
EN EN
EUROPEAN
COMMISSION
Brussels, 10.10.2019
SWD(2019) 375 final
COMMISSION STAFF WORKING DOCUMENT
Evaluation of the Union legislation on blood, tissues and cells
{SWD(2019) 376 final}
Table of Contents
LIST OF ABBREVIATIONS ............................................................................................. 1
GLOSSARY ........................................................................................................................ 2
1. INTRODUCTION ....................................................................................................... 7
1.1 Purpose of the Evaluation .................................................................................... 8
1.2 Scope of the Evaluation ....................................................................................... 8
2. BACKGROUND TO THE INTERVENTION ......................................................... 10
2.1 Concerns leading to the intervention ...................................................................... 10
2.2 Baseline and points of comparison ......................................................................... 16
2.2.1 Blood ................................................................................................................ 16
2.2.2 Tissues and Cells .............................................................................................. 17
2.2.3 Situation in Member States that joined after 2004 ........................................... 17
3. STATE OF PLAY ..................................................................................................... 19
3.1 The BTC Sector ...................................................................................................... 19
3.2 Transposition and updating of BTC legislation ...................................................... 21
3.3 Oversight functions at Member State level............................................................. 23
3.4 Monitoring Arrangements....................................................................................... 23
3.5 Support for implementation .................................................................................... 25
4. EVALUATION METHOD ....................................................................................... 26
4.1 Conducting the Evaluation...................................................................................... 26
4.1.1 Evaluation Roadmap ........................................................................................ 26
4.1.2 Stakeholder Consultation ................................................................................. 26
4.1.3 External Study .................................................................................................. 27
4.2 Limitations and robustness of the evaluation findings ........................................... 27
5. ANALYSIS AND ANSWERS TO THE EVALUATION QUESTIONS ................ 29
5.1 Relevance ........................................................................................................... 29
5.1.1 Evaluation question 1a): To what extent is the legislation sufficiently adapted
to, adaptable to, and up-to-date with scientific, technical and epidemiological
developments / innovation? ....................................................................................... 29
5.1.2 Evaluation question 1b) To what extent is the legislation adapted to other
changes in the sector such as commercialisation, internationalisation or other societal
changes? .................................................................................................................... 33
5.1.3 Evaluation question 1c) Are there any gaps in terms of substances of human
origin or activities that are not regulated by the Directives? .................................... 38
5.2 Effectiveness ........................................................................................................... 40
5.2.1 QUESTION 2: To what extent has the legislation increased the quality and
safety of blood and tissues and cells and achieved a high level of human health
protection? ................................................................................................................. 40
5.2.2 QUESTION 3: Has the legislation led to any unintended effects (positive or
negative)? .................................................................................................................. 48
5.2.3 QUESTION 4: What, if any, have been the barriers preventing effective
implementation of the legislation? ............................................................................ 49
5.2.4 QUESTION 5: Are the rules on oversight sufficient to address the increased
internationalisation? .................................................................................................. 51
5.2.5 QUESTION 6: What, if any, are the challenges to maintaining compliance
with the legislation? .................................................................................................. 52
5.2.6 QUESTION 7: To what extent, if any, has the legislation impacted on patient
access to blood, tissues and cells? ............................................................................. 53
5.3 Efficiency ................................................................................................................ 57
5.3.1 Evaluation question 8: How cost-effective has the application of the quality
and safety requirements in the legislation been for operators. Have the benefits
outweighed the costs? ............................................................................................... 58
5.3.2 Evaluation Question 9: Are there particular administrative or other burdens for
specific groups of operators, including downstream users of blood, tissues and cells
as starting materials for medicinal products? ............................................................ 61
5.4 Coherence ............................................................................................................... 64
5.4.1 Evaluation question 12a) To what extent is the legislation on blood and tissues
and cells consistent and coherent within its own provisions? ................................... 65
5.4.2 Evaluation question 12b) To what extent is the legislation coherent and
consistent with other relevant Union legislation? ..................................................... 66
5.4.3 Evaluation question 12c) Are the requirements of the Directives suitable when
blood, tissues and cells are used as starting materials for the manufacture of
medicinal products/medical devices? ........................................................................ 74
5.4.4 Evaluation question 12d): To what extent is the legislation coherent with other
relevant international / third country approaches to the regulation of the quality and
safety of blood and tissues and cells? ....................................................................... 76
5.5 EU Added Value ..................................................................................................... 78
5.5.1 Evaluation Question 13: To what extent has the legislative framework at EU
level added value to the regulation of blood and tissues and cells across the EU-28 in
a manner that could not have been achieved by measures taken at national or global
level? ......................................................................................................................... 78
5.5.2 Question 14. To what extent do stricter national measures pose an obstacle to
exchange of supplies between Member States? ........................................................ 80
6. CONCLUSIONS ........................................................................................................... 82
i) The legislation effectively increased the level of safety and quality of BTC across the
EU ................................................................................................................................. 82
ii) The current rules are no longer up to date with the dynamic BTC sectors .............. 82
iii) Key oversight principles are not sufficiently robust ............................................... 83
iv) Some citizen groups, such as donors and offspring are not adequately protected .. 84
v) The legislation does not keep pace with innovation ................................................. 84
vi) Requirements are insufficient to support sufficiency and a sustainable supply for all
BTC ............................................................................................................................... 85
Were possible areas for simplification identified? ....................................................... 86
Will the issues identified resolve or deteriorate over time? .......................................... 86
ANNEX I: PROCEDURAL INFORMATION ................................................................. 87
ANNEX II: TRANSMISSIONS OF INFECTIONS TO EU PATIENTS BY BLOOD
TRANSFUSION, TREATMENT WITH PLASMA DERIVED MEDICINAL
PRODUCTS AND TISSUES AND CELLS IN THE 1980 AND 1990S. ................ 88
ANNEX III: THE BACKGROUND TO THE REGULATION OF BLOOD AND
BLOOD COMPONENTS IN EUROPE ................................................................... 96
ANNEX IV: THE LEGAL BASIS AND THE LEGAL FRAMEWORK ADOPTED .. 102
Primary Legislation..................................................................................................... 102
Secondary legislation .................................................................................................. 102
Tertiary legislation ...................................................................................................... 104
ANNEX V: BASELINE .................................................................................................. 106
Part A - Blood and Blood components for transfusion ............................................... 106
Part B- Tissues and cells ............................................................................................. 112
ANNEX VI: CASES SENT BY NATIONAL COURTS TO THE EUROPEAN COURT
OF JUSTICE FOR A PRELIMINARY RULING ON QUESTIONS OF UNION
LAW RELATING TO THE BTC SECTORS. ....................................................... 118
ANNEX VII: WRITTEN PARLIAMENTARY QUESTIONS CONCERNING THE
BTC LEGISLATION SINCE 2002 ........................................................................ 124
ANNEX VIII: INTERPRETATION QUESTIONS ARISING IN THE NATIONAL
COMPETENT AUTHORITY (NCA) MEETINGS WITH THE COMMISSION . 127
ANNEX IX: LIST OF PUBLIC HEALTH PROGRAMME ACTIONS FUNDED TO
SUPPORT THE IMPLEMENTATION OF THE BTC LEGISLATION ............... 180
ANNEX X: ECDC – SUPPORT FOR EU ACTIVITIES ON SUBSTANCES OF
HUMAN ORIGIN 2012 – 2018 .............................................................................. 186
ANNEX XI: COLLABORATION WITH COUNCIL OF EUROPE ............................. 188
ANNEX XII: STAKEHOLDER CONSULTATION SYNOPSIS.................................. 189
1. Consultation Strategy .............................................................................................. 189
2. Consultation Activities and Key messages ............................................................. 192
2.1 Roadmap Feedback ........................................................................................... 192
2.2 Public Consultation ........................................................................................... 193
2.3 Targeted Stakeholder Consultation................................................................... 197
2.4 Stakeholder Event .............................................................................................. 201
3. Representativeness of Stakeholder Participation ................................................. 203
ANNEX XIII: EXAMPLES OF TECHNOLOGICAL CHANGES IN THE
PROCESSING OF TISSUES AND CELLS ........................................................... 207
ANNEX XIV: EMERGING/INCREASING COMMERCIAL ACTIVITIES ............... 209
ANNEX XV: A NON-EXHAUSTIVE LIST OF CLINICAL OUTCOME REGISTRIES
IN THE BTC SECTORS ......................................................................................... 210
ANNEX XVI: INCONSISTENCIES BETWEEN EU-LEGAL FRAMEWORKS ........ 212
1 | P a g e
List of abbreviations
ATMP
Advanced therapy medicinal products
BTC
Blood, tissues and cells
CoRE SoHO
Consortium of BTC representative societies
DLI
Donor Leukocytes for Infusion
EBA
European Blood Alliance
EBMT
The European Group for Blood and Marrow Transplantation
ECDC
The European Centre for Disease Prevention and Control
EHA
The European Hematology Association
EHC
The European Haemophilia Consortium
EPA
The European Plasma Alliance
ESHRE
The European Society for Human Reproduction and Embryology
FIODS
The International Federation of Blood Donor Organisations
FMT
Faecal Microbiota Transplants
GMP
Good manufacturing practices
GPG
Good Practice Guidelines
HIV
Human Immunodeficiency Virus
IHN
The International Haemovigilance Network
IPFA
The International Plasma Fractionation Association
IPOPI/PLUS
The International Patient Organisation for Primary Immunodeficiencies
ITE
Importing tissue establishments
IVF
In Vitro Fertilisation
MAR
Medically assisted reproduction
MSM
Men having sex with men
NAT
Nucleic Acid Test
OHSS
Ovarian Hyper stimulation Syndrome
PDMP
Plasma derived medicinal products
PMF
Plasma master file
PPTA
The Plasma Protein Therapeutics Association
PRF
Platelet Rich Fibrin
PRP
Platelet rich plasma
RAB
Rapid alert platform for human blood and blood components
RATC
Rapid alert platform for human tissues and cells
SAE
Serious adverse event
SAR
Serious adverse reaction
SARE
Serious adverse reactions and events
SoHO
Substances of Human Origin
TFEU
The Treaty on the Functioning of the European Union
VOC
Volatile organic compounds
VUD
Voluntary and unpaid donation
WMDA
World Marrow Donor Association
WNV
West Nile Virus

2 | P a g e
Glossary
Term
Definition
Advanced Therapy
Medicinal Products
An advanced therapy medicinal product
1
means any of the
following medicinal products for human use:
a gene therapy medicinal product as defined in Part
IV of Annex I to Directive 2001/83/EC,
a somatic cell therapy medicinal product as defined
in Part IV of Annex I to Directive 2001/83/EC,
a tissue engineered product as defined as containing
or consisting of engineered cells or tissues, and
presenting properties for or being used or
administered to human beings with a view to
regenerating, repairing or replacing a human tissue.
Allogeneic use
Cells or tissues removed from one person and applied to
another
2
.
Antibodies
Antibodies are immunoglobulins (Ig). They are large
proteins that are found in blood or other body fluids.
Antibodies are part of the immune system that identify and
neutralise foreign objects, such as bacteria and viruses.
Anticoagulant
An additive that stops or slows blood clotting.
Assisted Reproductive
Technology-ART
All treatments or procedures that include the in vitro
handling of human oocytes and sperm or embryos for the
purpose of establishing a pregnancy. This includes, but is
not limited to, IVF and transcervical embryo transfer,
gamete intra-Fallopian transfer, zygote intra-Fallopian
transfer, tubal embryo transfer, gamete and embryo
cryopreservation, oocyte and embryo donation and
gestational surrogacy. ART does not include assisted
insemination (artificial insemination) using sperm from
either a woman’s partner or sperm donor.
Autologous (transfusion,
donation or use)
Blood
3
: Autologous transfusion shall mean transfusion in
which the donor and the recipient are the same person and
in which pre-deposited blood and blood components are
used.
1
Regulation (EC) No 1394/2007 amending Directive 2001/83/EC and Regulation (EC) No 726/2004.
2
Directive 2004/23/EC.
3
Directive 2002/98/EC.

3 | P a g e
Tissues and cells
4
: Autologous use means cells or tissues
removed from and applied in the same person.
Basocellular Epithelioma
A common type of skin cancer.
Blood Establishments
Blood establishment shall mean any structure or body that
is responsible for any aspect of the collection and testing of
human blood or blood components, whatever their intended
purpose, and their processing, storage, and distribution
when intended for transfusion. This does not include
hospital blood banks
5
.
Bone marrow
See haematopoietic stem cells
Cytapheresis
A procedure for collecting particular cells from a donor or
patient.
Faecal microbiota
transplant – FMT
Fecal microbiota transplantation or FMT is the transfer of
fecal material containing bacteria and natural antibacterial
from a healthy individual into a diseased recipient.
Gametes
Sperm (spermatozoa) and eggs (oocytes).
Good Manufacturing
Practice
Good manufacturing practice shall mean the part of quality
assurance which ensures that products are consistently
produced and controlled to the quality standards appropriate
to their intended use
6
.
Haematopoietic stem cells
Cells in the bone marrow that produce new blood cells.
Haematopoietic stem cells are found in bone marrow and in
blood collected from the umbilical cord after the birth of a
baby. They can also be collected from a donor’s blood
stream if the donor is treated with particular hormones that
cause the cells to move out of the bone marrow into the
blood.
Haemoglobin
A protein found in the red blood cells that is responsible for
carrying oxygen around the body. Haemoglobin picks up
the oxygen in the lungs, and then releases it in the muscles
and other tissues where it is needed. Haemoglobin also
contains iron which is critical for it to work properly.
Haemovigilance
The set of surveillance procedures covering the entire blood
transfusion chain, from the donation and processing of
blood and its components, through to their provision and
4
Directive 2004/23/EC.
5
See: Directive 2002/98/EC.
6
Commission Directive 91/356/EEC.

4 | P a g e
transfusion to patients, and including their follow-up.
Immunodeficiency
A state in which the immune system's ability to fight
infectious disease and cancer is compromised or entirely
absent.
Immunoglobulin
Glycoproteins that are part of the immune system.
Imputability
The likelihood that a serious adverse reaction in a recipient
can be attributed to the tissue or cells applied or that a
serious adverse reaction in a living donor can be attributed
to the donation process
7
.
In vitro fertilisation (IVF)
An assisted reproductive technology (ART) procedure that
involves extracorporeal fertilisation.
Lymphocytes
White blood cells.
Medically assisted
reproduction (MAR)
Reproduction brought about through ovulation induction,
controlled ovarian stimulation, ovulation triggering, ART
procedures, and intrauterine, intracervical, and intravaginal
insemination with semen of donor.
Nucleic acid amplification
technique
A testing method to detect the presence of a targeted area of
a defined nucleic acid (e.g. viral genome) using
amplification techniques such as polymerase chain reaction
or transcription mediated amplification.
Partner Donations
The donation of reproductive cells between a man and a
woman who declare that they have an intimate physical
relationship
8
.
Plasma Derived Medicinal
Products
Medicinal products based on blood constituents which are
prepared industrially by public or private establishments,
such medicinal products including, in particular, albumin,
coagulating factors and immunoglobulins of human origin
9
.
Platelet
Platelets, also called thrombocytes, are a component of
blood whose function (along with the coagulation factors) is
to react to bleeding from blood vessel injury by clumping,
thereby initiating a blood clot.
Platelet Rich Fibrin (PRF)
A fibrin meshwork, in which platelet cytokines, growth
factors, and cells are entrapped and discharged after a
period and can serve as a resorbable film.
Platelet Rich Plasma
Plasma in which the donor’s platelets have been
7
See: Commission Directive 2005/61/EC implementing Directive 2002/98/EC.
8
Commission Directive 2006/17/EC.
9
Directive 2001/83/EC.

5 | P a g e
(PRP)
concentrated.
Reproductive cells
Sperm, eggs and embryos.
Same Surgical Procedure
Processing of human substances (blood, tissues or cells)
from a patient during surgery, within the surgical area.
Serious Adverse Event
Blood
10
: Any untoward occurrence associated with the
collection, testing, processing, storage and distribution, of
blood and blood components that might lead to death or
life-threatening, disabling or incapacitating conditions for
patients or which results in, or prolongs, hospitalisation or
morbidity.
Tissues and cells
11
: Any untoward occurrence associated
with the procurement, testing, processing, storage and
distribution of tissues and cells that might lead to the
transmission of a communicable disease, to death or
lifethreatening, disabling or incapacitating conditions for
patients or which might result in, or prolong, hospitalisation
or morbidity.
Serious Adverse Reaction
Blood
12
: An unintended response in donor or in patient
associated with the collection or transfusion of blood or
blood component that is fatal, life-threatening, disabling,
incapacitating, or which results in, or prolongs,
hospitalisation or morbidity.
Tissues and cells
13
: An unintended response, including a
communicable disease, in the donor or in the recipient
associated with the procurement or human application of
tissues and cells that is fatal, life-threatening, disabling,
incapacitating or which results in, or prolongs,
hospitalisation or morbidity.
Tissue Establishment
Tissue establishment means a tissue bank or a unit of a
hospital or another body where activities of processing,
preservation, storage or distribution of human tissues and
cells are undertaken. It may also be responsible for
procurement or testing of tissues and cells
14
.
Transmissible diseases
Comprises all clinically evident illnesses (i.e. characteristic
medical signs and/or symptoms of disease) resulting from
10
Directive 2002/98/EC.
11
Directive 2004/23/EC.
12
Directive 2002/98/EC.
13
Directive 2004/23/EC.
14
Directive 2004/23/EC.

6 | P a g e
the infection, presence and growth of microorganisms in an
individual or the transmission of genetic conditions to the
offspring. In the context of transplantation, malignancies
and autoimmune diseases may also be transmitted from
donor to recipient.

7 | P a g e
1. INTRODUCTION
In the European Union (EU), millions of blood donations are collected every year by
1,400 blood establishments, enabling the transfusion of millions of blood
components
15
,
16
. Plasma, a blood component, is also used for the manufacture of plasma
derived medicinal products (PDMP). Tissues and cells
17
are handled by over 4,000 tissue
establishments
18
and can also be the starting materials for the manufacture of medicinal
products and medical devices. Several of these substances are exchanged between
Member States.
Blood, tissues and cells (BTC) all come from the same source - donations from human
beings - either during life or after death. They are processed, tested and stored in blood
and tissue establishments before being supplied to hospitals and clinics. In most cases, no
alternative treatments exist to save or enhance human lives. However, these substances
can also cause adverse reactions in patients, including the transmission of disease.
To ensure high levels of public health protection for all stages of the process, the Blood
Directive (2002/98/EC) and the Tissues and Cells Directive (2004/23/EC) were adopted
in 2002 and 2004, respectively, laying down common (minimum) quality and safety
standards at Union level and aiming to facilitate increased exchange of these substances
between Member States. In this report, these two Directives are referred to jointly as ‘the
basic Acts’. Implementing legislation was also adopted to provide technical requirements
for both fields
19
. Since their adoption, some of the implementing Directives have been
amended (see Annex IV).
There has been significant scientific and technological development in the sector and
new risks of transmitting diseases, such as Zika, dengue fever and hepatitis E, have
emerged since the legislation was adopted, more than 15 years ago. The field has also
undergone organisational changes, including an increasing role of commercial players in
a traditionally non-profit sector with a high level of public sector involvement. No
evaluation of the basic Acts has taken place since their adoption. The European
Commission has published several implementation reports for each sub-sector, each
15
Red blood cells, platelets and plasma are components of a blood donation, and each can be transfused to patients.
16
DG SANTE website, introduction to the Commission’s work on blood.
17
Including corneas, bone, skin and heart valves for tissue replacement surgery, stem cells from bone marrow and cord
blood for transplantation and reproductive cells for medically assisted reproduction.
18
DG SANTE website, introduction to the Commission’s work on tissues and cells.
19
Blood: Commission Directive 2004/33/EC as regards technical requirements for blood and blood components,
Directive 2011/38/EU and Directive 2014/110/EU; Commission Directive 2005/61/EC as regards traceability
requirements and notification of serious adverse reactions and events; Commission Directive 2005/62/EC as regards
Community standards and specifications relating to a quality system for blood establishments; Tissues and cells:
Commission Directive 2006/17/EC as regards technical requirements for donation, procurement and testing;
Commission Directive 2006/86/EC as regards traceability requirements, notification of serious adverse reactions and
events and technical requirements for coding, processing, preservation, storage and distribution; Commission Directive
(EU) 2015/566 regarding the procedures for verifying the equivalent standards of quality and safety of imported tissues
and cells; Commission Decision 2010/453/EU on conditions of inspections; Commission Decision 2015/4460 on
agreements with tissue and cell coding organisations.
8 | P a g e
based on information provided by Member States. The most recent reports, published in
April 2016
20
,
21
highlighted a number of gaps and shortcomings.
1.1 Purpose of the Evaluation
The purpose of this evaluation is to provide a comprehensive assessment of the Union
legislation on BTC - the basic Acts and their implementing Directives, examining their
functioning across the EU. The European Commission’s evaluation criteria
22
, i.e.
effectiveness, efficiency, relevance, coherence and EU-added value, are assessed. In
particular, the evaluation assesses the extent to which the BTC legislation met its original
objectives and whether it remains fit for purpose.
1.2 Scope of the Evaluation
This evaluation covers the two basic Acts, Directives 2002/98/EC on blood and
2004/23/EC on tissues and cells, as well as their implementing Directives
5
in all EU
Member States from the date of their entry into force until the end of April 2019. For
those Member States that joined the Union after the entry into force of the Directives, the
evaluation covers the period from their date of accession.
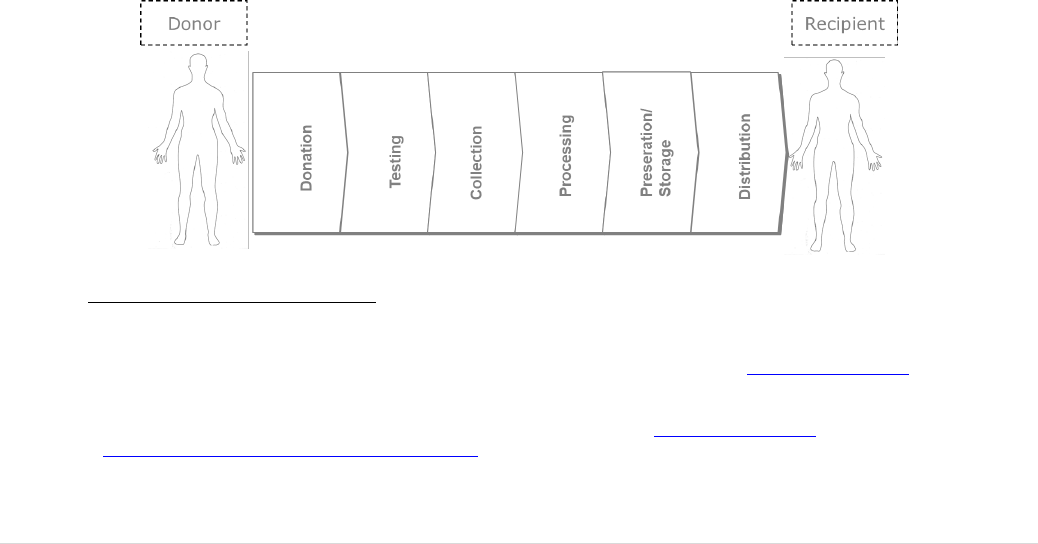
There are many commonalities between the two basic Acts. They have the same legal
basis
23
, similar generic oversight requirements and they aim to mitigate very similar risks
to safety and quality for all stages from donation to distribution for clinical use in a
patient (see Figure 1). Many professionals and authorities work across the two sub-
sectors. For these reasons, one evaluation was conducted to cover both legal frameworks.
FIGURE 1: THE STEPS FROM BTC DONATION TO CLINICAL APPLICATION
20
Report from the Commission to the European Parliament, the Council, the European Economic and Social
Committee and the Committee of the Regions on the Implementation of Directive 2002/98/EC and implementing
directives, setting standards of quality and safety for human blood and blood components - COM(2016) 224 final.
21
Report from the Commission to the European Parliament, the Council, the European Economic and Social
Committee and the Committee of the Regions on the Implementation of Directive 2004/23/EC and implementing
directives, setting standards of quality and safety for human tissues and cells - COM(2016) 223 final.
22
European Commission Better Regulation guidelines.
23
Article 168 (4) (a) of the Treaty on the Functioning of the European Union (TFEU).

9 | P a g e
The Human Organs Directive, adopted in 2010 (2010/53/EU), was excluded from the
scope of this evaluation, given that significant shortcomings had not been highlighted, as
they had for BTC, and the quality and safety provisions are significantly different and
less detailed
24
.
The regulations governing medicinal products, including advanced therapy medicinal
products (ATMPs) adopted in 2007
25
, and governing medical devices, adopted only in
2017
26
, are also excluded from the scope. However, the evaluation does cover the
coherence of the BTC legislation with these frameworks.
In the light of the significant and increasing international exchanges of some BTC with
third countries, the evaluation also considers coherence and similarities/differences with
relevant regulatory frameworks for BTC outside the EU. In particular, the equivalence of
safety and quality of BTC imported into the EU, mostly from the United States of
America (USA), is addressed.
The BTC sector is one where many ethical issues arise, ranging from debates
surrounding the use of technology for the creation of life to consent for donation after
death. EU legal competence to regulate this field is limited, by the Treaty on the
Functioning of the European Union (TFEU)
27
, to safety and quality. Decisions and
policies on the many ethical aspects remain at a Member State level, except where they
have an impact on safety and quality. Where Member States choose to allow particular
practices, such as testing or genetic manipulation of embryos, the safety and quality of
those activities are regulated by these Directives and are included in the scope of this
evaluation. Legal competence for issues related to the organisation of healthcare services
(including BTC services) also remains at the Member State level.
24
Human organs are transported directly from site of donation and procurement to site for transplantation. They do not
pass through dedicated establishments for processing, storage and/or distribution.
25
Regulation 1394/2007.
26
Regulation (EU) 2017/745.
27
Article 168 (4) (a) TFEU stipulates that the Union shall adopt measures setting high standards of quality and safety
of organs and substances of human origin, blood and blood derivatives; these measures shall not prevent any Member
State from maintaining or introducing more stringent protective measures.

