FDA recommends adopting a risk-based approach to
evaluating and managing immune responses to — or immunologically related adverse clinical
events associated with — therapeutic protein products that affect their pharmacokinetics,
pharmacodynamics, safety, and efficacy.
10
Immunogenicity tests should be designed to detect
ADA that could mediate unwanted biological or physiological consequences such as neutralizing
activity or hypersensitivity responses.
A. Assays for ADA Detection
Screening assays, also known as binding antibody assays, are used to detect antibodies that bind
to the therapeutic protein product. The specificity of ADA for the therapeutic protein product is
usually established by competition with a therapeutic protein in a confirmatory assay.
ADAs are
characterized further using titration and neutralization assays. Titration assays characterize the
magnitude of the ADA response. It is important to characterize this magnitude with titration
assays because the impact of ADA on pharmacokinetics, pharmacodynamics, safety, and
efficacy may correlate with ADA titer and persistence rather than incidence (Cohen and Rivera
2010). Neutralizing antibodies (NAbs) refer to those ADA with the ability to interfere with
interactions between the therapeutic protein product and its target. Neutralization assays assess
ADA for neutralizing activity. It is important to characterize neutralizing activity of ADA
because the impact of ADA on pharmacokinetics, pharmacodynamics, safety, and efficacy may
correlate with NAb activity rather than ADA incidence (Calabresi et al. 2007; Goodin et al.
2007; Cohen and Rivera 2010; Wang et al. 2016; Wu et al. 2016). Similarly, in some cases it
may be useful to establish NAb titers in addition to NAb qualitative results (for example, positive
or negative), depending on immunogenicity risk assessment. Additional characterization assays,
including isotyping, epitope mapping, and assessing cross-reactivity (for example, to endogenous
counterparts or to other products), may be useful.
The optimal time to design, develop, and validate ADA assays during therapeutic protein product
development depends on the risk assessment of the product (Mire-Sluis et al. 2004; Gupta et al.
2007; Shankar et al. 2008; Gupta et al. 2011). The sponsor should provide an immunogenicity
risk assessment as well as a rationale for the immunogenicity testing paradigm in the original
investigational new drug application (IND). FDA encourages sponsors to test samples during
9
Among other requirements, prescription drug labels must include information about the drug’s adverse reactions
(21 CFR 201.57(c)(7) and 21 CFR 201.57(a)(11)). Adverse reaction is defined in 21 CFR 201.57(c)(7) as “an
undesirable effect, reasonably associated with use of a drug, that may occur as part of the pharmacological action of
the drug or may be unpredictable in its occurrence.”
10
See the guidance for industry Immunogenicity Assessment for Therapeutic Protein Products.

Contains Nonbinding Recommendations
4
phase 1 and phase 2 studies using suitable screening, confirmatory, and in some instances
neutralization assays. Samples derived from pivotal clinical studies should be tested with fully
validated assays.
11
When immunogenicity poses a high clinical risk and real-time data
concerning subject responses are needed (for example, when there is an endogenous counterpart
with non-redundant function), FDA may request that assays suitable for their intended purpose
be developed before initiating clinical studies and that testing be performed in real time. In such
instances, timing and reporting of ADA assessment should be discussed with the Agency. In
other situations, the sponsor may store subject samples so they can be tested when suitable
assays are available. At the time of license application, the sponsor should provide data
supporting full validation of the assays (see section VIII). Recommendations regarding the
timing of ADA sample collection are provided in section VII.A.
12
B. Limitations in Comparing ADA Incidence Across Products
Results from assays for detection of ADA facilitate understanding of the immunogenicity,
pharmacokinetics, pharmacodynamics, safety, and efficacy of therapeutic protein products.
However, detection of ADA is dependent on key operating parameters of the assays; for
example, sensitivity, specificity.
13
Although information on ADA incidence is typically included
in the prescribing information under an Immunogenicity subsection of the ADVERSE
REACTIONS section, FDA cautions that comparison of ADA incidence across products, even
for products that share sequence or structural homology, can be misleading because detection of
ADA formation is highly dependent on the sensitivity, specificity, and drug tolerance level of the
assay. Additionally, the observed incidence of ADA is influenced by multiple factors including
method, sample handling, timing of sample collection, concomitant medications, and disease
condition. Therefore, comparing immunogenicity rates across therapeutic protein products with
structural homology for the same indication is unsound, even though fully validated assays are
employed. When a direct comparison of immunogenicity across different therapeutic protein
products that have homology — or across similar therapeutic proteins from different sources —
is needed, the comparison data should be obtained by conducting a head-to-head clinical study
from which samples obtained are tested using an assay demonstrated to have equivalent
sensitivity and specificity for antibodies against both therapeutic protein products.
The recommendations on assay development and validation provided in this guidance are based
on common issues encountered by the Agency upon review of immunogenicity submissions.
Sponsors should contact FDA for any product-specific advice, particularly for high-risk
products; for example, products with endogenous counterparts that have non-redundant
11
Pivotal clinical studies may be used to evaluate and establish the efficacy of the product.
12
See the guidance for industry Immunogenicity Assessment for Therapeutic Protein Products, where
immunogenicity risk assessment and mitigation considerations are covered in detail. Guidance on appropriate assay
development and validation for immunogenicity testing is also available in the ICH guidances for industry Q2A Text
on Validation of Analytical Procedures and Q2B Validation of Analytical Procedures: Methodology.
13
See the United States Pharmacopeia (USP) General Chapters 1106 Immunogenicity Assays — Design and
Validation of Immunoassays to Detect Anti-Drug Antibodies and 1106.1 Immunogenicity Assays — Design and
Validation of Assays to Detect Anti-Drug Neutralizing Antibody for a broader discussion of various assay types.

Contains Nonbinding Recommendations
5
function.
14
Assay designs for isotyping, epitope mapping, and cross-reactivity with endogenous
counterparts should be discussed with FDA. Other publications may also be consulted for
additional insight (Mire-Sluis et al. 2004; Gupta et al. 2007; Shankar et al. 2008; Gupta et al.
2011).
15
In general, FDA recommends that sponsors develop assays that are optimized for
sensitivity, specificity, selectivity, drug tolerance, precision, reproducibility, and robustness (see
sections IV.C through H).
IV. ASSAY DESIGN ELEMENTS
This section applies to all types of assays for detection of ADA, unless specified otherwise. The
bioanalytical scientist should evaluate the applicability of these factors and others based on
emerging science. FDA’s thinking on this matter may change as the science evolves.
A. Testing Strategy
1. Multi-Tiered Testing Approach
FDA recommends a multi-tiered ADA testing approach (see Appendix). In this paradigm, a
sensitive screening assay is initially used to assess clinical samples. To gain a more accurate
understanding of the natural history of the ADA response, the screening assay should be
sensitive and designed to detect low levels of low- and high-affinity ADA; for example, by
minimizing wash steps. However, in most cases it is not necessary to empirically determine the
affinity of antibodies that are detected by the initial screening assay. Samples testing positive in
the screening assay are then subjected to a confirmatory assay to demonstrate that ADAs are
specific for the therapeutic protein product. For example, a competition assay could confirm that
an antibody is specifically binding to the therapeutic protein product and that the positive finding
in the screening assay is not a result of non-specific interactions of the test serum or detection
reagent with other materials in the assay milieu such as plastic or other proteins.
Samples identified as positive in the confirmatory assay should be further characterized in other
assays, such as titration and neutralization assays. In some cases, assays to detect cross-
reactivity to other proteins, such as the corresponding endogenous protein, may be needed. For
example, assessment of cross-reactivity may be needed when the therapeutic protein product
belongs to a family of proteins with high homology and it is important to know whether other
family members are affected by ADA. Further, in some cases tests to assess the isotype of the
antibodies or their epitope specificity may also be recommended once samples containing
antibodies are confirmed as positive. Epitope specificity determination of the ADA response is
not frequently performed, although it is common to perform a more general assessment of
domain specificity for multi-domain products such as pegylated proteins, antibody-drug
conjugates, and bispecific antibodies (see section IV.A.3).
14
Ibid.
15
See the guidance for industry Immunogenicity Assessment for Therapeutic Protein Products.

Contains Nonbinding Recommendations
6
2. Immunoglobulin Isotypes or Subtypes
The initial screening assay should be able to detect all relevant immunoglobulin (Ig) isotypes.
For non-mucosal routes of administration and in the absence of a risk of anaphylaxis, the
relevant ADA isotypes are IgM and IgG. For mucosal routes of administration, IgA isotype
ADAs are also relevant.
16
Although FDA expects that all relevant isotypes be detected in
screening assays, it is not necessary that the screening assay establishes which isotypes are being
detected. For example, the bridging assay format can theoretically detect antibodies of most
isotypes but does not provide information on which isotypes are being detected.
17
In some circumstances the sponsor should develop assays that discriminate between antibody
isotypes. For example, for therapeutic protein products where there is a high risk for anaphylaxis
or where anaphylaxis has been observed, results from antigen-specific IgE assays may be
informative.
Assessment of ADA subtype may be informative in some situations. For example, the
generation of IgG4 antibodies has been associated with immune responses generated under
conditions of chronic antigen exposure, such as factor VIII treatment, and in erythropoietin-
treated subjects with pure red cell aplasia (Matsumoto et al. 2001; Aalberse and Schuurman
2002). Consequently, depending on the clinical concern, assessing for specific isotypes or
subtypes may be needed.
3. Domain Specificity
Some proteins possess multiple domains that function in different ways to mediate clinical
efficacy. An immune response to one domain may inhibit a specific function while leaving
others intact. FDA recommends that sponsors direct initial screening and confirmatory tests
against the whole therapeutic protein product. For multi-domain therapeutic protein products,
the sponsor may need to investigate whether the ADA binds to specific clinically relevant
domains in the protein. For example, to adequately understand the risk of ADA to subjects for
therapeutic protein products with modifications such as pegylation, sponsors should develop
assays to determine the specificity of ADA for the protein component as well as the modification
to the therapeutic protein product (Gorovits et al. 2014).
The domain specificity is generally assessed in ADA samples confirmed positive using the
whole molecule. Examination of immune responses to therapeutic protein products with
multiple functional domains such as bispecific antibodies may require development of multiple
assays to measure immune responses to different domains of the molecules (see section IV.L.4).
16
Mucosal routes of administration include oral, respiratory, vaginal, ocular, and rectal, where the drug is delivered
across a mucosal barrier.
17
Bridging assays may not be adequately robust for detecting IgG4 antibodies, which may underestimate the levels
of antibodies.

