
Application Note
Assembly and annotation of plastid genomes
using QIAGEN
®
CLC Genomics Workbench
Introduction
Chloroplast sequences are highly conserved, making
them very useful for taxonomic classification of plants.
Next-generation sequencing (NGS) allows cost-efficient
sequencing of whole chloroplast genomes. The arche-
typical chloroplast genome follows a distinct architecture:
A large single copy region, a small single copy region
and a pair of inverted repeats (IRs). The sequence of the
IRs is challenging to assemble from short reads alone,
so a strategy of combined long- and short-read hybrid
sequencing and assembly is usually preferred to obtain
the complete chloroplast genome sequence. Once the
genome sequence has been obtained, the next challenge
is annotation of the genes in the sequence.
In this application note, we describe how to assemble and
annotate plastid genomes using QIAGEN CLC Genomics
Workbench. We describe three distinct workflows. In the
first workflow, the chloroplast sequence is assembled
de novo using chloroplast reads extracted from whole
genome sequencing (WGS) data by first mapping the
reads to a related plastid reference. In the second workflow,
the chloroplast sequence is assembled from a sub-sampled
WGS dataset. This second workflow is suitable when no
related plastid genome is available. The third workflow
uses very long but low-fidelity reads for de novo assembly,
followed by contig polishing using short reads. This
workflow is used for species where long IRs are expected
to be present in the chloroplast genome.
We cover all the main steps required for efficient
chloroplast assembly:
1. Extracting plastid reads from the WGS data
2. Reducing sequencing datasets prior to de novo
assembly
3. De novo assembly of plastid genomes using long reads
4. Contig polishing using short reads
5. Transferring annotation from plastids of related plants
6. Validating and visualizing the newly assembled
chloroplasts
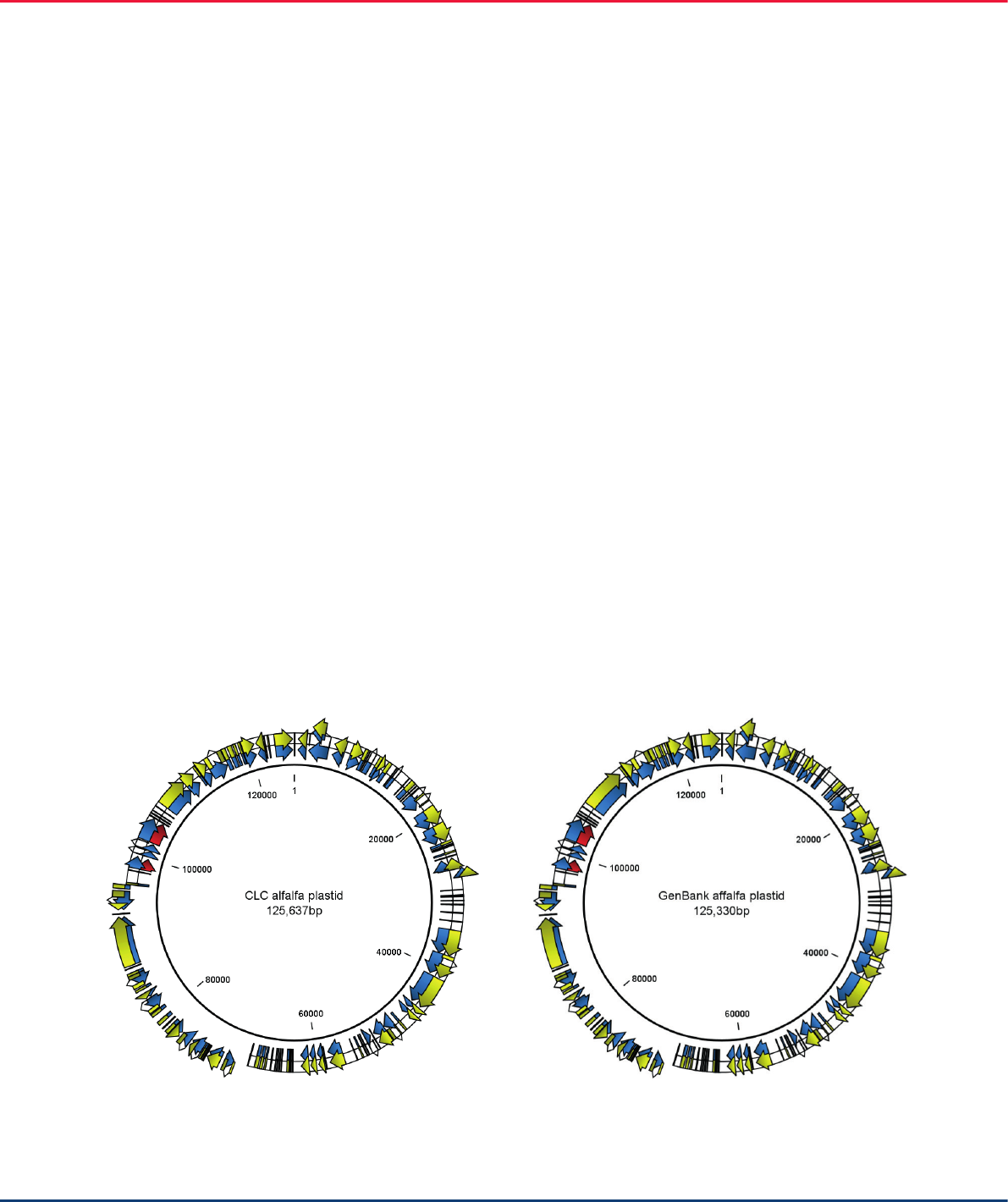
2 Assembly and annotation of plastid genomes using QIAGEN CLC Genomics Workbench 01/2022
Figure 1. QIAGEN CLC Genomics Workbench-assembled and annotated alfalfa chloroplast (left) and alfalfa chloroplast from a different cultivar,
imported from GenBank (right).
An important consideration when assembling plastid
genomes is the average length of reads used for
the assembly. Many plastids contain long IRs that
interfere with the accuracy and efficiency of assembly.
The plastid genomes themselves are usually between