10 | P a g e
2. BACKGROUND TO THE INTERVENTION
2.1 Concerns leading to the intervention
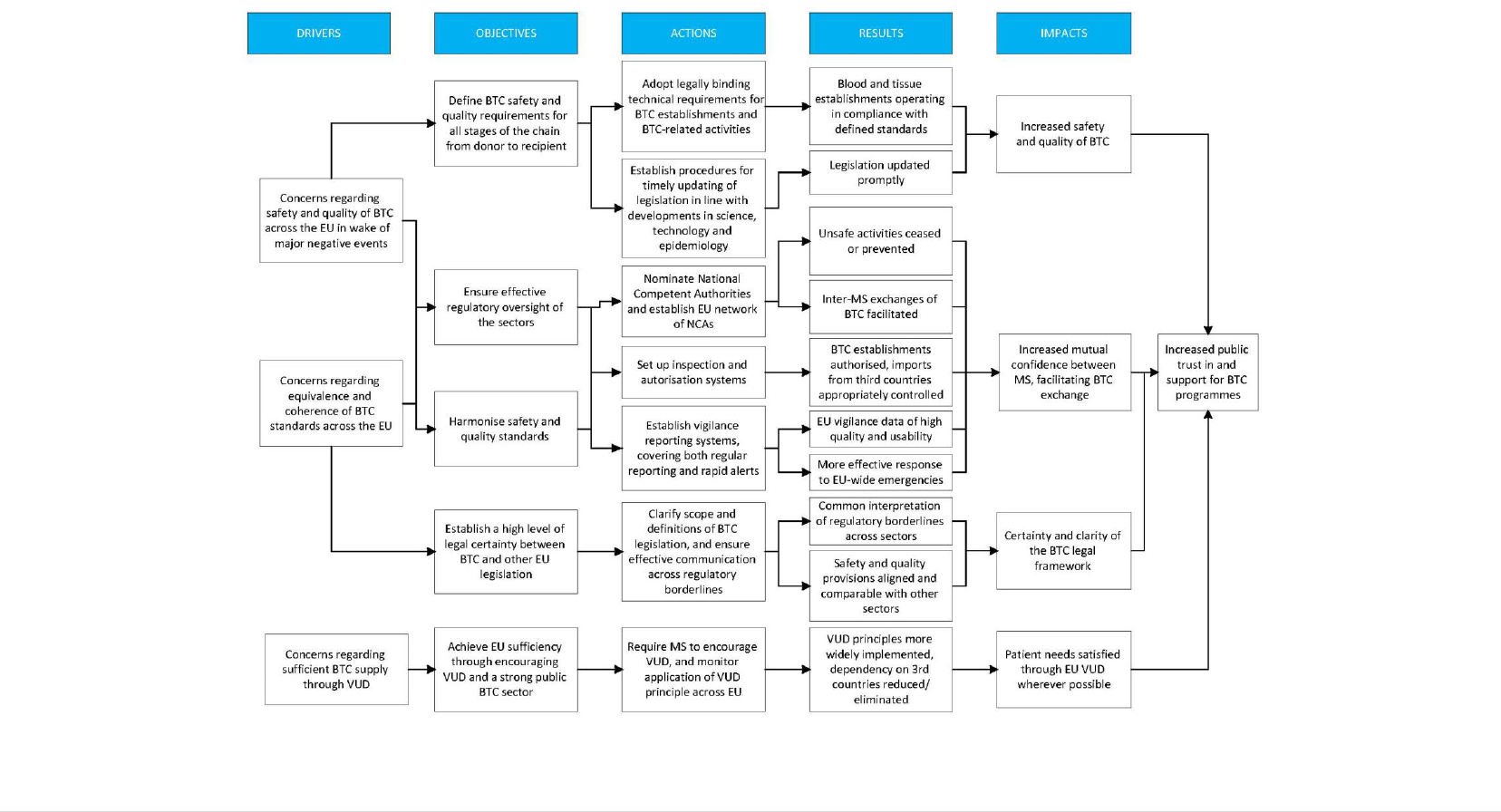
Three key concerns prompted the adoption of the blood, tissues and cells policy
intervention.
The first was the spread of infectious diseases by blood transfusion and treatment with
plasma-derived medicinal products, and the equivalent risks it posed for tissues and cells.
The second was the lack of equivalency and coherence of standards for BTC across
Member States. The third was the insufficiency of BTC supply, particularly blood and
plasma for the manufacturing of medicinal products, through voluntary and unpaid
donation (VUD).
Concern 1 - Widespread concerns due to disease transmission to
patients
The primary driver for taking action in the 80’s and 90’s, was the infection of tens of
thousands of patients across the EU with HIV, and later with hepatitis C, by the
transfusion of blood components and by treatment with plasma-derived medicinal
products. At least 20,000 transmissions of HIV by blood transfusion were recorded in
just seven Member States (Figure 2); the total number for the EU is likely to have been
significantly higher.
These events, with a high impact politically and socially, were widely reported in the
media and resulted in court cases and government inquiries in a number of Member
States. In the UK, Germany, Ireland and France, the cases were particularly public and
issues regarding compensation are still the subject of a legal inquiry in the UK today
28
(see also Annexes II and III where more detail is provided).
28
The UK legal inquiry on infected blood can be followed online.
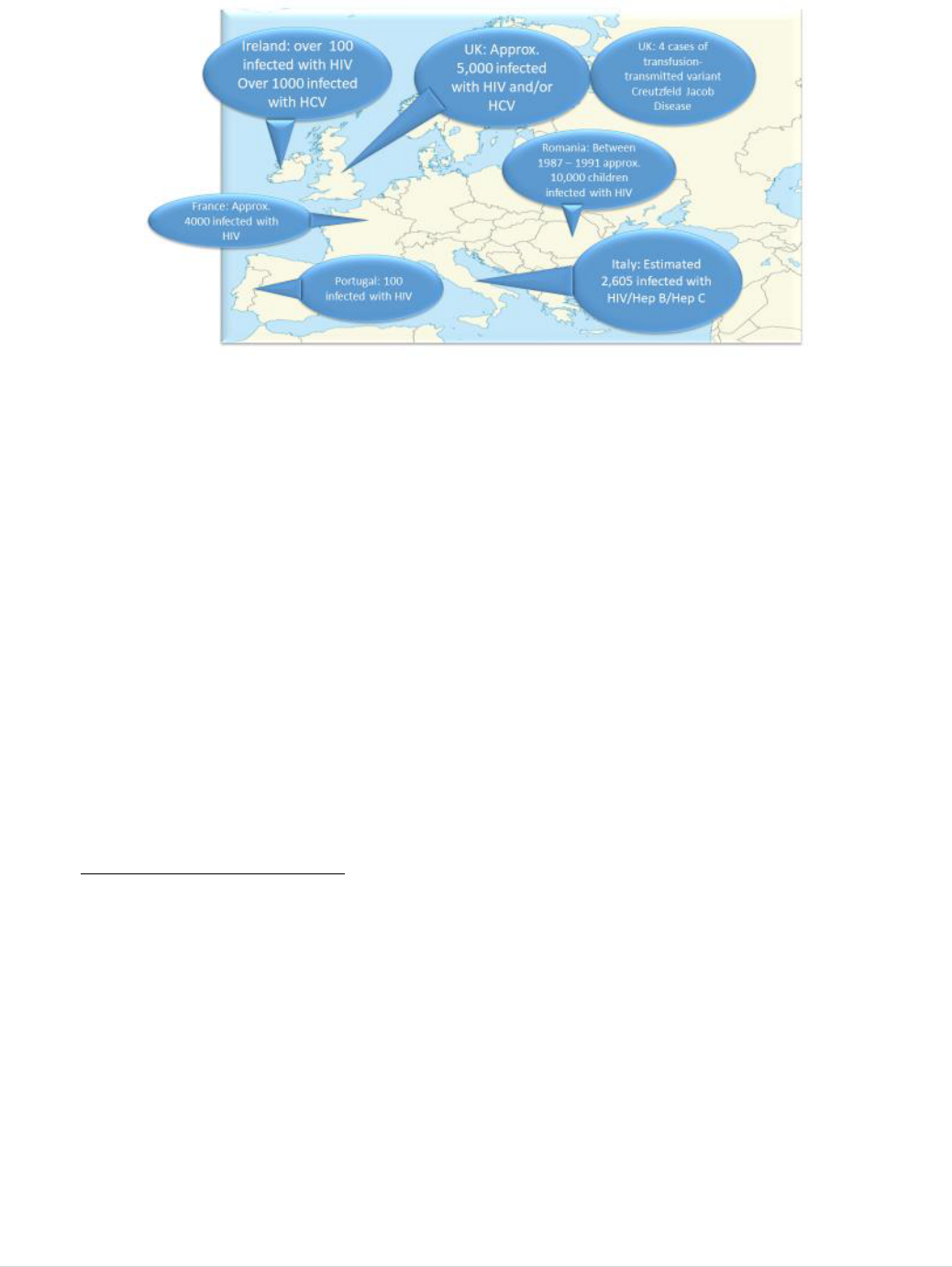
11 | P a g e
FIGURE 2: EXAMPLES IN THE HISTORY OF CONTAMINATED BLOOD AND BLOOD DERIVATIVES IN
EUROPE PRIOR TO THE ADOPTION OF THE EU BLOOD DIRECTIVE
While infectious transmissions on a scale comparable to blood did not occur for tissues
and cells, the equivalent risks to patients were indeed very real. A number of
scientifically documented sentinel events highlighted that HIV, hepatitis C or Creutzfeldt
Jacob disease transmissions by transplanted tissues and cells had occurred inside or
outside EU and in the latter case continue to emerge due to long incubation times
29
,
30
,
31
.
In the light of these disease transmission concerns, the Treaty of Amsterdam agreed in
1997
32
gives the Union the legal competence to set minimum quality and safety standards
for substances of human origin, while allowing Member States to take more stringent
measures.
The BTC intervention defined the EU safety and quality requirements for all stages of the
chain from donor to recipient (Objective 1- Safety and quality requirements) and aimed
to ensure effective regulatory oversight of the sectors (Objective 2- Oversight).
To achieve safety and quality of BTC (Objective 1), the following were put in place:
29
Simonds RJ et al. (1992) Transmission of Human Immunodeficiency Virus Type 1 from a seronegative organ and
tissue donor, NEJM 326 (11): 726-732.
30
Conrad EU et al.(2005) Transmission of the hepatitis C virus by tissue transplantation J Bone Joint Surg 77(2): 214-
224.
31
Over forty transmissions of Creutzfeldt Jacob disease by transplantation of highly processed dura mater (a tissue that
lines the skull) were detected from tissue processed in Germany and mostly exported to Japan. The long incubation
period for this prion disease means that new cases continued to be detected decades later in Japan, with the total
number of infected recipients reaching over 90 according to an update published by the US Centres for Disease Control
in 2008. CDC (2008) Update: Creutzfeldt Jacob disease associated with cadaveric dura mater grafts: Japan 1978 –
2008 MMWR 57(43): 1181.
32
(the now) Article 168 (4) TFEU: ‘By way of derogation from Article 2(5) and Article 6(a) and in accordance with
Article 4(2)(k) the European Parliament and the Council, acting in accordance with the ordinary legislative procedure
and after consulting the Economic and Social Committee and the Committee of the Regions, shall contribute to the
achievement of the objectives referred to in this Article through adopting in order to meet common safety concerns:
a) measures setting high standards of quality and safety of organs and substances of human origin, blood and blood
derivatives; these measures shall not prevent any Member State from maintaining or introducing more stringent
protective measures;’.
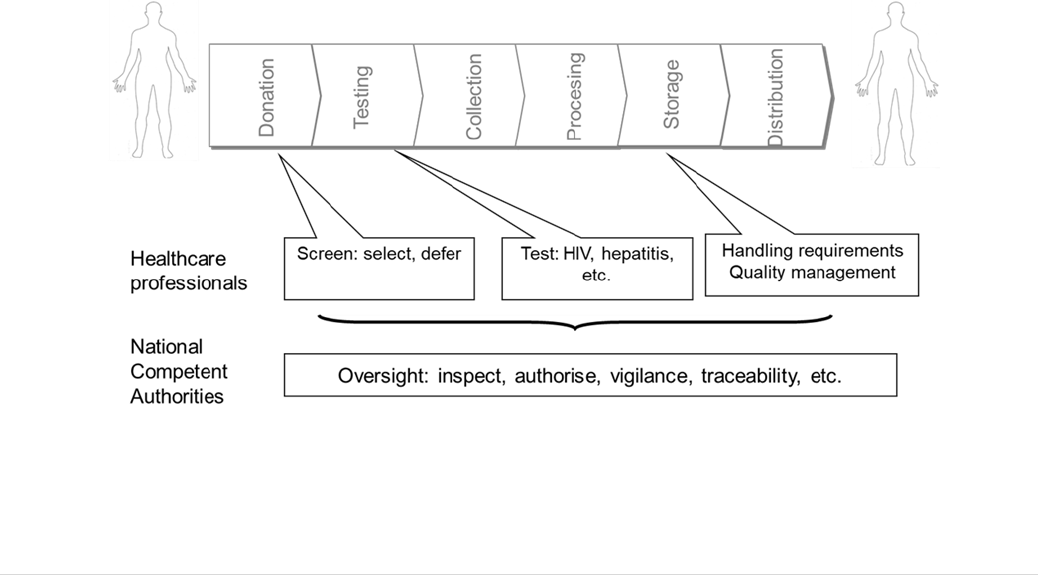
12 | P a g e
(i) a set of legally binding quality and safety requirements to address all activities from
donation to distribution, including screening, testing and handling requirements;
(ii) a set of legally binding requirements for blood and tissue establishments addressing
personnel, facilities, quality management etc. and
(iii) processes for the adaptation of the requirements in line with scientific,
technological and epidemiological changes.
To achieve an effective regulatory oversight of the sectors (Objective 2) the basic Acts
included:
(i) the establishment/nomination of Competent Authorities responsible for oversight at
Member State level;
(ii) establishment of a Competent Authority network at EU level;
(iii) set-up of inspection and authorisation systems and
(iv) set-up of vigilance systems (adverse reaction and event reporting and rapid alerts).
The national competent authorities in Member States are required to authorise blood and
tissue establishments, to inspect them at a 2-yearly frequency, to report annually to the
European Commission on serious adverse reactions (where a patient has been harmed),
to report in a similar way on serious adverse events (where an incident posed a risk of
harm to a donor or a patient), and to communicate with each other when more than one
Member State may be involved. For tissues and cells, the authorities must also ensure the
equivalence of imports from third countries in terms of quality and safety to those
provided under EU legislation through the authorisation of ‘Importing Tissue
Establishments’. Finally, Member States are obliged to put penalties in place for non-
compliance and to ensure appropriate data protection.
FIGURE 3: OBJECTIVES 1 AND 2 TO ACHIEVE SAFETY AND QUALITY FROM DONOR TO RECIPIENT AND
AND EFFECTIVE OVERSIGHT

13 | P a g e
To mitigate risk and prevent unsafe activities, the European Commission is required to
hold regular meetings with the competent authorities of the Member States to support the
network and facilitate the collection and publication of data and the operation of shared
platforms for information exchange (rapid alerts).
The implementation of the safety and quality and regulatory oversight objectives, and
associated actions, were expected to lead to: (a) increased safety and quality in the chain
from donor to recipient; (b) blood and tissue establishments operating in compliance with
the defined standards; (c) provisions updated promptly in line with technological
developments and new risks; (d) unsafe activities ceased or prevented; (e) vigilance data
feeding quality improvement and increased visibility of risks and (f) risks mitigated
through EU-wide communication and action.
Concern 2 - Lack of equivalency and coherence of standards across EU
Member States
Surveys of blood service organisation and practices across the EU provided evidence of a
wide variation in the standards of safety and quality being applied (see Annexe III).
Movement of plasma and of tissue and cells between Member States was seen as an
urgent problem to tackle because of the high frequency and volumes and concerns
regarding a lack of standardisation and organisational structures, respectively
33
.
Furthermore, Member States applied different rules for the different classification of
substances under national frameworks.
To address these concerns, the intervention aimed to achieve a degree of harmonisation
of safety and quality that facilitates inter-MS exchanges (Objective 3- harmonisation) but
also to stablish a high level of legal certainty at Union level (Objective 4- legal certainty).
The implementation across the EU of the actions for safety and quality requirements, and
for oversight (Objectives 1 and 2), also allow to achieve harmonisation of safety and
quality (Objective 3). Agreed technical requirements and oversight also facilitate the
inter-Member States exchanges, as long as the common minimum standards and the
common oversight obligations are met. These measures aimed to result in increased
mutual trust and confidence between MS, facilitating exchanges.
Legal certainty (Objective 4) was addressed by the scope and definitions provisions of
the basic Acts
34
and through effective communication between authorities for different
sectors within Member States. These aimed to ensure clarity across regulatory
borderlines where BTC are used to manufacture medicines or medical devices or where
the supply of critical devices impacts on the safety or supply of BTC.
33
European Group of Ethics Opinion on Ethical Aspects of Human Tissue Banking.
34
Articles 2 and 3 in both Acts.

14 | P a g e
Concern 3 - Insufficient supply of blood through VUD
Infectious transmissions from donations made by paid plasma donors in the United States
and imported to the EU (mainly to the UK) were reported in the public domain. These
imports, prompted by insufficiency of the local supply, were associated with significantly
higher risks of contamination with hepatitis and HIV
35
,
36
.
This concern led to a call for ensuring community sufficiency through Voluntary and
Unpaid Donations (VUD), a strategy that aimed at avoiding the inclusion of potentially
higher risk donors, motivated by payment, in the donor pool
37
.
Therefore, to achieve EU sufficiency through the encouragement of VUD and a strong
public sector was a priority (Objective 5 - sufficiency).
This was to be achieved by ensuring through legal provisions
38
that Member States
encourage VUD, satisfy patient needs through EU VUD wherever possible and maintain
a high level of safety.
The outcomes expected were good public willingness and awareness to donate
voluntarily, with common understanding of compensation and incentive concepts and a
decreased dependence on third country donations where higher risks might be accepted.
The above highlighted concerns were addressed
by the EU legislation for BTC, which
was adopted with two basic Acts, Directive 2002/98/EC
39
for blood and Directive
2004/23/EC
40
for tissues and cells. Figure 4 provides a summary of the concerns,
objectives, actions and intended outcomes of the intervention (the intervention logic).
The key reports and policy documents that highlighted the concerns, with their key
findings, are described in Annex III. Annex IV provides a full description of the legal
basis for the legislation and the provisions it includes.
35
Eastlund T. (1998) Monetary blood donation incentives and the risk of transfusion-transmitted infection.
Transfusion; 38: 874-82.
36
Van der Poel CL, Seifried E, Schaasberg WP. (2002) Paying for blood donations: still a risk? Vox Sang. 83: 285-
293.
37
Although donors were tested
for infectious HIV and hepatitis, individuals in the early stages of infection have not yet
produced antibodies to the virus (they are in the so-called ‘infectious window-period’) so their infectious status can be
missed by the anti-body tests used.
38
Article 20 in 2002/98/EC and Article 12 in 2004/23/EC.
39
Directive 2002/98/EC of the European Parliament and of the Council of 27 January 2003 setting standards of quality
and safety for the collection, testing, processing, storage and distribution of human blood and blood components and
amending Directive 2001/83/EC. (OJ L 33, 8.2.2003, p.30).
40
Directive 2004/23/EC of the European Parliament and of the Council of 31 March 2004 on setting standards of
quality and safety for the donation, procurement, testing, processing, preservation, storage and distribution of human
tissues and cells (OJ L 102, 7.4.2004, p. 48).
15 | P a g e
FIGURE 4: A SUMMARY OF THE CONCERNS, OBJECTIVES, ACTIONS AND INTENDED OUTCOMES OF THE INTERVENTION.
16 | P a g e
2.2 Baseline and points of comparison
The baseline used for the evaluation was the situation in fifteen Member States in the
period prior to the adoption of the basic Acts, which was complemented with information
from the 13 new Member States that joined the EU from 2004 onwards. There was no
Impact Assessment carried, out prior to the adoption of the legislation, that describes the
baseline. As many Member States did not have national reporting systems in place
41
,
precise baseline data are limited.
2.2.1 Blood
As early as 1994, the European Commission had raised concerns regarding blood safety
and sufficiency, noting that the donor selection process differed across the Community.
At that time, Directive 89/381/EEC required that testing of blood and plasma, when used
as starting materials for blood derived medicinal products, had to comply with the
recommendations of the Council of Europe, the WHO and the European Pharmacopoeia.
However, as this Directive did not apply to whole blood, to plasma or to blood cells of
human origin for transfusion, divergent testing requirements existed within the
Community for blood donations for transfusion and plasma donations for manufacture of
plasma derived medicinal products.
Licencing and accreditation of blood collection establishments differed widely across the
Member States. Many had no licencing requirements for the collection of blood or
plasma; no standard requirements for collection centres across the country; no routine
and/or unannounced inspections by national authorities nor peer inspections to ensure
that appropriate donor selection procedures were followed.
In 2001, voluntary non-remunerated blood donors were found only in five EU Member
States out of 13. In others, incentives, family replacement
42
and remuneration were
mechanisms used to encourage blood donation
43
. Haemovigilance was required by law in
only 11 countries, and, consequently, infectious transmissions during this time were
likely to have been underestimated.
The first Implementation Report
44
for Directive 2002/98/EC was published in 2006 and
indicated that seven Member States had organised inspections and control measures in
blood establishments in order to ensure compliance with the Directive’s requirements,
41
The situation varied depending on the type of substance but a recent survey of tissue and cell authorities indicated
that over a third of Member States did not have authorities in place for transplanted tissues and cells, and over half for
medically assisted reproduction, that could have gathered such reports. The survey was reported at a meeting of tissue
and cell competent authorities in May 2019.
42
The practice of asking family members to replace donations transfused to their relative by donating blood
themselves. This practice raised concerns from a safety perspective, as donors are not self-selected and, therefore,
might not reveal risk factors.
43
Mascaretti et al. 2004 Comparative analysis of national regulations concerning blood safety across Europe.
Transfusion Medicine, 14,105–111.
44
First report on the application of the Blood Directive from the European Commission, 19 June 2006.

17 | P a g e
confirming that this oversight had not been in place previously. Only nine reported that
donor selection procedures and donation deferral criteria were in place in blood
establishments for all donors of blood and blood components.
See Annex V, Part A, for a full description of the baseline for blood.
2.2.2 Tissues and Cells
Prior to the adoption of the directive there were shortcomings and differences in the
existing national rules, particularly in relation to donor selection and to the circulation of
tissues between countries, which were highlighted in 1998 by the European Group on
Ethics in Science and New Technologies
45
as well as by experts in the areas of organs,
tissues and cells in 2000
46
.
Health ministers from 11 EU Member States met in 2002 and supported a proposed
directive on the therapeutic use of human cells and tissues for transplantation. At that
time, only Spain, France, Belgium, and Denmark had specific legislation for tissue and
cell banks. Most EU countries had regulations only for transplantation of solid organs
47
.
Earlier publications from the UK had already raised concerns about the safety standards
of bone banking
48
,
49
. In addition, there was an increasing degree of uncontrolled tissue
movement between Member States and with third countries
50
.
Prior to the adoption of the basic Act on tissues and cells, or to accession of countries to
the EU, safety and quality rules and oversight were widely lacking
51
. By 2007, 11
Member States had not yet put inspection systems in pace and others had not yet
inspected all tissue establishments or put vigilance reporting procedures in place
52
.
See Annex V, Part B, for a full description of the baseline for tissues and cells.
2.2.3 Situation in Member States that joined after 2004
In three of the Member States that joined the EU after 2004 (Romania, Bulgaria,
Croatia), assessments during the accession process generally demonstrated limited or
45
European Group on Ethics in Science and New Technologies (European Commission). Ethical aspects of human
tissue banking.
46
At a meeting organised by the Portuguese Presidency of the EU, together with the European Commission described
in Sauer F et al. The Regulation of blood and tissues in the European Union. Pharmaceutical Policy and Law (2005) 6:
47-58.
47
Xavier Bosch. Health ministers support pan-European transplantation standards, The Lancet Vol. 359, ISSUE 9306,
P591, 16 February 2002.
48
Michaud RJ, Drabu KJ. Bone allograft banking in the United Kingdom. J Bone Joint Surg Br 1994;76:350.
49
Fehily D & Warwick RW. Safe tissue grafts: Should achieve same standards as for blood transfusion BMJ 1997;
314:1141.
50
Sauer F, Delaney F and Fernandez-Zinke E. The regulation of blood and tissues in the European Union.
Pharmaceuticals Policy and Law 6 (2005) 47-58.
51
Results of a Commission Survey presented to the tissue and cell competent authorities meeting in May 2019.
52
Summary Table of Responses from Competent Authorities: Questionnaire on the transposition and implementation
of the European Tissues and Cells regulatory framework, 06 February 2007.

18 | P a g e
absent oversight functions for BTC, such as an established competent authority,
inspections and authorisation activities. Vigilance programmes were not in place and
many had to upgrade facilities, equipment and quality management in their tissue and
cell services to meet the technical requirements for donation, testing, processing,
preservation, storage and distribution. At least three Member States (Romania, Bulgaria
and Lithuania), also had to change their donor base from paid or replacement donation
53
to VUD
54
,
55
.
Annex V provides more information on the situation prior to the adoption of the BTC
legislation.
53
The term replacement donation refers to systems where there is a request/obligation on the family of a patient
needing transfusion to donate blood in order to replace the blood used from the bank. This system is not considered
compatible with VUD and is considered to put too much pressure on donors and to risk untruthful donor risk
information.
54
WHO Europe (2007) Blood Services in South Eastern Europe – Current Status and Challenges.
55
Skarbalienė A Bikulčienė J (2016) Motivation and retention of voluntary, non-remunerated blood donors. Lithuanian
case. Health Policy and management 2016, 1(9).
19 | P a g e
3. STATE OF PLAY
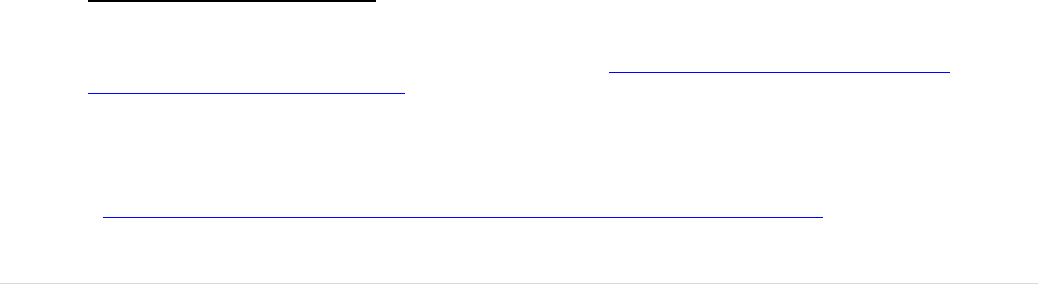
3.1 The BTC Sector
Blood, tissues and cells are collected, processed, stored, tested and supplied for human
application by 1,400 blood establishments
56
and over 4,000 tissue establishments
57
. The
blood sub-sector is broadly divided into blood or blood components for transfusion and
plasma for the manufacture of medicinal products. Tissue establishments are broadly
divided in three categories (see Figure 5).
FIGURE 5: THE THREE CATEGORIES OF TISSUE AND CELLS ESTABLISHMENTS
Figure 6 provides aggregated data for the BTC provided by BTC establishments in the
EU. Most blood establishments are national, regional or hospital based and are managed
by the public health sector or by non-profit organisations such as the Red Cross, while
the private sector plays an important role in the collection of plasma for the manufacture
of plasma derived medicinal products. Tissue establishments include banks of corneas,
bone, skin, heart valves, bone marrow and cord blood for transplantation, as well as
sperm banks and clinics for medically assisted reproduction (MAR). Tissue
56
Implementation report published in 2016.
57
See Tissue Establishment Compendium hosted by the European Commission.

20 | P a g e
establishments providing tissues and cells for transplantation are mostly public or non-
profit organisations while sperm banks and MAR clinics are both public and private.
Tissues and cells can also be used as starting materials for medicinal product
manufacture.
FIGURE 6: THE SCALE OF BTC ACTIVITY IN THE EU (DATA FROM 2018 OR MOST RECENT YEAR REPORTED)
Whole blood and blood components, such as platelets and red blood cells, have a limited
shelf life and are rarely exchanged between Member States, with the exception of
emergency or humanitarian situations. Plasma and plasma derived medicinal products
have a longer shelf life and, as plants manufacturing plasma derived medicinal products
exist only in twelve Member States, both plasma (the starting material) and the end
products are frequently exchanged across borders, within the EU and with third
countries. The EU is significantly reliant on importation of plasma from the United States
to meet the needs of patients in the EU for plasma derived medicinal products. The level
of inter-Member State exchange and global movement in general, varies depending on
the type of substance of human origin.
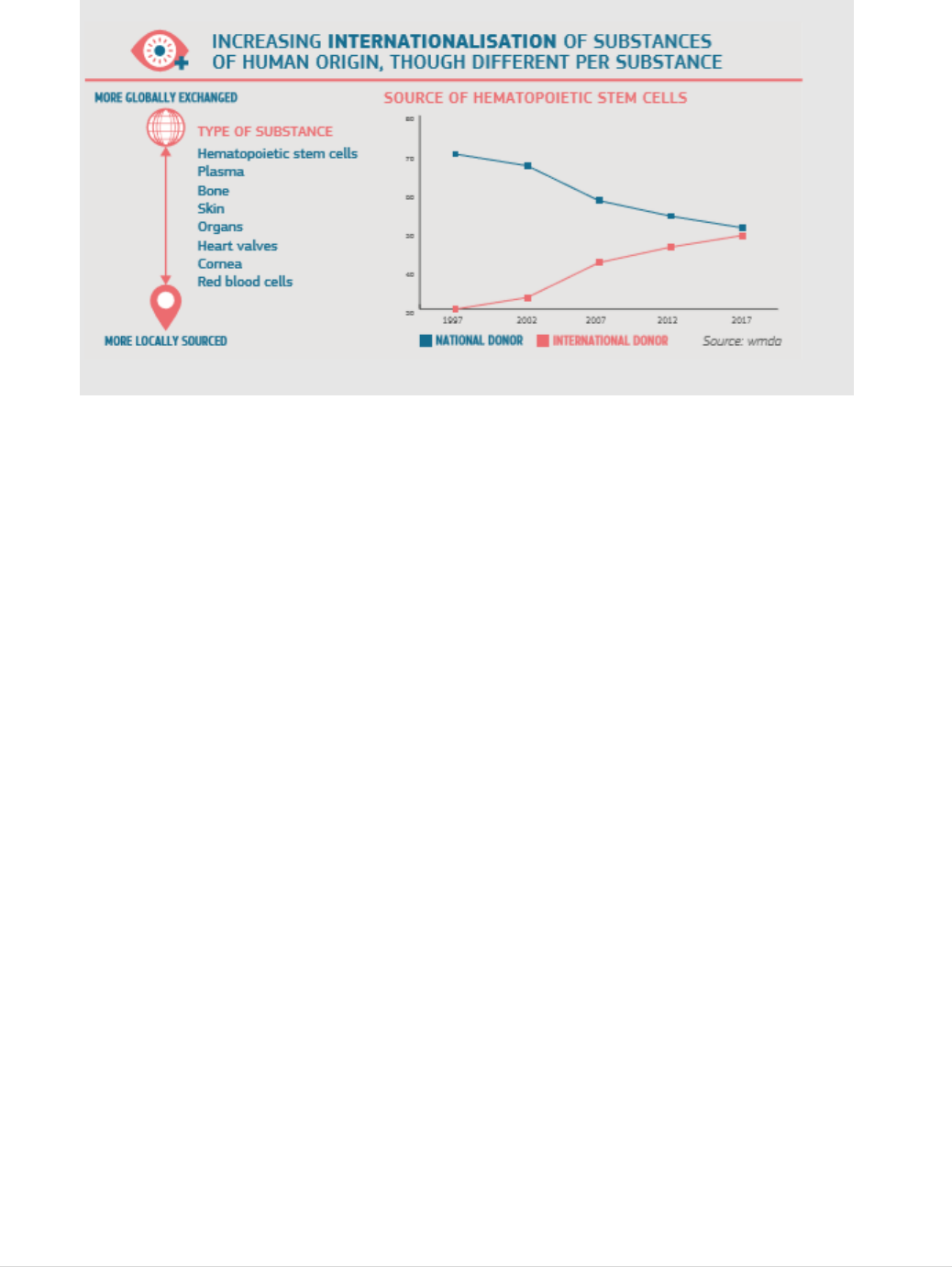
21 | P a g e
FIGURE 7: LEVELS OF INTERNATIONAL EXCHANGE VARY PER SUBSTANCES OF HUMAN ORIGIN
For tissues and cells, specifically, reports indicate significant volumes of human tissues
and cells being exchanged internationally (see Figure 7), with haematopoietic stem cells
(the stem cells in the bone marrow that make new blood cells) being the most exchanged
substance. It is difficult, however, to draw firm conclusions regarding the volume of
imports and exports of human tissues and cells due to the lack of mandatory reporting of
such information at national level and the absence of a harmonised framework for data
collection in the Member States. In addition, some Member States that do gather data on
a national level, and share it on a voluntary basis, do not distinguish between distribution
within the EU and import/export from/to third countries. It is clear, however, that large
quantities of bone and skin are imported from the United States, mostly from commercial
tissue banks, and that increasing numbers of bone marrow donations and other
haematopoietic stem cells circulate globally to facilitate high level donor to recipient
matching, a challenging requirement for successful transplantation in this sub-sector.
There is significant innovation in the sector, both in the way that BTC are processed in
establishments and the way they are used in patients. These innovative approaches are
commonly developed in the broad context of health service planning and they generally
improve patient access to new treatments.
3.2 Transposition and updating of BTC legislation
Most Member States transposed the basic Acts (2002/98/EC and 2004/23/EC) within the
defined deadlines in a complete and satisfactory manner. Between 2004 and 2006,
implementing Directives were adopted for both basic Acts (2004/33/EC, 2005/61/EC,
2005/62/EC for blood and 2006/17/EC and 2006/86/EC for tissues and cells). By 2008,
25 of the 27 Member States reported having transposed the basic Act on blood and all

22 | P a g e
three implementing Directives
58
. By 2009, 26 Member States had completely transposed
Directive 2004/23/EC, 2006/17/EC and 2006/86/EC.
As of February 2019, all current tissue and cell legislation, including more recent
implementing Directives
59
and Decisions
60
, has been transposed satisfactorily, with one
exception
61
. For blood, all legislation has been satisfactorily transposed
62
.
The powers given to the Commission to adapt technical requirements to scientific and
technical progress were used in a small number of specific cases
63
. In general, however,
the speed of technical development and of changing risks in the sectors proved
challenging and many changes are not reflected in the current provisions (see Section
5.1).
There have been some formal complaints to the Commission and a small number of court
cases linked to the legislation. The issues arising have mostly related to blood and blood
components. In particular, cases concerned restrictions applied at national level on the
purchase or import of plasma or plasma derived medicinal products. These restrictions on
imports were linked to the source of the plasma, with Member States that aim to achieve
sufficiency through VUD differentiating between plasma from unpaid donors and plasma
from donors compensated financially
64
.
The classification of certain substances as blood components or medicinal products has
also been the subject of legal discussions, largely resulting from different interpretations
of the scope of the basic act on blood and blood components (see Section 5.4).
Implementing Directive 2004/33/EC requires permanent deferral of prospective donors
whose sexual behaviour puts them at ‘high risk’ of acquiring severe infectious diseases
that can be transmitted by blood. The interpretation of ‘high risk’ led most Member
States to apply permanent exclusion of prospective male blood donors who have sex with
men (MSM). This led to complaints and one national court case that was referred to the
Court of Justice of the EU
65
. The Court ruled that permanent deferral was not a
58
Responses to Commission survey on the implementation of the blood legislation.
59
Blood: Directive 2016/1214 amending Directive 2005/62/EC on good practice guidelines; Tissues and Cells:
Directive 2015/566 on tissue and cell import, Directive 2015/4460 amending Directive 2016/86/EC on the Single
European Code.
60
Decision 2010/453/EU on conditions of inspections.
61
One failure to transpose an amendment to Directive 2006/17/EC (Directive 2012/39/EU) that has been referred to the
European Court of Justice (ECJ).
62
Apart from the most recent amendment, Directive (EU) 2016/1214 amending Directive 2005/62/EC, where 27
Member States have notified transposition and the verification of completeness is ongoing.
63
Blood: Directive 2011/38/EU amending Directive 004/33/EU regarding platelet pH values; Directive 2009/135/EC
amending Directive 2004/33/EC to allow donor eligibility derogations for the Influenza epidemic; Directive
2004/110/EU amending 2004/33/EC to allow testing for West Nile Virus. Tissues and cells: Directive 2012/39/EU
amending Directive 2006/17/EC to update HTLV and ART partner donor testing provisions.
64
Some Member States consider financial compensation as payment and, therefore, as inconsistent with the principle
of VUD described in Article 20 of the basic Act and aim to achieve national sufficiency through unpaid donation only.
Efforts to restrict national use of derived medicinal products to those manufactured from unpaid donors have been
challenged by industry that claims this approach contravenes the free market rules in place for medicinal products.
65
C-528/13, Geoffrey Léger v. Ministre des Affaires sociales, de la Santé et des Droits des Femmes et Établissement
français du sang.
23 | P a g e
proportionate measure. Many Member States have since amended their national rules to
accept MSM donors after one year, or less, since the last exposure to risk.
A full list of court cases, with brief descriptions, is at Annex VI.
The European Parliament also raised questions, mostly concerning the same issues as
those arising for blood in court cases. It also addressed issues such as plasma supply,
VUD and access to BTC treatments. A full list of Parliamentary Questions of relevance
to the BTC legislation is provided at Annex VII.
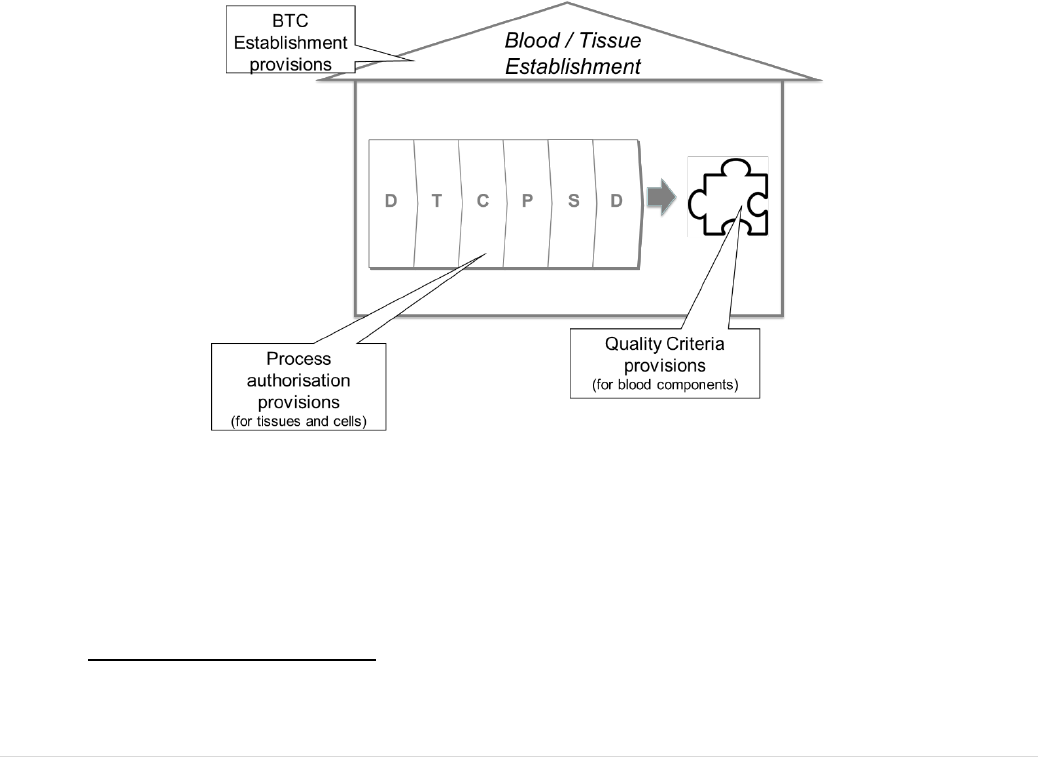
3.3 Oversight functions at Member State level
All Member States have designated one or more competent authorities to carry out the
oversight obligations of the BTC legislation
66
.
The types of organisation designated for
this role vary significantly (see Section 5.2.1.2). The competent authorities inspect and
accredit, designate, authorise or license the blood and tissue establishments. For tissues
and cells, this authorisation is complemented by provisions for authorisation of the
processes applied to the donations at the tissue establishment. For blood, on the other
hand, it includes verification of compliance with defined quality criteria for each blood
component prepared (Figure 8).
FIGURE 8: AUTHORISATION OF ESTABLISHMENTS AND PROCESSES.
3.4 Monitoring Arrangements
66
Study supporting the BTC evaluation, ICF, page 64.

