
Public
BSI Clinical Masterclass 2023
Session 1
The Clinical Evaluation Plan

Public
What can you expect from the Clinical Masterclass 2023 series?
5 sessions focusing on the best practice for detailing your key
clinical evaluation documents including:
- The Clinical Evaluation Plan
- The Clinical Evaluation Report
- The Post Market Clinical Follow Up Plan
- The Post Market Clinical Follow Up Report
- The Summary of Safety and Clinical Performance
At the end of these sessions, we will be providing you with a specific best practice
guide for documenting your clinical evaluation.
Public
Copyright © 2022 BSI. All rights reserved
3
Documenting a Clinical
Evaluation Plan. (CEP)
Annex XIV Part A

Public
Topics covered in the Clinical Evaluation Plan Session:
• The MDR Requirements
• Documenting a Clinical Evaluation Plan
• State of the art and defining objectives
• Legacy Device Clinical Evaluation Plans
• Clinical Development Plans
Copyright © 2022 BSI. All rights reserved
4
Public
What is the purpose of a Clinical Evaluation Plan?
• The clinical evaluation plan is the foundation of the overall
clinical evaluation process and provides the roadmap for the
process.
• The clinical evaluation plan sets out the required steps to
define the scope, the regulatory pathway and necessary
steps to gather the required clinical data in a methodological
and systematic approach for the device under evaluation.
‘Behind every good clinical evaluation report is a perfect
clinical evaluation plan’
Copyright © 2022 BSI. All rights reserved
5
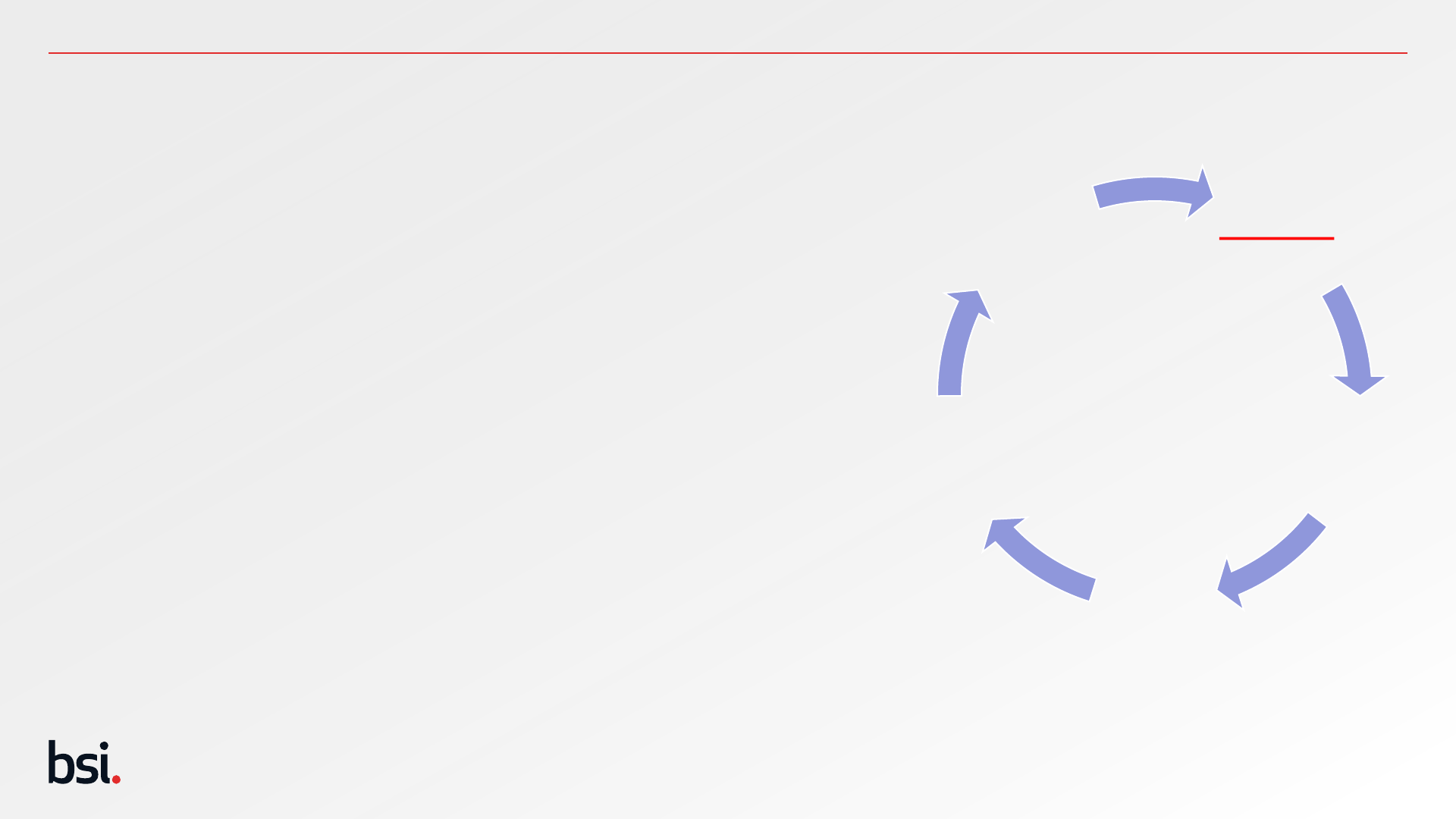
Establish
Identify
Appraise
Generate
Analyse