110–200 kb and the IRs can be between 10–30 kb
long. Because of these repeats, we need reads that are
long enough to be unambiguously placed in the contig.
Not all plants have long IRs, and the first and second
workflows are suitable in these cases. We use alfalfa
for the first two workflows. “Shorter” long NGS reads
suffice for plastid assembly in such species. For the third
workflow, we use a rice dataset. Rice plastid genomes
contains IRs that are approximately 20 kb long. The
long-read dataset used here for this workflow includes
reads that are up to 84 kb long.
Results
All three workflows produced fully assembled and
annotated plastid genomes. Figure 1 shows the
visualizations produced by QIAGEN CLC Genomics
Workbench for two different alfalfa chloroplasts.
The Workbench-assembled genome contains the
same number of annotations as the GenBank alfalfa
plastid reference. However, the Workbench-assembled
genome differs in length by about 300 nt compared
to the reference genome because it originates from a
different alfalfa cultivar.
Data
The data used for the assemblies of alfalfa chloroplast
(Workflows 1 and 2) are from Chen et al, 2020. The
alfalfa reads are available in the Sequence Read Archive
(SRA): A long PacBio
®
read dataset (SRR11285798),
and an Illumina
®
dataset (SRR9026574). The data
used for the assembly of rice chloroplast (Workflow 3)
is from a study by researchers at the University of
Arizona: A long read dataset (SRR10302209) and a
short read dataset (SRR10302299).
Assembly and annotation of plastid genomes using QIAGEN CLC Genomics Workbench 01/2022 3
The alfalfa plastid reference sequence used in
Workflow 1 has the GenBank accession NC_042841.1.
The rice plastid reference sequence used in Workflow 3
has GenBank accession NC_008155.1.
Workflow 1. Chloroplast assembly using a plastid reference from a related species
In this workflow, shown in Figure 2, we collected the
relevant plastid reads from the WGS long-read data
by mapping them to a related plastid reference. Then,
the reads were sub-sampled (to reduce the dataset)
and assembled de novo. The annotations were
transferred from a related plastid genome using the
Whole Genome Alignment Tool. The short-read plastid
reads were also mapped to the reference plastid
genome and sub-sampled. These short reads were only
used to evaluate the quality of the de novo assembled
plastid genome contig.
Figure 2. Chloroplast assembly using a related reference genome to extract the plastid reads from WGS datasets.