24 | P a g e
Implementation reports have been compiled by the Commission over the years, based on
questionnaires completed by Member State competent authorities. The most recent
reports were published in April 2016
67
,
68
. The gaps and shortcomings identified have
been explored in the evaluation and are described in Section 5 of this report.
Vigilance and surveillance programmes are one of the cornerstones of the safety and
quality framework, allowing the identification and detection of risks and the application
of corrective and preventive measures. Since 2008, in line with obligations defined in the
blood legislation
69
, the EU Member States and Iceland, Liechtenstein and Norway have
submitted to the Commission annual vigilance reports. The reports notify serious adverse
reactions (SAR) which occur in recipients of blood and blood components and serious
adverse events (SAE) which occur in the chain from donation to clinical application,
posing a risk of harm. An equivalent provision is included in the tissue and cell
legislation
70
. The Commission, in turn, must send to the competent authorities of the
Member States a summary of the reports received. Definitions for SAR and SAE are
provided in the legislation. The Commission has been working with the BTC competent
authorities over several years to standardise data collection procedures and to improve
both accuracy and comparability of the information submitted. The Commission provides
the Member States with a template, and guidance for its completion, and the summary
reports are published annually
71
. Serious adverse reaction (SAR) reports (per number of
recipients) have stayed relatively stable and low since the adoption of the Directives
(0.03 - 0.05 SAR per recipient for blood and 0.04 - 0.01 for tissues and cells). It is
reported that legislative provisions for vigilance and traceability have helped to prevent
harm to recipients
72
.
The BTC legislation obliges Member States to report to the Commission on a regular
basis on measures taken to encourage VUD and the Commission must inform the
European Parliament and the Council of any necessary further measure it intends to take
at EU level
73
. The Commission has fulfilled this obligation via a questionnaire survey of
Member States on the implementation of the principle of VUD for blood and blood
67
Report from the Commission on the implementation of the Directives 2002/98/EC, 2004/33/EC, 2005/61/EC and
2005/62/EC, 21 April 2016 (Blood);
Report from the Commission on the implementation of Directives 2004/23/EC, 2006/17/EC and 2006/86/EC (tissues
and cells), 21 April 2016.
68
Report from the Commission on the implementation of the Directives 2002/98/EC, 2004/33/EC, 2005/61/EC and
2005/62/EC, 21. April 2016 (Blood).
69
Article 8 of Directive 2005/61/EC provides that Member States shall submit to the Commission an annual report, by
30 June of the following year, on the notification of serious adverse reactions and events (SARE) received by the
competent authority using the formats in Part D of Annex II and C of Annex III.
70
Article 7 and Annexes III, IV and V of Directive 2006/86/EC.
71
The most recent published reports: Summary of the SARE Report for blood for 2017 and Summary of the SARE
Report for tissues and cells for 2017.
72
ICF study, pages 72-73.
73
Article 20 of Directive 2002/98/EC and Article 12 of 2004/23/EC.

25 | P a g e
components and for tissues and cells. The most recent surveys were conducted in 2014
and published in 2016
74
.
The principle of VUD for blood and blood components is recognised in all Member
States, albeit interpreted differently across the EU. It is common practice to provide
refreshments to donors (27 countries) and to give them small tokens such as pin badges,
pens, towels, t-shirts and mugs (24 countries). In around half of the Member States,
donors have their travel costs reimbursed and get time off work in the public and private
sector. In some Member States, donors receive a fixed payment that is not directly related
to actual costs incurred
75
; this is most common in those countries where large quantities
of plasma is collected by private sector companies.
For tissues and cells, the legislative provision also requires encouragement of VUD but is
slightly different to that for blood, allowing ‘compensation’, including for inconvenience.
Although the principle of VUD is mandatory in the large majority of the Member States,
its practical application varies across the EU. Many Member States allow payment of
sperm and egg donors at national level, considering it as compensation.
3.5 Support for implementation
A number of activities and initiatives have supported the implementation of the BTC
legislation since its adoption. The Expert Group of competent authorities responsible for
substances of human origin (CASoHO E01718) meets with the Commission in three
configurations, blood, tissues and cells and organs, once to twice a year each. The
meetings provide a useful forum for increasing standardisation and for joint working
76
.
The EU Public Health Programme has supported the implementation of the BTC
legislation through Joint Actions, Projects and Service Contracts. The outputs have
focused on those areas where legislation has been more challenging to implement or is
most open to interpretation
77
.
Expert support is also provided by the European Centre for Disease Prevention and
Control (ECDC), which was established in 2005 (after the adoption of the two basic BTC
Acts), which has access to the BTC Rapid Alert platforms
78
, and by the Council of
Europe, in particular through a series of Public Health Programme funded Grant
Agreements with defined areas of collaborative work
79
. Although not referenced in BTC
legislation (with the exception of the Council of Europe’s Good Practice Guidelines
74
Commission Staff Working Document on the implementation of the principle of voluntary and unpaid donation for
human blood and blood components and Commission Staff Working Document on the implementation of the principle
of voluntary and unpaid donation for human tissues and cells.
75
Notably Germany, Austria, Hungary and the Czech Republic.
76
A full list of interpretation issues raised in the group and their outcome is summarised at Annex VIII.
77
A full list of Public Health Programme actions in the BTC field is shown at Annex IX.
78
A full list of ECDC working in this field since 2012 is provided at Annex X.
79
Annex XI summarises the work of the Council of Europe in this field and lists the areas of formal collaboration
between DG Sante and the Council of Europe.

26 | P a g e
(GPG)
80
for Blood Establishments that are specifically referenced), the advice and
guidance of both bodies is widely used by BTC professionals and authorities.
4. EVALUATION METHOD
4.1 Conducting the Evaluation
4.1.1 Evaluation Roadmap
A Roadmap was published on 17 January 2017 for a four-week period and the feedback
received from 16 stakeholders was published
81
and taken into account in the conduct of
the evaluation.
4.1.2 Stakeholder Consultation
Stakeholder consultation was one of the key sources of evidence that was used to support
this evaluation. The level of stakeholder engagement in these activities was high and all
the stakeholders mapped in the Roadmap were reached, either through the public events
or by targeted activities (see Annex XII).
A Public Consultation was launched on 29 May 2017 and ran until 14 September 2017.
Submissions were received from 158 organisations and 43 citizens. A summary of the
outcome, together with the individual submissions, was published
82
. A Stakeholder Event
was held on 20 September 2017 in Brussels. The event attracted a high level of interest,
with over 200 stakeholders attending. A summary of the issues raised was provided in a
published report
83
.
Targeted Consultation was organised to fill any gaps in terms of stakeholders consulted
and to explore certain emerging issues in more depth.
Bilateral Meetings with key stakeholders allowed focused/specific input through
direct interaction. Meetings with relevant EU agencies and with third country
BTC Regulators were also held. Summary minutes were published on the DG
SANTE website.
Multi-lateral topic-specific meetings with selected stakeholders, including donor
and patients associations, industry and professionals working in the sector
84
80
Good Practice Guidelines in the Guide to the Preparation, Use and Quality Assurance of Blood Components 19th
Edition.
81
DG SANTE website.
82
Summary of Responses to the Public Consultation for the Evaluation of the Blood, Tissues and Cells Legislation by
the European Commission 2018.
83
Summary of the Blood, Tissues and Cells Stakeholder Event 20 September 2017.
84
https://ec.europa.eu/health/blood_tissues_organs/consultations/call_adhocstakeholdermeeting_en.

27 | P a g e
together with Member State authorities, were held when more in-depth analysis
of key emerging issues was required. These meetings, and the stakeholders
involved, are listed in Annex XII. Summary minutes were published on the DG
SANTE website.
A Synopsis report of Stakeholder Consultation activities and results is provided at Annex
XII. Numerous stakeholders were also engaged in the activities carried out as part of the
external study described below.
4.1.3 External Study
An external study
85
was commissioned to support this evaluation. This study was largely
desk-based, with a review of over 300 documents, including reports provided by the
Commission, relevant published literature and documents developed by other bodies
(professional associations, international organisations etc.). The contractor also organised
focus groups to address specific topics (including on medically assisted reproduction and
on coherence with other legal frameworks), interviews with experts and targeted surveys
to enhance their evidence gathering and analysis. The outcomes of the consultation
activities organised by the European Commission were also provided as material for use
in the study.
4.2 Limitations and robustness of the evaluation findings
Much of the evidence for the answers provided in this evaluation is considered robust.
There is a rich literature, as evidenced by the bibliography in the external study
supporting the evaluation, and stakeholders, from all sub-sectors, were motivated to share
detailed information and opinions. Many of the issues raised are well documented, either
in Commission monitoring reports, in professional publications or in technical meetings
with stakeholders, organised in the context of the evaluation. The same gaps and
shortcomings raised could be verified from multiple sources.
There were three important limitations to the evidence gathering exercise. Firstly, data
available on the situation in Member States prior to the adoption of the legislation, with
quantified indicators that could be compared to the current situation was limited. This
made measuring the impact of the legislation in a quantitative manner more challenging.
This was exacerbated by the heterogeneous situation across the EU at the time of
adoption in terms of measures already in place and administrative capacity for oversight.
A second important limitation was related to quantifying the costs incurred by the BTC
legislation for the assessment of efficiency. The majority of stakeholders impacted by the
legislation are working in public sector hospitals, clinics and centres often carrying out
85
Conducted by ICF Consulting and published together with this report.

28 | P a g e
many other functions apart from BTC activities. In this setting, typically, costs are poorly
quantified and are often not allocated specifically to the activities affected by this
legislation. Indeed, economic studies conducted for DG-SANTE in both main sub-sectors
were greatly limited by the low cost-awareness of these organisations and the scarcity of
robust cost data related to BTC-specific aspects of healthcare
86
. This factor was also
identified as a constraint by the contractor conducting the external study
87
. Where BTC
service provider costs are available, they relate to the full costs e.g. collection, testing and
processing of blood, without separating those elements that were required by the EU
legislation. Significant cost increments were incurred over the years since the adoption of
the EU legislation because measures were introduced when new technologies, such as
microbial inactivation processes, became available and were implemented despite not
being mandated by the EU legislation. The implementation of these measures made
service provision more expensive but the degree to which these costs were related to EU
legislation is often limited. Additionally, many regulatory costs are the result or more
stringent requirements introduced at a national level, such as more stringent donor testing
requirements, that are not separately identified by stakeholders.
A third limitation related to the limited reliable data available on donation rates, clinical
demand, sufficiency of supply and the extent of cross-border and third country
exchanges, particularly for tissues and cells. Some data from vigilance reporting
denominators was used, together with data gathered in an external study but the
completeness and coherence of the data is not fully reliable and this made it difficult to
assess the impact of some differences between Member States and confirm the degree of
Union sufficiency for some BTC.
Given these limitations, it was challenging to assess the overall regulatory impact and the
extent to which costs were proportionate to benefits. For the baseline situation,
documents published to support the Commission proposal were used for blood and, for
tissues and cells, a short retrospective survey of Member State competent authorities was
carried out. For efficiency, a qualitative approach was adopted. Specific concerns on
cost-effectiveness, as raised by stakeholders in the Public Consultation and by BTC
authorities, were listed. The efficiency analysis focused on those topics, where there was
a clear message that the benefits achieved by the provisions did not justify the costs
accrued.
86
Creative Ceutical Report, revised by the Commission to include stakeholders’ comments;
Economic landscapes of human tissues and cells for clinical application in the EU- Final Report Rathenau.
87
Study supporting the BTC evaluation, ICF, page 98.

29 | P a g e
5. ANALYSIS AND ANSWERS TO THE EVALUATION
QUESTIONS
This Section provides answers to the questions defined in the Roadmap for this
evaluation
88
under the European Commission’s standard five evaluation assessment
criteria.
5.1 Relevance
The legislation is not up-to-date with scientific, technological,
epidemiological and societal developments and is not flexible enough to
adapt provisions to such changes as they emerge.
The relevance assessment criterion was addressed by evaluation question 1, which was
sub-divided in 3 sub-questions.
5.1.1 Evaluation question 1a): To what extent is the legislation sufficiently
adapted to, adaptable to, and up-to-date with scientific, technical and
epidemiological developments / innovation?
SUMMARY ANSWER: Significant scientific and technological developments
have taken place since the legislation was adopted and many new epidemiological
risks have emerged. Despite some minor amendments to update provisions in line
with changing risks, legislation has not addressed most of the changes. As a result,
some potential safety and quality measures are not appropriately updated in the
legislation and, due to more stringent measures adopted by Member States to
address this, the legislation is no longer consistently applied. The consequence is
an insufficient achievement of the outcome planned for objective 1 (safety and
quality) or of the outcome of objective 3 (harmonisation).
5.1.1.1 The legislation has not kept pace with science and technology
The most significant scientific and technological developments from the point of view of
the BTC legislation fall under four main areas
89
:
88
Evaluation of Union legislation on blood, tissues and cells-Evaluation and Fitness Check (FC) Roadmap by the EC.
89
Additional, less significant, topics where developments have rendered the BTC legislation outdated, are addressed in
the Study supporting the BTC evaluation, ICF, Table 8, page 30.
30 | P a g e
New ways of testing for viruses that can be passed from donors to recipients
(transmissible viruses): Testing for transmissible viruses by more sensitive
technologies is now widely available and brings increased safety
90
. The BTC
legislation names specific infectious agents and the precise methodologies to be
used for testing
91
, which may no longer guarantee adequate protection. Many
Member States have therefore introduced more stringent testing requirements at
national level
92
, which may act as a barrier to exchange between them.
Significant developments in testing for genetic diseases: Extensive genetic
screening of gamete (sperm and egg) donors is now feasible, using panels of more
than 100 genes, and genetic testing of embryos prior to implantation is also
possible. Current legislation includes a generic provision for screening for genetic
conditions known to occur particularly frequently in the donor’s native
population, but no specific genetic testing strategies are defined for the gamete
donor population
93
. Thus, although genetic disease transmission is the primary
risk for medically assisted reproduction (MAR)
94
, and the consequences of a
serious transmission can have an impact on multiple children
95
, there are
divergent levels of safety across EU Member States with regards to donor
screening
96
.
Much innovation in BTC processing methodologies: This is true both for
blood, where many new blood components have been introduced into routine use,
and for tissues and cells. Pathogen reduction during blood processing, for
example, is now required by national legislation for some components in some
Member States
97
. For tissues and cells, each sub-sector has introduced innovative
processing and preservations methods. The blood Directive functions through a
precise quality specification for each individual blood component
98
, which no
longer ensure the safety and quality of components that are not included in the
90
W. K. Roth, M. P. Busch, A. Schuller et al. (2011) International Survey on NAT testing of blood donations:
expanding implementation and yield from 1999 to 2009. Vox Sang 102(1).
91
Directive 2002/98/EC Annex IV and Directive 2006/17/EC Article 4 and Annexes II and III.
92
See also the “Mapping of More Stringent Tissues and Cells Donor Testing Requirements - Mapping Exercise 2015”
and the "Mapping of More Stringent Blood Donor Testing Requirements - Mapping Exercise 2015" which showed that
the level of viral safety achieved across the EU is no longer standardised.
93
Directive 2006/17/EC Annex III 3.6.
94
Of the rapid alerts communicated between Member States in 2017, 15 of the total 18 alerts concerned genetic
conditions detected in a gamete donor or in a child born from donated gametes.
95
Danish Sperm Donor passed neurofibromatosis to five children BMJ 2012; 345:e6570.
96
For recent scientific studies recommending up-to-date testing approaches, see Harper JC et al. Recent developments
in genetics and medically assisted reproduction: from research to clinical applications. Eur J Hum Genet (2018) 26:12–
33; Henneman L et al. Responsible implementation of expanded carrier screening Eur J Hum Genet (2016) 24(6):e1-
e12; Edwards JG et al. Expanded carrier screening in reproductive medicine—points to consider. Obstet Gynecol.
2015; 125:653–62. See also Study supporting the BTC evaluation, ICF, page 49.
97
Pathogen reduction is a new technology that reduces the risk of viral and bacterial transmission by blood
components; See: Minutes of a meeting between the European Commission, Member State blood authorities and
stakeholders.
98
Directive 2004/33/EC Annex V: Quality and safety requirements for blood and blood components (as referred to in
Article 6).

31 | P a g e
original set of specifications
99
. The tissues and cells directive is more flexible, by
focusing on the authorisation of preparation processes at national level
100
, but
provisions are generic and lacking in requirements for the demonstration of
positive outcomes for patients
101
.
Novel clinical practices create new categories of patients and donors not
covered by existing legislation: Thus, for example, the storage of reproductive
tissue or gametes for later use by the same patient
102
does not fall easily into the
categories of “partner” or “non-partner” donation
103
, and is thus not adequately
regulated under the current safety provisions of the Tissues and Cells Directive
104
.
A number of other developments in clinical application of substances of human
origin have emerged and are seen as inadequately regulated (see Section 5.1.3 on
legislative gaps below).
5.1.1.2 The legislation has not kept pace with changing epidemiology of diseases
transmissible by BTC
Increased human travel, migration and global warming have contributed to substantial
changes in the risks of transmission of emerging infections by BTC in the EU, along with
other environmental and social factors. The infectious risks are addressed by testing or
donor deferral rules in the legislation. Since the BTC legislation was adopted, the sector
has faced risks from both viruses and parasites that were not present, or were present at
much lower levels, in the early 2000s
105
. Chikungunya, Ebola, Zika and hepatitis E
106
,
among others, have emerged as threats at different time points. Provisions in the current
blood legislation are quite specific and aimed at mitigating the risks that were current at
the time it was adopted. For example, tattooing posed a significant risk of infection with
viruses in the past but is now usually regulated and linked to very low risks of viral
infection.
99
There are 18 component specifications in Directive 2004/33/EC Annex II but 38 equivalent blood component
monographs in the regularly updated Council of Europe Guide to the preparation, use and quality assurance of blood
components 19th Edition.
100
Directive 2006/86/EC Annex II.
101
Ad-Hoc Meeting between Stakeholders and representatives members of the Competent Authorities on Substances of
Human Origin Expert Group (CASoHO E01718), 22 February 2017.
102
Oktay K (2017) Fertility Preservation in cancer patients. Oncology Times 39(4):1-8; Lallemant C Vassard D Nyboe
Andersen A et al. (2016) Medical and social egg freezing: internet-based survey of knowledge and attitudes among
women in Denmark and the UK Acta Obstet Gynecol Scand 95(12):1402-1410; Andersen CY and Kristensen SG
(2015) Novel use of the ovarian follicular pool to postpone menopause and delay osteoporosis Reproductive
Biomedicine 31, 128-131.
103
The term ‘partner donation’ in the tissue and cell legislation is defined in Directive 2006/17/EC as ‘the donation of
reproductive cells between a man and a woman who declare that they have an intimate physical relationship’. Non-
partner donation refers a donation by a person outside such a relationship.
104
Reference to supporting passages in external study.
105
In 2011, nine diseases transmitted by arthropods, were identified as posing an urgent communicable disease threat
to the safety of BTC in the EU, most of which were not addressed in the detailed provisions of the Directive
2004/33/EC. Report of an ECDC Expert Meeting.
106
ECDC has reported a 10-fold increase in hepatitis E infection from 2005 to 2015. One death from transfusion
transmitted hepatitis E was reported in the European Commission’s annual Blood SARE report 2016.
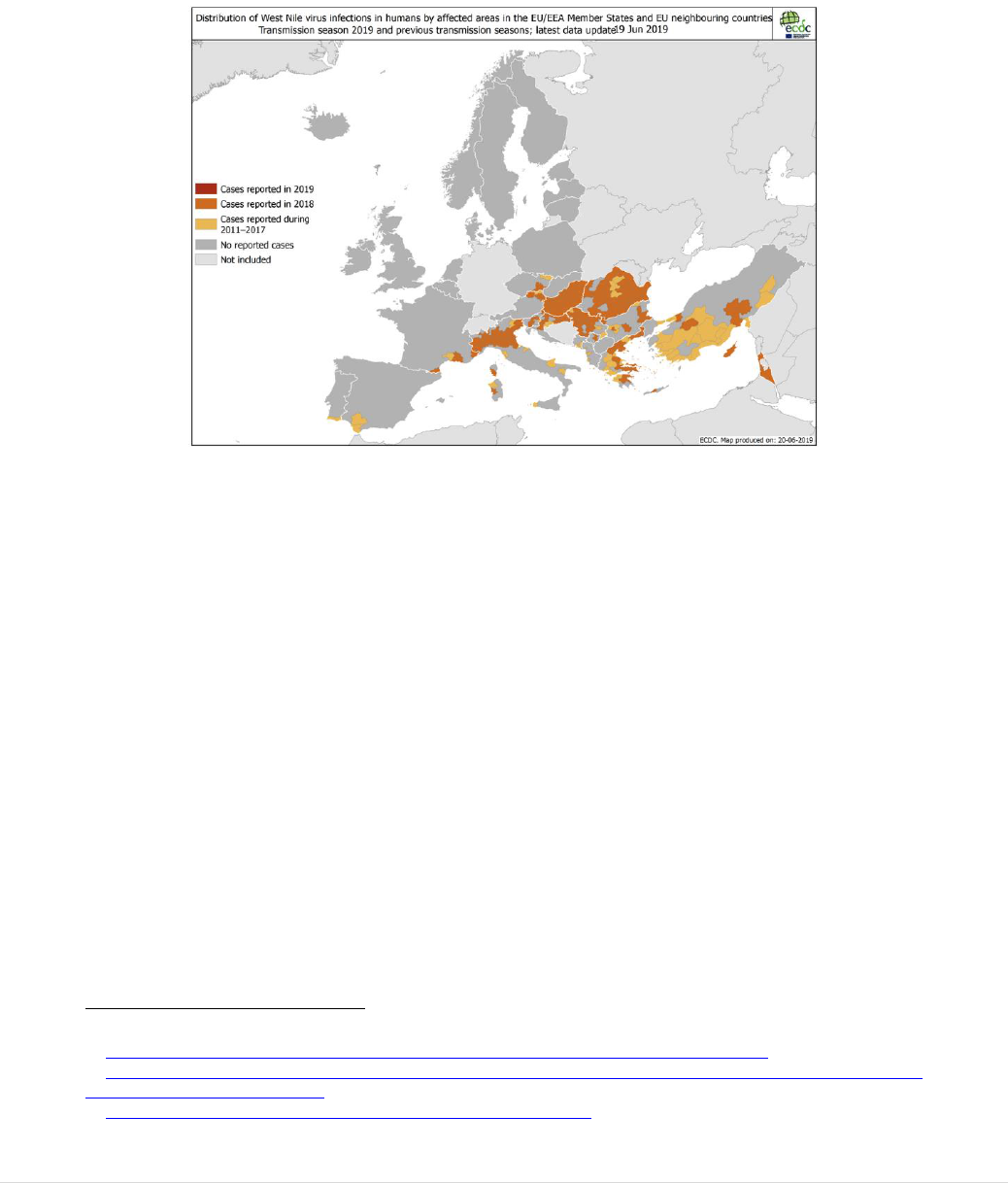
32 | P a g e
The example of West Nile virus (WNV): WNV has been in the EU since the early
2000’s, the distribution and number of cases reported has increased steadily as shown on
maps published by ECDC (see Figure 9). By 2014, validated WNV testing was available
and Directive 2004/33/EC was amended
107
to allow testing and avoid serious blood
shortages in those Mediterranean countries most affected.
FIGURE 9: INCREASING WEST NILE VIRUS THREAT FROM 2011 TO 2019
The amended provision called for the safest option by requiring individual donor sample
testing for WNV. Subsequently, it was concluded that the additional cost of applying the
revised provision was not robustly justified by the science and could be replaced with
testing in small pools of samples, with significantly lower cost implications (the added
cost of individual sample testing versus mini-pool testing was estimated at circa 2 million
Euros if applied to affected EU Member States), that would be equally effective
108
. The
European Blood Alliance (EBA) now requests a further update to the Directive.
Other examples include tattooing, endoscopic examination and acupuncture that
represented a significant risk of infectious disease transmission in the early 2000s but
have become safer and better controlled. The provisions are no longer considered by
many experts to reflect real risks and to cause, now, unnecessary donation losses
109
. The
EBA, expert committees of the Council of Europe and the main EU-level professional
associations all call for provisions with a more dynamic, risk-based approach to defining
best practice for donor selection
110
.
107
Directive 2014/110/EC.
108
Meeting of blood stakeholders, Member State Competent Authorities and DG SANTE in 2016.
109
Blood, tissue and cell donor selection criteria report: 2017 from The Advisory Committee on the Safety of Blood,
Tissues and Organs (SaBTO), UK.
110
European Blood Alliance- EBA fact sheet on Blood Donor Selection..

33 | P a g e
5.1.1.3 Challenge of responding effectively to frequently changing disease
transmission risks
There are two approaches to responding to these changing risks.
Regular updates of detailed legislative provisions: Both basic Acts empower
the Commission to lay down technical requirements and adapt them to scientific
and technical progress, through the adoption of autonomous acts and following
consultation of committees of Member State representatives
111
. The basic Acts
also provide for the use of an urgency procedure for amending measures relating
to donor eligibility and testing. These empowerments have been used by the
Commission to develop the technical Directives
112
, to make changes to these
technical provisions
113
and to address an urgency
114
. However, considering the
level of detail in the legislation and the frequency of these changes, often month
by month, it has been challenging to keep the legislation up to date.
Authoritative risk-based guidance: ECDC addresses the risk of transmission of
communicable disease by substances of human origin, as they emerge, in a timely
manner, with recommendations on testing and donor deferral (see Annex X and
published risk assessments
115
). However, reflecting the fact that ECDC did not
exist at the time the basic BTC Acts were adopted, there is no obligation in the
current legislation to take ECDC recommendations into account.
On this topic, there is a general call across professionals working in BTC establishments,
as well as the authorities that regulate them, for less detail in legislation, with clear
references to authoritative and regularly updated guidance to be taken into account when
setting rules for donor acceptance and testing.
5.1.2 Evaluation question 1b) To what extent is the legislation adapted to
other changes in the sector such as commercialisation, internationalisation or
other societal changes?
SUMMARY ANSWER: There have been important societal changes since the
legislation was adopted, with an increased level of commercialisation and
international exchange. The legislation was adopted at a time when the services it
regulates were largely organised from within local public health services and
commercial interests were limited. As a consequence, the clinical effectiveness and
donor protection provisions no longer address the risks associated with these
activities in a comprehensive manner, both for donors and for recipients (objective
1, safety and quality), or the sufficiency of supply (objective 5).
111
Referred to as the ‘Comitology’ procedure – see Articles 28 and 29 in both basic Acts.
112
Directives 2004/33/EC, 2005/61/EC, 2005/62/EC, 2006/17/EC, 2006/86/EC, (EU) 2015/566.
113
Directives 2011/38/EU, 2012/39/EU, 2014/110/EU, (EU) 2015/565, (EU) 2016/1214.
114
In response to a risk of blood shortages due to the Influenza A (H1N1) pandemic Directive 2009/135/EC.
115
See regularly updated Zika Virus Rapid Risk Assessment and other ECDC risk assessments.

34 | P a g e
5.1.2.1 Provisions in BTC legislation do not address risks associated with increased
commercialisation adequately
Article 12 of the basic Act for tissues and cells requires Member States to endeavour to
ensure that the procurement of tissues and cells is carried out on a non-profit basis. It also
requires Member States to provide guidelines restricting or prohibiting advertising the
need for or availability of human tissues and cells with a view to offering or seeking
financial gain or comparative advantage. The Act also includes a recital that calls for
Member States to encourage a strong public and non-profit involvement in the sector
116
.
The BTC Directives do not, however, prohibit the participation, per se, of the private
sector in the processes from donation to supply for clinical use. Although the commercial
sector already played a significant role in plasma collection and in the running of in vitro
fertilisation (IVF) clinics and sperm banks at the time that the legislation was adopted,
there has been a marked increase in commercial activity since then in many areas that
were previously predominantly, or entirely, run by the public sector. Annex XIV includes
examples of commercial BTC activities that have appeared, or significantly increased, in
all BTC sub-sectors since the legislation was adopted.
Under the TFEU, Member States are responsible for issues relating to the organisation of
health services. The issue of commercialisation is addressed, in this evaluation, only in
terms of any impact it might have on a sufficient supply of BTC at the required levels of
safety and quality. The impact of the increasing commercialisation described here
touches on three key safety, quality and sufficiency issues:
Unsubstantiated claims for clinical effectiveness: As the legislation has limited
provisions for demonstrating clinical effectiveness, commercial companies can
promote their products as ‘superior’ to the existing options for a particular clinical
application or as effective for a range of conditions, without a legal obligation to
justify such claims with robust clinical evidence. This could open the door to
increased safety risks with no corresponding benefit
117
.
Increasing commercial demand for donors: In the context of the limited
provisions in the legislation to protect and monitor donor health (see sub-Section
5.2.1.2), potential needs of commercial companies for increasing numbers of
donors could present risks to donor health that will not be adequately mitigated
by regulatory action. This risk is more important for donations that are more
invasive and imply greater risk to the donor, such as egg donation
118
.
Potential threats to the achievement of EU sufficiency of BTC: The
sustainability of supply might be threatened where the success of a commercial
116
Directive 2004/23/EC Recital 18.
117
This applies notably to the field of stem cells where claims for effectiveness are often unsubstantiated. See: Marks P
Witten C and Califf (2017) R. Clarifying Stem-Cell Therapy’s Benefits and Risks N Engl J Med 376; 11.
118
Pearson H (2006) Health effects of egg donation may take decades to emerge Nature 442: 607–608.