Public
The MDR Requirements of the Clinical Evaluation Plan
The MDR is prescriptive on the requirements of the CEP:
Copyright © 2022 BSI. All rights reserved
6
The CEP needs to identify
the general safety and
performance requirements
that require clinical data
The intended purpose of
the device
Intended target groups, clear
indications and contra-indications
Intended clinical benefits to patients
with relevant and specified clinical
outcome parameters
Methods used for qualitative and
quantitively aspects of clinical
safety to determine residual
risk/side effects
Parameters to be used to determine
State of the Art and acceptability of
benefit/risk for all indications
Benefit-risk issues relating to
specific components such as use
of pharmaceutical, non- viable
animal or human tissues
A clinical development plan….
Public
Where and how should I document my Clinical Evaluation Plan?
Copyright © 2022 BSI. All rights reserved
7
The Clinical
Evaluation
Plan
The clinical evaluation plan should be clearly highlighted/titled
within the technical documentation.
The plan can be presented either as a separate document or
as part of the clinical evaluation report. It is essential that
wherever the plan is documented the information is easily
identifiable, and the complete information is presented.

Public
Considerations when documenting the requirements.
Requirement: an identification of the general safety and performance requirements that require support from relevant
clinical data;
Copyright © 2022 BSI. All rights reserved
8
Devices shall achieve the performance intended by their manufacturer and shall be designed and manufactured in such a way that, during
normal conditions of use, they are suitable for their intended purpose. They shall be safe and effective and shall not compromise the
clinical condition or the safety of patients, or the safety and health of users or, where applicable, other persons, provided that any risks
which may be associated with their use constitute acceptable risks when weighed against the benefits to the patient and are compatible
with a high level of protection of health and safety, taking into account the generally acknowledged state of the art.
In eliminating or reducing risks related to use error, the manufacturer shall: (a) reduce as far as possible the risks related to the ergonomic
features of the device and the environment in which the device is intended to be used (design for patient safety), and (b) give
consideration to the technical knowledge, experience, education, training and use environment, where applicable, and the medical and
physical conditions of intended users (design for lay, professional, disabled or other users).
All known and foreseeable risks, and any undesirable side-effects, shall be minimised and be acceptable when weighed against the
evaluated benefits to the patient and/or user arising from the achieved performance of the device during normal conditions of use.