Contains Nonbinding Recommendations
7
B. Assay Cut-Point
The cut-point of the assay is the level of response of the assay that defines the sample response
as positive or negative. Information specific to establishing the cut-point for the respective assay
types is provided in sections V and VI. Establishing the appropriate cut-point is critical to
minimizing the risk of false-negative results.
The cut-point of the assay can be influenced by a myriad of interfering product or matrix
components.
18
These components should be considered early on in assay development when
defining the cut-point and are discussed in detail in section IV.K. Because samples from
different target populations and disease states may have components that can cause the
background signal from the assay to vary, different cut-points may be needed for discrete
populations.
Where feasible, the cut-point should be statistically determined using samples from treatment-
naïve subjects.
19
By performing replicate assay runs with these samples, the variability of the
assay can be estimated. The statistical approach employed to determine the cut-point may entail
various processes, such as removing statistical outliers from analyses, and using an approach to
account for pre-existing antibodies. During assay development, a small number of samples may
be used to estimate the cut-point.
The sponsor should consider the impact of statistically determined outlier values and true-
positive samples when establishing the cut-point. The sponsor should provide justification for
the removal of any data points, along with the respective method used to determine their status as
outliers. Sponsors should consult with FDA if there is a concern regarding the exclusion of
outliers.
Apparent positive values and samples may derive from the presence of pre-existing antibodies or
other serum factors in subject samples (Ross et al. 1990; Turano et al. 1992; Coutinho et al.
1995; Caruso and Turano 1997; van der Meide and Schellekens 1997; Boes 2000). Although
pre-existing antibodies to a variety of endogenous proteins are present in healthy individuals,
these can be much higher in some disease states. The sponsor should identify those samples with
pre-existing antibodies (for example, through competition with drug) and remove them from the
cut-point analysis. If subjects in the study have pre-existing antibodies, it may be necessary to
assign positive responses using a cut-point based on the difference between individual subject
results before and after exposure to identify subjects in whom ADA increases following
treatment, also known as treatment-boosted ADA. A common approach to evaluating treatment-
boosted ADA responses is to assess changes in antibody titers. If it is not possible to use the
methods described earlier in section IV.B for establishing the cut-point, sponsors should consult
with the Agency to explore alternative methods.
18
The term matrix when used in this guidance may include serum, plasma, saliva, etc.
19
Treatment-naïve subjects could be healthy individuals or a patient population not exposed to a therapeutic protein
product, depending on the stage of assay development or validation and the availability of samples. Sponsors should
provide justification for the appropriateness of the samples used.

Contains Nonbinding Recommendations
8
C. Sensitivity
1. Assay Sensitivity
Assay sensitivity is the lowest concentration at which the antibody preparation consistently
produces either a positive result or a readout equal to the cut-point determined for that assay.
The assays should have sufficient sensitivity to enable detection of ADA before they reach levels
that can be associated with altered pharmacokinetic (PK), pharmacodynamic (PD), safety, or
efficacy profiles. Assay sensitivity is assessed using positive control antibody preparations that
may not represent the ADA response in a specific subject. For example, positive controls are
frequently developed under conditions that enrich for high affinity antibodies. Such high affinity
positive controls may overestimate the sensitivity of the assay. Because of this, the assay
sensitivity determination contributes to the overall understanding of how the assay performs
rather than setting an absolute mass of ADA that will be detected in any given subject. Because
the measurement of assay sensitivity can be affected by onboard drug, it is also important to
determine assay sensitivity in the presence of the expected concentration of onboard drug (see
section IV.C.2).
20
FDA recommends that screening and confirmatory IgG and IgM ADA assays
achieve a sensitivity of at least 100 nanograms per milliliter (ng/mL) although a limit of
sensitivity greater than 100 ng/mL may be acceptable depending on risk and prior knowledge.
Traditionally, FDA has recommended sensitivity of at least 250 to 500 ng/mL. However, recent
data suggest that concentrations as low as 100 ng/mL may be associated with clinical events
(Plotkin 2010; Zhou et al. 2013). It is understood that neutralization assays may not achieve that
level of sensitivity. Assays developed to assess IgE ADA should have sensitivity in the high
picograms per milliliter (pg/mL) to low ng/mL range.
The sensitivity should be expressed as mass of antibody detectable/mL of undiluted matrix; for
example, plasma, sera, saliva. Assay sensitivity should not be reported as titer. Assay sensitivity
should be reported after factoring in the minimal required dilution (MRD). For example, an
assay with 50 ng/mL sensitivity and an MRD of 20 would be reported as 1000 ng/mL. Testing
of assay sensitivity should be performed with the relevant dilution of the same biological matrix
as will be used to test the clinical samples. For example, assay sensitivity should be determined
using the same anticoagulant as the diluent used with clinical samples.
During development, sensitivity may be assessed by testing serial dilutions of a positive control
antibody of known concentration, using individual or pooled matrix from treatment-naïve
subjects. The dilution series should be no greater than two- or threefold, and a minimum of five
dilutions should be tested. The sensitivity can be calculated by interpolating the linear portion of
the dilution curve to the assay cut-point.
A purified preparation of antibodies specific to the therapeutic protein product should be used as
the positive control to determine the sensitivity of the assay so that assay sensitivity can be
reported in mass units/mL of matrix. Positive control antibodies used to assess sensitivity can
take the form of polyclonal preparations affinity purified against the therapeutic protein product
or monoclonal antibodies (mAb).
20
See the USP General Chapters 1106 and 1106.1 for a discussion on Relative Sensitivity.

Contains Nonbinding Recommendations
9
During routine performance of the assay, a low positive system suitability control should be used
to ensure that the sensitivity of the assay is acceptable across assay runs. Additionally, the low
positive control should be consistently demonstrated as positive in both screening and
confirmatory tiers (see section IV.J.1). Both positive and negative controls are discussed in
detail in sections IV.J.1 and IV.J.2.
2. Drug Tolerance, Sensitivity, and Assay Suitability
The therapeutic protein product or its endogenous counterpart present in the serum may interfere
with the sensitivity of the assay. The assessment of assay sensitivity in the presence of the
expected levels of interfering therapeutic protein product, also known as the assay’s drug
tolerance, is critical to understanding the sensitivity and suitability of the method for detecting
ADA in dosed subjects.
21
FDA recommends that sponsors examine assay drug tolerance early in
assay development. The sponsor may examine drug tolerance by deliberately adding different
known amounts of positive control antibody into ADA-negative control samples in the absence
or presence of different quantities of the therapeutic protein product to determine whether the
therapeutic protein product interferes with ADA detection. Results obtained in the absence and
presence of different quantities of the therapeutic protein product under consideration should be
compared. Drug tolerance may be improved using approaches such as acid dissociation that
disrupt circulating ADA-drug complexes. The selectivity of the assay, the nature of the target,
and the type of positive control should be taken into consideration when developing the assay
because these factors impact the assessment of drug tolerance. For example, acid dissociation
may not be appropriate when antibodies are acid labile or the drug target is soluble. Interference
from the therapeutic protein product can be minimized by collecting subject samples at trough
drug levels. See section VII.A for recommendations regarding the timing of ADA sample
collection.
D. Specificity
Specificity refers to the ability of a method to exclusively detect the target analyte, in this case
the ADA.
22
Lack of assay specificity can lead to false-positive results, which could obscure
relationships between ADA generating immune response, pharmacokinetics, pharmacodynamics,
and clinical safety and efficacy measures. Demonstrating the specificity of antibody responses to
mAb, Fc-fusion proteins, and Ig-fusion proteins poses challenges because of the high
concentration of Ig in human serum. The assay should specifically detect anti-mAb antibodies
but not the mAb product itself, soluble drug target, non-specific endogenous antibodies, or
antibody reagents used in the assay. Similarly, for subject populations with a high incidence of
rheumatoid factor (RF), the sponsor should demonstrate that RF does not interfere with the
detection method or that the assay can differentiate between RF and specific antibodies. RF is
discussed in detail in section IV.L.2. In cases where ADA demonstrates cross-reactivity with
host cell proteins and other product-related impurities, the specificity of these reactions may need
further evaluation.
21
See the USP General Chapters 1106 and 1106.1.
22
Ibid.
Contains Nonbinding Recommendations
10
A straightforward approach to addressing specificity is to demonstrate that binding can be
blocked by soluble or unlabeled purified therapeutic protein product. One approach is to
incubate positive and negative control antibody samples with the purified therapeutic protein
product or its components under consideration. Inhibition of signal in the presence of the
relevant therapeutic protein product or its components indicates that the response is specific.
Establishing the specificity of multimeric antibodies such as IgM by competitive inhibition may
be difficult, so establishing assay capability for these circumstances requires careful development
or additional approaches. For ADA to mAb products, inclusion of another mAb with the same
Fc but different variable region can be informative. If the assay is specific and selective for
ADA to the therapeutic protein product being studied, generally the addition of that therapeutic
protein product or its components in solution will reduce the assay signal. Conversely, addition
of the therapeutic protein product or its components should have little effect on antibodies of
other specificities.
E. Selectivity
The selectivity of an ADA assay is its ability to identify ADAs specific to the therapeutic protein
product in the presence of other components in the sample. Assay results may be affected by
interference from the matrix or onboard therapeutic protein product. It is important to note that
most assay matrices contain significant amounts of proteins of various sizes and charges. Failure
to establish selectivity can contribute to non-specific signal, thereby obscuring positive results.
1. Matrix Interference
An important consideration is how the sample matrix (for example, plasma, serum, saliva) can
affect assay performance. Some degree of signal suppression is expected when comparing assay
performance in diluent versus matrix. Endogenous and exogenous components in a matrix may
influence assay results, and it is usually necessary to dilute subject samples for testing to
minimize such effects. The sponsor should define the matrix and dilution factor that will be used
for preparation of subject samples before performing validation studies assessing potential
interference of this matrix on assay results (see section IV.E.2 on MRD).
Various substances in the matrix, such as free hemoglobin (hemolysis), lipids (lipemia), bilirubin
(interus), and presence of concomitant medications, can interfere with assay results. For
example, the anticoagulants used during sample collection may have different effects on the
assay, potentially affecting the assay sensitivity. The sponsor may examine matrix interference
by spiking different known amounts of positive control antibodies in the presence or absence of
matrix. Comparing the recovery of ADA in buffer alone with that in the matrix can provide
input on the degree of interference from matrix components. Furthermore, such analysis may
guide decisions on the MRD recommended for sample testing. This information may be useful
to understanding assay sensitivity.
Buffer components that are chemically related to the therapeutic protein product may also cause
interference in the assay. For example, polysorbate is chemically similar to polyethylene glycol