35 | P a g e
company causes the public sector to withdraw, and results in a high dependence
on one or a small number of commercial suppliers
119
that may choose to supply
only the most profitable BTC
120
. The blood supply might also be threatened when
potential blood donors are attracted away from blood donation by compensation
given for plasma donation in the commercial sector
121
. In addition, there is a risk
that commercial entities might allocate and supply the final processed donation to
limited EU Member States or to third countries, where potential profits are
highest, putting EU sufficiency at risk, even in circumstances where EU donation
rates are high.
Summarising the impact of increased commercialisation, it can be said that the
aspirations of the Directives in relation to the establishment of BTC services as public
health activities organised on the basis of patient need are reflected in limited provisions
that were not designed for the level of commercialisation now seen.
5.1.2.2. BTC legislation does not fully facilitate the internationalisation needed for
the supply of certain BTC
It is notable that around half of all bone marrow transplants
122
performed in the EU are
donated by donors in another Member State or a third country, provided via a global
network of national and regional registries of potential donors. These registries are
accredited by the World Marrow Donors Association (WMDA)
123
that matches donors to
recipients and supports the movement of these cells around the world, while monitoring
traceability and vigilance. This activity has grown rapidly over the years since the EU
legislation was adopted as shown in Figure 10.
119
Mannis MJ, Sugar J (2018) Is This the Future of Eye Banking? Cornea (editorial) Cornea 37(7): 811-812.
120
Fact Sheet published by the European Blood Alliance.
121
Fact Sheet published by the European Blood Alliance.
122
Bone marrow or other sources of blood forming stem cells known as haematopoietic stem cells (HPC).
123
https://www.wmda.info.
36 | P a g e
FIGURE 10: INCREASING GLOBAL DONATION OF HAEMATOPOIETIC STEM CELLS (HPC) FROM BONE
MARROW (HPC MARROW), PERIPHERAL BLOOD STEM CELLS (HPC APHERESIS) AND CORD BLOOD
(HPC CORDS)
124
FOR TRANSPLANTATION GLOBAL DISTRIBUTION.
This international distribution is needed for these cells that require a high level of donor
to patient matching. Significant efforts by professional associations developing
accreditation programmes have contributed to improving harmonisation of the safety and
quality of donations supplied via these registries
125
. The World Marrow Donors
Association (WMDA) has indicated that the current provisions leave a gap with regards
to the role of national and international registries. They also point to the need for a global
approach to vigilance in their field where rare events would not emerge without the
collation of global data
126
. This is hampered to some extent, by EU vigilance definitions
that are not standardised with definitions internationally.
The increasing demand for plasma derived medicinal products also currently relies on
effective international exchange. This is addressed in Section 5.2.6.1.
5.1.2.3 Other Societal changes impact on the relevance of the BTC legislation
There have been other significant changes in society that have rendered specific
provisions no longer suitable for achieving the objectives of the intervention. These
include aging of the population, increasing demands for medically assisted reproduction
124
Data presented at a meeting of Stakeholders with Competent Authorities for tissues and cells and DG SANTE in
February 2017.
125
For more, see: Website European Society for Blood and Marrow Transplantation- JACIE Standards.
126
Punzel M. et al. Detection of hepatitis b virus DNA in the blood of a stem cell donor after granulocyte colony-
stimulating factor treatment, Hepatology. 2016 Nov; 64(5):1803-1805. doi: 10.1002/hep.28667. Epub 2016 Jul 9.

37 | P a g e
(MAR) including across borders, the use of the internet for ordering of BTC and the use
of BTC for cosmetic surgery (e.g. breast or penis augmentation). The implications of
these developments for the relevance of the legislation are discussed in the external study
that is published in parallel with this report
127
.
Two issues that have caused particular concern are highlighted here.
Culturally unacceptable terminology: Different technical provisions for many
aspects of the chain from donation to clinical application vary between the
‘partner donation’ and the ‘non-partner donation’ scenarios in the context of
MAR
128
. While the scientific basis of the different rules is accepted as rational,
the term ‘partners’ as defined in the current legislation (‘the donation of
reproductive cells between a man and a woman who declare that they have an
intimate physical relationship’)
129
is questioned. It does not reflect the use of the
term to also describe couples of the same gender in today’s society.
Permanent exclusion from blood donation of men having sex with men:
Blood legislation that requires the permanent deferral from donation of persons
whose sexual behaviour puts them at high risk of acquiring severe infectious
diseases that can be transmitted by blood
130
. Although ECDC has confirmed that
sex between men is the main mode of HIV transmission in the EU
131
, the risks of
HIV and hepatitis C transmission by donations from men having sex with men
(MSM) are significantly reduced by the more sensitive tests available today.
Thus, the interpretation that MSM implies a ‘high risk of acquiring severe
infectious diseases’, and therefore requires permanent deferral from blood
donation
132
, is no longer widely accepted as justified
133
and is seen by many as
discrimination. Many Member States now implement the provision by a less
stringent deferral for a limited period of time since the last exposure to risk
134
.
Application of the provision in its strictest interpretation has led to a national
court case being referred to the Court of Justice of the EU, to a number of
complaints from citizens and to 16 questions from the European Parliament (see
Annex VII). The variability across the EU, creates particular difficulties for the
supply of plasma to medicinal product manufacturing companies working
internationally
135
.
127
Study supporting the BTC evaluation, ICF, page 36-39.
128
Notably, annexes 1 to 4 in Directive 2006/17/EC.
129
Directive 2006/17/EC.
130
Directive 2004/33/EC Annex III 2.1.
131
Special report: HIV and men who have sex with men, by ECDC.
132
Annex III of Directive 2004/33/EC.
133
Sturrock BRH and Mucklow S (2018) What is the evidence for the change in the blood donation deferral period for
high-risk groups and does it go far enough? Clin Med (Lond). 2018 Aug; 18(4): 304–307.
134
Minutes of the blood CA meeting of November 2015.
135
Summary Minutes of the Meeting of the CA for blood 10/11 October 2018.

38 | P a g e
5.1.3 Evaluation question 1c) Are there any gaps in terms of substances of
human origin or activities that are not regulated by the Directives?
Summary Answer: Important technical, clinical and societal changes have taken
place that have left gaps in terms of the regulation of some substances of human
origin, that could effectively be covered by the BTC legislation. In the Online
Public Consultation, around half of the respondents stated that they were aware of
substances of human origin or activities that fall in regulatory gaps. The main
consequence is that the substances are regulated in different ways across Member
States, compromising the achievement of harmonisation, or they are not regulated,
with the consequence that none of the other objectives is achieved for these
substances.
5.1.3.1 New substances of human origin fall under the mandate of the Treaty on the
Functioning of the European Union (TFEU) but outside the BTC scope
At the time the BTC legislation was adopted, the defined scope in the two basic Acts
covered most of the human substances that were being used therapeutically and for
which the TFEU gave competence to the EU to regulate safety and quality, apart from
organs which were addressed in a third basic Act in 2010. A number of therapies have
emerged, and are now in routine use, that fall outside the scope of the BTC legislation
although they are substances of human origin and they imply similar risks to those
regulated by it. Three examples are described here.
Blood components used for purposes other than transfusion: These are
excluded from the scope of the blood legislation
136
. Serum eye drops are used in
ophthalmology and platelet rich plasma (PRP), platelet rich fibrin (PRF), and
others are routinely prepared in hospitals and used in a range of surgery types.
137
Apart from CE marking, when the preparation involves the use of devices, there
is no current EU regulation of these therapies that ensures safety and quality.
Faecal Microbiota Transplantation (FMT)
138
is a rapidly growing therapy that
aims to repopulate the gut microflora in patients with Clostridium difficile
infection, following bone marrow transplantation or for other indications. In a
meeting with the Member State competent authorities, the view was shared that
FMT does not meet the definition of tissues or cells in Directive 2004/23, but
does fall within the Treaty mandate of substances of human origin
139
. A recent
Commission survey of the EU tissue and cell authorities indicated that 13
136
Directive 2002/98/EC Article 2.
137
PRP treatments, prepared with medical device kits, are currently valued at $100 million globally for the medical
device industry, with Europe accounting for 25%. This value is expected to rise to almost 300 million by 2025
according to market research data provided by Medtech Europe.
138
FMT is a rapidly growing therapy that aims to repopulate the gut microflora in patients with Clostridium difficile
infection, following bone marrow transplantation or for other indications.
139
Minutes NCA meeting Tissues and Cells, December 2014.

39 | P a g e
Member States do not regulate FMT, 5 regulate as a medicinal product (non-
ATMP), 2 regulate under tissue and cell legislation and 1 regulates under Food
legislation
140
. This issue is also addressed in the external study
141
.
Donated human breast milk has nutritional properties, it is also used to enhance
immunity in preterm infants where the mother cannot breastfeed. In this case,
also, a view was shared in the network of competent authorities that the substance
falls under the Treaty legal mandate for substances of human origin but not
within the definitions of tissues and cells in the current legislation
142
. Some
Member States apply the tissue and cell legislation, others the food legislation
and, in some cases, breast milk banking services are provided by blood banks in
the EU and abroad
143
,
144
. At least 11 Member States currently do not regulate the
activity while in others it is regulated under food, tissue and cell, or other
frameworks
145
.
These substances carry risks, including disease transmission
146
,
147
,
148
, that could be
mitigated by application of the rules in the BTC legislation. There is consensus among
the Member State competent authorities that they should be regulated by provisions
equivalent to those existing for BTC
149
. In some cases, requests for clarification have
been made to the Commission (see Annex VIII). These cases have also been raised in
other fora, including the Online Public Consultation and by the European Medicines
Agency
150
as well as in the scientific literature
151
,
152
,
153
. There is general consensus that
EU regulation is needed so that recipients of these treatments are protected in the same
way as BTC recipients. It is notable that the Guide to the Safety and Quality of Tissues
and Cells, published by the EDQM (Council of Europe) now includes guidance
154
for
most of these autologous substances
155
. The consequence of the current situation is
widely differing regulatory approaches in Member States, and no regulation in some,
compromising the achievement of increased harmonisation.
140
Tissues and Cells competent authority meeting minutes May 2019 with survey results.
141
Study supporting the BTC evaluation, ICF, page 55.
142
Minutes of competent authority meeting Tissues and Cells, December 2014.
143
For more, see: Héma-Québe Website- Public Mothers’ Milk Bank.
144
For more, see: Banc de Sang Website- Banco de Leche Materna.
145
Tissues and Cells competent authority meeting minutes May 2019.
146
U.S. Food& Drug Administration. Information Pertaining to Additional Safety Protections Regarding Use of Fecal
Microbiota for Transplantation..
147
Keim SA, Hogan JS, McNamara KA et al. (2013) Microbial contamination of human milk purchased via the
Internet. Pediatrics 132(5):e1227-35.
148
GL Buser, S Mató, AY Zhang et al. (2017) Late-Onset Infant Group B Streptococcus Infection Associated with
Maternal Consumption of Capsules Containing Dehydrated Placenta — Oregon, 2016 MMWR 66(25): 677-678.
149
Minutes of tissues and cells competent authority meeting May 2019.
150
Summary minutes of the meeting between EC and EMA Sept 2018.
151
MB Smith, C Kelly and EJ Alm (20 February 2014) How to regulate faecal transplants. For medical use, human
stool should be considered a tissue, not a drug Nature, 506: 290-291.
152
J Chisholm, B Vontigerstrom, P Bedford et al. (2017) Workshop to address gaps in regulation of minimally
manipulated autologous cell therapies for homologous use in Canada Cytotherapy 19: 1400–1411.
153
M Ratner (2014) Fecal transplantation poses dilemma for FDA. Nature 32(5):401-402.
154
This guidance is not binding in the EU or in Council of Europe Member States.
155
Guide to the quality and safety of organs for transplantation, EDQM.

40 | P a g e
5.1.3.2 The exclusion of tissues and cells taken from patients and returned to them
during the ‘Same Surgical Procedure’ leaves some processed substances
unregulated.
The basic Act for tissues and cells specifically excludes tissues and cells procured and
returned to the patient during the ‘same surgical procedure’
156
. At the time the legislation
was adopted, this exclusion aimed to avoid intervening in the practice of clinicians that
remove, for example, a piece of bone or a blood vessel from one part of the body of a
patient and use it to reconstruct another part of the body. The impact of this exclusion,
however, has been to leave a number of processes now carried out in hospitals and clinics
unregulated at EU level. An example is the separation of adipose cells from adipose
tissue by centrifugation, with return to the patient for reconstruction of tissue defects, or
for cosmetic purposes.
5.2 Effectiveness
This criterion was addressed by six evaluation questions. Questions 2 to 6 relate to the
achievement of safety and quality, with a level of harmonisation that facilitates inter-
Member State exchanges (Objectives 1, 2 and 3), while question 7 relates to the
achievement of sufficient supplies to meet patient needs (Objective 5). The achievement
of legal certainty (objective 4) is addressed in the coherence Section (5.4).
The evidence points to significant improvements in safety and quality of BTC
and improved human health protection. However, shortcomings in relation to
ensuring the safety of some citizen groups were identified, particularly BTC
donors, and provisions for sufficiency and oversight are not adequately
robust.
5.2.1 QUESTION 2: To what extent has the legislation increased the quality
and safety of blood and tissues and cells and achieved a high level of human
health protection?
SUMMARY ANSWER: In general, the legislation has achieved important
improvements in the safety and quality of BTC across the EU, with oversight
established in all Member States. There has been no major secondary spread of
disease by BTC since its adoption, despite a number of new emerging infectious
risks during this period, and there is evidence of overall trust in the sector.
156
Directive 2004/23/EC Article 2 (2a).

41 | P a g e
However, the oversight provisions are broad and generic, with the result that there
have been widely varying approaches to the set-up of regulatory oversight across
Member States. Thus, some competent authorities are fully independent of the
sector they regulate and have developed specialised expertise, while others risk
conflict of interest or have limited technical expertise.
Additionally, EU level vigilance monitoring does not provide comparable data
from Member States due to provisions that are not adequately clear and are,
therefore, implemented differently. Shortcomings are identified in the provisions
for health protection of some citizens, particularly BTC donors. Finally, provisions
for authorisation of processing facilities and processing methods are not sufficient
to ensure a high level of BTC quality and a demonstrably beneficial outcome in
patients.
As a consequence, the legislation cannot fully ensure achievement of objective 1
(safety and quality), 2 (effective oversight) and objective 3 (a degree of
harmonisation).
5.2.1.1 Significant improvements in Safety and Quality of BTC
In the Online Public Consultation, the great majority of stakeholders from all categories
of respondent, including blood and tissue establishments, authorities and industry,
considered the impact of the legislation to have been positive, increasing safety and
quality to some extent, or to a great extent (see Annex XII). A Special Eurobarometer
published in 2015 with responses from 27,868 EU citizens indicated a high level of
confidence of EU citizens in the safety of the systems that supply BTC; 81% would
accept being treated with one or more human substance
157
. The improvements are linked
particularly to the following consequences of the legislation.
Safety and quality rules and oversight are in place. Thus, blood and tissue
establishments that organise donation, procurement, testing, processing, storage
and distribution of BTC across the EU must comply with legally binding rules.
Member States have nominated competent authorities that verify this compliance
through a series of oversight functions including inspection, authorisation and
vigilance.
Disease transmissions from donors to recipients are at a very low level.
Despite the emergence and re-emergence of many infectious agents since the
adoption of the BTC legislation, the level of transmission by these secondary
routes has been kept to an extremely low or negligible number of isolated
cases
158
,
159
.
157
An earlier Eurobarometer specifically addressing blood donation and transfusion (2009, survey of 26,788 citizens)
indicated that 57% of respondents considered that blood transfusions were safer that 10 years earlier.
158
Annual EU Vigilance reports for BTC indicate that the risk of transmitting viral infection by transfusion in the EU is
now less than 0.000001.
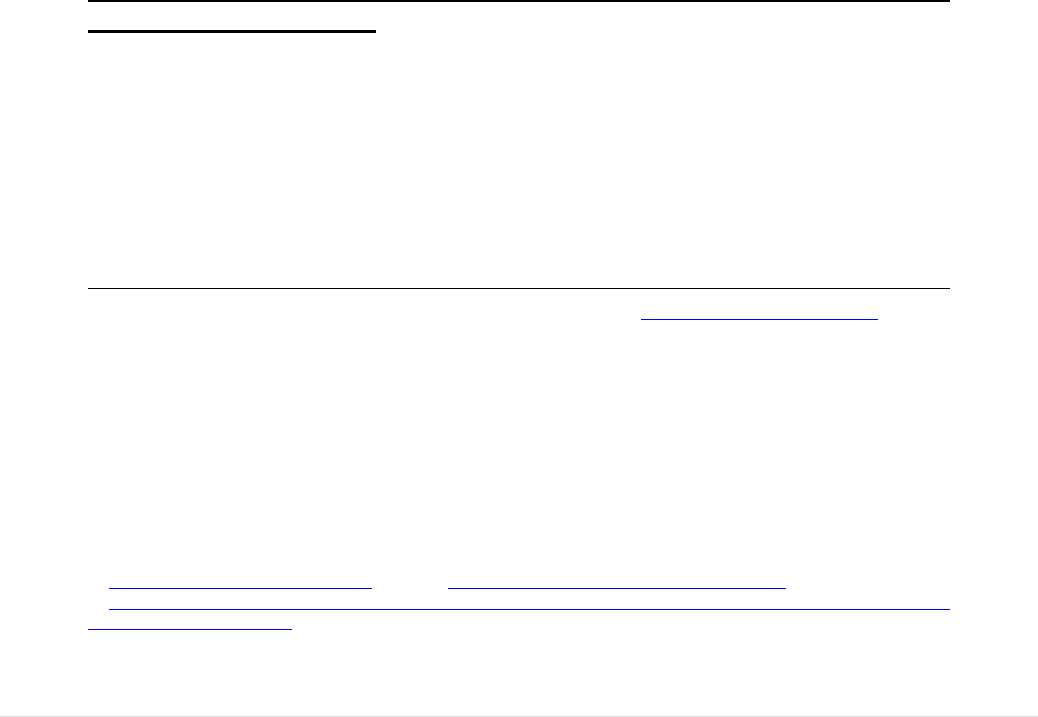
42 | P a g e
Quality Management is well established in blood and tissue establishments.
Risks to BTC recipients, other than disease transmission from donors, include
contamination with bacteria or fungi during donation, processing or storage,
cross-contamination from other donations, poor quality in relation to the
clinically required characteristics or loss of traceability and mix-ups. These risks
are reduced through the application of legally required quality management
160
.
The facilities in which blood and tissue establishments operate across the EU
have generally improved significantly
161
and the quality management rules have
been strengthened over time
162
. Member States report high levels of compliance
with the quality and safety rules, as verified through inspection and
authorisation
163
.
Rapid Alerts communicated between Member State authorities mitigate risk.
To facilitate Member States in effectively communicating rapidly with each other
to mitigate risk associated with BTC distributed across borders, the Commission
hosts rapid alert platforms. They are used routinely to communicate
epidemiological risks and quality defects and allow Member States to modify
donor selection or testing practices, conduct recalls or follow up patients as
appropriate
164
. There were 21 alerts launched on blood and 18 on tissues and cells
during 2017.
5.2.1.2 Provisions are not specific enough to ensure common and robust
implementation of oversight
The two basic Acts include provisions for the establishment of oversight as the key tool
for verifying safety and quality
165
. Although the establishment of oversight is seen as a
major achievement of the legislation, a number of shortcomings have emerged that may
limit its effectiveness.
159
No virus transmissions by transplanted tissues and cells were recorded in the most recent EU SARE report for
tissues and cells.
160
For blood, Chapter IV of 2002/98/EC and implementing Directive 2005/62/EC as amended by (EU) 2016/1214 and
for tissues and cells, Chapter IV of Directive 2004/23/EC and implementing Directive 2006/86/EC.
161
This is confirmed by professionals that contribute to the updating of EDQM (Council of Europe) safety and quality
guides. For tissues and cells, they consider that the application of clean room standards, even higher than those defined
in the EU directives are necessary and are routinely provided across the EU for some substances. Such standards are
now reflected in those guides.
162
With the most recent initiative being the adoption of Good Practice Guidelines (GPG) developed jointly by the
Commission and EDQM (Council of Europe) for Blood Establishments and referenced in a recent amendment to an
implementing Directive (EU) 2016/1214. Those guidelines are broadly based on the EU Guidelines for Good
Manufacturing Practice for the manufacture of medicinal products. An equivalent GPG, with a similar focus is
currently under development at EDQM for tissue establishments, indicating a similar trend in that sector.
163
Commission Implementation Report 2016 and Tissues and Cells Implementation Report 2016.
164
Rapid Alert system for human Tissues and Cells (RATC) and for human Blood and Blood Components (RAB)-
Summary of 2017 activities.
165
Article 4 paragraph 1 in both Directive 2002/98/EC and Directive2004/23/EC.

43 | P a g e
Independence and technical expertise of competent authorities not fully
ensured: In contrast to other regulatory frameworks
166
, the BTC Directives do
not specify generic oversight principles to be followed by competent
authorities
167
. Widely varying approaches to the set-up of BTC authorities, with
the functions being carried out by medicinal product authorities in some Member
States, BTC specialist authorities in others and national or regional health
administrations in still others are in place. Consequently, enforcement powers,
levels of independence from the sector and from government, as well as technical
competencies vary significantly.
Inspections are conducted differently across the EU: Both basic Acts include
provisions for the inspection of establishments working in this sector
168
.
Inspections of blood and tissue establishments are carried out in variable ways
across the EU
169
,
170
. Some are conducted by generalist inspectors, with
backgrounds in quality management and inspection of other health product
related sectors such as pharmaceuticals. Others are conducted by specialists from
the BTC sector with high-level technical knowledge in the specific sub-sector
they are inspecting. Still others are conducted by general health inspectors that
are working on a local or regional basis, inspecting a wide range of health
facilities from operating rooms to hospital canteens. A Decision adopted by the
Commission on the conditions of inspection for tissues and cells
171
, and a number
of Public Health Programme actions on this topic (see Annex IX), have brought a
degree of improvement and harmonisation, with some common guidance and
training. However, significant differences between national inspection systems
persist. The variability may impact on inter-Member State confidence in
oversight, particularly in the absence of any provision for verification of
equivalence in inspection approaches across the EU. The experiences of EU level
inspection system auditing in the food and medicinal product sectors are
examples where this challenge has been overcome in other areas of EU
oversight
172
.
166
Article 126 (b) of Directive 2001/83/EC on medicinal products specifies requirements for independence and lack of
conflict of interest for competent authority personnel. Articles 35 (2) and 38 of Medical Device Regulation 2017/745
provide for similar principles of independence and transparency of authorities Article 37 of Food Regulation 178/2002,
also defines an independence principle for Member State authorities.
167
This issue is addressed in the external study supporting the BTC evaluation, ICF, page 78.
168
Directive 2002/98/EC Article 8 and Directive 2004/23/EC Article 7.
169
This has been documented in Commission Implementation Reports.
170
Survey conducted in a Health Programme project. Fehily D, Delvecchio C, Di Ciaccio P et al. (2007): “The Eustite
project: working towards harmonised implementation of European regulation of tissues and cells”. Organs, Tissues and
Cells 2007; 10(1): 31-36.
171
Commission Decision 2010/453/EU.
172
In those frameworks, there are various mechanisms for verifying their effectiveness, such as EU level auditing of
inspectorate systems. Examples are Article 79 of Regulation 536/2014 (in force but not applicable yet) that provides
for the Commission to conduct controls in order to verify whether Member States correctly supervise compliance with
provisions for clinical trials. Similarly, Articles 45(1) and (2) of the Food Regulation 882/2004 provide for
Commission verification of Member State control systems.

44 | P a g e
Divergent approaches to preparation process authorisation: Almost every
type of BTC is subjected to a manipulation process in the blood or tissue
establishment, before being supplied to clinicians for use. In this case, also, there
is wide variability in the procedures for application and approval of preparation
processes and variable quality criteria applied across the EU, sometimes leading
to a lack of mutual acceptance of authorisations between Member States
173
. Many
authorities review detailed dossiers and require clinical outcome data as part of
their authorisation of preparation processes, while others apply a minimal
approach. Stakeholders and authorities agree that process authorisation needs to
be strengthened and should, in many circumstances, include requirements for
review of clinical outcome data
174
,
175
,
176
,
177
,
178
.
A general consensus on this topic
has emerged for both blood and tissues and cells with a common view that the
gap in the current legislation makes the achievement of objective 1 (quality and
safety) particularly challenging from the point of view of ‘quality’.
Variable approaches to reporting of serious adverse events and reactions:
The legislation obliges authorities to ensure that serious adverse reactions and
events are notified to them. They submit summaries of the notified cases to the
Commission each year (see legal provisions described in Annex IV). The EU
wide annual vigilance reporting obligation has brought a much-valued
aggregation of data and information, supporting policy development at national
and EU level. However, the provisions are generic, leaving room for variable
interpretations. The challenges include agreeing on the level of seriousness of
adverse events and reactions that should trigger reporting. For example, 62% of
the serious adverse events reported for blood in the EU in 2016 were reported by
just two Member States. Standardising the reporting of denominator data, to
allow meaningful comparisons between Member States and across the EU over
time, has also been challenging. For skin grafts, for example, some report by
number of packets while others report by area of skin in centimetres squared. In
addition, the provisions do not address all areas of risk to citizens, with limited or
unclear reporting requirements for adverse reactions in donors
179
and in offspring
of medically assisted reproduction (MAR). These latter issues are further
173
This was highlighted in a survey conducted as part of the Joint Action VISTART on vigilance and inspection in
BTC establishments and confirmed in a subsequent survey by the Joint Action GAPP on BTC preparation process
authorisation.
174
In the field of MAR, for example, key stakeholders stated: ‘The ultimate measure of quality in [Assisted
Reproduction Technology} ART should be considered as 'live births per treatment initiated'.
175
Clinical outcome is also monitored in the medium-to-long term for many other tissues and cells (see Annex XV).
176
Many professionals conduct patient studies that come close to, or comply fully with, requirements for clinical trials
on medicinal products.
177
The main consortium of BTC representative societies (CoRE SoHO) has stated that it considers that clinical
outcome monitoring should be regulated, in particular for novel and more complex processing methods.
178
BTC authorities are working together in a Health Programme Joint Action to explore how to improve practices in
preparation process authorisation, including by reviewing clinical outcome data.
179
Inconsistent voluntary reporting by Member States. Although 23 Member States voluntarily reported donor
reactions in the 2017 exercise for blood, 50% of the reactions were reported by one Member State.

45 | P a g e
described under 5.2.1.2 below. These shortcomings limit the potential for learning
from adverse incidents.
Vigilance is enhanced by provisions for Member States to rapidly inform each
other of incidents implying risks for patients in more than one Member State. This
communication is facilitated by a Rapid Alert platform hosted by the
Commission. This platform is used routinely to communicate epidemiological
risks and quality defects and allow Member States to modify donor selection or
testing practices, conduct recalls or follow up patients as appropriate, to minimise
the impact of unexpected risks
180
.
The platform is much appreciated by the Member States although there continues
to be debate regarding what should be the criteria for triggering an alert and how
much information should be shared outside the authority network. These issues
have been raised at meetings of competent authorities and a Public Health
Programme Joint Action that explored current vigilance programmes for BTC
181
.
Alerts related to BTC are often of relevance to other regulated sectors. For
example, defects in medical devices can have significant impacts on the safety
and quality of BTC as donor blood samples are tested with CE marked in vitro
diagnostic kits and BTC are processed and/or stored in certified medical devices.
In addition, BTC can be used for the manufacture of medicinal products and
medical devices (see Section 5.4 on Coherence). Alerts in the communicable
disease field can be of key importance to the selection and testing of donors of
BTC. In this context, lacking provisions for ensuring effective communication
with alert systems in different but related sectors are seen as a shortcoming.
Effective responses when there is an adverse outcome depend on robust
traceability of BTC
182
,
183
. Collaborating coding standards organisations have
highlighted that the current provisions lack a requirement for regular testing of the
traceability systems in place, to verify that all distributed BTC can be traced to
the patients in whom they were used
184
.
5.2.1.3 Provisions do not adequately protect all affected citizens
The evaluation has identified some provisions that are limited or absent in terms of
protecting the safety of all those citizens affected by the chain from donation to clinical
use (see Figure 11).
180
Rapid Alert system for human Tissues and Cells (RATC) and for human Blood and Blood Components (RAB)-
Summary of 2017 activities.
181
See: Website VISTART.
182
The tissue and cell legislation includes specific provisions for coding, in recognition of the degree to which these
substances are exchanged across Member States.
183
Directive 2004/23/EC, Article 25 and Directive 2015/566.
184
Minutes of meeting between DG SANTE and representatives of the coding standards organisations ICCBBA and
Eurocode 3 October 2018.
46 | P a g e
FIGURE 11: INADEQUATE PROTECTION OF ALL AFFECTED CITIZENS IN THE BTC CHAIN
Firstly, provisions to protect BTC donors from risks associated with donation are not
seen as adequate. In the aftermath of the infectious disease transmission crises that
preceded the adoption of the BTC legislation, it is not surprising that the legal provisions
in the Directives focused very much on safety for recipients, and therefore on the safety
of the BTC themselves. Donating BTC might also, however, put donors at risk,
particularly when their health is already compromised in some way or when the donation
requires some form of (hormonal) treatment, as in egg donation and peripheral blood
stem cell donation
185
, or an invasive procedure, as in egg donation and bone marrow
donation
186
. Even without hormonal treatment or an invasive procedure, high frequency
donation of blood or plasma might cause a donor to have diminished reserves of
Haemoglobin and/or other proteins. Iron depletion is particularly important in young
blood donors, pre-menopausal female blood donors, frequent blood donors and those
blood donors whose haemoglobin levels are close to the minimum for eligibility. In spite
of these risks, the current legislation includes only limited donor protection provisions,
many of which are not sufficiently specific or are no longer adequate.
In both BTC basic Acts reporting donor reactions is mandated, as part of vigilance, only
when the safety or quality of the donated substance itself has been compromised; this is
very rare. The great majority of donor reactions are, therefore, not reportable according
to the legislation, even in cases of donor death. Reporting of these cases at national level
varies considerably across Member States, as evidenced in the voluntary reporting
currently conducted in the EU exercises on serious adverse reactions and events
(SARE)
187
. In the most recent SARE reports
188
, 7,658 reactions in blood donors were
185
A commonly used alternative to bone marrow donation involves treating donors with a hormone called Granulocyte
Colony Stimulating Factor (G-CSF) that releases granulocytes and stem cells from the bone marrow into the blood
stream allowing them to be collected from the blood stream by apheresis in a manner similar to the collection of
plasma or platelets. Donors of eggs are stimulated with Follicle Stimulating Hormones over a 10 day period to cause
many follicles to produce eggs and allow the collection of multiple eggs at one time.
186
Bone marrow donation involves the use of needles to withdraw liquid marrow from both sides of the back of the
pelvic bone and egg donation involves collection by a transvaginal surgical aspiration procedure.
187
The annual collation of Serious Adverse Reactions and Events at EU level for blood and for tissues and cells.