Public
Tips when documenting the GSPRs that will require clinical data.
- Document the GSPRs clearly with consideration of a table format to identify and where possible
explain the rationale for clinical data.
- This rationale will help understand the thought process and can assist the Notified Body in
determining whether the manufacturer has adequately identified all the applicable GSPRs that
require support from relevant clinical data for the device under assessment.
Copyright © 2022 BSI. All rights reserved
9
GSPR #
Requirement
Justification
14.5
Devices that are intended to be
operated together with other
devices or products shall be
designed and manufactured in
such a way that the interoperability
and compatibility are reliable and
safe.
The implantable device may be
used with other manufacturer
configurations. The is a lack of data
on longer term safety and
performance for these
combinations. Longer term clinical
data from investigations is required
to support claims of compatibility
with other devices for the lifetime.

Public
Tips when documenting the GSPRs that will require clinical data.
- Avoid generalised statements covering all GSPRs. – This potentially demonstrates that the
manufacturer is not applying an appropriate thought process to the device under evaluation.
- Understand the definition of Clinical Data – Article 2(48)
- Clinical data is not:
- Animal studies
- Toxicology testing
- Bench testing
- Expert opinion not related to clinical experience
- Compliance with CS or standards
- Clinical experience not published in peer-reviewed or scientific literature.
Copyright © 2022 BSI. All rights reserved
10

Public
Considerations when documenting the Intended Purpose.
Requirement: a specification of the intended purpose of the device;
Intended Purpose:
Copyright © 2022 BSI. All rights reserved
11
means the use for which a device is intended according to the data supplied by the manufacturer on the label, in
the instructions for use or in promotional or sales materials or statements and as specified by the manufacturer in
the ‘intended purpose’ clinical evaluation; (Article 2 (12))
In the clinical evaluation planning phase, it can be difficult to determine your intended purpose before your
collection of clinical data.
The intended purpose should be reflective of what you intend the device to achieve.
N.B: Intended purpose is synonymous with intended use (MDCG 2020-6 (1)) and is different to a device’s
indication.

Public
Considerations when documenting the Intended Purpose
Requirement: a specification of the intended purpose of the device;
The following information should be considered within the Intended purpose of the device:
✓ exact medical indications (if applicable)
✓ name of disease or condition/ clinical form, stage, severity/ symptoms or aspects to be
✓ treated, managed or diagnosed
✓ patient populations (adults / children / infants, other aspects)
✓ intended user (use by health care professional / lay person)
✓ organs / parts of the body / tissues or body fluids contacted by the device
✓ duration of use or contact with the body
✓ repeat applications, including any restrictions as to the number or duration of reapplications
✓ contact with mucosal membranes/ invasiveness/ implantation
✓ contraindications
✓ precautions required by the manufacturer
✓ single use / reusable
✓ other aspects
Copyright © 2022 BSI. All rights reserved
12
Example
- The Notified Body device is a permanent implant situated in either the right or left atrium, intended to treat the symptoms of
stage 4 heart failure and unstable angina, in adults over the age of 80 years, by improving coronary vasodilatation and
should only be implanted by those trained in interventional cardiology procedures with on-site surgical facilities.
- The device is contraindicated in patients with a systolic blood pressure <90mmHg. Patients should be suitable for
anticoagulation therapy to be eligible for implant.

Public
Tips when documenting the Intended Purpose within the CEP
• The intended purpose of the device should be
clear and unambiguous. Statements that are
vague or nebulous will invite scrutiny from the
Notified Body and so it is important to be
concise, specific and accurate.
• Ensure that, in all instances in which the
intended purpose is cited within the technical
documentation, the wording exactly matches
what is stated within the CEP.
• If during the clinical evaluation process, the
intended purpose changes then it is important to
document this and explain the rationale for
changing the intended purpose of the device.
Copyright © 2022 BSI. All rights reserved
13