Contains Nonbinding Recommendations
11
(PEG) and therefore may interfere in the detection of anti-PEG antibodies. The chemical
composition of the buffer should be carefully considered during assay development.
2. Minimal Required Dilution
Matrix components can contribute to non-specific signal, thereby obscuring positive results.
Therefore, there is frequently a need to dilute subject samples to maintain a reasonable ability to
detect ADA. Multiple definitions of MRD have been proposed, including the sample dilution
that yields the highest signal-to-noise ratio; the sample dilution that results in a signal closest to
assay diluent; and the sample dilution that results in the highest signal to variability ratio (Mire-
Sluis et al. 2004).
23
Sponsors may use any of these definitions, but for the purposes of
calculating assay sensitivity and titer, the MRD should take into consideration the final dilution
of the sample in the assay, which typically ranges from 1:5 to 1:100 (that is, 1/5 to 1/100).
FDA recommends that sponsors determine the MRD from a panel of appropriate number of
samples from treatment-naïve subjects. Determination of MRD usually involves serially diluting
treatment-naïve ADA-negative samples, as well as testing known amounts of purified antibody
at high, medium, and low concentrations in serially diluted matrix in comparison to the same
amount of positive control antibody in diluent. This ensures a reasonable signal-to-noise ratio
throughout the range of the assay. The MRD should be calculated using an appropriate number
of individual serum samples. The appropriate number of samples will depend on various factors,
including the variability of the individual samples; however, at least 10 samples are frequently
recommended (Mire-Sluis et al. 2004).
24
Although the MRD ultimately selected by the sponsor will depend on the assay design and
subject population, FDA recommends that MRD not exceed 1:100. Higher MRD may result in
false-negative responses. However, in some instances higher MRD may be required, and the
overall effect of such MRD on assay sensitivity and immunogenicity risk assessment should be
considered.
F. Precision
Precision is a measure of the variability in a series of measurements for the same material run in
a method. Results should be reproducible within and between assay runs to assure adequate
precision.
25
Demonstrating assay precision is critical to the assessment of ADA because assay
variability is the basis for determining the cut-points and ensuring that low positive samples are
detected as positive. To provide reliable estimates, the sponsor should evaluate both intra-assay
(repeatability) and inter-assay (intermediate precision) variability of assay responses. In cases
23
Ibid.
24
Ibid.
25
For more information on precision, see the guidance for industry Bioanalytical Method Validation. Also see the
USP General Chapters 1106 and 1106.1.

Contains Nonbinding Recommendations
12
where a floating cut-point is needed, inter-assay precision may be calculated using normalized
values.
G. Reproducibility
Reproducibility is an important consideration if an assay will be run by two or more independent
laboratories during a study, and a sponsor should establish the comparability of the data
produced by each laboratory.
26
Comparable assay performance, including sensitivity, drug
tolerance, and precision, should be established between laboratories.
H. Robustness and Sample Stability
Assay robustness is an indication of the assay’s reliability during normal usage
27
and is assessed
by the capacity of the assay to remain unaffected by small but deliberate variations in method
and instrument performance that would be expected under relevant, real-life circumstances in
routine laboratory practice. For example, changes in temperature, incubation times, or buffer
characteristics such as pH and salt concentration can all impact assay results. The complexity of
bioassays makes them particularly susceptible to variations in assay conditions, and it is essential
to evaluate and optimize parameters such as cell passage number, incubation times, and culture
media components. The sponsor should examine robustness during the development phase, and
if small changes in specific steps in the assay affect results, precautions should be taken to
control that step. Some aspects of robustness may be included in the assay validation exercise
(see section VI.A). Because it is generally not feasible to establish the stability of subject
samples, FDA recommends storing subject samples in a manner that preserves antibody
reactivity at the time of testing. FDA recommends that sponsors minimize freeze-thaw cycles by
appropriately aliquoting subjects’ samples because freezing and thawing such samples may also
affect assay results. However, studies evaluating short-term stability, including, as relevant,
freeze-thaw cycle and refrigerator- and room-temperature stability of positive control antibodies,
may be useful.
I. Selection of Format
Different assay formats and instrumentation are available that can be used for detection of ADA.
These include, but are not limited to, direct binding assays, bridging assays, and soluble-phase
binding assays; for example, radioimmunoprecipitation assay. Each assay format has advantages
and disadvantages, including throughput, sensitivity, selectivity, dynamic range, ability to detect
various Ig isotypes, ability to detect rapidly dissociating antibodies, and availability of reagents.
Bridging assay formats may be subject to false-negative results when the antigen (for example,
PEG) has repetitive motifs. One of the major differences between these assay formats is the
number and vigor of washes, which can influence assay sensitivity. Epitope exposure is also
26
For more information on reproducibility, see the guidance for industry Bioanalytical Method Validation. Also,
see the USP General Chapters 1106 and 1106.1; the USP General Chapter 1225 Validation of Compendial
Procedures; and the ICH guidance for industry Q2B Validation of Analytical Procedures: Methodology.
27
For more information on robustness, see the ICH guidance for industry Q2B Validation of Analytical Procedures:
Methodology. Also see the USP General Chapters 1106 and 1106.1.