47 | P a g e
reported by 23 countries but 50% of these were reported by a single Member State.
Similarly, 700 reactions in tissue and cell donors (including partner and non-partner
gamete donors) were reported voluntarily by 19 countries, while 9 countries did not
report any. These data indicate that voluntary reporting is not fully effective, despite the
important frequency of donor reactions.
Protection prior to and following donation is, thus, a concern across the sectors, among
both professionals and authorities. At a meeting with key blood stakeholders
189
and
blood competent authorities, the topic of donor protection was explored in depth and
robust evidence was presented to confirm that this issue is of high priority and that
current provisions are not adequate
190
. An equivalent meeting of the key professional
organisations in the tissue and cell sub-sector with authorities in that field identified the
same concern, in particular for egg donation
191
and for donation of blood stem cells from
bone marrow or blood
192
. In 2010, a Directive on organ donation and transplantation was
adopted
193
. This Directive includes provisions for the protection of living organ donors,
for their follow-up post-donation and for the reporting of adverse reactions in donors,
regardless of the impact on the quality or safety of the donated organ. These provisions,
adopted on the same legal basis as the BTC Directives, reflect an objective of achieving
safety for donors as well as recipients and a high level of human health protection. The
major concerns for protecting recipients from disease transmission risk following the
HIV and hepatitis C crises of the 1980s and 1990s are now counter-balanced by a
concern for the protection of donors.
Children born from sperm, egg or embryo donation were identified as a second group
of citizens that is inadequately protected by the current provisions. The provisions for
reporting transmissions of genetic conditions in offspring via vigilance programmes are
unclear. The definition of ‘serious adverse reaction’ refers to outcomes in ‘recipients’,
not clearly taking into account the offspring resulting from MAR using donated sperm or
eggs. While at least one large sperm bank supplying across the EU argues that serious
genetic conditions inherited by children from a sperm donor do not meet the definition of
188
Summary of the 2017 SARE Report on Blood and Summary of the 2017 SARE Report on Tissues and Cell.
189
All EU level organisation representing blood establishments, plasma collectors and plasma fractionators were
present.
190
Minutes of the Ad-Hoc Meeting between Stakeholders and representatives members of the Competent Authorities
on Substances of Human Origin Expert Group (CASoHO E01718) 22 February 2017.
191
To protect non-partner gamete donors, the European Society for Human Reproduction and Embryology (ESHRE)
proposed consideration of the following actions: to limit the number of oocyte donations per donor; to define strict
rules on economical compensation (cross-border differences); to require reporting of complications of egg retrieval,
including Ovarian Hyper-stimulation Syndrome (OHSS, a medical condition that can occur in women following
hormonal stimulation of egg growth) in non-partner donations, at an EU level and to improve traceability in cross-
border MAR, eliminating the possibility of direct distribution of gametes to patients.
The equivalent European Society in the field of haematopoietic stem cells (The European Society for Blood and
Marrow Transplantation, EBMT) stressed that their donors undergo a medical procedure with no benefit to themselves
and noted in particular that children are increasingly donating for siblings and should be robustly protected from risks
of donation. They consider that all incidental findings should trigger proper care and counselling. See Minutes of the
Ad-Hoc Meeting between Stakeholders and representatives members of the Competent Authorities on Substances of
Human Origin Expert Group (CASoHO E01718) 22 February 2017.
192
Ad-Hoc Meeting between Stakeholders and representatives members of the Competent Authorities on Substances of
Human Origin Expert Group, February 2017 .
193
Directive 2010/53/EU.

48 | P a g e
a serious adverse reaction in the legislation and should not be communicated in the EU
rapid alert platform
194
, this view is not supported by the major professional association
for MAR
195
,
196
or by the competent authorities
197
,
198
. In addition, the professional
association also considers that the absence of mandatory requirements for monitoring the
health of children born through MAR is an important gap in the legislation
199
.
The protection of recipients is strong but provisions are too rigid. Apart from the
outdated provisions to prevent disease transmission from donors, described in the
Relevance Section (5.1), some technical provisions for the facilities in which BTC are
processed are also unclear and give rise to different standards applied in Member States
to prevent contamination or cross-contamination during processing. This is a particular
issue for tissues and cells
200
. For almost all tissues and cells, there is exposure to the
processing environment during processing. For this reason, the legislation includes
provisions for the air quality of processing areas, making reference to Annex 1 of the
Good Manufacturing Practice (GMP)
201
standards for processing of sterile medicinal
products, but with less demanding air quality specifications, reflecting the specificities of
this sector
202
. These provisions have resulted in improvement of the quality in tissue and
cell processing facilities across the EU, to a common minimum standard, but they are
also seen as problematic. By setting fixed minimum requirements, they are now seen to
allow the processing of some tissues and cells in environments that might not be clean
enough when the risks associated with the particular process and the particular mode of
clinical application are significant. In contrast, they are considered to be too stringent for
situations where microbial contamination does not represent a significant risk to
recipients. The latter situation is addressed under Efficiency (see Section 5.3) as the costs
of providing processing rooms that are classified according to GMP are significant.
5.2.2 QUESTION 3: Has the legislation led to any unintended effects
(positive or negative)?
SUMMARY ANSWER: Unintended effects have been relatively minor and
included increased bureaucracy and administrative costs at both the authority and
the professional level, as well as some challenges for smaller blood and tissue
194
Annex to Submission to the Online Public Consultation by Cryos International, Denmark.
195
Summary of the Blood, Tissues and Cells Stakeholder Event 20 September 2017.
196
Meeting between the Commission and the European Society for Human Reproduction and Embryology 3 May
2018.
197
Meeting of Tissue and Cells competent authorities 20-21 June 2018, item 9.2.2.
198
The results of a ‘neighbour check’ survey by Denmark were reported in the meeting of Tissues and Cells competent
authories 13-14 May 2019, item 5.3.4.
199
Meeting with stakeholders and competent authorities on 22 February 2017, item 5.
200
Blood is processed in closed blood bag systems that protect the substance from the risks of contamination from the
environment or cross-contamination from other donations and, therefore, provisions for facilities where blood and
blood components are less critical.
201
EudraLex Volume 4 of "The rules governing medicinal products in the European Union".
202
Specifically, Directive 2006/86/EC includes a provision (Annex II) that tissues and cells must be processed in an
area equivalent to a grade A with a background of at least grade D, as a minimum. A number of exceptions are noted
where a lower standard can apply.

49 | P a g e
establishments and some shortages of particular BTC in a very few specific cases
in particular Member States.
Negative side effects of the BTC legislation are elaborated under the Efficiency Section
(5.3) and relate to the burden of reporting requirements, the need for some smaller
establishments to consolidate and the loss of donations and supply in some very limited
situations. The many positive effects of the legislation are seen to have been intended.
5.2.3 QUESTION 4: What, if any, have been the barriers preventing
effective implementation of the legislation?
SUMMARY ANSWER: Some broad, or missing, definitions in the BTC
legislation have created barriers to effective implementation due to divergent
interpretation at Member State level. Views vary in particular on the application of
the principle of Voluntary Unpaid Donation. This has limited the achievement of
harmonised standards and hence of inter-Member State exchange. In addition, both
authorities and establishments are hampered by limited resources to support
implementation.
5.2.3.1 Variable interpretations of certain key definitions
A number of shortcomings of the current legislation relate to different interpretations of
definitions but are addressed under other criteria in this report. For example, the terms
‘inspection’ (see Efficiency Section - 5.3), ‘Serious Adverse Reaction’ (see question 2,
above), the term ‘blood establishment’ and ‘partner’ have all been subject to differing
interpretations, with practical consequences for the way the rules are applied and the
sector is regulated
203
.
5.2.3.2 The principle of Voluntary unpaid donation is applied in different ways
One of the key concepts in the legislation is that of ‘voluntary unpaid donation’ (VUD).
VUD is not defined specifically in either of the basic Acts. Directive 2004/23/EC on
tissues and cells, however, provides more detail than the blood basic Act, including that
compensation to make good expenses and inconveniences related to the donation may be
acceptable
204
.
Member States have taken different approaches to interpreting what VUD means. The
differences in purchasing power between Member States may contribute to the context
whereby a measure is considered a “compensation” in one country and is viewed as an
203
A number of other definitions, e.g. ‘storage’, ‘distribution’, ‘transport’, ‘tissue’, ‘cells’ and ‘medical practitioner’,
have led to discussions at the meetings of competent authorities due to differing interpretations, which are further
described in Annex VIII and in the Study supporting the BTC evaluation, ICF, page 88.
204
Directive 2004/23/EC Article 12, paragraph 1.

50 | P a g e
“incentive” in another. To fulfil their legal obligations
205
, Member States report every
three years to the Commission on the measures taken with regard to VUD
206
,
207
. These
reports indicate that all Member States consider that they comply with the principle, even
though some pay fixed or variable amounts to egg and plasma donors and others offer as
much as a day of paid leave from work for blood donation. Some consider the payment
of egg donors to be reasonable compensation
208
while others see it as trafficking
209
.
The issue arises mostly in the areas of plasma and egg donation, two of the substances
that are moved frequently between Member States or are the subject of cross-border
treatment. The consequences have been important, with some Member States putting up
barriers to the movement of BTC, notably plasma collected by the private sector, where
they consider that it has not been collected in compliance with the VUD principle. In
addition, some Member States do not permit private plasma collectors to run donation
programmes for this reason, limiting the volumes of plasma collected and contributing to
the reliance on the US for this substance. Restrictive practices, related to VUD, have led
to complaints and court cases (see Annex VI) and questions in the European
Parliament
210
.
On 11 September 2012, the European Parliament published a Resolution
211
calling for a
more stringent approach to the implementation of the VUD principle for tissues and cells,
raising particular concerns particularly on egg donation and calling for donor protection
measures
212
.
Work carried out at the Council of Europe
213
and by the Nuffield Council of Bioethics in
the UK
214
is moving forward current thinking on how to approach and apply the concept
of VUD and proposing definitions that they consider easier to apply consistently.
205
Directive 2004/23/EC Article 12, paragraph 1 and 2002/98/EC Article 20, paragraph 2.
206
The Commission must inform the Parliament and the Council of any necessary further measures it intends to take.
The Commission fulfils this obligation by publishing the results of a VUD survey of Member States and providing it to
the Parliament and the Council.
207
Commission Staff Working Document on the implementation of the principle of voluntary and unpaid donation for
human blood and blood components and Commission Staff Working Document on the implementation of the principle
of voluntary and unpaid donation for human tissues and cells.
208
V. Pavone “50% of European egg donation happens in Spain. Why?”- International Medical Travel Journal .
209
B.Jones “Human egg-trafficking scam uncovered in Romania”-BioNews .
210
Of the 79 Questions tabled in the European Parliament (since the BTC legislation was adopted - see Annex VII), 12
have pointed to concerns regarding the interpretation of the VUD principle.
211
European Parliament resolution of 11 September 2012 on voluntary and unpaid donation of tissues and cells
(2011/2193(INI)).
212
The resolution addressed implementation of the current rules but also called for a revision of main Act on tissues
and cells and of the Regulation on Advanced Therapy Medicinal Products, to bring them in line with the organ
Directive adopted in 2010
as regards provisions on VUD.
213
Guide for the implementation of the Principle of Prohibition of Financial Gain with respect to the human body and
its parts, as such, from living or deceased donors.
214
“Human Bodies: Donation for Medicine and Research”.

51 | P a g e
5.2.3.3 Limited resources hamper good and consistent implementation
Both authorities and establishments report that limited resources have presented barriers
to the implementation of the legislation. This issue is addressed under Efficiency (see
Section 5.3).
5.2.4 QUESTION 5: Are the rules on oversight sufficient to address the
increased internationalisation?
SUMMARY ANSWER: Some aspects of increasing internationalisation are
considered not to be adequately addressed by the legislation. Activity data
reporting requirements are not sufficient to allow for a clear picture regarding the
volumes of tissues and cells imported to the EU, exported from the EU or
exchanged between Member States. Provisions are lacking to regulate direct online
distribution, for mutual recognition agreements with third countries and for
addressing the role of donor registries in ensuring safety and quality of cells that
are distributed internationally. This impacts negatively on the achievement of
oversight (objective 2), but also on the achievement of community sufficiency
(objective 5).
The BTC legislation aimed to facilitate exchanges between Member States by
establishing equivalent levels of quality and safety across the EU. BTC imported from
outside the EU should be demonstrated to be of equivalent safety and quality. This is
specifically regulated for tissues and cells
215
. Despite these provisions, some gaps and
inadequacies have been identified, which are relevant in the context of increased
internationalisation
216
and commercialisation (see Section 5.1.2.1 and 5.1.2.2).
5.2.4.1 No clear picture of international exchange of BTC due to limited monitoring
provisions
There are limited provisions for data reporting, which means that, while certain general
trends are evident, it is challenging to have a precise and clear EU picture of volumes of
imports or exports for all BTC, particularly for tissues. Neither do most authorities have a
clear overview of the level of dependence on third countries, on the level of community
sufficiency, or on the numbers of donors moving between Member States or entering the
EU to donate.
There is a consensus that certain data reporting should be mandated at EU level
217
. This
would allow authorities to monitor import and export and assess whether international
activities are threatening EU supply or creating high dependency on other continents. In
215
Directive 2015/566 includes provisions for the authorisation of individual tissue establishments to carry out the
imports, including the assessment of equivalence.
216
See the external Study supporting the BTC evaluation, ICF, pages 50-52.
217
A meeting of EU professional experts and authorities, convened by the Council of Europe and DG SANTE in 2017
and reported on at the Meeting of Tissue and Cell competent authorities on June 21-22 2018, item 7.

52 | P a g e
addition, it would ensure transparency to citizens and provide comparable denominators
to better interpret vigilance reports.
Such monitoring provisions would also allow authorities to assess the development of
online distribution of tissues and cells (particularly sperm and bone for dental
applications) which has raised safety and quality concerns, particularly linked to
traceability and vigilance
218
.
5.2.4.2 Barriers to international exchange may hamper access to BTC
While high levels of export from, or import to, the EU might imply risks to the
achievement of EU sufficiency, international exchange is essential in some cases. This
applies, for example, to ensuring that patients have access to BTC that are not (or not
sufficiently) available in the EU, such as plasma and certain types of tissues, and to
facilitating high level genetic matching between donors and recipients for bone marrow
and other types of haematopoietic stem cells (HPC) transplants.
For tissues and cells, the import Directive
219
places the obligation on tissue
establishments, specifically and individually authorised as Importing Tissue
Establishments, to verify equivalent safety and quality for imported tissues and cells.
This differs from other sectors, where EU level agreements with third countries facilitate
and simplify the international exchange of products such as medicinal products and
medical devices. International stakeholders, particularly tissue establishments, report
significant challenges when trying to export to the EU
220
.
Regarding exchange within the EU, the possibility for Member States to add more
stringent requirements to the minimum requirements foreseen in the EU BTC legislation,
although clearly permitted by the Treaty, is also reported as a barrier. This is further
elaborated under the Section on EU added value (5.5).
5.2.5 QUESTION 6: What, if any, are the challenges to maintaining
compliance with the legislation?
SUMMARY ANSWER: The challenges raised in response to question 4, above,
are equally relevant to this question along with all of the rapidly changing factors
that are outlined under the Relevance and the Coherence criteria. The most
important of these are the limited resources at authorities and establishments, the
rapidly changing technological and epidemiological landscape and the borderlines
with other regulatory frameworks.
218
Notably the direct ordering and distribution of bone to clinicians (see Annex VIII, part 2, item 9) and of sperm to
individuals has raised concerns among the tissue and cell authorities and, for sperm, has led to changes in Danish
national law (see Annex VIII, part 2, item 30).
219
Directive 2015/566.
220
Summary of the Blood, Tissues and Cells Stakeholder Event 20 September 2017.

53 | P a g e
5.2.6 QUESTION 7: To what extent, if any, has the legislation impacted on
patient access to blood, tissues and cells?
SUMMARY ANSWER: In general, while sufficiency is defined by demand and
supply, legal provisions for achieving the sufficiency objective are limited. The
supply of blood and blood components for transfusion is generally sufficient and
demand for red blood cells is decreasing. Some BTC-specific negative impacts on
supply have resulted from technical provisions in the legislation that are no longer
considered to be scientifically justified. The most important sufficiency challenges,
however, relate to the EU reliance on imports for plasma for the manufacture of
plasma derived medicinal products and for some tissues, and to a lack of provisions
for ensuring sufficiency in emergency situations.
The supply situation in the EU is different for different types of BTC:
The supply of blood and blood components for transfusion generally meets
demand in the EU although shortages can be associated with seasonal factors,
usually affecting particular Member States, or regions of Member States, such as
the loss of donations due to an increased risk of West Nile Virus infection in
summer in some Mediterranean countries. In general, the demand for red blood
cells is decreasing due to changing clinical practice, particularly the
implementation of an approach known as Patient Blood Management than can
significantly reduce the exposure of patients to the risks of transfusion
221
,
222
,
223
.
There is a significant shortage of plasma for the manufacture of plasma derived
medicinal products that is increasing with increasing demand, with a consequent
reliance on import from the United States (see more detail below).
There appears to be a high level of importation of some tissues for transplantation
(see more detail below).
Bone marrow, and other types of haematopoietic stem cells, are exchanged
internationally to achieve the necessary high level of donor to recipient matching
and shortages tend to be associated with the need for matching for certain ethnic
groups.
In the field of medically assisted reproduction, access to donated eggs and sperm
can be limited by national rules that are beyond the competence of the EU in this
field.
221
M Mueller, H Van Remoortel, P Meybohm et al. Patient Blood Management Recommendations from the 2018
Frankfurt Consensus Conference JAMA.2019;321(10):983-997.
222
Building national programmes of Patient Blood Management (PBM) in the EU- A Guide for Health Authorities, by
the EC March 2017.
223
Supporting Patient Blood Management (PBM) in the EU- A Practical Implementation Guide for Hospital, by the EC
March 2017.

54 | P a g e
5.2.6.1 Shortages due to limited provisions for ensuring sufficiency
Although sufficiency of supply was a key objective, provisions to ensure it are very
limited, focusing only on the need to encourage VUD and recitals on achieving
community sufficiency through VUD. This Section describes the two important areas
where indications are that the EU has not achieved sufficiency and is reliant on imports
to meet patient needs. The lack of EU-level mandatory provisions for monitoring supply,
demand, import/export and inter-Member State exchanges, makes it challenging to
understand the extent to which these flows are the result of marketing activities by the
commercial sector (see Section 5.1.2.1) or genuine shortages of BTC for essential
procedures.
This issue could be seen in comparison to other regions (e.g. the US and Japan) where
legal provisions support the achievement of sufficiency. Examples include obligations
for monitoring supply to facilitate taking pre-emptive action, requirements to report to an
authority when donations fall below a certain level or blocking of export when local
patient needs have not been met.
The first important case of insufficient supply concerns plasma where the EU is highly
dependent on imports from the US. Plasma is the starting material for the manufacture of
a range of medicinal products
224
. While in the EU, the number of private plasma
collection centres
225
increased from 37 in 2005 to 103 in 2016
226
, this is far from
sufficient to keep up with increasing demand for manufacturing of plasma derived
medicines. According to the non-profit plasma manufacturing association, the
International Plasma Fractionators Association (IPFA), 8 million additional plasma
collections would need to be organised in the EU in order to supply all EU patients with
the plasma derived medicines needed. The private sector plasma manufacturing
association, PPTA (Plasma Protein Therapeutics Association), in a detailed response to
the Online Public Consultation, highlights that EU supplies only meet around 60% of the
overall demand for plasma for the manufacture of medicinal products in Europe, with the
difference being primarily imported from the US
227
. The US is the main global source for
plasma, supplying the EU and – to a lesser extent – other areas. Data from PPTA shows
that the global volume of plasma collected for the manufacture of medicinal products
increased by 269% between 1990 and 2014
228
, and with an average annual growth rate of
224
Plasma derived medicines are proteins, such as immunoglobulins and clotting factors that are derived (essentially
filtered or purified) from the liquid plasma. Around 70% of patients with primary immunodeficiencies (PID) need
lifelong treatment with immunoglobulins (Ig) and have no alternative treatment available. Patients with haemophilia
and similar disorders also rely on a robust supply of clotting factors.
225
Plasma for medicinal product manufacture can be separated from donated whole blood or collected directly from the
donor using a machine that separates the blood cells and returns them to the donor (the process is known as apheresis).
The majority of apheresis donation in the EU is organised by the private sector in Germany, Austria, Hungary and the
Czech Republic but the public sector blood services and the Red Cross are also running and developing their
plasmapheresis programmes. According to ICF ref (European Commission, 2018d) there is a significant variability of
supply across the EU.
226
Meeting of the Competent Authorities on blood and blood components 22-23 June 2017.
227
PPTA submission to the Online Public Consultation, Annex II.
228
Plasma Protein Therapeutics Association-Vision paper.

55 | P a g e
over 9%, the sector is expected to more than double between 2015 and 2023
229
. Data
provided in an article by The Economist
230
shows that in 2014, the US transferred 15.9
million litres to Europe, 0.083 to Canada, 0.055 to Latin America, 0.074 to Asia-Pacific
and 0.001 to the Middle East and Africa.
All stakeholders (public, private, blood and plasma collectors and plasma derived
medicinal product manufacturers) recognise this dependency and point to an important
risk to the continuity of the supply if there were a supply interruption from the US
231
.
The situation is underlined by stakeholders, in the context of the steadily increasing
global demand for these products, particularly immunoglobulins. The US FDA has
recently raised concerns regarding increased demand and shortages of immunoglobulins
in the US
232
that might exacerbate the impact of the EU dependence on that region. The
issue has also raised concerns in the European Parliament, where the ENVI Committee is
discussing a Union Act proposed by a group of cross-party MEPs
233
that calls on the
Commission to revise the blood legislation in order to address plasma sufficiency.
PPTA considers that co-existence between the public sector, with voluntary unpaid non-
compensated donations, and the private sector, with voluntary unpaid but compensated
donations would facilitate efforts to increase plasma collection across the EU
234
. The
European Blood Alliance, in its position paper on the subject
235
, calls for increasing the
efficiency of apheresis plasma collection in blood establishments and for reducing the
wastage of plasma separated from whole blood, while maintaining a principle of non-
payment.
The Council of Europe, with the support of the European Commission, organised a
dedicated symposium early 2019
236
that resulted in a set of recommendations for action
by blood services, national authorities, companies and international institutions to
address the situation. Key recommendations of relevance to the EU legal framework
relate to the need for stronger donor protection and vigilance measures, in order to allow
for a safe increase in collection of plasma within the EU. Other points pertaining to blood
legislation relate to monitoring of supply trends (such as in the US and Japan), the free
market of plasma and plasma derived medicinal products (import, export, local supplies)
and improving the interaction between the blood and pharmaceutical frameworks.
The second area where there appears to be significant import from the USA is less
robustly documented and concerns the import of tissues for transplant, particularly
229
Allied Market Research (2018).
230
The Economist (2018). Bans on paying for human blood distort a vital global market.
231
Ad-Hoc Meeting between Stakeholders and representatives members of the Competent Authorities on Substances of
Human Origin Expert Group (CASoHO E01718), 22 June 2017.
232
U.S. FDA Website - Information on Immune Globulin (Human) Product Shortage.
233
Any Member of the European Parliament may table a proposal requesting the Commission to propose a Union act
(a new act or the amendment of an existing act) on the basis of the right of initiative granted to Parliament under
Article 225 TFEU.
234
Meeting between the European Commission and PPTA on 19 June 2018.
235
European Blood Alliance fact sheet on European self-sufficiency for blood components and plasma for
fractionation.
236
EDQM Website.

56 | P a g e
bone for dental and orthopaedic procedures but also skin and ocular tissue. An external
study on the economic landscape of tissues and cells in the EU points to high quantities
of surplus musculoskeletal tissues, some corneal grafts and specific sizes of heart valves,
being exported to Europe
237
. This study also indicated that this level of importation might
impact the viability of smaller and more local tissue establishments. Data from the
Eurocet database, and quoted in that study, indicates that almost 25% of distributed
musculoskeletal tissue distributed in the EU in 2012 was imported
238
. Eurocet data
published for 2017 indicates that this proportion fell to 11% of distributed
musculoskeletal tissue
239
, but the data should be seen as indicative as not all Member
States send data to this platform and some send incomplete data. This importation is also
referred to in a report of US tissue banking activity
240
based on a survey of USA tissue
banks in 2015, where the UK and Germany are listed as countries importing tissues from
a high percentage of US banks.
5.2.6.2 Negative impact on supply due to some specific technical provisions
In general, technical provisions in the legislation did not cause shortages. However, there
are a small number of reports of reduced access in specific situations.
Cornea collection rates and available supply were reported as negatively
impacted in some Member States
241
due to a provision that blood samples for
donor testing be taken from deceased donors within 24 hours of death
242
. Many
have pointed to an absence of scientific evidence to support this provision and to
publications that indicate that blood samples taken later are adequate for reliable
donor testing
243
,
244
.
The plasma industry considers that there is considerable plasma wastage,
particularly plasma separated from whole blood. This results from:
o some eligibility criteria for whole blood donation that are unjustified for
the donation of plasma for the manufacture of medicinal products
245
;
237
Study of the economic landscape for tissues and cells, commissioned by the European Commission and published in
2015, page 12.
238
Study of the economic landscape for tissues and cells, commissioned by the European Commission and published in
2015, Table 17, p77. The table quotes data from the Eurocet database showing that of 222,925 musculoskeletal grafts
distributed in the EU in 2012, 53,879 were imported, mainly through the UK and Germany.
239
Eurocet Website.
240
USA Department of Health and Human Services The 2012 and 2015 National Tissue Recovery through Utilisation
Survey Report, p78-79.
241
Summary Minutes of the meeting between the Committee (Executive Board) of the European Eye Bank Association
and DG SANTE B4, 26 January 2018.
242
Annex II paragraph 2.4 Directive 2006/17/EC.
243
Edler E, Wulff B, Schroeder A-S et al. (2011) A prospective time-course study on serological testing for human
immunodeficiency virus, hepatitis B virus and hepatitis C virus with blood samples taken up to 48 h after death.
Journal of Medical Microbiology: 60, 920–926.
244
Meyera T, Polywkaa S, Wulff B (2012) Virus NAT for HIV, HBV, and HCV in Post-Mortal Blood Specimens over
48 h after Death of Infected Patients – First Results. Transfus Med Hemother 2012; 39:376–380.
245
The issue of applying more liberal donor eligibility criteria for plasma only donors has also been raised by the
private sector representatives of the industry.

57 | P a g e
o the need to apply the pharmaceutical standard of good manufacturing
practice (GMP) immediately after collection of whole blood if the plasma
is to be used for the manufacture of plasma derived medicinal products
(the blood legislation applies only as far as collection and testing is
concerned
246
) even though the plasma is stored in a closed system;
o Differing interpretations of VUD and deferral of prospective male donors
who have sex with other men (MSM) requirements (see Annex VIII) lead
to some plasma not being used for the manufacture of medicinal
products.
5.2.6.3 Provisions are not foreseen to ensure supply in emergency situations
National or regional shortages due to mosquito-borne disease outbreaks in Southern
Member States
247
, where it was necessary to defer donors in line with the EU
legislation
248
, and interruptions to the supply of critical medical devices (in particular, in
vitro diagnostics
249
) have caused concern in recent years
250
. There are no specific legal
provisions to support inter-Member State exchanges or to other local supply
sustainability during crises. On one occasion, (H1N1 epidemic in 2009) the
empowerment provided for in Article 29 of Directive 2002/98/EC (urgency procedure)
was used to introduce a rapid amendment
251
to allow for temporary derogations from EU
donor eligibility requirements to mitigate the risk of blood shortage. Emergency planning
to ensure sustainability of the blood supply has emerged as a key priority for
authorities
252
and for stakeholders in the blood
253
field.
5.3 Efficiency
Assessing cost effectiveness has been particularly challenging in this evaluation, given
that no specific cost impact analysis was carried out in the early 2000s and data, even on
current costs, are very difficult to define precisely. In this largely public sector, costs are
often not analysed by specific activity and are merged with other healthcare provision
246
Article 2, paragraph 1 of Directive 2002/98/EC.
247
As early as 2012, the spread of West Nile Virus was causing concern for Greece, Italy, Romania and France who
began work on preparedness planning to protect the blood supply from the impact of the outbreak.
248
Directive 2004/33/EC.
249
A recent withdrawal of Syphilis testing kits due a defect caused significant supply problems in some Member
States.
250
For example the minutes of a meeting of competent authorities for blood and blood components on 22-23 June 2017
noted a communication from the European Blood Alliance indicating supply difficulties for many EU blood
establishments following withdrawal of a defective text kit for syphilis and Romania and Bulgaria reported a blood
supply crisis caused by a recall of a test kit for hepatitis C.
251
Directive 2009/135/EC.
252
Meeting of Competent Authorities for blood 27-28 February 2018.
253
Ad-Hoc Meeting between Stakeholders and representatives of members of the Competent Authorities on Substances
of Human Origin Expert Group (CASoHO E01718), 10 October 2018.

58 | P a g e
costs. These challenges were also identified by the external contractor and are described
in the published study
254
.
Generally, there is a general consensus among stakeholder groups that costs
were justified by benefits for patients
255
. There are a few exceptions related
to particular technical provisions and the rules regarding frequency of
inspection.
5.3.1 Evaluation question 8: How cost-effective has the application of the
quality and safety requirements in the legislation been for operators. Have
the benefits outweighed the costs?
SUMMARY ANSWER: There is a consensus among operators that the benefits
for patients of implementing the legislation have outweighed the costs for
operators. However, some specific exceptions to this emerged, for certain technical
provisions and certain sub-sectors.
In the course of the online public consultation for this evaluation, evidence of
unjustified costs did not emerge as a major challenge to the implementation of the
legislation
256
. The professionals and authorities responding to the Online Public
Consultation pointed to some specific costs as unjustified, or partially justified.
Similar findings are reported in the independent study published together with this
report
257
. This Section addresses those specific provisions where costs incurred
were considered to be unjustified.
5.3.1.1 The costs of implementing some blood donor eligibility criteria are not
justified by improved safety
As described under Relevance (Section 5.1), some specific donor eligibility
requirements
258
imply costs but no longer bring additional safety. For example, tattooing,
endoscopic examination and acupuncture now carry less risk of disease
transmission
259
,
260
. Age and haemoglobin donation limits are also questioned by experts
in the sector
261
,
262
. The provisions result in unjustifiably lost donations and additional
254
External Study for the BTC Evaluation, ICF, pages 98-99.
255
The exceptions to this were one large sperm bank and a small number of national associations from one country that
considered the legislation to be inapprropriate for medically assisted reproduction.
256
See published Summary of Responses to the OPC, pages 13 – 15.
257
External Study for the BTC Evaluation, ICF, page 100.
258
Defined in Directive 2004/33/EC, Annex III and in Directive 2006/17/EC. Annexes I and III.
259
UK Advisory Committee on the Safety of Blood, Tissues and Organs (SaBTO), Donor Selection Criteria report
(2017).
260
Borra et al (2016) (2016) Blood donor deferral: time for change? An evidence-based analysis. International Journal
of Clinical Transfusion Medicine, 4: pp.55–66.
261
Borra et al (2016) (2016) Blood donor deferral: time for change? An evidence-based analysis. International Journal
of Clinical Transfusion Medicine, 4: pp.55–66.