Public
Considerations when documenting Intended Target groups
Requirement: — a clear specification of intended target groups with clear indications and contra-indications;
Copyright © 2022 BSI. All rights reserved
14
‘Patients’ ‘Users’
Gender, Age, Stage/severity
of disease, mobility
Healthcare professionals, nurses,
scientists, doctors, stage of
seniority, sub-specialities

Public
Indications
‘indication’, ‘indication for use’: refers to the clinical condition that is to be diagnosed, prevented, monitored, treated,
alleviated, compensated for, replaced, modified or controlled by the medical device. It should be distinguished from
‘intended purpose/intended use’, which describes the effect of a device. All devices have an intended purpose/intended use.
(MDCG 2020-6 Section 1)
Copyright © 2022 BSI. All rights reserved
15
When considering writing indications for the device, they should be thought of
as a checklist of eligible criteria that qualifies the patient to receive the device.
This means they should be specific and unambiguous.
Not all devices have an indication, but these are typically devices such as
sterilisers or disinfectants. Any absence of an indication should always be
strongly justified

Public
Poll Question
Copyright © 2022 BSI. All rights reserved
16
Q: Can the indications be identical
to the intended purpose?
• Yes
• No

Public
Poll Question
Copyright © 2022 BSI. All rights reserved
17
Q: Can the indications be identical
to the intended purpose?
• It Depends…

Public
Contraindications
Tell when a device should not be used (contraindications). Contraindications are
conditions under which the device should not be used because the risk of use
clearly outweighs any possible benefit. There may be persons in whom the device
should not be used because of their health status. For example, the device may be
contraindicated for pregnant women.
Contraindications typically are where this is clear evidence of known harm
to patients/users.
Warnings and precautions tell the reader about hazards, other than those that are
contraindications to device use. Warnings and precautions provide information on
how to avoid these hazards, i.e., sources of harm in the use of the device.
Guidance on Medical Device Patient Labelling; Final Guidance for Industry and FDA Reviewers Document issued on:
April 19, 2001
Copyright © 2022 BSI. All rights reserved
18

Public
Considerations when documenting the clinical benefit
Requirement: a detailed description of intended clinical benefits to patients with
relevant and specified clinical outcome parameters;
Copyright © 2022 BSI. All rights reserved
19
• The MDR requires that manufacturers describe the intended clinical benefits to patients in the CEP. In many cases,
these will align with the intended purpose statement.
• However, manufacturers can sometimes have difficulty expressing the clinical benefits as benefits afforded to the
patient. It is useful therefore to ask the following questions:
• How does the use of the device improve the health of the patient?
• What is the positive outcome of using the device, from the perspective of the patient?
‘clinical benefit’ means the positive impact of a device on the health of an individual, expressed in terms of a
meaningful, measurable, patient-relevant clinical outcome(s), including outcome(s) related to diagnosis, or a positive
impact on patient management or public health; (Article 2(53))

Public
Considerations when documenting the clinical benefit
Further, the MDR expects that manufacturers specify the relevant outcome parameters that enable them to
demonstrate that these benefits are delivered by the device. It can be thought of in the following way:
If device x is going to have positive outcome y on the patient, what aspect of y can be measured to confirm that
the outcome is achieved?
Copyright © 2022 BSI. All rights reserved
20
By way of example, a relevant outcome parameter for an orthopaedic device could
be an improvement in mobility score reported at x weeks post-surgery.