Map long reads to a related plastid
Extract and sample mapped reads
De novo assemble the long reads
Align the de novo assembled plastid contig
with a related plastid, shift the start position
and transfer the annotation
Map short reads to a related plastid
Extract and sample mapped reads
Map short reads to de novo
assembled contigs
Call variants on mapped reads to
confirm the contig quality
Import a related plastid genome with annotation
Import long and short NGS reads

4 Assembly and annotation of plastid genomes using QIAGEN CLC Genomics Workbench 01/2022
Data preparation
The first step in all three workflows was to import a
related plastid genome with annotation from GenBank.
We imported directly to QIAGEN CLC Workbench
using the “Download from GenBank” importer. Both
the long and short NGS reads were imported using
the Workbench “Short Read Archive” importer. The
long reads were mapped using “Map Long Reads
to Reference” under the “Long Read Support” folder
(Figure 3). The short reads were mapped using
“Map Reads to Reference” under the “Resequencing
Analysis” folder (Figure 4).
The plastid read-mapping coverage was excessive
for both datasets: 28,000x coverage for long reads
and 15,000x coverage for short reads. Both sets
of mapped reads were extracted using the “Extract
Reads” tool under the “Utility Tools” folder. For efficient
assembly and further data analysis, the reads were
sampled using the “Sample Reads” utility tool (Figure 5)
to significantly reduce the amount of data. For
the PacBio dataset, we down-sampled to 5,000
reads representing approximately 500x coverage
of the plastid genome, and, for the Illumina data,
we down-sampled to 100,000 reads representing
approximately 120x coverage.
De novo assembly
The alfalfa reads used in this workflow are high-fidelity
PacBio reads. The reads are between 10 and 17 kb
in length. This length is sufficient to easily assemble
chloroplasts without long IRs. With a large quantity of
high-fidelity reads, we found the best word size was
28 rather than the default of 13. Running the “De
Novo Assemble Long Reads” tool (Figure 6) with this
configuration produced a single circular contig of
125,637 nucleotides (Figure 7).
Figure 4. The short read mapping tool.
Figure 5. Tools used to extract reads from mappings to sample reads.
Figure 6. The “Long Read Support” tools with the “De Novo Assemble
Long Reads” tool selected.
Figure 7. The single circular contig of 125,637 nucleotides produced by
the “De Novo Assemble Long Reads” tool.
Figure 8. The “Basic Variant Detection” tool under the “Variant
Detection” folder.
Figure 3. The long read mapping tool.

Assembly and annotation of plastid genomes using QIAGEN CLC Genomics Workbench 01/2022 5
Validation of the assembly quality
In the next step, we confirmed that the assembled
contig was free of errors by mapping Illumina reads to
it and calling variants. The short reads were mapped
using the “Map Reads to Reference” tool under the
“Resequencing Analysis” folder (Figure 4). The variants
were called using the “Basic Variant Detection” tool
under the “Variant Detection” folder (Figure 8). No
significant variants were found. The highest variant
frequency found was 19% in a homopolymeric area.
The validation we performed here confirms that the de
novo assembled contig is of a high quality and does
not contain assembly errors.
Whole genome alignment and transfer
of annotations
In this step, we annotated the de novo assembled
contig by aligning it to a related chloroplast reference
genome and transferring its annotations to the new
contig.
The “Create Whole Genome Alignment” tool does
not just align the genomes but also shifts the contig’s
start position relative to a reference genome. It can
also transfer the reference genome annotations to the
newly assembled contig. The tool can be found under
the “Whole Genome Alignment” folder (Figure 9).
These tools are available when the Whole Genome
Alignment plugin is installed.
In the settings dialog, it is important to select “Rearrange
contigs” and “Copy annotations from reference”. The
“Genetic Code” option should be set to “11” for plastid
genomes (Figure 10).