59 | P a g e
resources dedicated to explaining to donors why their donation cannot be accepted and to
recruiting replacement donors. It was not feasible to quantify these costs in the evaluation
but published evidence suggests that, despite the many different reasons for donor
exclusion (donor or patient safety, product quality, feasibility of the collection, etc.), no
justifying scientific evidence was available for 60% of the top 30 reasons for deferral
263
.
It is suggested that many measures used to increase safety in blood banking cost
significantly more than the standard cost-effectiveness threshold of $50,000 per quality-
adjusted life year (QALY)
264
,
265
. Donor eligibility criteria were not subjected to
cost/benefit assessment when introduced and some of them now imply cost
inefficiencies.
5.3.1.2 Certain donor testing provisions bring unjustified costs
Most donor testing provisions are fully in line with, or are less stringent than, existing
professional standards and their costs are not disputed. This is not the case, however, for
certain situations and sub-sectors. The unjustified donor testing provisions that have been
most evident during this evaluation concern the testing of sperm and egg donors and the
testing provisions for West Nile Virus in blood donors. The issues are summarised in
Table 1.
TABLE 1: DONOR TESTING INEFFICIENCIES IDENTIFIED IN THE PUBLIC CONSULTATION
Provision
In-efficiency
Blood samples must be obtained at
the time of each non-partner
gamete donation
266
Non-partner gamete donors make multiple donations over weeks
and the donations can be quarantined frozen before release for use.
Repetitive testing for each donation is costly without benefit in
terms of safety
267
.
Donations from donors who visited
an area with West Nile Virus
should not be accepted unless an
individual sample is tested for
WNV by nucleic acid technology
(NAT)
268
Individual (ID-)NAT tests cost 7 Euro more per donation tested
compared to applying an (MP-) NAT test in pooled plasma
samples from multiple donations
269
, while the MP NAT tests
provide a comparable level of safety
270
,
271
.
262
Stainsby D, Butler M. Recommendations for the Removal of the Upper Age Limit for Regular Whole Blood and
Component Donors. Joint United Kingdom (UK) Blood Transfusion and Tissue Transplantation Services Professional
Advisory Committee; 2008.
263
Borra et al (2016) Blood donor deferral: time for change? An evidence-based analysis. International Journal of
Clinical Transfusion Medicine, 4: pp.55–66.
264
The quality-adjusted life year or quality-adjusted life-year (QALY) is a generic measure of disease burden used in
economic evaluation to assess the value for money of medical interventions. A cost–effectiveness ratio, i.e. EUR
spent/QALY gained, represents the magnitude of additional health gained per additional unit of resources spent of an
intervention or policy option.
265
Neumann PJ, Cohen JT, Weinstein MC. Updating cost-effectiveness–the curious resilience of the $50,000-per-
QALY threshold. N Engl J Med. 2014; 371(9):796–797.
266
Point 4.2 of Annex III of Directive 2006/17/EC.
267
As discussed in the NCA meeting tissues and cells of November 2017 and June 2018.
268
Directive 2014/110/EU.
269
A study by the European Blood Alliance demonstrated that the cost of this provision could reach an additional 2
million Euros if all at-risk donors are tested across the EU by individual sample testing rather than in pools of samples
from six donors (MP-NAT). Analysis presented at meetings of blood competent authorities and stakeholders.

60 | P a g e
5.3.1.3 The costs of compliance with the processing facility requirements are not
justified by increased levels of safety for some tissue and cell types
Air quality standards for the processing of tissues and cells should comply with certain
standards referenced to GMP for medicinal products
272
. The application of the minimum
requirement is considered overly stringent where the risks associated with contamination
and cross-contamination during processing are extremely low to negligible
273
,
274
due to
both the length of time of exposure to the processing environment and the mode of
application to the patients.
Providing a processing environment with a classified air quality is one of the most costly
measures provided for in the legislation, sometimes reaching €1 million or higher to
install, depending on the size and the cleanliness grade, and significant subsequent
maintenance costs
275
,
276
. The provisions are not cost efficient in those situations where
the processing environment does not impact in a proportionate way on the safety of the
tissues and cells provided for clinical application.
5.3.1.4 Traceability requirements for tissues and cells are cost-effective only for
tissue establishments that were not already using an established coding standard.
The Single European Code (SEC) introduced in 2015 aims to ensure the traceability of
tissues and cells intended for human application. The legislation brought increased
transparency on the regulatory status of tissue establishments providing tissues and cells
and a harmonised code to support traceability of tissues and cells across the EU. The
external study indicates that for stakeholders that were already complying with an
international coding standard, a significant portion of establishments, this provision
brought costs associated with IT upgrades and labelling changes that did not bring
additional benefits over the existing systems
277
leading to cost inefficiencies in the
system.
On the other hand, it provided a cost-effective solution for those tissue establishments
that did not already have a coding system in place. A survey conducted by the
270
ECDC has confirmed that an equivalent level of safety can be achieved by testing at the beginning of a donation
period and again at the end, with quarantine of the donations until final negative results are recorded. ECDC
assessment requested and reported at 2 meetings of blood competent authorities in February 2017 and November 2017.
271
The US FDA allows testing of pooled samples and recommends that blood services establish a threshold of West
Nile virus activity at which they should switch to individual sample testing.
272
EudraLex Volume 4 “Good Manufacturing Practice (GMP) guidelines”.
273
Mortimer D (2005) A critical assessment of the impact of the European Union Tissues and Cells Directive (2004)
on laboratory practices in assisted reproduction. Reproductive BioMedicine Online Vol 11. No 2: 162–176.
274
Summary Minutes of the Meeting between the European Society for Human Reproduction and Embryology
(ESHRE) and DG SANTE B4 03 May 2018.
275
Study by the Rathenau Instituut, 2015, Economic landscapes of human tissues and cells for clinical application in
the EU.
276
Study supporting the BTC evaluation, ICF page 103.
277
Summary Minutes of the Meeting between representatives of ICCBBA and Eurocode and DG SANTE B4, 03
October 2018.

61 | P a g e
Commission in 2011-2012
278
, before the adoption of the EU coding legislation, revealed
that most Member States had a unique code for each donation being assigned
predominantly by the tissue establishments and not, therefore, providing uniqueness
nationally or in the EU.
5.3.2 Evaluation Question 9: Are there particular administrative or other
burdens for specific groups of operators, including downstream users of
blood, tissues and cells as starting materials for medicinal products?
SUMMARY ANSWER: For most operators, the administrative burdens were not
highlighted as unjustified by the benefits. However, limited burdens were identified
for some stakeholders, particularly downstream manufacturers of products made
from blood, tissues or cells.
5.3.2.1 Some donor eligibility provisions are not justified for manufacturing of
plasma-derived medicinal products
Manufacturers of PDMPs report facing burdens associated with donor eligibility
provisions that are not justified for plasma donated for this purpose because of the
subsequent manufacturing steps applied, including microbial inactivation (see Section
5.2.6.2).
279
The cost of donor tests that do not add safety when the plasma is used for
medicinal product manufacture implies an unjustified burden for these stakeholders.
5.3.2.2 Costs of complying with BTC import eligibility provisions are challenged by
ATMP developers as inefficient
Directive 2015/566 on verifying equivalent standards of quality and safety of imported
tissues and cells (i.e. from outside the EU) requires Member States to authorise certain
tissue establishments as ‘importing tissue establishments’ (ITEs). These establishments
must then verify equivalent quality and safety of the tissue and cells to be imported. For
imported tissues or cells that are destined for manufacture of Advanced Therapy
Medicinal Products (ATMP), the ITE must verify the equivalence of the donation,
procurement and testing steps.
This is seen as a burden by EU ATMP manufacturers that must find an authorised ITE,
which is willing to be contracted by them, to carry out this verification. Alternatively,
they must apply for authorisation themselves as an ITE under the tissues and cells
278
Annex 1: Individual country responses to the survey on the implementation of the EU Tissues and Cells Directives
conducted in 2012 and based on 2011 information.
279
An illustrative example provided was the requirement for Human T cell lymphotropic virus (HTLV I and II) testing,
which International Plasma Fractionators Association (IPFA, public sector) stated can be transmitted by blood
transfusion but all risk is removed during the process of plasma derived medicinal product manufacture. See summary
minutes of the Meeting between International Plasma Fractionators Association (IPFA) and DG SANTE B4, 17 April
2018.

62 | P a g e
legislative framework, an additional regulatory burden. Both of these options are seen as
unjustified by the benefits. The burden is exacerbated by divergent donor testing and
eligibility requirements in many Member States caused to the implementation of more
stringent national provisions (see Section 5.1). This issue was raised by ATMP
stakeholders in the Stakeholder Event
280
, in the Public Consultation
281
and in the External
Study.
282
Medicinal product authorities have also raised concerns regarding the burden associated
with the need to apply the donation, procurement and testing provisions of the tissue and
cell legislation for autologous ATMP treatments
283
.
5.3.3 Evaluation Question 10: To what extent has the legislation resulted in
cost implications for hospitals/patients using/receiving blood, tissues and
cells?
SUMMARY ANSWER: It was not possible to disentangle costs for BTC from
overall costs of hospital treatments using BTC. However, stakeholders did not raise
concerns related to additional costs for hospitals for receiving BTC that are in
compliance with EU legislation.
Challenging to estimate additional costs for hospitals. The mechanisms for funding
BTC supply in the EU are regulated at national level and vary considerably between
Member States
284
. Where hospitals are charged, the prices have increased over time but it
has not been possible to separate the increased costs caused by the legislation and those
that were the result of other factors such as increasing costs of materials, testing etc.
Neither during meetings and events with stakeholders in the course of the evaluation, nor
in the Online Public Consultation were costs to hospitals/patients, incurred by the EU
legislation, raised as being prohibitive or unjustified. This is likely to be because costs for
BTC are usually integrated in costs for procedures such as surgery or transplantation and
represent a small portion of the overall intervention cost.
280
Report of the BTC Stakeholder Event, 20 September 2017.
281
18 of 112 responding organisations considered that the legislation, introduces significant or major inefficiencies or
unjustified burdens when ensuring the safety and quality of medicinal products manufactured from BTC.
282
Key stakeholders, such as the Alliance for Regenerative Medicine and the International Society for Cell Therapies,
raised this concern. Study supporting the BTC evaluation, ICF, page 119.
283
Minutes of a Joint meeting between Tissue and Cell competent authorities, medicinal product authorities and
members of the Committee for Advanced Therapies.
284
They range from supply to hospitals/clinics free of charge by establishments that are centrally funded, to cost
recovery by public sector organisations (with fees set nationally or by the establishments themselves), or supply by
private sector companies (with prices set independently).

63 | P a g e
Limited evidence of unjustified costs for patients. In these sectors, it is rare for
patients to pay specifically for the BTC that are used in their treatment, unless they are
being treated privately and in that case, the cost of the BTC is usually incorporated in the
overall treatment cost. The issue of costs for patients has not emerged in the evaluation
apart from in the particular case of couples having medically assisted reproduction
treatment with the use of their own sperm or eggs where the cost burden for patients was
resolved by an amendment
285
.
5.3.4 Evaluation Question 11: To which extent does the oversight required
by regulatory bodies pose a burden to public authorities (has the burden
been proportionate to achieving the original oversight objectives of the
legislation?)?
SUMMARY ANSWER: In general, significant costs were associated with putting
the required oversight functions in place, particularly in those Member States that
did not have them before the legislation was adopted or before they joined the EU.
However, the burden on authorities is largely justified by the benefits, with the
exception of the fixed rule on inspection frequency. The latter emerged as being
over-burdensome without proportionate benefits.
5.3.4.1 Provision for a fixed 2-yearly inspection frequency is costly and does not
verify compliance in the most efficient and effective way
Both basic Acts include provisions for the conduct of inspection and control measures at
blood and tissue establishments at 2-yearly intervals
286
. In the absence of any definition
for ‘inspection’ or ‘control measure’, most Member States interpret the provision as a
requirement to visit each establishment and conduct an on-site inspection, although a
Commission Decision published in 2010 provides for the conduct of a desk based
inspection of tissue establishments in some circumstances
287
. Given that there are 5,400
BTC establishments in the EU and that inspections involve an average of 2 inspectors for
2-3 days, with additional days allocated to review of documentation prior to inspection
and reporting writing following inspection, the investment in this activity is very
considerable. In general, it brings a much valued assurance of compliance with the
legislative provisions and opportunities for improvement where shortcomings are
observed.
285
Testing costs are often borne by the patients themselves and were significant when the patients had to be tested at
each gamete collection. Directive 2006/17/EC was amended in 2012 Directive 2012/39/EU to make the provision less
stringent, allowing testing to be performed up to 3 months before the first collection of gametes and to remain valid for
up to 24 months. It is estimated that more than 160 million Euros a year was saved. See Hughes, C., Grundy, K.,
Emerson, G., and Mocanu, E. (2011). Viral screening at the time of each donation in ART patients: is it justified?
Human Reproduction, 26(11): pp.3169–3172.
286
Directive 2002/98/EC Article 8, paragraph 2 and Directive 2004/23/EC Article 7, paragraph 3.
287
Commission Decision 2010/453/EU.

64 | P a g e
However, the fixed 2-yearly frequency has arisen as an important inefficiency in the
oversight system. It is noted that some establishments are very small with limited
activities, for example mobile blood collection sites
288
, while others are large and
complex. Some perform very well while others have more frequent non-compliances and
adverse incidents. Inspectorates, already resource limited, must return to inspect small
and/or well performing establishments that imply low risk for donors or patients, rather
than investing their time and resources in visiting establishments with identified risk, or
high impact on patients, more frequently or in other activities. There is general consensus
among BTC authorities that a fixed 2-yearly frequency is not cost efficient
289
,
290
. The
European Medicines Agency has issued a guidance document on the use of a Risk Based
Approach
291
to inspection scheduling. Inspections in other regulated sectors, such as
medicinal products and medical devices, and in the BTC sector in the US, are usually
scheduled on the basis of risk assessment.
5.4 Coherence
The coherence of the BTC legislation was addressed by evaluation question 12, with 4
sub-parts focusing on different aspects of coherence. This evaluation looked at the
coherence of the BTC legislation with other Union-level regulatory frameworks such as
medical devices and medicinal products, but did not evaluate those other frameworks.
There are some technical inconsistencies between the different Directives in
the field of substances of human origin. More importantly, the degree of
interdependence between the BTC legislation and other EU regulatory
frameworks is sometimes challenging and, in some cases, the application of
the BTC safety and quality requirements when BTC are used as starting
material for products regulated under other frameworks may be a
shortcoming.
While most BTC based products and substances fall clearly into one or other
regulatory framework (BTC, medicinal products, medical devices), there are
a small number of cases at the borderlines where classification is more
challenging. This is illustrated by some uncertainty and differences between
Member States in the classification and regulatory frameworks applied for
the same substances. In addition, the criteria and mechanisms for defining
288
See Annex VIII Part 1 Number 4.
289
Summary Minutes of the Meeting of the Competent Authorities on Blood and Blood Components 11/12 November
2015.
290
Although it has been difficult to precisely establish the cost implications of this rule, it is estimated that the cost
across the EU for BTC inspections runs to many millions of euros every year. Study supporting the BTC evaluation,
ICF, Annex 6, page 217.
291
Published on EMA’s secure platform for Medicinal Product Authorities.

65 | P a g e
whether the full scope of the BTC legislation is applicable are sometimes a
subject of discussion.
5.4.1 Evaluation question 12a) To what extent is the legislation on blood and
tissues and cells consistent and coherent within its own provisions?
SUMMARY ANSWER: There is broad internal consistency although some
testing and process authorisation requirements differ without a justifiable rationale.
The concept of voluntary unpaid donation is described differently in the two Acts
and traceability for tissues and cells is more stringently regulated than for blood.
The latter is justified by the greater degree of exchanges of tissues and cells
between Member States.
5.4.1.1 Different descriptions of the voluntary unpaid donation concept
Provisions on voluntary unpaid donation (VUD) are included in both the blood and tissue
and cell basic Acts but with different descriptions.
The tissue and cell Act specifies that ‘donors may receive compensation, which is
strictly limited to making good the expenses and inconveniences related to the
donation’, leaving to the Member States to define the conditions for
compensation (Article 12). As a result, the compensation of oocyte donors with
sums up to, and beyond, 1,000 Euros is common and sperm donors are
compensated with much smaller amounts in most Member States
292
.
No such provision for compensation is included in the blood Act.
It is noted however that the degree of inconvenience and the time required for different
types of donation differ considerably, some, such as oocyte or bone marrow donation
requiring hormonal treatment and a significant intervention while others imply minimal
discomfort (see Section 5.2).
5.4.1.2 Some technical differences in safety and quality provisions
A series of less significant technical differences between the blood and the tissue and
cells Directives has emerged, such as donor testing for specific diseases (e.g., syphilis),
traceability and import rules, authorisation of preparation processes, requirements for
quality systems and reporting adverse reactions (see Annex XVI, Table 1). The latter is
largely justified by the greater degree of inter-Member State exchanges of tissues and
cells.
292
For example, the Human Fertilisation and Embryology Authority (HFEA-UK) defines expense limits of 35£ for
sperm donors and 750£ for egg donors, per donation.

66 | P a g e
Apart from raising doubts regarding the need for the provision in one legislation if it is
not needed in the other where the risks should be similar, these differences may also have
an impact in a small number of cases where it was unclear whether a substance falls
under the blood or the tissue and cell framework. The most important of these related to
Donor Lymphocyte Infusions (DLI)
293
, used to increase success of bone marrow
transplants for blood cancer patients. Justifiable arguments can be made for applying
either the blood or tissues and cells legislation. This can lead to Member State questions
on whether the appropriate safety and quality requirements are applied for DLI that are
exchanged cross-borders following collection in another Member State.
5.4.2 Evaluation question 12b) To what extent is the legislation coherent and
consistent with other relevant Union legislation?
SUMMARY ANSWER: Some provisions, such as those for VUSD and donor
protection, are stronger in the organ legislation compared to the BTC legislation.
Issues related to the borderlines with medicinal products and medical devices were
highlighted. A lack of regulatory clarity may result in disincentives for BTC
establishments to invest and innovate.
5.4.2.1 Some incoherence with Legislation on Organs
The Directive on organ donation requires Member States to ‘ensure’ (rather than to
‘encourage’) the principle of Voluntary Unpaid Donation. Financial incentives or
benefit for the donor are to be avoided. However, as in the tissue and cell legislation, but
unlike the blood legislation, compensation of living organ donors is permitted for
expenses and loss of income (Article 13). This more binding requirement is in line with
the EU Charter of Fundamental Rights
294
, which entered into force in 2010 with the
TFEU. Some public sector blood and tissue stakeholders are therefore calling that this
more stringent application of the principle of VUD is also introduced in the BTC
framework.
Considerably more stringent provisions are in place to ensure living organ donor
protection compared to donors of BTC, including the need for registers of donors and
donor follow-up at a national level. Finally, the provisions extend to the oversight
(including authorisation) of clinical transplantation centres, a scope that is significantly
broader than that in place for BTC. Concerns on the absence of such provisions in the
BTC framework for donor protection and for clinical follow-up are brought forward
under the Effectiveness Section (see Section 5.2).
293
Although the cells are separated from blood, they are usually collected from bone marrow (or peripheral blood stem
cell) donors and applied to patients as a support to a transplant of those cells, which are regulated under the basic act
for tissues and cells (see Annex VIII, Part 1, item 3 and 21 and Part 2, item 1).
294
Article 3 of the Charter calls for “the prohibition on making the human body and its parts as such a source of
financial gain”.

67 | P a g e
In this context, there have been only a small number of borderline cases. Most notable,
was the case of hand or face transplantation. From an anatomical point of view, these
involve the transplant of a number of combined tissues in a single complex structure.
Following extensive discussions with both networks of organs authorities and of
tissue/cell authorities, the Member State authorities agree that the donation and
transplantation process that must be followed, without the need for a tissue
establishment, makes the organ legislation more appropriate than the tissue and cell
legislation to regulate this activity (see Annex VIII, part 2, item 9).
There also are some technical inconsistencies between the BTC and organs legislation.
In general, the regulatory provisions of the organ Directive
295
are less detailed than those
in the BTC legislation. For example, there is no EU-wide reporting of adverse outcomes
required and oversight and quality management provisions are not as detailed. These
differences are partly justified by a context where organ donation and transplantation are
temporally closely associated, without the possibilities for complex processing or storage
and by the high benefit to risk ratio in this field.
There are recent calls for closer collaboration between organ and BTC competent
authorities on some of these technical requirements, in particular for vigilance
reporting
296
.
5.4.2.2 Coherence with other Legislation in the field of Health – medicinal products
Legal provisions: There is a direct link between the BTC directives and the medicinal
product legislation. According to Article 2(1) of the Blood Directive, it applies to
collection and testing of human blood and blood components whatever their intended
purpose and to their processing storage and distribution when intended for transfusion.
This means that the entire path from donation to supply for clinical use is regulated by
the Blood Directive if the final use is for transfusion. When plasma is used for
manufacturing of medicinal products only the first steps, collection and testing, are
regulated by the blood legislation while those steps from manufacturing onwards are
regulated by the medicinal product legislation
297
.
Similarly, for tissues and cells for human application, the first steps, procurement and
testing, are regulated by the Tissue and Cell Directive (Article 2(1)), whatever the
intended use. The subsequent steps are also regulated by the Tissue and Cell Directive as
long as the tissues and cells are not used for the manufacture of products that are covered
by other Directives. This means that tissues and cells used for manufacturing of
medicinal products are regulated by the medicinal product legislation for the steps from
manufacturing and onwards. In this case, tissue and cell based medicinal products that
have been substantially manipulated or are not intended to be used for the same essential
295
Directive 2010/53/EU on standards of quality and safety of human organs intended for transplantation.
296
In their June 2018 meeting, organs Competent Authorities reiterated an interest in having an expert network within
the existing SoHO Vigilance expert sub-group to focus on organ vigilance.
297
Directive 2001/83/EC.

68 | P a g e
function in the recipient are regulated by the Advanced Therapy Medicinal Product
(ATMP) legislation since 2007
298
.
Classifying a substance/product as a BTC or as a medicinal product or establishing which
of the respective legal framework applies is primarily a Member State responsibility.
While most BTC based substances/products fall clearly into either the medicinal or BTC
legal framework, the evaluation suggests that in some cases it is challenging to decide on
classification and determine which legislation applies. This Section describes some of the
challenges this has created for different stakeholder groups including BTC
establishments, stakeholder associations
.
and national competent authorities
299
,
300
.
Extent of problem: While most of the BTC and BTC based products fall clearly within
one legal framework or the other, the evaluation has shown that on some occasions the
same substances have been subject to divergent classifications in different Member
States
301
,
302
, as illustrated in a 2019 survey of EU Member States tissue and cell
authorities
303
. This has led to the application of different legal regimes for the same
substance in different Member States.
One example of divergent classification pointed out by EATCB
304
, the main
representation of EU replacement tissue banks, is a skin cell based suspension used for
treating wounds, injuries and skin diseases, for which 9 Member States classify the
activity as BTC and 7 Member States as medicinal product (ATMP or hospital
exemption)
305
. Other examples of divergent classifications identified in a survey from
April 2019 relate to substances like liver cells, fat cells or platelet rich plasma, serum eye
drops or amniotic membrane (for more details, see Annex XVI).
306
The challenges created by such divergent classifications were raised several times in the
course of the BTC evaluation (Online Public Consultation
307
, Commission Stakeholder
event
308
, ad hoc meetings with stakeholders
309
) by various stakeholders including BTC
public authorities, practitioners and manufacturers from both BTC and medicinal sectors.
298
Regulation (EC) 1394/2007.
299
External Study supporting the BTC evaluation, ICF, p. 121-122 (study to be published together with the SWD).
300
Commission Summary of Responses to the Public Consultation for the Evaluation of the Blood, Tissues and Cells
Legislation, p. 17-18.
301
Forgo N and HildebrandtM (2013) Joint conference on the impact of the EU legislation on therapeutic advance.
Cytotherapy 15: 1444-1448.
302
Pearce KM, Hildebrandt M, Greinix H et al. (2014) Regulation of advanced therapy medicinal products in Europe
and the role of academia.
303
See Annex XVI to this SWD.
304
Presentation to the meeting of the network of National Competent Authorities on Tissues and Cells.
305
See Annex XVI to this report.
306
It should be noted that CAT make their recommendations on classification on the basis of individual product
manufacturing processes and not on categories of products.
307
20 organisations responding to the Commission Public Consultation for the Evaluation of the Blood, Tissues and
Cells Legislation considered that there were substantial inconsistencies with medicinal products legislation. These
included large EU level professional associations, national level BTC professional associations, BTC and medicinal
product competent authorities, BTC establishments and Council of Europe (EDQM), (Commission Summary of
Responses to the Public Consultation for the Evaluation of the Blood, Tissues and Cells Legislation, p. 16-18).
308
Summary of the Blood, Tissues and Cells Stakeholder Event organised by the European Commission Services (DG
SANTE) on 20 September 2017 in the context of the BTC evaluation, p. 9.

69 | P a g e
The Council of Europe (EDQM) refers to inconsistencies and ambiguities in the scope
and regulatory borderlines that lead to confusion and hamper oversight and patient
access
310
. The Common Representation for Substances of Human Origin (CoReSoHO),
the main sector organisation that represents blood and non-reproductive tissue
establishments, identified the need for a clear definition of criteria for the classification
of tissues and cells, in order to avoid that the same product is considered a tissue/cellular
therapy in one Member State, and a medicinal product (ATMP) in another
311
.
Public
authorities from three large Member States mentioned inconsistencies, gaps and double
status of products in their contributions to the public consultation
312
. Furthermore, the
challenges were highlighted by the External Study conducted to support the
Evaluation
313
.
Consequences: While it is difficult to quantify precisely the impact of specific
classification decisions in borderline cases and divergent classifications across Member
States, the problem is closely linked with the very different requirements under the two
legal frameworks, reflecting proportionately the different risks associated with BTC
versus manufactured product.
Organisations representing tissue establishments have indicated that they hesitate to enter
into the development of new processes when it is not clear what legal requirements apply
and, thus, what level and type of investments might be needed. This could affect
innovation in the BTC field
314
,
315
,
316
,
317
. This is exacerbated when these establishments
have invested already to perform activities which have traditionally been carried out by
BTC establishments and have been regulated under the BTC legislation for many years,
and that since the introduction of the ATMP Regulation (in 2007) have been reclassified
as Medicinal Products in some Member States
318
,
319
.
In addition, the Danish Ministry of Industry considered that the challenge related to
classification uncertainty could create administrative burden or additional investment
(resource and time) for innovative health companies, and ultimately may result in costs
and delays to patients’ accessing new innovative therapies
320
. The concerns on patient
309
DG SANTE and stakeholder meetings in 2017-2018. See for instance (i) the meeting with Consortium of SoHO
societies; (ii) the meeting with the European Eye Banking Association .
310
Submission to the OPC by Council of Europe/EDQM with file title CoE-EDQM.pdf and its annex.
311
Submission to the OPC by the Common Representation for Substances of Human Origin (CoReSoHO).
312
Submission to the OPC by the Federal Ministry of Health (DE), as well as by two public authorities not consenting
to publish their submissions.
313
Study supporting the BTC evaluation, ICF, page 121 and following.
314
DG SANTE meeting with Consortium of SoHO societies of 14 March 2017 (European Blood Alliance, European
Society for Blood and Marrow transplantation, European Association of Tissue Banks and European Association of
Eye Banks).
315
Summary of the Blood, Tissues and Cells Stakeholder Event organised by the European Commission Services (DG
SANTE) on 20 September 2017 in the context of the BTC evaluation, p. 8ff.
316
Response by the European Society of Blood and Marrow Transplantation (EBMT) to the IMI consultation on
Advanced Therapies Concept Paper, 2016.
317
Economic landscapes of human tissues and cells for clinical application in the EU (2015) Rathenau, p 225-226.
318
Submission to the OPC by the Common Representation for Substances of Human Origin (CoReSoHO).
319
Economic landscapes of human tissues and cells for clinical application in the EU (2015) Rathenau, p 27-8.
320
Proposals for simplification of EU-Legislation Prepared jointly by the Danish Minister for Business, Industry and
Financial Affairs and the Danish Business Forum for Better regulation March 2019, p. 4.

70 | P a g e
access are supported by national competent authorities for BTC
321
and are also
highlighted by professionals
322
,
323
,
324
,
325
,
326
.
Moreover, where it happens that Member States classify the same product in divergent
ways, this could create challenges to inter-MS exchanges. Although many of the
substances/products at the current borderline are autologous, and cross-border exchange
is not frequent at this stage, this is likely to change as innovation in both the BTC and the
ATMP field is expected to move forward
327
. The situation can also create challenges for
import from third countries that wish to supply to multiple Member States.
Causes of the problem: When establishing the applicability of acts other than the BTC
directives, divergent classification may occur due to different national interpretation of
certain key definitions and requirements:
Some stakeholders, including the Ministry of Health of Germany and the
Establissement Francais de Sang, as well as the external study supporting
this evaluation, mentioned the lack of clarity on the term ‘prepared
industrially or manufactured by a method involving an industrial
process’
328
,
329
, which is a determining factor for whether a product, fall
within the scope of the medicinal products legislation
330
. This lack of
clarity is problematic because of the direct link between the two legal
frameworks as explained above.
Also the term ‘substantial manipulation’, relevant to determine whether
the ATMP Regulation
331
is applicable or not, has been subject to variable
interpretation
,
as seen in the examples listed in Annex XVI. For example,
processing such as enzymatic isolation of cells have been considered
substantial in some Member States and non-substantial in others
Moreover, the term ‘used for the same essential function’ has sometimes
proved difficult to interpret
332
,
333
,
334
,
335
and can result in identical
321
Summary reports of the Tissue and Cell Competent Authority meeting of 13-14 May 2019.
322
Meeting with the Common Representation of SoHO Associations, 14th of March 2017.
323
Meeting with the International Society for Cell Therapy (ISCT) 14 June 2018.
324
Meeting with the Executive Board of the European Eye Bank Association.
325
Pirnay JP, Vanderkelen A, De Vos D et al. (2013) Business oriented EU human cell and tissue product legislation
will adversely impact Member States’ health care systems.
326
Dimitropoulos et al. (2016), Burn Patient care lost in good manufacturing practice. Annals of Burns and Fire
Disaster, XXIX, 2, p111.
327
Economic landscapes of human tissues and cells for clinical application in the EU (2015) Rathenau, p219 ff.
328
Submission to the OPC by the Federal Ministry of Health (DE) and the Etablissement Francais de Sang (FR).
329
ICF report supporting this evaluation, p 120-121.
330
Art 2 of 2001/83/EC.
331
The ATMP Regulation 1394/2007 includes a non-exhaustive list of ‘non-substantial’ manipulations, which are
governed by BTC rules, see Annex 1 of the Regulation. For non-listed, including new processes, a case-by-case
decision needs to be taken whether they are to be considered substantial or not.
332
Chabannon C, Caudnay-Rigot O, Faucher C et al. (2016) Accreditation and regulations in cell therapy. ISBT
Science Series 11(1): 271-276.
333
Izeta A, Herrera C, Mata R et al. (2012) Cell-based product classification procedure: What can be done differently
to improve decisions on borderline products? Cytotherapy 18:809-815.
334
Cuende N, Herrera C and Keating A (2013) When the best is the enemy of the good: the case of bone-marrow
mononuclear cells to treat ischemic syndromes. Haematologica 98(3): 323-324.
335
ICF report supporting this evaluation, p 121-122.

71 | P a g e
substances prepared through similar processes to be subject to different
safety and quality requirements
336
.
Mechanisms to support Member States to apply the criteria and definitions in a common
way exist, but are seen by some stakeholders as lacking coherence
337
.
One response to this issue comes from the mandate entrusted to EMA by the ATMP
Regulation to produce non-binding ‘scientific recommendations’ in relation to
classification issues regarding medicinal products based on the advice of the Committee
on Advanced Therapies established under the ATMP regulation. However, the
Committee functions by responding to queries concerning specific medicinal products
submitted by the substance/product developer
338
, and only assists in determining whether
a specific medicinal product falls, on scientific grounds, within the definition of an
ATMP, and not with providing indications of what the product is if it is not an ATMP.
The Committee’s recommendations to applicants are issued on the basis of the criteria
illustrated in a Reflection Paper
339
issued in 2015 and their summaries are published.
Tissue and Cell authorities discuss borderline issues on occasions in their regular
meetings (Competent Authorities on Substances of Human Origin Expert Group,
CASoHO E01718), but have no legal mandate to make formal recommendations to
Member States in view of ensuring a uniform interpretation of the complex legislative
framework.
The potential benefits of a cross-sector multi-disciplinary forum covering all innovative
health frameworks, including ATMP, for reaching common recommendations on
classification across this borderline has been highlighted in a REFIT Platform proposal
by Denmark in 2019
340
and by BTC competent authorities
341
.
5.4.2.3 Coherence with other Legislation in the field of Health – medical devices
As described above, the scope of the tissue and cell legislation covers all the steps from
donation to supply for clinical application for human tissues intended for human
application unless they are used to manufacture products that are covered by other
legislation, in which case the tissue and cell legislation covers only donation,
procurement and testing.
336
Submission to the OPC by the European Directorate of Quality of Medicines/Council of Europe (EDQM/CoE) and
the International Society for Cell Transplants (ISCT).
337
The submissions received in the Commission public consultation for the Evaluation of the Blood, Tissues and Cells
Legislation from stakeholders and citizens cf. the submission from the International Society for Cell Therapy (ISCT),
European Eye Bank Association (EEBA), Common Representation for Substances of Human Origin (CoReSoHO),
EDQM/Council of Europe.
338
Art 17.1 of Regulation (EC) N° 1394/2007.
339
Reflection paper on classification of ATMP (2015).
340
Proposals for simplification of EU-Legislation Prepared jointly by the Danish Minister for Business, Industry and
Financial Affairs and the Danish Business Forum for Better regulation March 2019, p. 4.
341
Summary reports of the Tissue and Cell Competent Authority meeting of 13-14 May 2019.