Public
Tips when documenting the clinical benefits
✓ The intended target group may be specific in the case of some devices and broad in the case of others. In
either case, it is important that manufacturers demonstrate an awareness of the groups of patients that will
benefit from the use of the device.
✓ When appropriate the target population should also be clarified considering, gender, age, co-morbidities and
other clinical aspects of which the data reflects.
✓ Users of the device should also be defined. This should be accompanied with the expected education, grade
and experience of the users that can use the device safely as demonstrated by the clinical data.
Consideration should also be given when devices are to be used under certain supervision.
✓ Where appropriate, the specification should include details such as the grade/stage of disease that the device
is indicated for, as well as any limitations that apply. For example, there may be (sub)groups of patients for
which use of the device would not be appropriate and this should be clearly stated within the CEP.
✓ Where the use of a device by particular group(s) and/or circumstances is deemed hazardous, the
manufacturer is expected to include a list of contra-indications along with warnings and precautions.
Copyright © 2022 BSI. All rights reserved
21

Public
Tips when documenting the clinical benefits
✓ When clinical benefit scoring systems are to be used, where possible these should ideally be validated
national/international agreed scoring systems. E.g. WHOQOL
✓ For many devices, the relevant outcome parameters will be obvious. However, for some devices - for
example, those used for imaging patients for a variety of purposes - it may be challenging for manufacturers
to define relevant outcome parameters. In this case, other performance parameters may be indirectly related
to patient benefit. Where the clinical benefit is not directly afforded to the patient, manufacturers should
clearly state this and provide a justification as to why the specified clinical performance parameters were
selected.
Copyright © 2022 BSI. All rights reserved
22

Public
Considerations when documenting aspects of safety
Requirement: a specification of methods to be used for examination of qualitative and
quantitative aspects of clinical safety with clear reference to the determination of residual risks and side-effects;
▪ The MDR expects manufacturers to specify, within the CEP, the methods they will use to evaluate the risks posed by use
of the device. It is important, therefore, to describe in detail the methods by which information relating to the risks
posed by use of the device will be gathered and analysed – both qualitatively and quantitatively.
▪ This requirement is related to the risk assessment, which should include risks that are identified as part of the clinical
evaluation of the device. Manufacturers should include details of how they intend to identify clinical risks as part of the
clinical evaluation and should also make clear their intention to determine the residual risks (i.e., post-mitigation) and
side-effects associated with the device.
Copyright © 2022 BSI. All rights reserved
23

Public
Considerations when documenting the parameters based on State of the Art .
Requirement: an indicative list and specification of parameters to be used to determine, based on the state of the art in
medicine, the acceptability of the benefit-risk ratio for the various indications and for the intended purpose or purposes of
the device;
Copyright © 2022 BSI. All rights reserved
24
‘state of the art’: IMDRF/GRRP WG/N47 provides the following definition:
Developed stage of current technical capability and/or accepted clinical practice in regard to products,
processes and patient management, based on the relevant consolidated findings of science, technology and
experience.
Note: The state-of-the-art embodies what is currently and generally accepted as good practice in technology
and medicine. The state-of-the-art does not necessarily imply the most technologically advanced solution. The
state-of-the-art described here is sometimes referred to as the “generally acknowledged state-of-the-art’
Reproduced from MDCG 2020-6 (1. Definitions)