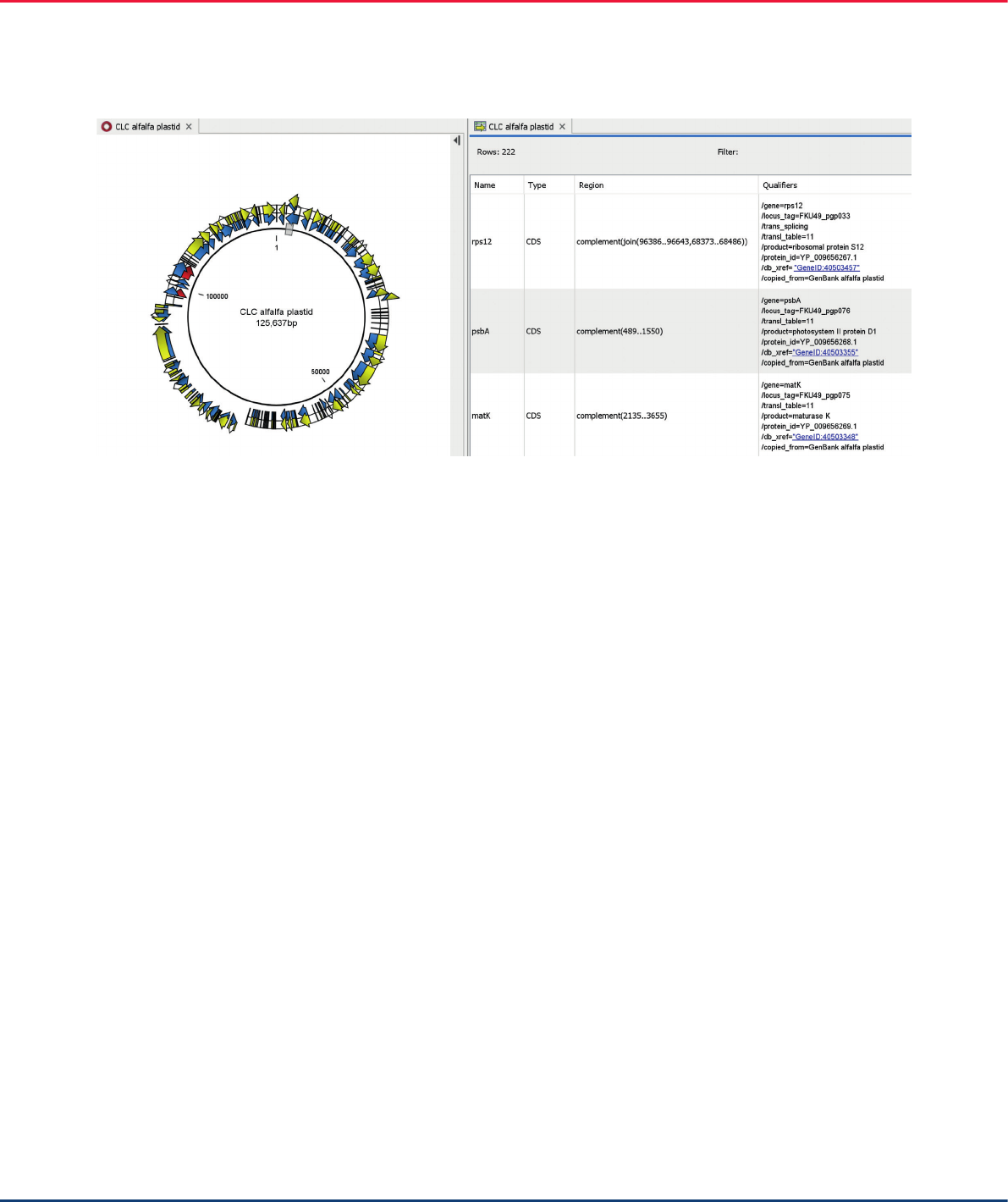
The resulting newly assembled contig contained the
same number of annotations as the GenBank alfalfa
reference genome. The annotations can be displayed
in tabular or graphical views (Figure 11).
Figure 9. The “Create Whole Genome Alignment” tool.
Figure 10. The recommended parameters for transferring plastid annotations.
6 Assembly and annotation of plastid genomes using QIAGEN CLC Genomics Workbench 01/2022
Workflow 2. Chloroplast assembly without a reference plastid
In this section, we describe a workflow for assembling
plant plastids using the same NGS data but without
extracting the plastid reads from WGS data. Instead,
we went directly to reducing the WGS data by
sampling the reads. The assembly workflow is shown
in Figure 12.