Contains Nonbinding Recommendations
13
important to consider because binding to plastic or coupling to other agents (for example,
fluorochrome, enzyme, or biotin reporters) can result in conformational changes of the antigen
that can obscure, expose, modify, or destroy relevant antibody binding sites on the therapeutic
protein product in question.
J. Selection of Reagents
Many components of the assays for ADA detection may be standard or obtained from
commercial sources; for example, microtiter plates. Other components, however, including
positive control antibodies, negative controls, and system suitability controls, may need to be
generated specifically for the assay. Qualification and stability of critical reagents is important
for ensuring consistent assay performance.
1. Development of Positive Control Antibodies
Sponsors may use the same or different positive control antibodies to develop, validate, and
monitor system suitability during routine assessment of assay performance. For system
suitability controls, a positive control antibody, either mono- or polyclonal, used at
concentrations adjusted to ensure assay sensitivity and detect hook effects, should be included.
28
Different approaches may be used to generate a positive control. Most frequently, positive
control antibodies are generated by immunizing animals in the absence or presence of adjuvants.
FDA recommends that positive control antibodies generated by immunizing animals be affinity
purified using the therapeutic protein product. This approach enriches the polyclonal antibody
preparation for ADA, which enables a better interpretation of sensitivity assessment results. The
selection of animal species when generating positive control antibodies should be carefully
considered. For example, if an anti-human Ig reagent will be used as a secondary reagent to
detect antibodies in subjects, the positive control antibodies and quality control (QC) samples
ideally should be detectable by that same reagent. When the positive control antibody is not
detectable by that same reagent (for example, if the positive control is generated in a rabbit and a
different secondary reagent is needed to detect the positive control antibody), a positive control
antibody for the secondary reagent used to detect human antibodies in the subject samples also
should be included in the assay to ensure that the reagent performs as expected. In some
instances, the sponsor may be able to generate a positive control antibody from subjects’
samples.
29
Such subject-derived positive controls can be very valuable but are generally not
available in early trials. Alternatively, individual mAb or panels of mAb may be used as positive
control antibodies. For therapeutic mAb, the sponsor should select a positive control antibody
that binds to the variable region of the therapeutic mAb. Sponsors should discuss with FDA
alternative approaches to assay development and validation in the rare event that a sponsor is not
able to generate a positive control antibody.
28
Hook effects are a reduction in signal that may occur because of the presence of a high concentration of a
particular analyte or antibody and may cause false-negative results.
29
Proper informed consent from patients is needed and should be planned ahead.
Contains Nonbinding Recommendations
14
Once a source of a positive control antibody has been identified, the sponsor should use that
source to assess assay performance characteristics such as sensitivity, selectivity, specificity,
drug tolerance, and reproducibility. FDA recommends that sponsors generate and reserve
positive control antibody for use as a quality control or system suitability control during routine
performance of the assay. For assay development and validation, dilutions should generate high,
intermediate, and low assay signal values. The intermediate value is useful for assessing
precision during assay validation. This is recommended even for development of qualitative
assays to understand whether assay performance is acceptable across a broad range of antibody
concentrations. Intermediate-value QC samples for detection of ADA are generally not needed
for monitoring system suitability during routine assay performance.
2. Development of Negative Controls
FDA recommends that sponsors establish a negative control for validation studies and subject-
sample testing. In this regard, a pool of sera from an appropriate number of treatment-naïve
subjects can serve as a negative control. Importantly, the value obtained for the negative control
should be below but close to the cut-point determined for the assay in the subject population
being tested. Negative controls that yield values far below the mean value derived from
individual serum samples used to establish the cut-point may not be useful in ensuring proper
assay performance.
When possible, negative control samples should be collected from treatment-naïve subjects with
the medical condition being studied and should include subjects with similar gender, age, and
concomitant medications so that the sample matrix is representative of the study population.
Control samples should be collected and handled in the same manner as study samples with
respect to, for example, type of anticoagulant used, volume, and sample preparation and storage
because these pre-analytical variables can impact the performance of control samples in the
assay. It is frequently the case that such control samples are not available for use during
development or pre-study validation exercises. In those situations, it is acceptable to use
purchased samples or samples from healthy donors, but important parameters of assay
performance such as cut-point, sensitivity, and selectivity should be confirmed when samples
from treatment-naïve subjects from the appropriate target population become available. If cut-
point and selectivity differ when negative controls from different populations are used, re-
evaluating other assay parameters (for example, sensitivity) may be needed.
3. Controlling Non-Specific Binding
Every test component, from the plastic of the microtiter plates to the developing agent, can affect
assay sensitivity and non-specific binding. One of the most critical elements is the selection of
the proper assay buffer and blocking reagents used to prevent non-specific binding. The sponsor
should carefully consider the number and timing of wash steps as well as the detergents added to
the assay buffer (for example, blocking or wash buffer) to reduce background noise while
maintaining sensitivity. A variety of proteins can be used as blocking reagents to provide
acceptable signal-to-noise ratio. However, these proteins may not all perform equivalently in
specific immunoassays. For example, they may not bind well to all types of solid phases or may
show unexpected cross-reactivity with the detecting reagent. Therefore, the sponsor may need to
Contains Nonbinding Recommendations
15
test several blocking agents to optimize assay performance. Moreover, including uncoated wells
is insufficient to assess non-specific binding. Rather, determining the capacity of ADAs to bind
to an unrelated protein of similar size and charge that may be present in the sample may prove to
be a better test of binding specificity.
K. Reporting Results for Qualitative and Quasi-Quantitative Assays
Several approaches may be used to report positive antibody responses, and the appropriateness of
the approach used should be evaluated on a case-by-case basis. The most common approach is
qualitative, with subjects reported as having a positive or negative antibody response.
For subjects who are confirmed to be ADA positive, determining antibody levels can be
informative because it allows for stratified assessment of ADAs and their impact on safety and
efficacy. Positive antibody levels may be evaluated using a titer. Reporting levels of antibodies
in terms of titers is appropriate and generally understood by the medical community. Most
frequently titer is determined from the reciprocal of the highest dilution that gives a value at or
just above the cut-point of the assay. Alternatively, titer may be determined by extrapolating the
dilution to the assay cut-point using the linear portion of the dose response curve. All sample
dilutions, such as the MRD and acid dissociations, should be factored into the calculations of
titers and provided when reporting titers.
When reporting results for neutralization assays, values may also be reported as amount of mass
units of therapeutic protein product neutralized per volume serum with the caveat that these are
arbitrary in vitro assay units and cannot be used to estimate in vivo availability of the therapeutic
protein product.
Unless the assay method used allows for independent determination of mass per volume of
undiluted matrix, antibody levels reported in mass units are generally not acceptable. This is
because the mass unit estimations are based on interpolation of data from standard curves
generated with a positive control antibody, and parallelism between the positive control and test
article cannot be assumed. Furthermore, even if parallelism between the positive control and test
article is demonstrated, the absolute mass units cannot accurately be calculated because the
samples are likely to contain different populations of antibodies. Thus, FDA does not consider it
necessary or desirable for the sponsor to report subject antibody results in terms of mass units
unless (1) the results are determined by quantitative means or (2) a universally accepted and
accessible source of validated antibody is available as a control and parallelism between the
dilution curves of the control antibody and subject samples has been demonstrated.
L. Other Considerations for Assay Development
A myriad of factors can affect the assessment of ADA levels, such as subject-sample variability;
therapeutic protein product-dose response of the cells used to generate the standard curve in a
cell-based neutralization bioassay; affinity and avidity of the ADA; and concentration of
competing product in confirmatory assays. Accounting for such factors is important to
understand and analyze assay variability and avoid errors. Common factors that should be
considered include the following:
Contains Nonbinding Recommendations
16
1. Pre-Existing Antibodies
Pre-existing antibodies may have clinical effects that affect the efficacy of the therapeutic protein
product being tested. An alternative to the qualitative screening assay approach may be needed
to assess the quantity and quality of ADA when pre-existing antibodies are present. For
example, testing samples for an increase in ADA using a semi-quantitative assay such as a
titration assay (see sections V.C and VI.D) can provide information on the impact of a
therapeutic protein product on product immunogenicity that is not provided by a qualitative
assay. When there are pre-existing antibodies and the titer of antibodies increases after exposure
to the therapeutic protein product, they can be reported as treatment-boosted to differentiate them
from treatment-induced antibody titers. For example, a boosted ADA response may be defined
as a titer that is two dilution steps greater than the pre-treatment titer, when twofold dilutions are
used to determine the titer.
2. Rheumatoid Factor
Measuring immune responses to therapeutic protein products that possess Fc regions, such as
mAb and Fc-fusion proteins, may be particularly difficult when RF is present in the matrix. RF
is generally an IgM antibody that recognizes IgG, although other RF Ig specificities have been
noted. Consequently, RF will bind Fc regions, making it appear that specific antibody to the
therapeutic protein product exists. Several approaches for minimizing interference from RF have
proven useful, including treatment with aspartame (Ramsland et al. 1999) and careful
optimization of reagent concentrations so as to reduce background binding. When examining
immune responses to Fc-fusion proteins in clinical settings where RF generates false-positive
results during development, FDA recommends developing an assay specific for the non-Fc
region of the proteins rather than against the intact biotherapeutics.
3. Monoclonal Antibodies
Technologies reducing the presence of non-human sequences in mAb, such as chimerization and
humanization, have reduced but not eliminated ADA. In these cases, the immune responses are
directed largely against the variable regions of the mAb (Harding et al. 2010; van Schouwenburg
et al. 2014). The assays that can detect the reactivity against variable regions are considered
more appropriate to evaluate the potential impact of antibodies against mAb-based therapeutics
in subjects. If the Fc region is engineered or bound to another molecule, an assay that
characterizes this response may be needed.
4. Conjugated Proteins
Antibody-drug conjugates (ADCs) are antibodies conjugated with small molecule drugs, so they
represent a classic hapten-carrier molecule. Therefore, the immunogenicity assays should
measure the responses to all components of the ADC therapeutic protein product, including the
antibody, linker-drug, and new epitopes that may result from conjugation. When ADCs need to
be labeled for immunogenicity assays, the conjugation should consider the potential for
increased hydrophobicity of the labeled molecules because they may cause aggregation. The
Contains Nonbinding Recommendations
17
stability and solubility of these capture reagents should be adequately characterized (see
section IV.A.3).
V. ASSAY DEVELOPMENT
Information specific to the development of respective assay types is provided in sections
A through D below. These sections supplement the information provided in section IV that is
relevant to all assay types.
A. Development of Screening Assay
Based on the multi-tiered approach discussed previously in section IV.A, the first assay to be
employed for detection of ADA should be a highly sensitive screening assay that detects low-
and high-affinity ADA. Approximately 5 to 10 individual samples may be used to estimate the
cut-point early in assay development; however, this may need to be adjusted when treatment-
naïve samples from the target population become available. A low but defined false-positive rate
of approximately 5% is desirable for the initial screening assay because it maximizes detection of
true positives. Subsequent assays can be employed to exclude false-positive results when
determining the true incidence of immunogenicity.
B. Development of Confirmatory Assay
Because the screening assay is designed to broadly detect the presence of antibodies that bind
product in serum samples with a defined false-positive rate of approximately 5%, FDA
recommends that the sponsor develop assays to confirm the binding of antibodies that are
specific to the therapeutic protein product. Implementation of a suitable confirmatory assay is
important to prevent data on ADA false-positive subjects from confounding the analyses of the
impact of ADA on safety and efficacy.
1. Selection of Format for Confirmatory Assay
It is expected that the selected confirmatory assay will have similar sensitivity to the screening
assay, with the caveat that the assay false-positive rates are different, but have higher specificity
and at least as good selectivity to identify any false-positive samples. The method and
instrument platform selected may be similar to or different from those used for the screening
assay. Frequently, both screening and confirmatory assays use the same method and instrument
platform. In such cases, the sensitivity of each assay should be determined in mass units and
confirmed using system suitability controls to ensure that the assay is sensitive to the presence of
binding antibody. When using a binding competition assay, the concentration of competing
product should be optimized to confirm the presence of antibodies throughout and above the
range of the assay.