Public
State of the Art & Defining Objectives
25
Understanding the safety and performance profile of similar devices from State of the Art allows the manufacturer to develop an acceptable safety
and performance profile for the device under evaluation. This allows the manufacturer to compare its data against those other technologies to
confirm its safety and performance is equal or better than those available devices and ultimately its right to have a position on the market.
State of the Art Results should drive the
Safety and Performance objectives for the
device under evaluation
Results of SoTA Search
Risk identified - Thrombosis at 12 months – 6-9%
Performance identified – Patency at 5 years – 82-86%
3 Stents identified from State-of-the-Art Search
Stent under Evaluation
Objectives for Device under Evaluation
Safety Objective - Thrombosis at 12 months – < 9%
Performance Objective – Patency at 5 years – >82%
Public
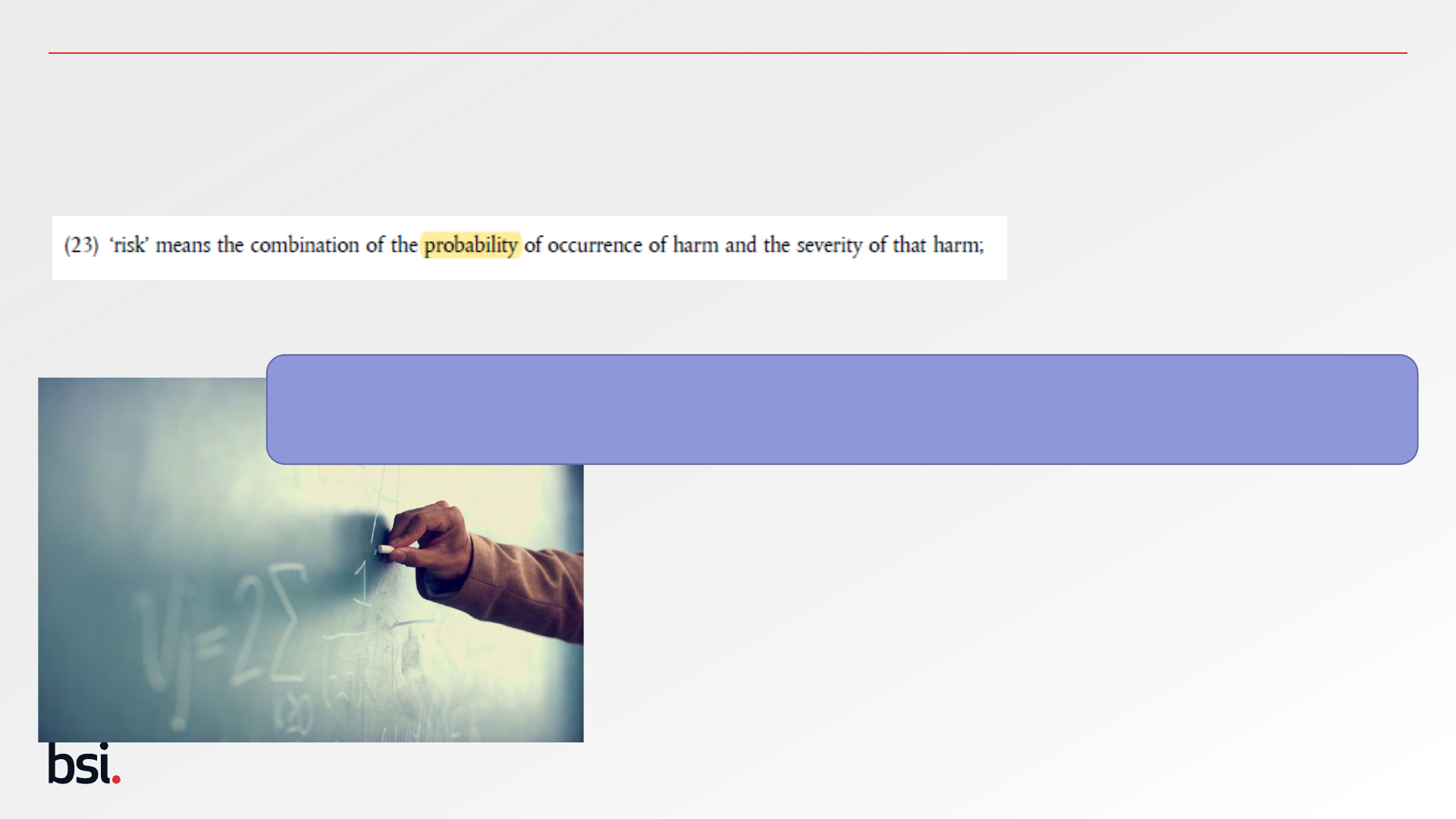
MDR 2017/745 Article 2 (23) – Safety Objectives
26
The term risk includes the words probability, so there is always a need for quantification.
Public
Safety Objectives
27
Safety Objectives should be quantified
according to;
1. magnitude, (extent, amount, intensity)
this should be measurable and patient
group specific
2. Probability based on patient
populations should be considered.
Statements to reflect general
population may not be appropriate.
3. Duration - Think quantity!
Outcomes of clinical evaluation activities the input for Risk
Management.
Clinical risks are identified through:
• literature reviews
• Clinical investigations,
• PMCF related activities
Outcomes of Risk Management Activities the input for clinical
evaluation.
Clinical risks are identified:
During development process from clinical experts
Relating to foreseeable misuse (or assessing questions of
EN ISO 14971 Annex C)
Post Market Surveillance Activities