Data preparation: Sampling
The purpose of sampling is to reduce the number of
nuclear genomic reads. Plant genomes usually contain
plastid-related fragments in their nuclear chromosomes.
To prevent nuclear homologs of plastid sequences from
being included in the plastid assembly, we reduced the
data set size, which reduced both the number of nuclear
and plastid reads. Because there is a significantly
smaller quantity of plastid-related nuclear reads, they
were reduced enough to prevent their incorporation
into the plastid assembly.
Despite the small genome size of chloroplasts, the
chloroplast-originating reads usually comprise 5–6%
of WGS data from green plant tissues. This high
percentage results of the fact that a green cell can
contain a few hundred chloroplasts, each with their
own genome. In this dataset, we had approximately
28,000x coverage for the plastid genome. We reduced
this excessive coverage not just to suppress nuclear
homologs, but also to prevent erroneous assembly
caused by systemic sequencing errors. These errors
become apparent at excessively high coverage. We
sampled 2% long reads (about 110K reads) from the
original data set using the “Sample Reads” tool in the
“Utility Tools” folder (Figure 5).
Figure 11. Annotation displayed in graphical 9 (left) and tabular (right) views.

Assembly and annotation of plastid genomes using QIAGEN CLC Genomics Workbench 01/2022 7
De novo assembly
The “De Novo Assemble Long Reads” tool (Figure 6)
with the default parameters assembled these
approximately 110,000 long reads into over 2,000
contigs (Figure 13).
The second longest contig in the table was circular, as
is expected for a chloroplast contig. This contig also
had the same number of nucleotides as the previously
assembled contig from the first workflow. We expected
the correctly assembled plastid contig to have
disproportionally high coverage, as there are hundreds
of copies of plastid genome in each cell. To confirm
that the candidate contig had the highest coverage, we
mapped the long reads back to all contigs. This was
done using the “Map Long Reads to Reference” tool
(Figure 3). As expected, the candidate contig had an
average coverage of over 600x, which is significantly
higher than any other contigs in the mapping table that
are presumably contigs originating from the nuclear
genome (Figure 14).
Figure 13. Contigs assembled from the reduced WGS dataset.
Figure 14. The mapping coverage information for the de novo assembled
contigs. The longest circular contig is selected.
Figure 12. Chloroplast assembly using sampled reads from WGS datasets.
Sample long reads
De novo assemble long reads,
map long reads back to contigs
Align the de novo assembled plastid contig
with a related plastid, shift the start position
and transfer the annotation
Sample short reads
Map short reads to de novo
assembled contigs
Call variants on mapped reads to
confirm the contig quality
Import long and short NGS

8 Assembly and annotation of plastid genomes using QIAGEN CLC Genomics Workbench 01/2022
Validating the assembly quality
The assembly quality validation was performed as
described for Workflow 1, by mapping a subset of
short reads and calling the variants. No variants were
found, which confirmed that the contig is of high quality
and does not contain assembly errors.
Annotating the longest circular contig
The annotation was performed as described for
Workflow 1. We used the “Create Whole Genome
Alignment” tool (Figure 9) to transfer the annotation
from a related plastid genome. The tool produced the
same annotations as shown in Figure 11 for the plastid
assembled with Workflow 1.
Two other options for annotating newly assembled
plastid genomes are to search for coding sequences
using the “Find Open Reading Frames” tool and then
annotate these with the “Annotate with DIAMOND”
and “Annotate with BLAST” tools in the Microbial
Genomics Module (Figure 15).
Figure 15. “Annotate with DIAMOND” and “Annotate with BLAST” tools
in the Microbial Genomics Module.
Figure 16. Chloroplast assembly using long low-fidelity reads.
Map long reads to a related plastid
Extract and sample mapped reads
De novo assemble the long reads and
polish the contigs with short reads
Align the de novo assembled plastid contig
with a related plastid, shift the start position
and transfer the annotation
Map short reads to a related plastid
Extract and sample mapped reads
Map short reads to de novo
assembled contigs
Call variants on mapped reads to
confirm the contig quality
Import a related plastid genome with annotation
Import long and short NGS reads
Assembly and annotation of plastid genomes using QIAGEN CLC Genomics Workbench 01/2022 9
Workflow 3. Chloroplast assembly of a data sets containing long inverted repeats
Finally, we describe a workflow for assembling plant
plastid genomes with long IRs. For correct assembly
of these plastid genomes, a portion of the long reads
should be longer than the length of the repeats. This
workflow (Figure 16) is similar to Workflow 1, but with
an additional step for polishing the contigs created
using long but imperfect reads. The short Illumina reads
are used in the polishing step.