Contains Nonbinding Recommendations
18
2. Cut-Point of Confirmatory Assay
If a competitive inhibition format is selected, a recommended approach to determining the cut-
point uses the data from the signal generated by antibody-negative treatment-naïve subject
samples in the presence of the competitor, which is usually the therapeutic protein product. In
this case, the amount of therapeutic protein product used to establish the cut-point should be the
same as the amount of therapeutic protein product that will be used as a competitive inhibitor in
the assay. However, this approach may not be appropriate when dealing with samples where pre-
existing antibodies are present in the treatment-naïve population. In those cases, the sponsor
should exclude true positives from the cut-point assessment. In rare cases when baseline
negative samples are not available, sponsors may evaluate changes in titer or use an orthogonal
method to confirm samples that screen positive.
C. Development of Titration Assay
In subjects that have pre-existing ADA, treatment-boosted ADA responses may be identified by
post-treatment increases in titer. A cut-point for defining the treatment-boosted responses should
be determined. For example, a boosted ADA response may be defined as a titer that is two
dilution steps greater than the pre-treatment titer, when twofold dilutions are used to determine
the titer. If titer is established by extrapolating the dilution curve to the assay cut-point,
treatment-induced responses may be determined using estimates of assay variability.
D. Development of Neutralization Assay
In vitro neutralization assays indicate the potential of ADA to inhibit the therapeutic activity of
the product. Such NAb can interfere with the clinical activity of a therapeutic protein product by
preventing the product from reaching its target or by interfering with its pharmacologic activity
such as receptor-ligand interactions. The testing method selected to assess neutralizing potential
for ADA-positive samples should be based on the mechanism of action of the therapeutic protein
product.
30
In selected cases, where there is a highly sensitive PD marker or an appropriately
designed PK assay or both that generate data that inform clinical activity, it may be possible to
use these in lieu of a NAb assay. This determination should be done in consultation with the
Agency.
1. Selection of Format for Neutralization Assay
Two approaches have been used to measure NAb activity: cell-based bioassays and non-cell-
based competitive ligand binding assays. Selection of the appropriate assay format depends on
various factors (Wu et al. 2016). These factors include, but are not limited to, the mechanism of
action of the therapeutic protein product and the selectivity, sensitivity, precision, and robustness
of the assay. In general, FDA recommends that neutralization assays use a cell-based bioassay
format. Depending on the therapeutic protein product’s mechanism of action, there may be
alternative strategies for assessing neutralizing activity. For example, ligand binding assays may
be appropriate for antagonistic mAbs or receptor Fc fusion proteins that bind and inhibit the
30
See footnote 13.
Contains Nonbinding Recommendations
19
target; however, alternative strategies to assess neutralizing activity should be discussed with the
Agency before implementation.
Different cellular responses may be measured in these bioassays, such as phosphorylation of
intracellular substrates, calcium mobilization, proliferation, and cell death. In some cases,
sponsors have developed cell lines to express relevant receptors or reporter constructs. When
therapeutic protein products directly stimulate a cellular response, the direct effect of NAb on
reducing bioactivity in the bioassay can be measured. When therapeutic protein products
indirectly impact cellular activity (for example, by blocking a receptor-ligand interaction), the
indirect effect of the NAb on restoring bioactivity in a bioassay can be measured. Some
bioassays have significant variability and a limited dynamic range for their activity curves. Such
problems can make development and validation of neutralization assays difficult.
There are cases when non-cell-based assay formats, such as ligand binding assays or enzyme
activity assays, may be used (Wu et al. 2016). One such case is when sufficiently sensitive or
selective cell-based bioassays cannot be developed. Another case is when the therapeutic protein
product does not have a cell-based mechanism of action; for example, enzyme therapeutic
protein products that do not require cellular uptake. Sponsors should discuss using ligand
binding assays with FDA in such cases.
2. Activity Curve of Neutralization Assay
Generally, the neutralization bioassays use a single concentration of therapeutic protein product
with a single dilution of antibody. Consequently, the sponsor should choose a therapeutic
protein product concentration whose activity readout is sensitive to inhibition. Dosing cells in
the lower part of the dose response curve may not allow for enough dynamic range in the
response to meet neutralization thresholds. If the assay is performed at concentrations near the
plateaus of the dose-response curve, marked “No” in Figure 1 below, it may not be possible to
discern NAb-positive samples with low amounts of NAb. FDA recommends that the
neutralization assay be performed at therapeutic protein product concentrations that are on the
linear range of the curve, marked “Yes” in Figure 1.
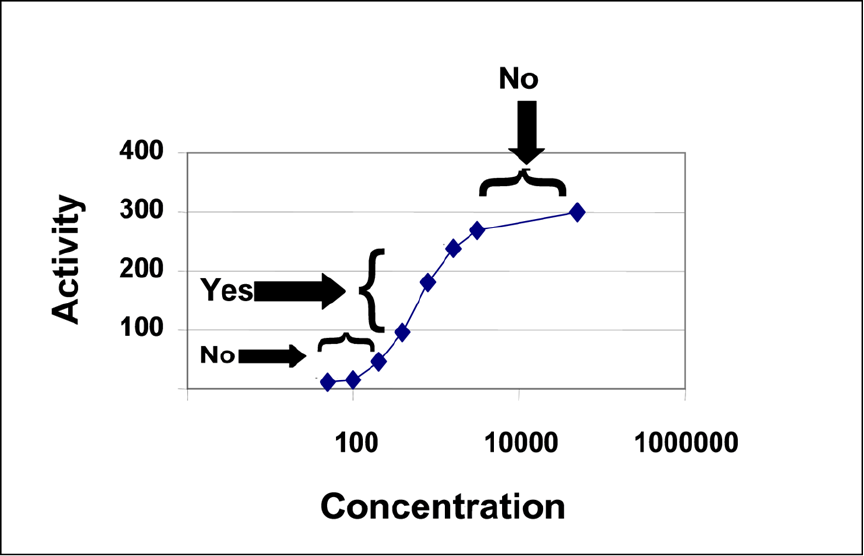
Contains Nonbinding Recommendations
20
Figure 1. Activity Curve for a Representative Therapeutic Protein Product
The x-axis (Concentration) indicates a concentration of the therapeutic protein product, and the
y-axis (Activity) indicates resultant activity; for example, the concentration of cytokine secretion
of a cell line upon stimulation with the therapeutic protein product. The curve demonstrates a
steep response to a therapeutic protein product that plateaus at approximately 300. The “No”
arrows indicate a concentration of a therapeutic protein product that may be inappropriate to use
in a single-dose neutralization assay because it would represent a range of concentrations where
the activity induced by the therapeutic protein product would be relatively insensitive to
inhibition by NAb. The “Yes” arrow represents a range of concentrations on the linear part of
the curve where the activity induced by the therapeutic protein product would be sensitive to
neutralization by antibody.
3. Considerations for Matrix Interference for Neutralization Assay
The matrix can interfere with neutralization assays, particularly as matrix components may
enhance or inhibit the activity of a therapeutic protein product in bioassays. For example, sera
from subjects with particular diseases may contain elevated levels of one or more cytokines that
might serve to activate cells in the bioassay. This could obscure the presence of NAb by
increasing the response to the original stimulatory factor or therapeutic protein product.
Therefore, the sponsor should understand matrix effects in these assays and choose a cell line
that is specifically activated by the therapeutic protein product. Alternatively, the interfering
factors can be inhibited or depleted by using a specific antibody or a cell line that specifically
responds to drug treatment. Enriching the ADA from matrix samples may be appropriate for
Contains Nonbinding Recommendations
21
these types of situations. However, this approach may result in the loss of NAb and,
consequently, will require careful examination and validation by the sponsor.
4. Cut-Point of Neutralization Assay
As with all assays, the cut-point should be determined based on the assay variability established
using samples from treatment-naïve subjects. If neutralization assays are performed on samples
that tested positive in screening and confirmatory assays, a 1% false-positive rate is usually
acceptable. In the rare cases when the neutralization assay is used for screening, a 5% false-
positive rate should be used (see section VI.B.2). If the degree of sample variation makes it
difficult to assess NAb activity, other approaches may be considered, but should be discussed
with FDA before implementation. Alternatively, exploring other assay formats that lead to less
variability and provide a more accurate assignment of cut-point may be necessary. Most
frequently fixed cut-points are established for NAb assays where, depending on the mechanism
of action of the drug, a threshold percent inhibition or stimulation of signal is established, but
floating cut-points may be used. See section IV.B for general information on assay cut-point.
5. Additional Considerations for Neutralization Assay
Because neutralization assays are most commonly performed only on samples that are confirmed
to have antigen-specific ADA, confirmatory approaches are not usually necessary. However,
because of the complexity of bioassays, in some cases confirmation of assay specificity may be
useful in determining whether subjects have mounted a true NAb response. The sponsor should
consider the following approaches:
a. Unrelated inhibitory molecules may cause neutralizing activity, and sometimes it may
be unclear whether the observed neutralizing activity is caused by neutralizing
antibodies or by other inhibitory molecules. Test results from baseline pre-exposure
samples may be informative. When there is concern that there is non-specific
inhibition, antibody depletion assays should be performed to evaluate whether the
neutralizing activity is truly caused by ADA and not caused by other inhibitory
molecules.
b. Cell lines may be responsive to multiple stimuli other than the therapeutic protein
product under study. In such cases, the presence of NAb can be examined in the
presence of the therapeutic protein product, which should be blocked by a specific
NAb response, versus alternative stimuli, which should not be blocked by a specific
NAb response.
c. The matrix contains components such as soluble receptors or endogenous product
counterparts that may yield false results in the neutralization assay. In such instances,
adding test matrix samples directly to the bioassay in the absence of therapeutic
protein product, or blocking the matrix factor, if known, is useful in understanding
assay results.

Contains Nonbinding Recommendations
22
d. The presence of onboard drug should also be considered when designing
neutralization assays, particularly when drugs with a long half-life are used.
VI. ASSAY VALIDATION
Assay validation is a process of demonstrating, through specific laboratory investigations, that
the performance characteristics of the ADA assay employed are suitable for its intended use.
The extent of validation depends on the stage of product development and the risks of
consequences of immunogenicity to subjects associated with the therapeutic protein product. For
most products, a partial validation involving assessments of assay sensitivity, specificity,
precision, cut-point, and drug tolerance — with less emphasis on robustness, reproducibility, and
stability — may be adequate for the earlier stages of clinical development such as phase 1 and
phase 2 studies. High-risk products may require full validation before any clinical studies.
However, as stated in section VI.A, fully validated assays should be used for testing samples
from pivotal and postmarketing studies.
Information specific to validation of respective assay types is provided in sections VI.B through
VI.E. These sections supplement information relevant to all assay types provided in sections IV
and VI.A.
A. General Considerations for Assay Validation
Samples derived from pivotal studies should be tested with fully validated assays. At the time of
license application, the sponsor should provide data demonstrating that the assays are fully
validated. Validation includes assessments that demonstrate that an assay used for measurement
of ADA is suitable for the intended purpose. The fundamental parameters for validation include
(1) cut-point, (2) sensitivity and drug tolerance, (3) specificity and selectivity, (4) precision,
(5) reproducibility
31
when relevant, (6) robustness of some assay features, and (7) in-use stability
of critical reagents.
Determination of cut-point is a fundamental aspect of assay validation. Balanced study designs
should be used for cut-point determination. If plate homogeneity of response is not
demonstrated, alternative plate layouts should be used during cut-point determination. If
treatment-naïve samples from the appropriate subject population are not available for the pre-
study validation exercise, alternative samples may be used.
32
Frequently, these are samples from
commercial sources. When alternative samples are used to determine the cut-point in the
validation exercise, the cut-point should be confirmed once samples from the appropriate
population are available; for example, samples from treatment-naïve subjects that are collected,
handled, and stored under study conditions. If the cut-point established using matrix samples
31
Reproducibility (also called cross-validation) is needed when more than one laboratory will be used to assess
samples.
32
Treatment-naïve subjects could be healthy individuals or a patient population not exposed to the therapeutic
protein product, depending on the stage of assay development or validation and on the availability of samples.
Sponsors should provide justification for the appropriateness of the samples used.