72 | P a g e
The 2017 Medical Device Regulation
342
explicitly excludes tissues and cells, from its
scope except for:
Products manufactured utilising derivatives
343
of non-viable
344
tissues or cells of
human origin.
Products that combine a medical device with non-viable tissues or cells or their
derivatives. Any device which, when placed on the market or put into service,
incorporates, as an integral part, non-viable tissues or cells of human origin or
their derivatives that have an action ancillary to that of the device shall be
assessed and authorised in accordance with the Medical Device regulation. In that
case, only the provisions for donation, procurement and testing laid down in
Directive 2004/23/EC shall apply. However, if the action of those tissues or cells
or their derivatives is principal and not ancillary to that of the device, and the
product is not governed by the Medical Device Regulation, the product falls also
for its processing, storage and distribution under the rules for tissues and cells.
For the safety and performance of the device part the relevant general safety and
performance requirements set out in the Medical Device Regulation shall
apply
345
.
Despite the further clarity brought by the Medical Device Regulation, a 2019 survey
amongst tissue and cell competent authorities indicated divergence in Member State
classification. While most countries regulate demineralised bone, decellularised heart
valves and decellularised skin as tissues and cells, demineralised bone was reported in
one country to be regulated as medical device (see Annex XVI, Table 3).The question
concerning whether tissues that do not contain living cells should now to be subject to
the new medical device regulatory framework has been raised
346
. Tissue and Cell
competent authorities consider them to be appropriately regulated under the tissues and
cells legislation, without major concerns on safety and quality.
Given that many tissue products currently supplied under the tissue and cell legislative
framework are non-viable, and that it is not the intention of the Medical Device
Regulation to cover tissues and cells which fall within the scope of Directive
2004/23/EC, a clear and consistent understanding of the borderline between the two sets
of legislation concerning non-viable tissues and cells and their derivatives is important
for all involved.
In recent years, a wide range of new bedside equipment allows BTC collection and
processing at the patient’s side. These processes fall outside the current BTC legislative
342
Regulation (EU) 2017/745.
343
In Article 2 (17) of Regulation (EU) 2017/745 ‘Derivative’ is defined as meaning ‘a ‘non-cellular substance’
extracted from human or animal tissue or cells through a manufacturing process. The final substance used for
manufacturing of the device in this case does not contain any cells or tissues’.
344
In this context non-viable means not living.
345
Annex I of Regulation (EU) 2017/745.
346
This topic is under discussion in the Borderline & Classification subgroup of the Medical Devices Co-ordination
Group.

73 | P a g e
scope. They include equipment for the collection and reinfusion of blood during surgery
to devices for the preparation of platelet rich plasma or stem cells from adipose tissue. In
the course of the evaluation, both authorities and professionals have raised the need for
BTC regulatory oversight of these processes due to the specificity of that legislation for
ensuring safety and quality of processed tissues and cells. While the equipment itself is
normally subject to the Medical Device Regulation, there is no regulation to ensure
safety and quality of BTC produced with this equipment. Solutions would need to be
found in cases where tissues or cells are processed at the bedside in a manner that might
require an authorisation from a tissue and cell authority, without impact on the surgical
procedure itself. This is not possible under the current legislation due to the exclusion of
autologous tissue and cell processes carried out in the ‘same surgical procedure’
347
.
The materials and equipment used in the process from donation to supply of BTC,
including donor testing, are almost all certified medical devices. The critical importance
of medical devices at almost every step in the collection, testing, processing, storage and
distribution has been highlighted when there have been interruptions to the supply of a
particular device due to a defect
348
. This has happened in recent years in relation to
haemoglobin testing devices used on donors, in vitro diagnostic test kits for infectious
disease testing of donor blood samples and culture media used to culture embryos in
assisted reproduction centres (see also Section 5.2.6.3). Such sudden interruptions have
for example required the French national blood service to switch test-kits overnight, with
the help of the UK national service, in order to allow for continued supply of safe blood.
Ensuring sustainability of supply, particularly for blood and blood components, has
emerged as a priority that is not adequately addressed in the current BTC legislation,
particularly in circumstances of epidemiological or other crises. Discussions have taken
place on this topic, with competent authorities, with EDQM (Council of Europe) and
with the medical device industry. They indicate that any initiative to ensure the
sustainability of the BTC supply chain must include effective cross-sector planning,
monitoring and vigilance actions related to the critical medical device supply
349
. It needs
to be noted that the Medical Device framework itself does not address criticality or
shortages of devices.
5.4.2.4 Coherence with other Legislation in the field of Health – communicable
diseases
In 2004, the EU adopted a Regulation on communicable diseases
350
, establishing the
European Centre for Disease Prevention and Control (ECDC). A subsequent EU
Decision
351
provides for a network for the epidemiological surveillance of communicable
347
Article 2, paragraph 2(a) 2004/23/EC.
348
See example in the study supporting the BTC evaluation, ICF, page 124.
349
Summary Minutes of the Ad-Hoc Meeting between Stakeholders and representatives of members of the Competent
Authorities on Substances of Human Origin Expert Group (CASoHO E01718) 10 October 2018.
350
Regulation (EC) No 851/2004.
351
Decision
1082/2013/EU on serious cross-border threats to health.

74 | P a g e
diseases, operated by ECDC, and for the “Early Warning and Response System”
(EWRS)
352
that ensures rapid inter-Member State communication of emerging
communicable disease threats. In the Online Public Consultation for this evaluation, a
proportion of respondents (21% for blood and 27% for tissues and cells) indicated their
view that there are minor or significant inconsistencies with that legislation, reflecting a
view that the surveillance requirements are not adequately reflected in the BTC
legislation.
5.4.2.5 Coherence with other EU Legislation
The Charter of Fundamental Rights of the European Union was adopted in 2007
353
and
includes a provision that the human body as such shall not be a source of financial gain,
as well as one prohibiting eugenic practices, in particular those aimed at the selection of
persons. A proportion of respondents to the Online Public Consultation (35% for blood
and 27% for tissues and cells) considered that there are minor or significant
inconsistencies between the Charter and the BTC legislation, reflecting a view that these
issues are not adequately addressed for BTC.
5.4.3 Evaluation question 12c) Are the requirements of the Directives
suitable when blood, tissues and cells are used as starting materials for the
manufacture of medicinal products/medical devices?
SUMMARY ANSWER: The requirements are generally adequate although some
donor eligibility provisions are inappropriate and the need to import tissues and
cells via authorised tissue establishments emerged as an unjustified burden for
ATMP developers.
As described in 5.4.2.2 and 5.4.2.3, BTC can be the starting material for other categories
of health products, including plasma-derived medicinal products, advanced therapy
medicinal products (ATMP) and medical devices. See Figure 12.
352
See also: Commission Implementing Decision (EU) 2017/253.
353
Charter of Fundamental Rights of the European Union.
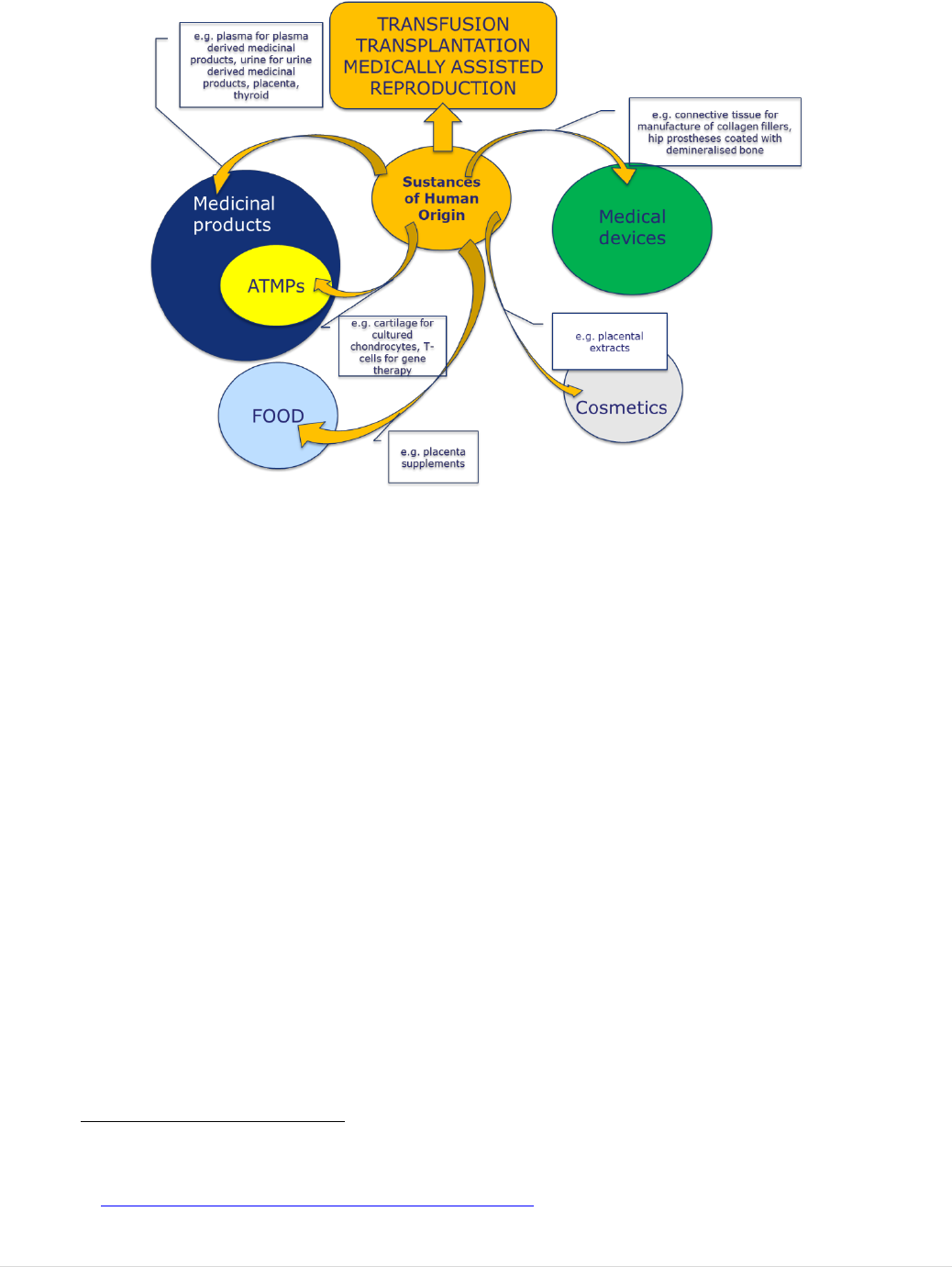
75 | P a g e
FIGURE 12: BTC AS STARTING MATERIALS FOR MANUFACTURED PRODUCTS REGULATED UNDER OTHER
LEGISLATIVE FRAMEWORKS
354
This situation sometimes raises the question whether the provisions for
collection/donation/procurement and testing of BTC are adequate when the BTC are
subsequently used for manufacturing under these other legal frameworks. This aspect of
coherence was raised in the evaluation in relation to both plasma-derived medicinal
products and to ATMPs.
As explained in the Efficiency Section (see Section 5.3), for plasma-derived medicinal
products, donated plasma is subjected to a process that includes many microbial
inactivation steps and that completely removes any cells. In this context, many of the
donor eligibility and testing provisions are seen as unjustifiably stringent due to the
significantly lower risk in the final product
355
.
For ATMPs, in contrast, the donor selection and testing provisions in the current
legislation might not be stringent enough, as it is rarely possible to sterilise those
products and they might, in the future, be supplied to high numbers of recipients from a
single donor. In addition, ATMP developers have pointed to challenges when importing
tissues and cells into the EU as starting materials. In this situation, the tissue and cell
legislation requires import via an authorised importing tissue establishment
356
. This
prevents the ATMP developer from importing directly to their facility (See Section
5.3.2.2).
354
Human tissues and cells are specifically excluded as ingredients in the manufacture of cosmetics in the EU.
However, products are advertised online that are based on extracts from BTC.
355
See Annex VIII Part 1, number 11 and 13.
356
Submission of the Alliance of Regenerative Medicine to the OPC.

76 | P a g e
For medical devices tissue and cell authorities should be consulted during the
authorisation of medical devices containing tissues or cells or their derivatives. These do
not extend to vigilance and traceability activities following the clinical application of the
device. There are no provisions in the BTC legislation to ensure appropriate
communication across the regulatory borderlines for ensuring vigilance, traceability or
efficacy/performance of devices manufactured with human tissues and cells as a starting
material. It is noted that in some Member States
357
, this issue is solved because the
medical device, medicinal product and BTC authorities can ensure appropriate
communication and collaboration, in several cases by being all within one organization.
5.4.4 Evaluation question 12d): To what extent is the legislation coherent
with other relevant international / third country approaches to the regulation
of the quality and safety of blood and tissues and cells?
SUMMARY ANSWER: There is general consistency between the EU BTC
requirements and those of the US as the key country with whom BTC are
exchanged. However, some BTC are classified differently in different jurisdictions,
making international exchange more difficult. In contrast to the EU,
recommendations on classification, in other jurisdictions, involves discussions in
cross-sectoral committees. In addition, BTC vigilance requirements are more
comprehensive in the EU than in other jurisdictions.
In the course of the evaluation, the Commission held discussions with a number of key
international regulators and stakeholders to explore differences in legislative provisions
and regulatory approaches in this field. The most important of these is the FDA/CBER in
the United States
358
, because of the large quantities of plasma and tissues that are
exported from there to the EU. While there are many commonalities between the legal
provisions and regulatory approaches of the other jurisdictions that were explored, there
were some differences of interest identified.
The Commission collaborates with the European Directorate for Quality Management
(EDQM) of the Council of Europe that publishes regularly updated guidance on quality
and safety of blood, tissues and cells
359
. The guidance is widely accepted in the EU and
the other Council of Europe Member States as representing best practice in this sector.
EDQM commits to ensure that their guidance does not contradict the provisions of the
EU BTC legislation. In just one case, good practice guidelines (GPG) for blood
establishments, a Directive was amended to require that Member States take the EDQM
guidance into account
360
. In general however, as described in Section 5.1, while the
357
In NL and UK, these authorities have common fora to discuss topics of common interest. In some countries, like FI
or IE, the authorities for BTC, medical devices and medicinal product are within one organisation.
358
Meeting between the Commission and FDA/CBER to discuss the EU BTC evaluation.
359
EDQM Guide for tissues and cells; EDQM Guide for blood.
360
Commission Directive (EU) 2016/1214 of 25 July 2016 amending Directive 2005/62/EC as regards quality system
standards and specifications for blood establishments.

77 | P a g e
EDQM guidance has been regularly updated to reflect important developments in the
area, the EU legislation has remained more static and in some cases even outdated,
resulting in some degree of incoherence internationally.
5.4.4.1 Some diverging areas of BTC regulation between the US and the EU
The FDA is the sole regulatory entity with the authority for regulating blood and blood
components, cell, tissue and tissue-based products (HCT/Ps) in the U.S. FDA does not
classify HCT/Ps. Developers make their determination on classification and FDA
provides recommendations or binding decisions if asked. HCT/Ps are regulated only by
the Public Health Service Act, with traditional, minimally processed substances falling
under the lighter rules in Section 361 (comparable with the EU TC framework). The
legal basis for this regulation is limited to the prevention of communicable disease
transmission, a scope that is narrower than the legal basis for regulation in the EU. More
than minimally manipulated substances fall under the more stringent rules of Section 351
as well as those of 361
361
(comparable with the EU ATMP framework).
Further criteria cause certain HCT/Ps, including some that are combined with handling
agents or other synthetic substances, to be classified as medical devices. The US
classifies some BTC differently from the EU
362
.
Tissue and cell stakeholders find the differences in classification to be a burden that
makes export to the EU more difficult
363
,
364
. This is in particular relevant for
demineralized bone supplied by US companies and for bone marrow, and other types of
haematopoietic stem cells, which are often exchanged globally.
FDA discusses classification of tissue and cell based products across the different sectors
in a Tissue Reference Group and publishes common recommendations on classification
that provide clarity and certainty to stakeholders in the U.S.
The regulatory authorities in Korea, Japan and Australia have an approach similar to the
U.S. This contrasts with the EU where, as described above, each sector has its own
advisory body, with the exception of the BTC sector.
361
Although the extent of manipulation is the key criterion, other criteria also apply. To fall under 361 alone, they need
to (1) be minimally manipulated, (2) be intended for homologous use only (based on the manufacturers objective
intent), (3) not be combined with another article and (4) not have a systemic/ metabolic effect or be dependent on the
activity of a living cell, unless for autologous or reproductive use. See 21 CFR 1271.10(a).
362
Such as unrelated haematopoietic stem cell transplants, encapsulated pancreatic islet cells and demineralised bone
combined with handling agents.
363
Summary of the Blood, Tissues and Cells Stakeholder Event 20th September 2017.
364
Summary Minutes of the Meeting between American Association of Tissue Banks (AATB) and the European
Commission (DG SANTE B4) 9 March 2018.

78 | P a g e
5.4.4.2 Use of different types of guidance allow FDA to keep technical specifications
up to date
As noted under the Relevance criterion, it is challenging for authorities to keep the many
specific technical requirements in the BTC legislation up-to-date and in line with
changing technologies and risks.
To address this same challenge, FDA uses different types of guidance:
FDA regularly publishes ‘guidance for industry’ to recommend measures for
achieving compliance with binding legislation and regulations; such guidance
itself is not legally binding but is widely followed. A key advantage of issuing
guidance is that its development takes less time, on average 6-12 months
(compared to at least 12 months for a change to regulations), and therefore allows
for more flexible updating of technical specifications.
When needed, FDA/CBER can also rapidly issue guidance for immediate
implementation, with the submission of public comments permitted any time after
issuance of the guidance. In 2016, for example, the risks posed by Zika virus
(ZIKV) were addressed mostly through guidance for immediate implementation.
It is also notable that documents developed by professional associations (e.g.
guidance from the American Association of Blood Banks are formally recognised
by FDA as an acceptable approach to meet FDA requirements. This collaboration
has worked well for standards developed by the association on a donor history
questionnaire and a Circular of Information required by legislation to be issue to
hospitals together with blood components for transfusion.
5.5 EU Added Value
This assessment criterion is addressed by two evaluation questions addressing overall
added value at the EU level and the impact of more stringent requirements at national
level.
In general, the Directives improved the quality and safety of BTC in a manner
that would not have happened, or would have happened more slowly, without
EU legislation. However, more stringent national requirements, although
permitted by the Treaty, limit the EU added value, particularly in terms of
exchanges between Member States.

79 | P a g e
5.5.1 Evaluation Question 13: To what extent has the legislative framework
at EU level added value to the regulation of blood and tissues and cells
across the EU-28 in a manner that could not have been achieved by
measures taken at national or global level?
SUMMARY ANSWER: The Directives improved safety and quality across the
EU, through defining minimum safety and quality requirements and putting
oversight in place. This achievement would have taken considerably longer, would
have developed in divergent ways bringing inconsistent levels of safety and quality
and might not have happened in some Member States, without EU legislation.
5.5.1.1 Safety and quality rules and BTC oversight were introduced in many
Member States
The situation regarding safety and quality rules, as well as oversight, varied considerably
across the EU prior to the adoption of the legislation. A number of the economically
stronger Member States had robust rules and oversight in place, particularly those that
had needed to respond to transfusion-transmitted epidemics of HIV and hepatitis (see
Section 2.1). Many others, however, and all of those that joined the EU after the adoption
of the legislation, had few, or no, safety and quality rules and little oversight in place and
relied on the professionals to follow international safety and quality standards. Annex V,
describes the baseline situation, referring to a number of key source documents. A recent
retrospective survey of tissue and cell authorities demonstrated that, prior to the adoption
of the 2004/23/EC, or to their accession to the EU, 7 Member States did not have binding
rules for safety and quality of replacement tissues and 8 did not have equivalent rules for
haematopoietic stem cells or for MAR. Similarly, 12 had no regulatory oversight for
MAR, while 7 and 8 had no oversight for replacement tissues and haematopoietic stem
cells, respectively
365
.
The early implementation reports
366
and discussions in Public Health Programme funded
actions carried out after the adoption of the legislation, pointed to intense activity across
the EU in the establishment of new inspection and vigilance systems. Professionals saw,
and still see, the initiative as one that increased, or standardised, quality and
safety
367
,
368
,
369
,
370
. It appears that many, or even most, blood and tissue establishments
had to improve their facilities and their quality management systems.
365
Minutes of tissue and cell competent authority meeting May 2019.
366
First report on the application of the Blood Directive June 2006;
Summary Table of Responses from Competent Authorities: Questionnaire on the transposition and implementation of
the European Tissues and Cells regulatory framework February 2007.
367
European Blood Alliance (20 Book 1 Safety by Regulation (2013).
368
Burgermeister J (2004) Doctors hail new EU directive on tissues and cells. BMJ 328:10.
369
Governing the Human Body: Examing EU Regulatory Developments in Relation to Substances of Human Origin
(2005) Journal of Social Welfare and Family Law, 27:3-4.
370
Hartshorne G (2005) Challenges of the EU ‘tissues and cells’ directive. Reproductive Biomedicine Online 18
August 2005.

80 | P a g e
5.5.1.2 Significant added value of EU-wide vigilance, traceability and networking
Significant added value is seen from the increased networking and collaboration between
Member States, resulting from the implementation of the EU provisions, in a number of
areas.
The regular meetings of authorities (Expert Group CASoHO E01718) have
facilitated discussion, and helped to find solutions, for many issues, as
demonstrated in Annex VIII
371
.
The provisions for reporting serious adverse reactions and events on an annual
basis to the Commission has brought the added value of an aggregated view of
safety, allowing a more accurate estimation of risk.
The Single European Code for tissues and cells has improved traceability of
tissues and cells as they circulate in the EU, with public transparency on the
authorisation status of tissue establishments
372
.
The legislation has provided an impetus for significant levels of EU financial
support for implementation through the Public Health Programme. Apart from the
practical outputs of the actions: inspection guidance, training for professionals
and inspectors, common vigilance tools, IT platforms etc., these projects and Joint
Actions have brought positive outcomes in terms of building and sharing strong
expertise across the EU. See Annex IX.
The organisations responding to the Online Public Consultation confirmed these positive
findings
373
.
5.5.2 Question 14. To what extent do stricter national measures pose an
obstacle to exchange of supplies between Member States?
SUMMARY ANSWER: More stringent national requirements, permitted by the
Treaty, emerged as a factor posing barriers to effective sharing of BTC within the
EU and with third countries. These are put in place, to a significant extent due to
outdated technical provisions and important developments in the sector.
371
The expert group of competent authorities has established BTC expert sub-groups focusing on Vigilance practices
(Vigilance Expert Subgroup), Inspection practices (Inspections Expert Subgroup) and traceability/coding practices
(Coding Expert Sub-group – for tissues and cells only).
372
See the EU compendium of authorised tissue establishments.
373
See the summary results of the Public Consultation.

81 | P a g e
5.5.2.1 More stringent national requirements pose barriers to inter-Member State
exchanges of BTC
The Treaty
374
gives competence to the EU for the adoption of safety and quality
standards but allows Member States to put in place more stringent requirements to
protect their citizens. Many Member States have chosen that option, in particular to
address important developments in the sector and the difficulties of updating the EU
provisions quickly, as well as to reflect geographical differences in transmissible disease
risk. This was particularly highlighted in a mapping exercise, conducted by the
Commission services in 2015, on more stringent donor testing measures required or
recommended for BTC donors in Member States. The mapping exercise was published
for blood
375
and for tissues and cells
376
and indicates that the level of viral safety
achieved across the EU is no longer standardised. This has been highlighted as limiting
EU added value by professionals in the field
377
,
378
by third country organisations that
export to the EU
379
and by researchers wishing to use BTC in multi-country clinical
trials
380
. In the Public Consultation, 30% of responding organisations indicated that more
stringent national requirements pose significant obstacles to inter Member State
exchanges.
374
Article 168 (4)(a) TFEU.
375
"Mapping of More Stringent Blood Donor Testing Requirements - Mapping Exercise 2015".
376
“Mapping of More Stringent Tissues and Cells Donor Testing Requirements - Mapping Exercise 2015”.
377
Summary Minutes of the Meeting between the International Society for Cell Therapy and DG SANTE B4 14 June
2018.
378
Summary Minutes of the Meeting between the Board of the European Association of Tissue Banks and DG SANTE
B4 18 September 2018.
379
Summary Minutes of the Meeting between American Association of Tissue Banks (AATB) and the European
Commission (DG SANTE B4) 09 March 2018.
380
Internal Commission discussions with the Directorate General for Research.

82 | P a g e
6. CONCLUSIONS
This evaluation evaluated the BTC legislation, adopted in 2002 for blood and 2004 for
tissues and cells, according to the 5 criteria of effectiveness, relevance, efficiency,
coherence and EU-added value. It builds on a Public Consultation, an external study,
literature review, expert interviews and scientific documents, as well as a wide range of
meetings with key stakeholders and National Competent Authorities. The two main
limitations for this exercise were low data availability on the situation in Member States
prior to the adoption of the legislation, and difficulties to quantify costs incurred by the
BTC legislation.
i) The legislation effectively increased the level of safety and quality of BTC
across the EU
Common minimum safety and quality requirements were mandated in all Member States
and oversight was established to ensure inspection, authorisation and vigilance, bringing
significant changes compared to the situation prior to the adoption of the BTC
legislation. Despite a number of new disease outbreaks since the early 2000s,
transmissions of infections by BTC have been maintained at low to negligible levels.
This has restored public trust in the BTC sector and its oversight, which had been
negatively impacted by large-scale transmission of infectious diseases such as HIV and
hepatitis by BTC in the 80’s and 90’s.
These safety and quality objectives remain relevant. However, there have been
significant developments since the legislation was adopted. Some contributed to
improvements in safety and benefits for patients, but others brought new risks. This
partly puts the future relevance and effectiveness of the legal frameworks into question.
What follows are areas where the evaluation found the legislation to have shortcomings
or gaps.
ii) The current rules are no longer up to date with the dynamic BTC sectors
Many of the detailed and prescriptive safety and quality requirements are no longer
adequate to address fully the challenges associated with rapid technological, scientific
and epidemiological developments.
For example, many requirements for BTC donor testing in the current legislation are out
of date following the development of more sensitive tests for HIV and hepatitis. The
current required tests provided for in the BTC legislation no longer reflect current best
practice. In addition, the current donor eligibility rules no longer address many of the
important risks for ensuring safety. For example, current donor acceptance requirements
do not fully take into account (re-)emerging diseases (such as Zika, hepatitis E, malaria

83 | P a g e
or West-Nile Virus). Similarly, current criteria for the quality verification of the BTC
applied to patients are either lacking or are reported not to reflect current clinical uses
and processing methods. Some Member States have added new, more stringent safety
measures, which can sometimes reduce the relevance of those already provided for in the
BTC legislation or render them unnecessary. While this is possible under the Treaty, it
has resulted in divergence and unequal protection of patients across the EU.
In addition, some new therapies, such as faecal microbiota transplants (FMT) and
donated breast milk, have developed since the BTC legislation was adopted and are not
covered by the definitions and scope of the current BTC legislation. This has resulted in
divergent classifications and regulatory approaches, in different Member States, to ensure
safety and quality for these treatments.
In line with these findings, many of the EU requirements no longer reflect best practice
and guidance issued by authoritative expert bodies in the field, including the Council of
Europe or the European Centre for Disease Prevention and Control (ECDC).
The approach in the current legislation is that requirements should be updated in line
with technical and scientific progress through the comitology procedure. However, it has
proven resource intensive and challenging due to the very rapid and multiple changes in
the field in combination with the current rather specific technical requirements. The
efficiency assessment shows that the application of many outdated donor selection
provisions now imply costs that are not proportionate to the benefits in terms of safety
for recipients.
iii) Key oversight principles are not sufficiently robust
One of the main findings of the effectiveness assessment is that several oversight
requirements in the BTC legislation are not adequately specified to ensure robust
oversight of the BTC sectors. These shortcomings relate to a lack of generic principles
that would ensure independence of the competent authorities, from BTC establishments
and a lack of provisions for verification of effective implementation of oversight
functions in Member States. The shortcomings are common across the BTC sub-sectors.
The shortcomings concern the oversight provisions relating to vigilance, inspections and
authorisations. Requirements that emerge as being overly general, leaving room for
different interpretations and variable implementation across the EU, resulting in unequal
levels of protection for EU donors and patients. For example, inspection and
authorisation are carried out differently, and reporting of adverse incidents and alerts is
inconsistent, across Member States, limiting the possibilities for aggregation of data and
evaluation of risk. Provisions for fixed frequency inspection are also seen as cost
inefficient. The lack of common requirements for reporting data on numbers of
donations, clinical applications and international exchanges limit the potential for data
aggregation for policymaking and sufficiency monitoring. In addition to compromising

84 | P a g e
the effectiveness of legislative framework, these shortcomings reduce its potential EU
added value.
The limited specifications for oversight are particularly relevant in the context of an
increased number of commercial actors to be authorised and a high degree of innovation
to be authorised, as shown in the assessment of relevance.
iv) Some citizen groups, such as donors and offspring are not adequately
protected
Another finding of the effectiveness assessment is that, while the current provisions
focus on the safety of transfusion, transplantation and medically assisted reproduction
recipients, there are limited provisions to protect the health of other specific groups of
citizens, notably BTC donors or offspring of medically assisted reproduction treatments.
There are insufficient measures in place to protect BTC donors, since it is not required to
report serious adverse reactions that affect donors unless the quality or safety of the
donated substance has been affected, even in cases of donor death. Although many
Member States do report donor reactions to the EU level on a voluntary basis, this is not
consistently done. Protecting donors is considered key to maintaining high levels of
safety and quality of the donated BTC, as well as high levels of public confidence in the
system. Donor protection and public confidence are critical to ensure sustained levels of
donation, in particular where there is an increase in demand and therefore also in
expectations to donate.
Children born from sperm, egg or embryo donation were highlighted, by the medically
assisted reproduction sector, to be inadequately protected by the current provisions on
safety and quality, compromising overall effectiveness. While there are requirements to
protect women from diseases possibly transmitted through donated gametes or embryos,
there are no requirements to follow-up on children born from these donations and very
limited requirements for testing gamete donors for genetic conditions. This sub-sector is
growing rapidly so the shortcomings highlighted are more relevant today than they were
in the past.
v) The legislation does not keep pace with innovation
This evaluation has revealed a high level of innovation in the BTC sector over the past 15
years. The evidence suggests that this is likely to continue or increase. New ways to
collect, prepare, store and apply BTC to patients can bring significant health benefits,
usually in a cost-effective manner that achieves wide patient access. But these
developments also need to be enframed by adequate legislation. Increasing
commercialisation of some innovations, sometimes with unsubstantiated claims for
clinical benefits, can exacerbate the challenge.