Public
Safety Objectives.
The main task of the clinical evaluation activities is now to strengthen the confidence of these
assumptions through the targeted identification of clinical data e.g. search terms or safety
endpoints which relate to specific clinical risks.
28

Public
MDR 2017/745 – Measurable Objectives.
29
Clinical Performance
Objectives look at ‘clinical
benefit’ and here is the
expectation that clinical
benefit is measurable and
meaningful.
Article 2 (53)

Public
Performance Objectives
Performance objectives should be clinically meaningful:
• Positive impact on clinical outcomes such as reduced probability of
adverse events or improvement of impaired body function,
• Patients Quality of Life (freedom from symptoms). Considering international
recognized scoring systems
• Outcomes related to Diagnosis (Earlier detection, Sensitivity and specificity
of diagnosis)
• Positive impact from diagnostic devices on clinical outcomes,
(Pharmacological intervention sooner).
• Public Health Impact (Ability of a diagnostic tool to prevent spread of
disease).
• Valid Surrogate endpoints can be used if there is a validated predictor. (e.g.
measurements of biochemical markers)
30
Performance Objectives should be
quantified according to;
1. magnitude, (extent, amount intensity)
this should be measurable and patient
specific
2. Probability based on patient
populations should be considered.
Statements to reflect the general
population may not be appropriate.
(Think Meaningful!)
3. Duration - Think quantity!

Public
Considerations when documenting specific components
Requirement: an indication how benefit-risk issues relating to specific components such as use of pharmaceutical, non-
viable animal or human tissues, are to be addressed; and
Copyright © 2022 BSI. All rights reserved
31
Some medical devices feature hazardous components such as pharmaceutical, viable animal or human tissues. Since the
action(s) of these may need to be considered separately from the overall device, the manufacturer should explain how
they intend to assess the benefits and risks posed by these components as part of the overall clinical evaluation of the
device. Where the long-term exposure of the drug or material in its particular application is not fully known, this may
also point to a requirement for follow-up studies within the PMCF Plan (Annex XIV, 6.1a)
Public
Copyright © 2022 BSI. All rights reserved
32
Clinical Evaluation Plans for
Legacy Device
MDCG 2020-6 Appendix A

Public
Expectations of clinical evaluation plans for legacy devices
Copyright © 2022 BSI. All rights reserved
33
There is no justification for an absence of a CEP
for a legacy device – remember the CEP is the
foundation of your clinical evaluation and this is a
continuous process.
MDCG 2020-6 Appendix II provides the minimum
information expected for legacy device CEP.
Remember this is for devices that have not had
design modifications or expansion of indications.
This list can form the titles for the various
sections of your CEP, allowing the notified body
to locate information easily and quickly.
Don’t forget the clinical development plan!
Public
Copyright © 2022 BSI. All rights reserved
34
Documenting a Clinical
Development Plan.
Annex XIV Part A (1) (a) Indent 8.