Contains Nonbinding Recommendations
23
from the treatment-naïve population is significantly different from that obtained during assay
validation, the cut-point should be amended. The cut-point established using the appropriate
samples should be used to determine whether study samples are positive for ADA.
For validation of the fundamental assay parameters, FDA recommends that inter-assay precision
be evaluated on different days and by different analysts using the same instrument platform and
model, although different instruments should be used to include all sources of variability. This
design results in at least six independent determinations for each sample. Intra-assay precision
should be evaluated with a minimum of six independent preparations of the same sample per
plate independently prepared by the same analyst. Alternatively, in assays with low throughput
(for example, titer assays), when it may not be possible to run six independent preparations of the
same sample on a plate, intra-assay precision should be evaluated with a minimum of three
independent preparations of the same sample per plate and at least nine total independent
preparations of the same samples. Samples should include negative controls and positive
samples whose testing yields low, intermediate, and high values of the assay dynamic range. In
general, the intra-assay and inter-assay precision as expressed by percent coefficient of variation
(%CV) is expected to be lower than 20%. However, it may be higher in some assay formats
such as cell-based assays. In cases where intra-assay or inter-assay precision has a %CV greater
than 20%, sponsors should consider the need to refine the assay parameters to optimize the assay
precision to the extent possible or provide justification to explain why higher %CV should be
acceptable. For negative controls, a larger %CV is acceptable. The sponsor should evaluate
inter-instrument and inter-operator precision when relevant.
Specific parameters may need to be validated depending on the method, technology, or
instrument platform used for the assay. For example, surface plasmon resonance assays should
be validated for surface stability upon regeneration, and criteria should be set for baseline
performance of the chip. The sponsor should examine robustness during the development phase
and determine whether aspects of assay robustness should be validated. For example, the
efficiency and stability of labeled reagents and incubation times and temperature should be
established.
33
When changes are made to a previously validated method, the sponsor should exercise judgment
as to how much additional validation is needed. During a typical product development program,
a defined ADA assay may undergo modifications. Occasionally, samples may need to be re-
tested with the optimized validated assay. Therefore, provisions should be made to preserve
sufficient sample volume under conditions that allow for re-testing until the assays have been
completely validated and evaluated by the Agency.
34
33
A reagent is considered labeled if it is conjugated or fused to a moiety that will aid in its capture or visualization;
for example, conjugation to biotin, streptavidin, or a fluorochrome. An unlabeled reagent is a reagent (for example,
a drug) that is not labeled.
34
See the guidance for industry Bioanalytical Method Validation for different types and levels of validation. Also,
see the USP General Chapters 1106 and 1106.1.

Contains Nonbinding Recommendations
24
B. Validation of Screening Assay
1. Sensitivity of Screening Assay
All the general considerations for assay validation discussed previously apply to validation of
screening assay. As noted earlier, the sensitivity is particularly important in the initial screening
assay because these results dictate the further analysis of the sample.
2. Cut-Point of Screening Assay
The cut-point should be determined statistically with an appropriate number of treatment-naïve
samples, generally around 50, from the subject population. Each sample should be tested by at
least two analysts on at least three different days for a total of at least six individual
measurements. One approach that allows for high assurance of a 5% false-positive rate is to
apply a 90% one-sided lower confidence interval for the 95
th
percentile of the negative control
population (Shen et al. 2015). This will assure at least a 5% false-positive rate with a 90%
confidence level. This approach improves the probability of the assay identifying all subjects
who may develop antibodies. When using the approach published by Shen et al., the reportable
value for each sample should be the average of the six measurements. The statistical method
used to determine the cut-point should be based on the statistical distribution of the data. For
example, the 95
th
percentile of the normal distribution is estimated by the mean plus 1.645
standard deviation. Other approaches may be used for estimating 95
th
percentile, including the
use of median and median absolute deviation value instead of mean and standard deviation.
The mean response of negative control samples may be constant or may vary between assays,
plates, or analysts. When the mean varies between assays, plates, or analysts but the variance
around the mean is constant, a normalization factor can be statistically determined and applied
in-study. This is known as a floating cut-point and is the most common type of cut-point used.
For normally distributed data, when the mean is constant, a cut-point may be established during
assay validation that can be applied to the assay in-study. This is known as a fixed cut-point.
The use of a fixed cut-point is discouraged because it does not allow for the possibility that
negative control means may vary in-study. When both the mean and variance vary, a cut-point
may need to be established for each assay, plate, or analyst. This is known as a dynamic cut-
point. However, this approach is frequently not practical because of the need to have more
replicates of the negative control. When a dynamic cut-point is indicated, further assay
development should be considered instead of using a dynamic cut-point.
C. Validation of Confirmatory Assay
Confirmatory assays should be fully validated in a manner similar to screening and neutralization
assays. As a scientific matter, the studies to validate the assay will depend on the assay format
and instrumentation chosen. If these assays are based on competition for antigen binding
35
by
the antibodies in subject samples and the measurement is loss of response, it is critical to identify
35
Competition for antigen binding refers to a competition assay where the ability of antigen-specific antibodies to
bind to either labeled or plate-bound antigen is inhibited by unlabeled or soluble antigen.

Contains Nonbinding Recommendations
25
the degree of inhibition or depletion that will be used to ascribe positivity to a sample. In the
past, fixed percentages of binding reduction were used, but these numbers were often arbitrary
and are unlikely to be relevant for all assays. FDA recommends establishing a cut-point based
on the assessment of the binding changes observed in negative control samples that are known to
lack the antibodies when competing antigen is added. FDA also recommends that the sensitivity
of the confirmatory assay be demonstrated using a low concentration of the positive control
antibody.
One approach for the estimation of the confirmatory assay cut-point is to use an 80% to 90%
one-sided lower confidence interval for the 99
th
percentile. Because the purpose of this assay is
to eliminate false-positive samples arising as a result of non-specific binding, it is adequate to
use a 1% false-positive rate for the calculation of the confirmatory cut-point. The use of tighter
false-positive rates such as 0.1% is not recommended, but may be acceptable for larger studies.
See section IV.B for general information on assay cut-point.
The confirmatory assay format is frequently a competiton assay in which a competitor, usually
an unlabeled therapeutic protein product,
36
is added to the reaction mixture to inhibit ADA
binding to the capture reagent for the assay. For this assay format, the same concentration of
unlabeled therapeutic protein product should be added to the negative control samples when
determining the confirmatory cut-point.
D. Validation of Titration Assay
The principles of assay validation described in section VI.A apply in general to validation of
titration assays. The cut-point of the titration assay may be the same as or different from that of
the screening assay. For example, the United States Pharmacopeia recommends establishing a
titration assay cut-point when the signal from the assay diluent or matrix causes higher results
than the screening assay cut-point because of a blocking effect of serum or if samples at a
dilution higher than the MRD do not generate consistently negative results, usually, when the
screening cut-point falls on the lower plateau of the positive control dilution curve.
37
When a
titration assay specific cut-point is used, it should be validated. When the titration assay is not
used for screening, the cut-point may be established using a 0.1% false-positive rate. When the
titration assay is used for screening (for example, when the subject population has a high
incidence of pre-existing ADA), the cut-point should be established using a 5% false-positive
rate.
E. Validation of Neutralization Assay
A minimum of 30 samples tested on at least 3 different days by at least two analysts should be
used to determine the cut-point, using suitable statistical methods (see section V.D.4).
The assessment of assay sensitivity can be affected by the kind of positive control that is used
(for example, mAb or polyclonal antibody), how variable the assay is, and how the assay cut-
36
See footnote 33.
37
See the USP General Chapter 1106.
Contains Nonbinding Recommendations
26
point is determined. Nevertheless, it is important to evaluate assay sensitivity during the
validation exercise.
The positive control for neutralization assays can be either monoclonal or affinity purified
polyclonal antibodies. Further, if an affinity purified polyclonal positive control antibody
preparation is used, it is likely that only a portion of the antibodies are neutralizing, which can
make the assay appear less sensitive.
Sponsors should validate assay specificity for cell-based neutralization bioassays. As mentioned,
for cells that may be responsive to stimuli other than the specific therapeutic protein product, the
ability to demonstrate that NAb only inhibit the response to the therapeutic protein product and
not the response to other stimuli is a good indication of assay specificity. In such studies, FDA
recommends that the other stimuli be employed at a concentration that yields an outcome similar
to that of the therapeutic protein product. The sponsor should also confirm the absence of
alternative stimuli in subject serum (see sections IV.C through E).
Cell-based neutralization bioassays frequently have reduced precision when compared to ligand
binding assays because biologic responses can be inherently more variable than carefully
controlled binding studies. When assay precision is poor, the sponsor may consider performing
more replicates for assessment of precision and assessment of subject responses than for the
screening assay (see section IV.F).
When cells at the low, middle, and high range of the allowed passage numbers and the cell
density and cell viability are used, additional assay performance parameters should be
established. This is frequently done during assay development and may not be part of the
validation exercise (see section IV.H).
VII. IMPLEMENTATION OF ASSAY TESTING
A. Obtaining Subject Samples
FDA recommends that sponsors obtain pre-treatment samples from all subjects. Because there is
the potential for pre-existing antibodies or confounding components in the matrix, understanding
the degree of reactivity before treatment is essential. The sponsor should obtain subsequent
samples, with the timing depending on the frequency of dosing. Optimally, samples taken 7 to
14 days after the first exposure can help elucidate an early IgM response. Samples taken at 3 to
6 weeks after the first exposure are generally optimal for determining IgG responses. IgA
responses may peak earlier than IgG responses, at around 2 to 3 weeks after antigen exposure
(Schütz et al. 2013; Macpherson et al. 2008). For individuals receiving a single dose of a
therapeutic protein product, these time frames may be adequate. However, for subjects receiving
a therapeutic protein product at multiple times during the trial, the sponsor should obtain samples
at appropriate intervals throughout the trial and obtain a sample approximately 30 days after the
last exposure. For products with long half-lives, samples should be obtained approximately five
half-lives after last exposure. When there is a high risk of serious consequences from ADAs,
sponsors should plan to collect samples from subjects until ADAs return to baseline levels.