Data preparation: selecting long chloroplast
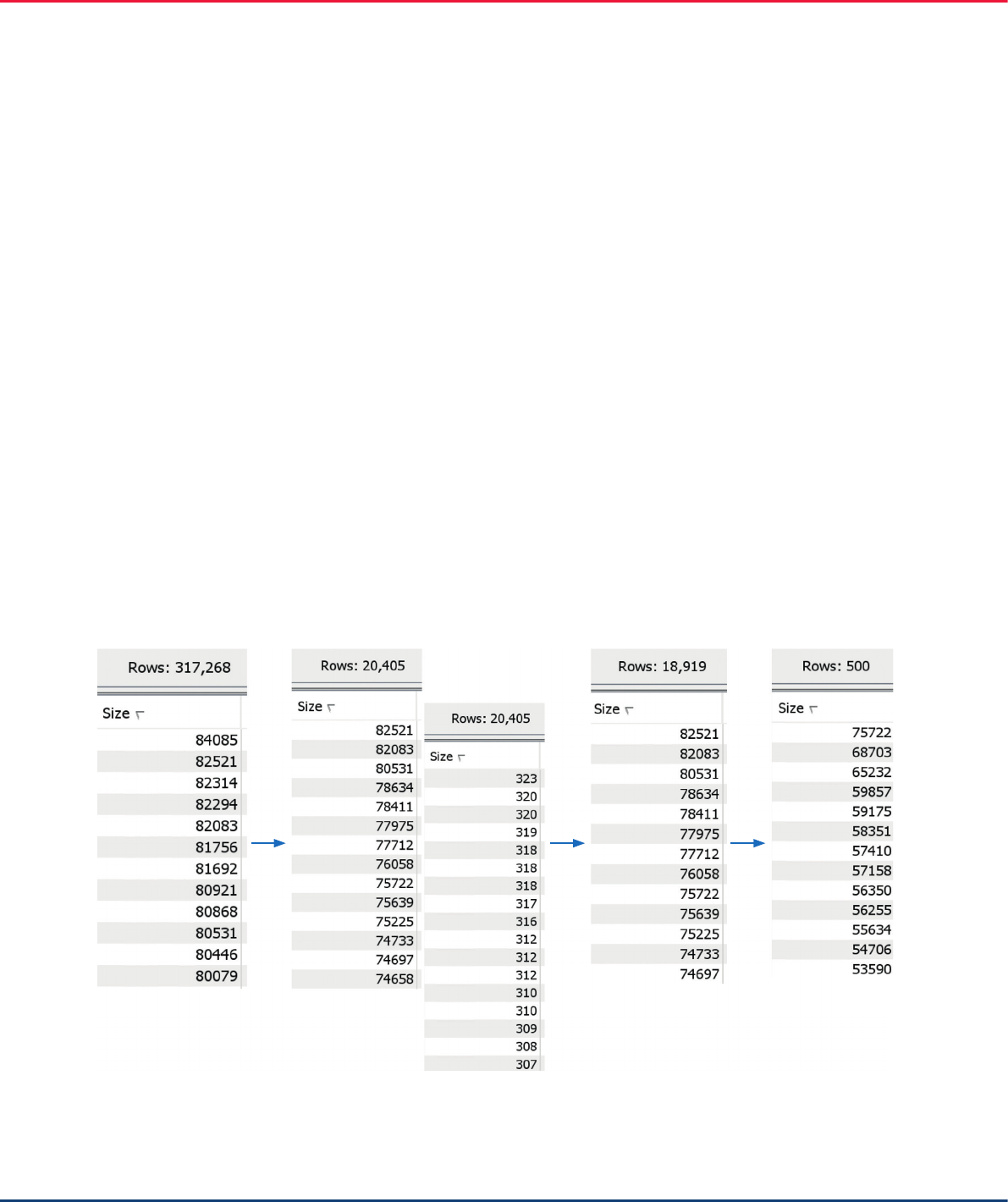
reads for assembly
The long reads used here were the low fidelity PacBio
reads up to 84 kb in length, which are sufficient to
span the 20 kb long rice plastid IRs. There were over
300,000 of these long PacBio reads. They were
mapped to a related plastid (NC_008155.1). This
resulted in about 20,000 mapped reads, which were
then extracted from the mapping (see Figures 3 and 5
for the tools used). In this dataset of long reads, there
were erroneously short reads, which we excluded from
further processing by removing reads under 2 kb in
length. From the resulting set of approximately 19,000
reads, we used 500 randomly sampled reads for de
novo assembly (Figure 17).