Public
The Requirement (MDR 2017/745)
35
Public
Clinical Development Plan in 1 Slide (Annex XIV Part A)
36
*Off label studies are not PMCF studies and are subject to the same scrutiny and processes
of the Competent Authority as non-CE marked investigations.
The clinical development plan
defines how you will collect
sufficient clinical data for later
clinical evaluation. It is the
first step of the overall clinical
evaluation plan.
The Clinical Development Plan
should indicate potential
acceptance criteria.
This may include exploratory
investigations, first-in-man
studies, feasibility and pilot
studies, to confirmatory
investigations; an outlook for
possible PMCF activities is
also possible at this stage.
CDP
List all investigations/studies
performed from pre-clinical to
post market studies, detailing
the expected outcomes of
these investigations.
If these outcomes are not
reached what decisions and
actions are required to fulfil
those unanswered questions.
This may also include
information where the
manufacturer intends to
perform clinical
investigations* ‘off-label’ to
expand the indications of the
medical device in the future.
Public
Copyright © 2022 BSI. All rights reserved
37
Documenting a Clinical
Development Plan for New
MDR Devices.
Annex XIV Part A (1) (a) Indent 8.

Public
Key Components of a CDP
Prospective Patients
Scientific Rationale for Development
Commercial Rationale for Development
Clinical Data (Emphasis on Clinical Investigations)
Strategic Planning
38
CDP

Public
Scientific Rationale for Development
Unmet Clinical Needs/Expansion of Clinical
Indications
Limitations of Current Therapies/Diagnostics
Drug/Device Interaction
39

Public
Commercial Rationale for Development
Unmet Market Need(s) – Unmet Clinical Need(s)
Market Size Assumption and Projections
International Considerations
Competitive Situation
40

Public
Clinical Investigations
41
Consider Compliance of CIP to ISO14155 Annex A & MDR Annex XV
Clearly Document within the CDP:
• Study design.
• Devices identified. – Think Accessories!!!
• Patient population.
• Patient numbers.
• Objectives and endpoints.
• Length of follow up and intervals.
• Study locations.
MDCG 2020-13 encourages NBs to review all this information. MDCG 2019-9 also provides a list of detailed information in relation to reporting clinical investigations.
Consider potential deviations.
The MDCG have published a
Clinical Investigations Reporting
Template (MDCG 2021-8)

Public
Strategic Planning
42
- Milestones
- Longer term plans (PMCF)
- Market Access (Controlled Roll Out?)
- No-go Criteria
- Risk assessment & Contingency Plans
- Regulatory Aspects

Public
Poll Question
Copyright © 2022 BSI. All rights reserved
43
Q: Is a Clinical Development Plan
Required for Legacy Devices?
• Yes
• No

Public
Poll Question
Copyright © 2022 BSI. All rights reserved
44
Q: Is a Clinical Development Plan
Required for Legacy Devices?
• Yes
• No

Public
Considerations
The Clinical Development Plan is a critical document for those wishing to gain consultation from the
Expert Panels as described in Article 61 (2) – Class III and Class IIb Rule 12 Administer or Remove
Medicinal Substances.
Consultation Process is likely to be available 2023 for a limited number of devices – Further
information is likely early 2023.
45

Public
Copyright © 2022 BSI. All rights reserved
46
Documenting a Clinical
Development Plan for
Legacy Devices.
Annex XIV Part A (1) (a) Indent 8.

Public
Legacy Devices and the CDP
47
ALL manufacturers are required to document a clinical
development plan to meet the requirements of MDR Annex XIV
Section 1a.
Premarket elements of the plan as described in the final indent of
MDR Annex XIV Section1a (first-in-man studies, feasibility and
pilot studies) are not generally relevant to legacy devices which
are unchanged in design or indications.
However, the context for the plan as described in indents 1-7 and
the basis for the PMCF as described in indent 8 of MDR Annex
XIV Section 1a are considered relevant and necessary for
demonstration of compliance to the MDR.

Public
Next Session Slide:
Copyright © 2022 BSI. All rights reserved
48
Next Session: Wednesday 25
th
January 2023
Clinical Evaluation Report Part I
How to document:
✓ Device Description
✓ Equivalence
✓ Similar Device Data
✓ Clinical Claims
✓ Literature Searches
✓ Updates and Competency

Public
End slide
Copyright © 2022 BSI. All rights reserved
50