Contains Nonbinding Recommendations
27
Obtaining samples at a time when there will be minimal interference from the therapeutic protein
product present in the matrix is essential. A sponsor should consider the therapeutic protein
product’s half-life and dosing regimen to help determine appropriate times for sampling. This is
especially important for mAb products because these products can have half-lives of several
weeks or more and, depending on the dosing regimen, the therapeutic mAb itself could remain
present in the serum for months. Under circumstances when testing for IgE is needed, the timing
of sample collection should be discussed with FDA.
If therapeutic protein product-free samples cannot be obtained during the treatment phase of the
trial, the sponsor should take additional measures to ensure that the assay is sensitive in the
presence of expected onboard drug; and samples should be obtained after an appropriate washout
period, generally five half-lives. Obtaining samples to test for meaningful antibody response can
also be complicated if the therapeutic protein product in question is itself an immune
suppressant. In such instances, the sampling schedule should be adjusted in accordance with the
immunosuppressant regimen, to the extent possible.
Samples to determine serum concentrations of the therapeutic protein product should be obtained
at the same time as immunogenicity samples. Testing such samples can provide information on
whether the therapeutic protein product in the samples is interfering with ADA testing and
whether ADA is altering the therapeutic protein product’s pharmacokinetics. It is important that
study subjects be properly consented to allow for continued testing until ADAs reach baseline
and samples are available to confirm or requalify assays as needed. It may also be useful to
consent subjects to allow for sample use in assay development and control.
B. Concurrent Positive and Negative Quality Controls
If the sponsor completes the proper validation work and makes the cut-point determinations, the
immunogenicity status of subjects should be straightforward to determine. However, positive
control and quality control (QC) samples are critical and should be run concurrently with subject
samples. We recommend that these samples span a level of positivity with QC samples having a
known negative, low, and high signal in the assay. More important, the QC samples should be
diluted in the matrix in which subject samples will be examined. For example, the QC sample
should be diluted in the same anticoagulant as the subject samples. For the low-positive QC
sample, we recommend that a concentration be selected that, upon statistical analysis, would lead
to the rejection of an assay run 1% of the time. In this way, the sponsor ensures that the assay is
performing as expected and that subject samples are correctly evaluated. If the assay is subject
to a prozone effect, the concentration of high-positive QC samples should be set to monitor
prozone effects.
38
FDA also recommends that these QC samples be obtained from humans or animals possessing
antibodies that are detected by the secondary detecting reagent to ensure that negative results that
might be observed are truly caused by lack of antigen reactivity but not caused by failure of the
38
Prozone effects, also referred to as hook effects, are a reduction in signal that may occur because of the presence
of a high concentration of a particular analyte or antibody and may cause false-negative results.