Figure 17. Selection of long reads for de novo assembly.
Original dataset
of long reads
Reads mapped to
related plastid
Mapped reads
>2 kb in length
Randomly sampled
reads for de novo
assembly

10 Assembly and annotation of plastid genomes using QIAGEN CLC Genomics Workbench 01/2022
De novo assembly and polishing with
high-quality short reads
Using the 500 long reads extracted in the previous
step, we used the “De Novo Assemble Long Reads”
tool (Figure 6) with a word size of 18 to assemble a
single circular contig of 134,674 nucleotides (Figure 18).
For the polishing step, two million short reads were
sampled from the WGS Illumina dataset. This dataset
contained paired-end Illumina reads approximately
151 nucleotides long. They were then mapped to the
related plastid (NC_008155). The mapped reads
(approximately 100,000) were extracted with the
“Extract Reads” tool (Figure 5) and used to polish the
de novo assembled contig. Polishing was performed
using the “Polish with Reads” tool, found under the Long
Read Support Folder (Figure 6). After running this tool,
the final size of the contig was reduced in length by
about 200 nucleotides (Figure 19).
Rice chloroplast assembly validation
and annotation
The assembly was validated and annotated
as described in Workflow 1 in the “Validation of
the assembly quality” section. The annotation was
transferred as described in the “Whole genome
alignment and transfer of annotations” section using
the “Create Whole Genome Alignment” tool (Figure 9).
Figure 18. The single circular contig produced by the long-read assembler.
Figure 19. The single circular contig after polishing.

Assembly and annotation of plastid genomes using QIAGEN CLC Genomics Workbench 01/2022 11
Summary
This application note describes three different
workflows for plastid assembly using QIAGEN
CLC Genomics Workbench. The choice of tools and
workflows depends on the structure of the plastid in the
species of interest, as well as the type of sequencing
data. Assembling plastids with long IRs requires reads
that are long enough to span the repeats. Such long
reads are usually of low fidelity and the assemblies
require polishing. Assembling plastids without long
IRs can be achieved using “shorter” high-fidelity
long reads and does not require contig polishing.
Another step we emphasize is the reduction of NGS
datasets before assembling plastids. We describe
different de novo assembly workflows with and without
preselection of chloroplast reads from whole genome
sequencing data.

1126623 01/2022
QIAGEN CLC Genomics products are intended for molecular biology applications. These products are not intended for
the diagnosis, prevention or treatment of a disease.
For up-to-date licensing information and product-specific disclaimers, see the respective QIAGEN OmicSoft Land product
website. Further information can be requested from [email protected] or by contacting your local account
manager at [email protected].
Reference
Chen, H., Zeng, Y., Yang, Y. et al. (2020) Allele-aware chromosome-level
genome assembly and efficient transgene-free genome editing for
the autotetraploid cultivated alfalfa. Nature Communications 11 , 2494.
https://doi.org/10.1038/s41467-020-16338-x
Trademarks: QIAGEN
®
, Sample to Insight
®
(QIAGEN Group); Illumina
®
(Illumina, Inc); PacBio
®
(Pacific Biosciences of California, Inc.). Registered names, trademarks, etc. used in this
document, even when not specifically marked as such, may still be protected by law.
PROM-19783-001 1126623 01/2022 © 2022 QIAGEN, all rights reserved.
Ordering www.qiagen.com/bioinformatics Technical Support digitalinsights.qiagen.com/support Website digitalinsights.qiagen.com
Learn more and request a trial at digitalinsights.qiagen.com/GXWB.