Contains Nonbinding Recommendations
28
secondary reagent. This issue is not a problem for bridging assays where labeled antigen is used
for detection.
C. Confirmation of Cut-Point in the Target Population
Samples from different populations can have different background activity in ADA assays.
Similarly, the background activity can change when samples used to determine the cut-point
during assay validation were not obtained and handled in a manner that represents how samples
will be obtained and handled in-study. Therefore, it is necessary to confirm that the cut-point
determined during assay validation is suitable for the population being studied. A sufficient
number of samples from the target population should be used, and justification for the number
used should be provided. If sufficient numbers of samples are not available, agreement with the
Agency should be sought for the number of samples to be used.
VIII. DOCUMENTATION
Currently the data relevant to the assessment of immunogenicity are dispersed throughout
different locations of the eCTD. To facilitate the clinical development of therapeutic biologics,
we recommend a life-cycle management approach to immunogenicity through the creation of an
integrated immunogenicity summary report that sponsors begin populating early in therapeutic
protein product development and update at regular intervals as the individual product clinical
program progresses through IND stages into the BLA and even postapproval stages. We
recommend that the document be arranged into distinct sections to be populated with stage-
appropriate information as it becomes available, including (1) Immunogenicity Risk Assessment,
(2) Tiered Bioanalytical Strategy and Assay Validation Summaries, (3) Clinical Study Design
and Detailed Immunogenicity Sampling Plans, (4) Clinical Immunogenicity Data Analysis, and
(5) Conclusions and Risk Evaluation and Mitigation Strategies (REMS).
For the BLA file, we recommend that the applicant provide brief summaries of the
immunogenicity results in relevant places in eCTD section 2.7. Clinical Summary and the full
report in section 5.3.5.3 Reports of Analysis of Data from More than One Study.
39
This
Integrated Summary of Immunogenicity should provide the following:
a. Immunogenicity Risk Assessment: This section should provide a concise
immunogenicity risk assessment specific to the therapeutic protein product.
40
This
section should include discussions on therapeutic protein product quality-related factors
39
See the FDA website for further information on eCTD submissions, available at
https://www.fda.gov/Drugs/DevelopmentApprovalProcess/FormsSubmissionRequirements/ElectronicSubmissions/u
cm153574.htm. For more information about the agreed-upon common format for the preparation of a well-
structured Efficacy section of the CTD for applications that will be submitted to regulatory authorities, see the ICH
guidance for industry M4E: The CTD — Efficacy. For more information on how sponsors and applicants must
organize the content they submit to the Agency electronically for all submission types under section 745A(a) of the
Federal Food, Drug, and Cosmetic Act, see the guidance for industry (and the technical specification documents it
incorporates by reference) Providing Regulatory Submissions in Electronic Format — Certain Human
Pharmaceutical Product Applications and Related Submissions Using the eCTD Specifications.
40
See the guidance for industry Immunogenicity Assessment for Therapeutic Protein Products.
Contains Nonbinding Recommendations
29
and how these may impact the immunogenic potential of the therapeutic protein product;
subject-related factors, including a discussion on how likely is the subject population and
clinical indication to result in immunogenic responses to the therapeutic protein product;
and a section on trial design-related factors, as well as a discussion of any strategies or
clinical study conditions implemented to manage the immunogenic response to the
therapeutic protein product.
b. Tiered Strategy and Stage-Appropriate Bioanalytical Assays: This section should
provide a summary of the immunogenicity assessment strategies used during each phase
of the clinical program and a characterization for the various methods that were
developed throughout the program. In addition, this section should provide links to the
method development and validation reports for the pivotal clinical studies supporting the
application.
c. Clinical Study Design and Sampling Strategy: This section should include the
immunogenicity sampling plan(s) for all clinical studies that had an immunogenicity
assessment performed. This section should also include sampling time points for
immunogenicity and pharmacokinetics of the therapeutic protein product, where
applicable.
d. Clinical Immunogenicity Data Analysis: This section should provide summary results of
immunogenicity analyses for all clinical studies having an immunogenicity component,
including the results of linear or non-linear correlation analyses between ADA status and
titers with PK, PD, efficacy, and safety (adverse event) data. This section should include
drug levels measured in the samples tested for ADA and should trace drug product lots
used in the individual clinical studies. Discussion should examine the impact of any pre-
existing antibodies or treatment-boosted or treatment-induced antibodies on
pharmacokinetics, pharmacodynamics, efficacy, and safety of the therapeutic protein
product.
e. Conclusions and REMS, if applicable: This section should discuss how therapeutic
protein product immunogenicity affects the safety and efficacy of the therapeutic protein
product for the subject population. In addition, consideration should be given to how
therapeutic protein product immunogenicity will be monitored in the postmarketing stage
and how this will be incorporated into any planned risk evaluation and mitigation
strategies. Lastly, a discussion should be provided regarding life-cycle management of
approved immunogenicity assays, including an assay requalification schedule and assay
transfer to contract testing laboratories for postmarketing surveillance.
Contains Nonbinding Recommendations
30
REFERENCES
Aalberse, R. C. and J. Schuurman (2002). “IgG4 breaking the rules.” Immunology 105(1): 9–19.
Boes, M. (2000). “Role of natural and immune IgM antibodies in immune responses.” Mol
Immunol 37(18): 1141–1149. doi: 10.1016/S0161-5890(01)00025-6.
Calabresi, P. A., G. Giovannoni, C. Confavreux, S. L. Galetta, E. Havrdova, M. Hutchinson, L.
Kappos, D. H. Miller, P. W. O'Connor, J. T. Phillips, C. H. Polman, E. W. Radue, R. A. Rudick,
W. H. Stuart, F. D. Lublin, A. Wajgt, B. Weinstock-Guttman, D. R. Wynn, F. Lynn, and M. A.
Panzara (2007). “The incidence and significance of anti-natalizumab antibodies: results from
AFFIRM and SENTINEL.” Neurology 69(14): 1391–1403.
doi: 10.1212/01.wnl.0000277457.17420.b5.
Caruso, A. and A. Turano (1997). “Natural antibodies to interferon-gamma.” Biotherapy 10(1):
29–37.
Cohen, B. A. and V. M. Rivera (2010). “PRISMS: the story of a pivotal clinical trial series in
multiple sclerosis.” Curr Med Res Opin 26(4): 827–838. doi: 10.1185/03007991003604018.
Coutinho, A., M. D. Kazatchkine, and S. Avrameas (1995). “Natural autoantibodies.” Curr Opin
Immunol 7(6): 812–818.
Goodin, D. S., E. M. Frohman, B. Hurwitz, P.W. O'Connor, J. J. Oger, A. T. Reder, and J. C.
Stevens (2007). “Neutralizing antibodies to interferon beta: assessment of their clinical and
radiographic impact: an evidence report: report of the Therapeutics and Technology Assessment
Subcommittee of the American Academy of Neurology.” Neurology 68(13): 977–984.
doi: 10.1212/01.wnl.0000258545.73854.cf.
Gorovits, B., E. Wakshull, R. Pillutla, Y. Xu, M. S. Manning, and J. Goyal (2014)
“Recommendations for the characterization of immunogenicity response to multiple domain
biotherapeutics.” J Immunol Meth. 408: 1–12. doi: 10.1016/j.jim.2014.05.010.
Gupta, S., V. Devanarayan, D. Finco, G. R. Gunn, S. Kirshner, S. Richards, B. Rup, A. Song,
and M. Subramanyam (2011). “Recommendations for the validation of cell-based assays used for
the detection of neutralizing antibody immune responses elicited against biological therapeutics.”
J Pharm Biomed Anal 55(5): 878–888. doi: 10.1016/j.jpba.2011.03.038.
Gupta, S., S. R. Indelicato, V. Jethwa, T. Kawabata, M. Kelley, A. R. Mire-Sluis, S. M.
Richards, B. Rup, E. Shores, S. J. Swanson, and F. Wakshull (2007). “Recommendations for the
design, optimization, and qualification of cell-based assays used for the detection of neutralizing
antibody responses elicited to biological therapeutics.” J Immunol Methods 321(1-2): 1–18.
doi: 10.1016/j.jim.2006.12.004.
Contains Nonbinding Recommendations
31
Harding, F. A., M. M. Stickler, J. Razo, and R. B. DuBridge (2010). “The immunogenicity of
humanized and fully human antibodies: residual immunogenicity resides in the CDR regions.”
MAbs 2(3): 256–265.
Macpherson A.J., K. D. McCoy, F. E. Johansen, and P. Brandtzaeg (2010). “The immune
geography of IgA induction and function.” Mucosal Immunol 1(1): 11–22.
doi: 10.1038/mi.2007.6.
Matsumoto, T., M. Shima, K. Fukuda, K. Nogami, J. C. Giddings, T. Murakami, I. Tanaka, and
A. Yoshioka (2001). “Immunological characterization of factor VIII autoantibodies in patients
with acquired hemophilia A in the presence or absence of underlying disease.” Thromb Res
104(6): 381–388.
Mire-Sluis, A. R., Y. C. Barrett, V. Devanarayan, E. Koren, H. Liu, M. Maia, T. Parish, G.
Scott, G. Shankar, E. Shores, S. J. Swanson, G. Taniguchi, D. Wierda, and L. A. Zuckerman
(2004). “Recommendations for the design and optimization of immunoassays used in the
detection of host antibodies against biotechnology products.” J Immunol Methods 289(1-2): 1–
16. doi: 10.1016/j.jim.2004.06.002.
Plotkin, S. A. (2010). “Correlates of protection induced by vaccination.” Clin Vaccine Immunol
17(7): 1055–1065. doi: 10.1128/CVI.00131-10.
Ramsland, P. A., B. F. Movafagh, M. Reichlin, and A. B. Edmundson (1999). “Interference of
rheumatoid factor activity by aspartame, a dipeptide methyl ester.” J Mol Recognit 12(5): 249–
257.
Ross, C., M. B. Hansen, T. Schyberg, and K. Berg (1990). “Autoantibodies to crude human
leucocyte interferon (IFN), native human IFN, recombinant human IFN-alpha 2b and human
IFN-gamma in healthy blood donors.” Clin Exp Immunol 82(1): 57–62. doi: 10.1111/j.1365-
2249.1990.tb05403.x.
Schütz, K., R. G. Hughes, A. Parker, I. Quinti, V. Thon, M. Cavaliere, M. Würfel, W. Herzog, J.
E. Gessner, and U. Baumann (2013). “Kinetic of IgM and IgA antibody response to 23-valent
pneumococcal polysaccharide vaccination in healthy subjects.” J Clin Immunol 33(1): 288–296.
doi: 10.1007/s10875-012-9792-y.
Shankar, G., V. Devanarayan, L. Amaravadi, Y. C. Barrett, R. Bowsher, D. Finco-Kent, M.
Fiscella, B. Gorovits, S. Kirschner, M. Moxness, T. Parish, V. Quarmby, H. Smith, W. Smith, L.
A. Zuckerman, and E. Koren (2008). “Recommendations for the validation of immunoassays
used for detection of host antibodies against biotechnology products.” J Pharm Biomed Anal
48(5): 1267–1281. doi: 10.1016/j.jpba.2008.09.020.
Shen, M., X. Dong, and Y. Tsong (2015). “Statistical evaluation of several methods for cut-point
determination of immunogenicity screening assay.” J Biopharm Stat 25(2): 269–279.
https://doi.org/10.1080/10543406.2014.979196.
Contains Nonbinding Recommendations
32
Turano, A., A. Balsari, E. Viani, S. Landolfo, L. Zanoni, F. Gargiulo, and A. Caruso (1992).
“Natural human antibodies to gamma interferon interfere with the immunomodulating activity of
the lymphokine.” Proc Natl Acad Sci U S A 89(10): 4447–4451.
van der Meide, P. H. and H. Schellekens (1997). “Anti-cytokine autoantibodies: epiphenomenon
or critical modulators of cytokine action.” Biotherapy 10(1): 39–48.
van Schouwenburg, P. A., S. Kruithof, C. Votsmeier, K. van Schie, M. H. Hart, R N. de Jong, E.
L. van Buren, M. van Ham, L. Aarden, G. Wolbink, D. Wouters, and T. Rispens (2014).
“Functional analysis of the anti-adalimumab response using patient-derived monoclonal
antibodies.” J Biol Chem 289(50): 34482–34488. doi: 10.1074/jbc.M114.615500.
Wang, Y. C., J. Wang, Y. YiHon, L. Zhou, L. Fang, and H. Y. Ahn (2016). “Evaluating and
reporting the immunogenicity impacts for biological products – a clinical pharmacology
perspective.” AAPS J 18(2): 395–403. doi: 10.1208/s12248-015-9857-y.
Wu, B., S. Chung, X. R. Jiang, J. McNally, J. Pedras-Vasconcelos, R. Pillutla, J. T. White, Y.
Xu, and S. Gupta (2016). “Strategies to determine assay format for the assessment of
neutralizing antibody responses to biotherapeutics.” AAPS J 18(6): 1335–1350.
doi: 0.1208/s12248-016-9954-6.
Zhou, L., S. A. Hoofring, Y. Wu, T. Vu, M. Peiming, S. J. Swanson, N. Chirmule, and M.
Starcevic (2013). “Stratification of antibody-positive subjects by antibody level reveals an
impact of immunogenicity on pharmacokinetics.” AAPS J 15(1): 30–40. doi: 10.1208/s12248-
012-9408-8.
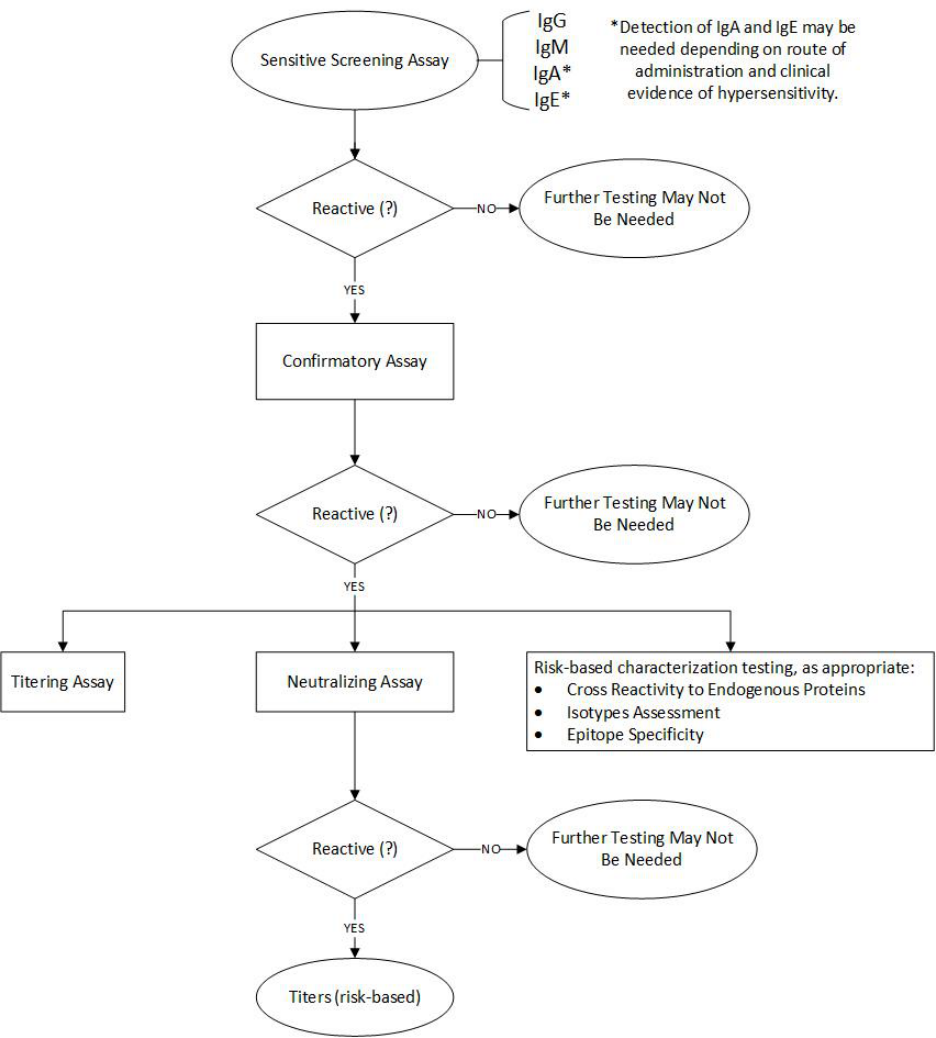
Contains Nonbinding Recommendations
33
APPENDIX: MULTI-TIERED APPROACH TO ANTI-DRUG ANTIBODY TESTING