85 | P a g e
The effectiveness assessment pointed to a gap in provisions for authorisation of novel
BTC processing methods, including patient follow-up, monitoring of clinical outcome
and demonstration of clinical effectiveness, which are now too limited to ensure high
quality and effective use of BTC. For example, blood components can be prepared in
new ways, without clinical studies to demonstrate equivalent or improved functioning of
the component in the recipient. Professionals and some authorities have put more
stringent authorisation measures in place, resulting in divergent levels of safety and
effectiveness for patients across Member States.
Facilitating innovation also requires legal clarity and good interplay with other legislative
frameworks, particularly those regulating medicinal products and medical devices. The
coherence assessment has shown that, while most BTC and BTC-based products fall
clearly into one or other regulatory framework, there are sometimes difficulties in
defining some of the borderlines, particularly for the more novel BTC described in the
relevance assessment. Some criteria are interpreted differently and the EU level support
mechanisms for classification lack a cross-sectoral forum for classification discussions
and advice. This causes some uncertainty for producers and users on the applicable
regulatory frameworks in different Member States. Organisations representing blood and
tissue establishments reported that this uncertainty can create disincentives to embark on
potentially costly innovation.
The coherence assessment also pointed to an increasing number of innovations where
medical devices are used at the bedside or in the operating theatre to prepare BTC from a
patient for immediate application to the same patient, following a processing step. While
the exclusion, from the current legislation, of tissues and cells that are collected and used
within the ‘same surgical procedure’ is important for unprocessed BTC, it limits the
effective oversight of some promising autologous BTC therapies where processing is
carried out near to the patient.
Many blood and tissue establishments also pointed to insufficient collaboration between
authorities responsible for different legal frameworks when BTC become starting
materials for medicinal products or medical devices. This concerns the need to define
appropriate donor eligibility criteria where BTC are to be used for the manufacture of
batches of manufactured products. Effective cross-sectoral communication for vigilance
reporting in these circumstances is also seen as important for the facilitation of
innovative developments based on BTC as starting materials.
vi) Requirements are insufficient to support sufficiency and a sustainable supply
for all BTC
The effectiveness assessment also underlined that the current legislation is limited in
provisions that help achieve sufficiency, although one of the objectives for adopting the
legislation was indeed to ensure sufficiency and continuity of supply. The relevance
assessment shows a rise in commercialisation and internationalisation of some BTC, in
particular plasma used to manufacture medicinal products. While the number of private

86 | P a g e
plasma collection centres in the EU has increased in recent years, this is far from
sufficient to keep up with increasing demand for plasma derived medicines. This has led
to high dependency on import of plasma from the US.
Crises that might threaten the donation and supply of BTC, such as epidemiological
outbreaks, have highlighted the even greater importance of ensuring sufficiency as part of
contingency planning for emergencies. The current legislation does not address this topic
and there are divergent approaches across Member States. While some Member States
have preparedness plans to manage a supply interruption, others do not. This makes
intra-Member State collaboration more complex in cases of need, and limits the potential
EU added value.
Were possible areas for simplification identified?
This evaluation identified the following areas for possible simplification: the oversight
for the blood, tissue and cell sector, the approach to updating of technical requirements,
the classification of novel therapies that might also involve other legal frameworks, the
reporting requirements for professionals and authorities and inspection frequency.
Will the issues identified resolve or deteriorate over time?
Many of the gaps and shortcomings identified in this process and described in this report
have been evident for some years and both professionals and authorities have initiated
voluntary collaborative actions to address them, with support from the EU Public Health
Programme and collaboration with expert bodies such as ECDC and the Council of
Europe.
Trends identified in this evaluation may increasingly challenge access to safe blood,
tissue and cell therapies, of high quality, for EU citizens.

87 | P a g e
Annex I: Procedural information
1. Lead DG/ Planning
The Directorate General for Health and Food Safety (DG SANTE – Unit B4) is the lead
DG for this evaluation. (PLAN/2016/154).
2. Organisation and timing
The initiative is a mixed ex-post Evaluation. An Interservice Steering Group (ISSG) was
established in October 2016 and met regularly to provide input at key stages of the
process. The following services were represented in the ISSG:
SG (Secretariat-General);
LS (Legal Service);
GROW (Internal Market, Industry, Entrepreneurship and SMEs);
RTD (Research and Innovation);
JUST (Justice and Home Affairs).
The Roadmap was published on 7 January 2017 and was followed by an Online Public
Consultation, which ran from 29 May until 14 September 2017. From the second half of
2017 until the second half of 2018 independent supporting study was undertaken by an
external contractor.
3. Exceptions to the better regulation guidelines
None
4. Evidence, sources and quality
This evaluation report is supported by the following sources of evidence:
Study supporting the evaluation of the EU legislation on Blood and Tissues and
Cells (SANTE/2017/B4/010)
Submissions to the Online Public Consultation and the summary report of these;
Inputs during the September 2017 Stakeholder Event and the summary report of
this event;
Meetings with key stakeholders and the minutes of these meetings;
Meetings of the BTC national competent authorities and the minutes of these
meetings.
Additional sources and literature used are referenced in the annex to the study listed
above.

88 | P a g e
Annex II: Transmissions of infections to EU patients by blood
transfusion, treatment with plasma derived medicinal products and
tissues and cells in the 1980 and 1990s.
This Annex provides details on the transmissions and sentinel events that occurred across
Europe and raised concerns regarding the safety of BTC.
Blood and plasma derived medicinal products
1. United Kingdom:
Transmission of HIV and Hepatitis to Haemophilia Patients:
In the 1970s and 1980s over 4,500 people with haemophilia and other bleeding disorders
were multiply-infected with HIV, Hepatitis B and C and a range of other blood-borne
viruses. Over 3,000 people have since died and of the 1,243 people known to be infected
with HIV less than 250 are still alive
381
.
Factor concentrates, such as Factor VIII for treatment of haemophilia A were, in the
1970’s, a revolutionary new treatment allowing patients for the first time to be treated
prophylactically, to reduce the likelihood of bleeds and the resulting joint damage. These
new treatments, however, were produced using a process, which involved pooling human
blood plasma from up to 40,000 donors and concentrating it to extract the required factor.
Blood products were known to transfer viruses such as Hepatitis and this risk was vastly
increased when they were pooled using the new techniques. This risk was further
exacerbated when supplies of UK produced factor concentrates were not sufficient to
cope with NHS demand, and products were increasingly imported from the United
States. In the US, high-risk paid donors were used, as well as using blood collected in
prisons increasing the risk of contamination with blood-borne viruses
An independent inquiry under Lord Archer reported in 2009 and made strong
recommendations but as an independent inquiry it was unable to insist on testimonies
from key individuals and organisations. In 2008, the Scottish Government set up a public
inquiry under Lord Penrose; no culpability was assigned. In 2018, the Langstaff Inquiry
was commissioned by the Government and began taking evidence from victims. Its work
is currently ongoing
382
,
383
.
Transmission of vCJD:
Variant Creutzfeldt–Jakob disease (vCJD) is a fatal neurological disease, caused by the
same agent (abnormal variant of prion protein) as bovine spongioform encephalopathy
(BSE or ‘mad cow disease’) in cattle and caused by eating beef from affected animals. It
381
Putting an end to the sequelae of contaminated blood transfusions. The Lancet Haematology. Volume 5, Issue 6,
PE232, June 2018. , June 01, 2018Pe232, June 01, 2018.
382
https://www.bbc.com/news/health-45591584.
383
https://haemophilia.org.uk.

89 | P a g e
was first identified in the UK in 1996. By the end of 2012 there had been 174 cases in the
UK, peaking in 2000.
Four cases of transfusion-transmitted vCJD infection have been identified to date, from
three apparently healthy donors who later developed vCJD. All occurred with non-
leucodepleted red cells donated before 1999. Three of the four recipients died of vCJD a
few years after the implicated transfusion. The fourth recipient died of unrelated causes
but had abnormal prion protein in the spleen at post-mortem examination (significance
uncertain). There are still many uncertainties around the pathogenesis and epidemiology
of vCJD and no practical screening test for blood donors has yet been developed. The
vCJD risk-reduction measures introduced in the UK include importation of plasma for
fractionated blood products (1998), leucodepletion of all blood components (1999),
importation (and viral inactivation) of fresh frozen plasma for all patients born on or after
1 January 1996 (when dietary transmission of vCJD is assumed to have ceased) (2002),
exclusion of blood donors who have received a blood transfusion in the UK since 1980
(2004) and importation of solvent detergent plasma for adult patients undergoing plasma
exchange for thrombotic thrombocytopenic purpura (2006).
Countries outside the UK, including the USA, Canada, New Zealand, Australia, Hong
Kong and several European countries including Germany, Switzerland, Austria and Eire
have taken the precautionary step of excluding blood donors who have spent more than a
defined period living in the UK between 1980 and 1996 in order to avoid the risk of
vCJD entering the blood transfusion chain.
2. Ireland
Hepatitis C Infection of Women who received Anti-D Immunoglobulin
Anti-D immunoglobulin, a product made from donated blood, was given to new mothers
whose own blood type was Rhesus Negative but who gave birth to Rhesus Positive
babies. It was administered to safeguard the health of future babies the woman might
have as some of the Rhesus Positive could have passed through the placenta into her
bloodstream and she would have developed antibodies to it. Those antibodies, left in her
system, could seriously damage or kill the foetus in a future pregnancy.
In the 1960s, around 40 babies a year died in this way so the development of the anti-D
treatment was considered a welcome breakthrough and its use became normal practice in
maternity hospitals in the 1970s.
In 1994 the Blood Transfusion Service Board (BTSB) became aware of the
contamination of two batches of Anti-D from two separate donors, one in 1977 and the
other in 1992. It was thought that Anti-D from these batches had been administered to
over 1000 women. The Finlay Tribunal of Inquiry was set up in 1996 and concluded the
contamination of the anti-D batches could have been prevented. The Hepatitis-C

90 | P a g e
Compensation Tribunal, set up on a transitional basis in 1995, was formally established
in 1997 as a result of the findings
384
.
On foot of the tribunal report, a criminal investigation began into the BTSB. Three senior
employees were arrested in 2003 and two were charged with causing grievous bodily
harm to Anti-D recipients, but one died before his case could come to court and the other,
who instigated lengthy legal challenges to her arrest, finally had all charges against her
withdrawn in 2009 due to the death of witnesses.
The Hepatitis-C Compensation Tribunal paid out around €310m in 1,539 awards to
women, their spouses, and children in its first seven years. In total it has paid out almost
€1bn in awards and legal costs associated with more than 4,500 claims. There are still
around 800 claims to be dealt with and further transmission could yet be confirmed
385
.
Infection of Haemophiliac Patients with HIV and Hepatitis C from Blood Products:
In May 2000, the Lindsay Tribunal of Inquiry was established to investigate the infection
of an estimated 252 haemophiliacs with HIV and/or hepatitis C from contaminated blood
products. At the time the tribunal began its work, seventy-eight haemophiliacs had died
from illnesses related to their infection. It emerged that 104 individuals had been infected
with HIV prior to 1985. These patients had received contaminated Factor IX (produced
nationally by the Blood Transfusion Service) and commercial (non-heat treated) batches
of Factor VIII. Factor IX product was prepared from donations prepared before the
introduction of routine HIV antibody testing. The commercial product (Factor VIII) was
purchased from the USA and was prepared from paid blood donors in the USA where
there was a much higher risk of HIV/AIDs at the time. In addition, these products were
not heat treated, a process which was known at the time to inactivate such viruses. The
Inquiry was critical of the decision making process undertaken by Clinicians and Senior
Scientific Staff at the BTSB in relation to the production, purchasing and continued use
of these products despite the emerging knowledge of HIV/Aids infection in the
Haemophiliac Community in the 1980s. A Compensation Court was also established as a
result of the findings of this Inquiry and it is estimated that over €1bn has been awarded
to date as a result of state failings regarding the safety of blood and blood products
386
,
387
.
3. Romania
HIV Infection of Children from ‘unscreened and untested’ blood microinfusions
In 1989, a dramatic epidemic of nosocomial HIV infection was discovered
predominantly among orphans and hospitalized children in Romania, infected through
transfusions of unscreened blood (micro infusions of unscreened and untested blood
administered to counteract malnourishment and anaemia) and injections with improperly
384
The Finlay Report.
385
https://www.irishexaminer.com/viewpoints/analysis/anti-d-scandal-was-a-bloody-disgrace-259488.html.
386
Report of the Tribunal of Inquiry into the Infection with HIV and Hepatitis C of Persons with Haemophilia and
Related Matters..
387
https://www.irishtimes.com/news/health/victim-of-contaminated-blood-products-was-awarded-2-96m-1.3251084.

91 | P a g e
sterilized equipment. 94% of initial cases identified were in children under the age of
1
388
.
A combination of factors, from failure of screening blood to lack of communication
about the risks to physicians and citizens – caused HIV to spread rapidly among children,
resulting in an unprecedented epidemic. By the year 2000, 60% of Europe's pediatric
HIV/AIDS cases were registered in Romania, mostly in infants living in public
institutions. In mid-2002, 12,559 cases of HIV were registered in Romania (9936 in
children), of which 2699 were already deceased. From 2002 onwards, with the assistance
of the WHO, a number of government programmes were established to help tackle this
epidemic by preventing the spread of further infection and also by providing appropriate
treatment to those children already infected.
4. France
HIV Infection by blood transfusion and treatment with plasma derived medicinal
products.
In total, it is estimated that approximately 4000 individuals in France may have been
infected with HIV during a period of time between the late 1970s and early 1980s. Of
these, some 1,250 haemophiliacs are believed to have been infected. Of these, 400 have
died.
Errors identified included the collection of blood in prisons and in public places
frequented by drug addicts; the neglect of a memorandum on how to prevent
contamination by blood transfusion, for more than a year and delays in the decision to
treat blood by heating. It was inferred that the French Government did not introduce
routine screening for HIV in a timely manner and delayed the commercial availability of
a blood test used in the United States in favour one being developed in France.
As a result of these events, a former Prime Minister, a former Social Affairs Minister,
and a former Secretary of State for Health were brought before the Court of Justice, a
special tribunal created in 1993 in France to judge members of the government for crimes
or misdeeds allegedly committed during the performance of their duties
389
. They were
required to answer to charges of involuntary homicide and involuntarily compromising
the well-being of others. The Prime Minister and Social Affairs Minister were acquitted
but the former Health Minister was convicted for his role in the contamination. In
addition, in 1993, the former Head of the National Blood Bank was sentenced to four
years in prison, and the Head of the Blood Transfusion Research Bureau was jailed for
two years. Two others were given suspended sentences
390
.
Tissues and Cells
388
Dente K, Hess J. Pediatric AIDS in Romania--a country faces its epidemic and serves as a model of success.
MedGenMed. 2006; 8(2):11. Published 2006 Apr 6.
388
Popovici F. et al. Acquired Immunodeficiency in Romania. The Lancet. Volume 338, ISSUE 8768, P645-649,
September 14, 1991.
389
Ex-Ministers to face trial in French Blood Scandal BMJ. 1998 Aug 1; 317(7154): 302.
390
Scandal Over Tainted Blood Widens in France.

92 | P a g e
While tissue and cell transplantation has not been associated with the large scale
transmission of infectious diseases that has occurred in the transfusion of blood
components and the administration of blood derived medicinal products, viral, bacterial,
and fungal infections have been transmitted via transplantation of tissue allografts such
as bone, skin, cornea, and heart valves, and cells such as islets, hematopoietic stem cells,
and semen
391
. Single donors may provide allografts for >100 organ and tissue recipients;
each allograft carries some, largely unquantifiable, risk of disease transmission which
highlights the need for tight quality and safety criteria such as appropriate donor
screening and testing
392
The following are a number of key case studies reported in
literature and in the media concerning the transmission of infectious diseases as a result
of tissue and cell transplantation.
Case Study 1: HIV Transmission by Bone in Germany
393
.
In 1997 it was reported that a leading transplantation centre in Germany was ordered to
pay compensation to a 58 year old man who developed AIDS after receiving a bone graft
from a donor who was a drug misuser. In January 1985 the man, who had a clavicle
fracture, received a transplanted lyophilised bone chip from a donor who was a drug
misuser, and who had died from drug overdosage. The donor was not tested for HIV. At
the time there were only around 150 cases of AIDS in Germany. At least two more
patients, out of 12 who received bone transplants from the same donor, later died from
AIDS. The Superior Court of Hanover decided there was no doubt that the plaintiff had
been infected with HIV via the bone graft. Although the judges conceded that in 1985 it
was not known that viral diseases could be transmitted by lyophilised bone grafts, the
court insisted that the donor’s drug addiction should have been a warning signal to
doctors.
Case Study 2: Transmission of HIV-1 from a sero-negative Organ and Tissue Donor in
the USA
394
.
In 1991, a woman whose only risk factor for HIV-1 infection was the receipt of a bone
allograft in December 1985 was identified by the health department in her state as being
infected. Subsequent investigation revealed that the donor was a 22-year-old HIV-1—
seronegative man who died after being shot in the head in October 1985, that 4 solid
organs and 54 other tissues had been distributed from the donor, and that a total of seven
of the recipients (4 solid organ recipients and 3 fresh frozen bone recipients) were
infected with HIV-1.
Case Study 3: Transmission of hepatitis C virus to several organ and tissue recipients
from an antibody-negative donor in the USA
395
.
391
Eastlund T. 1995. Infectious disease transmission through cell, tissue, and organ transplantation: reducing the risk
through donor selection. Cell Transplant Sep-Oct;4(5):455-77.
392
Fishman JA, Greenwald MA, Grossi PA. 2012. Transmission of infection with human allografts: essential
considerations in donor screening. Clin Dis Infect Sep; 55(5):720-7.
393
Karcher HL. HIV transmitted by bone graft. BMJ. 1997; 314(7090):1300.
394
Simonds RJ et al. 1992 Transmission of Human Immunodeficiency Virus Type 1 from a Seronegative Organ and
Tissue Donor. N Engl J Med 1992; 326:726-732.

93 | P a g e
In 2002, hepatitis C virus (HCV) transmission through tissue transplantation had been
rarely reported, however it was possible that a donor with undetected viremia may infect
several recipients. In June 2002, in the US, a patient developed acute symptomatic
Hepatitis C shortly after transplantation of a patellar tendon with bone allograft. The
donor had died in 2000 and his premortem serum had tested negative for anti-HCV (2
nd
Generation ELISA test). Ninety-one tissues or organs had been recovered from the
donor. The donor was anti-HCV-negative but was HCV RNA-positive (genotype 1a).
Forty persons received transplants from this donor during a 22 month period. Five
persons were HCV-infected before transplantation or had a genotype other than 1a, and 5
persons had no post-transplantation serum specimens available. Of the remaining 30
recipients, HCV infection occurred in 8 recipients: 3 of 3 organ recipients, 1 of 2
saphenous vein recipients, 1 of 3 tendon recipients, and 3 of 3 tendon with bone
recipients. These 8 recipients had viral isolates genetically related to those of the donor.
No cases occurred in recipients of skin (n = 2), cornea (n = 1), or irradiated bone (n =
16).
Comment: In this instance as with the previous case, it was likely that both donors were
in the ‘window period’ of infection before the development of detectable antibodies to
HIV and HCV. Cases such as these demonstrate not only the possibility of the spread of
infection to numerous recipients from a single donor but also of the need to ensure that
appropriate testing protocols using the most up to date (but validated) test methods are
implemented for both organ and tissue donors.
Case Study 4: Creutzfeldt-Jakob Disease associated with Dura Mater Graft processed in
Germany and transplanted in Japan
396
.
During 1975–2008, a total of 132 cases of dura mater graft-associated Creutzfeldt-Jakob
disease (dCJD), a fatal neurodegenerative disease caused by replicating, transmissible
prion proteins, had been identified in Japan and accounted for >60% of patients
worldwide with dCJD. This relatively high number of cases was most likely related to
the increased use in Japan of the primary vehicle of transmission, Lyodura brand
cadaveric dura mater grafts produced before May 1987 (After May 1987, the
manufacturer changed its production process to reduce the risk for prion transmission). It
had been identified that the manufacturer had previously mixed dura from multiple
donors during batch processing of single lots and sterilized the grafts with gamma
irradiation, a procedure that does not inactivate prions. It was also reported that the
company did not maintain records identifying donors, so they could not be traced. It is
estimated that 20,000 persons received a Lyodura graft each year during 1983–1987 in
Japan. During 2008–2017, an additional 22 dCJD patients, with onset from 1985 through
2016, were identified in Japan, resulting in 154 dCJD patients in Japan. No new dCJD
patient whose surgery occurred after 1993 has been identified. However, the latency
395
Tugwell BD et al. 2005. Transmission of Hepatitis C Virus to Several Organ and Tissue Recipients from an
Antibody-Negative Donor. Ann Intern Med;143:648–654.
396
Ae R, Hamaguchi T, Nakamura Y, et al. Update: Dura Mater Graft-Associated Creutzfeldt-Jakob Disease - Japan,
1975-2017. MMWR Morb Mortal Wkly Rep. 2018;67(9):274–278. Published 2018 Mar 9.
94 | P a g e
period is now known to be at least 30 years and because of the known potential for even
longer latency periods for prion diseases, this outbreak is likely to continue.
Data from the WHO Notify Library of adverse occurrences with Medical Products of
Human Origin
The Notify Library is a joint global initiative, co-sponsored by the World Health
Organization (WHO) and the Italian National Transplant Centre (CNT) as its
Collaborating Centre that supports the sharing of published vigilance information for
teaching purposes and for greater public transparency on the use of MPHO. The library
aims to be comprehensive, describing all types of reactions or events that might have
teaching value and assist in the estimation of risk in relation to what is known as
MPHO’s (Medicinal Products of Human Origin) including blood for transfusion and
tissues and cells for transplantation
397
.
The Notify Library is a publically accessible database of adverse outcomes collected and
analysed by dedicated editorial groups of international experts, regulators and clinicians.
The database is not a vigilance reporting programme but a collection and review of
information identified primarily by literature review (published articles in scientific
journals and/or books) although case reports from regulatory or professional vigilance
programmes (grey literature) are also considered for inclusion. For each adverse
occurrence type, at least one reference source is cited and the project’s collaborating
international experts provide a structured analysis.
A review of the records relating to viral infection of recipients of human tissues and cells
for transplantation in this database revealed the following number of references. Note:
These numbers to not relate to number of recipients but to the number of records
contained within the Notify Library (which is not exhaustive) and some records refer to
multiple recipients.
Replacement Tissues:
Musculoskeletal
Skin
Cardiovascular
Tissue
Ocular
Nerve
HIV
6
1
-
-
-
Hep B
1
-
1
2
-
Hep C
3
-
2
-
-
HTLV
1
-
-
-
-
CMV
-
3
-
2
-
Rabies
-
-
2
2
-
397
www.notifylibrary.org.
95 | P a g e
HSV
-
-
-
1
-
EBV
-
-
-
-
1
Cells:
Haemopoietic Stem
Cells
Pancreatic
Islets
Reproductive
Cells
HIV
1
-
2
Hep B
1
-
2
Hep C
-
-
-
HTLV
1
-
-
CMV
4
1
-
Parvovirus
3
-
-
Dengue
2
-
-
EBV
4
-
-
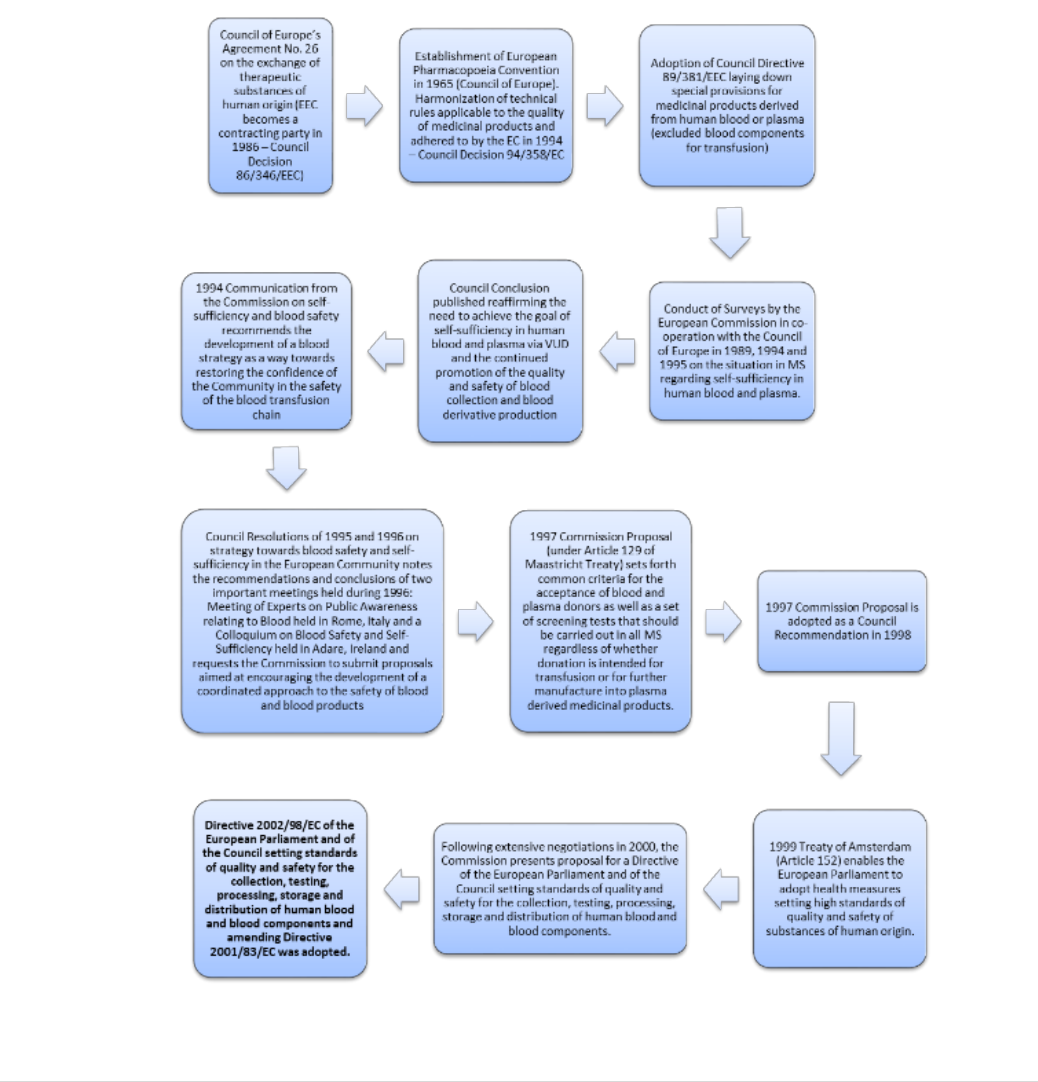
96 | P a g e
Annex III: The Background to the Regulation of Blood and Blood
Components in Europe
The adoption of the basic Act on blood and blood components in 2002 was preceded during the
previous decades by a series of initiatives and reports at European level. These are summarised
in the figure below and described in the subsequent text.

97 | P a g e
In 1958, the Council of Europe’s Agreement No. 26
1
on the exchange of therapeutic substances
of human origin became the starting point for cross-border activities in this field. Its main
purpose was to facilitate exchanges of human blood and its derivatives between Member States
of the Council of Europe in cases of urgent need and under the expressed condition that no profit
was made. In 1986, the European Community became a contracting party to this agreement as
outlined in Council Decision 86/346/EEC
2
.
Part 2 of the Council of Europe’s Agreement No. 26 outlined special provisions (basic quality
and safety criteria) for the collection and preparation of whole human blood, human red cell
concentrate, dried human plasma and derivatives such as human albumin, human plasma protein
fraction, human immunoglobulin, specific immunoglobulins, fibrinogen, coagulation factor VIII
and coagulation factor IX.
A further significant step in this field was the establishment of the European Pharmacopoeia
Convention, which was signed in 1964 under the aegis of the Council of Europe. It aimed to
harmonise the official technical rules applicable to the quality of medicinal products and
substances, including blood, and was adhered to by the European Community in June 1994 as
outlined in Council Decision 94/358/EC
3
.
The European Community had, in 1965, included human blood in the definition of medicinal
products (Council Directive 65/65/EEC
4
). In the late 1970’s and through the 1980’s, the reports
of blood contaminated with HIV were undermining the public’s confidence in the blood supply
and public health efforts were ongoing in trying to prevent the transmission by blood and other
substances of human origin of infectious agents. This led to the adoption of Council Directive
89/381/EEC
5
, which laid down special provisions for medicinal products derived from human
blood or plasma. This Directive included the concept of Community self-sufficiency in human
blood and plasma through the encouragement of voluntary, non-remunerated donations when
intended to be used for the production of plasma derived medicinal products but still excluded in
its scope blood, plasma and blood cells when used for transfusion.
In 1989, the Commission in cooperation with the Council of Europe undertook a survey
6
about
the situation in the MS regarding self-sufficiency in human blood and plasma. This was followed
by two additional surveys in 1994
7
and 1995
8
. The Commission’s resulting report
9
to the Council
on the state of Community self-sufficiency resulted in the Council adopting conclusions
10
in
which the need to achieve the goal of Community self- sufficiency in human blood and plasma
via voluntary unpaid donations through co-operation between Member States was reaffirmed and
the continued promotion of the quality and safety of blood collection and blood-derivative
production was inter alia agreed.
The Council's continuing concern about the quality, safety and efficacy of blood and blood
products resulted in it requesting, at its meeting of 13 December 1993, the Commission to
prepare a report related to the legal provisions and current practices in the Member States on the
collection, control, and treatment of blood and the distribution and trade in blood and blood

98 | P a g e
products with a view to proposing common safety criteria. Immediately following this meeting,
the Commission invited Member States to provide their legal regulations and administrative
provisions in this area
11
.
The 1994 Communication from the Commission on blood safety and self-sufficiency
11
was an
important report, which drew upon the responses provided by the Member States in written
submissions and during meetings of experts on self-sufficiency and blood safety convened by the
Commission. It addressed issues raised by the Ministers for Health, and provided, as a
background, a review of progress towards blood self-sufficiency in the Community as well as
coverage of this area by existing Community rules and regulations. It specified the need for
Community action in this area particularly in the context of the Council's Resolution concerning
the field of public health and the Commission Communication on AIDS and certain other
communicable diseases. It recommended the development of a blood strategy as a way towards
restoring the confidence of Community citizens in the safety of the blood transfusion chain and
fostering the goal of self-sufficiency.
Council Resolution of June 1995 on blood safety and self-sufficiency in the community
12
agreed
on the need to define a strategy for reinforcing trust in the safety of the blood-transfusion chain
and promoting self-sufficiency in the Community and outlined a number of potential activities
for the Commission to continue collaboration on at Member State level. These activities included
the development of policies and agreed procedures in the donor-selection process among blood-
collection establishments, the implementation of efficient, validated and reliable screening tests,
the development and use of quality-assessment criteria and good practices regarding the
collection, processing and transfusion of blood and blood products and patient follow-up
procedures, development of a haemovigilance system on the basis of existing networks for the
collection of epidemiological data related to the blood-transfusion chain, encouragement of
health professionals to make optimal use of blood and blood products, the establishment of basic
criteria for inspection and training of inspectors, and the dissemination to the public of
information on blood and blood products and on collection, processing and transfusion
procedures, taking into account socio-cultural differences.
Council Resolution of November 1996 on a strategy towards blood safety and self-sufficiency in
the European Community
13
took note of the conclusions and recommendations agreed at two
important meetings held during 2006; the first a meeting of experts on public awareness relating
to blood held in Rome in April and the second, a colloquium on blood safety and self-sufficiency
held at Adare, Ireland, in September. In particular, it agreed that the conclusions and
recommendations of the Adare colloquium provide for concrete steps in order to realise the
strategy called for in previous resolutions towards promoting blood safety, in particular to
reinforce trust and confidence in the safety of the blood-transfusion chain, and towards
promoting self-sufficiency in the Community.
The Council requested that steps be taken to ensure that rapid progress is made in taking forward
this work and accordingly invited the Member States to review their policies, procedures and

99 | P a g e
programmes aimed at ensuring the safety of the blood-transfusion chain and invited the
Commission to submit proposals as a matter of urgency in support of the actions of the Member
States, with a view to encouraging the development of a coordinated approach to the safety of
blood and blood products.
In 1997, the Commission took at major step forward when it published its first proposal
14
under
Article 129 (Paragraph 4a and 5) of the Maastricht Treaty setting forth common criteria for the
acceptance of blood plasma donors as well as a set of screening tests that should be carried out in
all Member States, whether the donation was intended for transfusion or for further
manufacturing into plasma derived products. This proposal was subsequently adopted as a
Council Recommendation
15
in 1998.
In 1999, Article 152 of the Amsterdam Treaty enabled the European Parliament and Council to
adopt health measures setting high standards of quality and safety of substances of human origin
without preventing Member States from introducing more stringent measures.
The Commission presented its proposal
16
for a Directive of the European Parliament and of the
Council setting standards of quality and safety for the collection, testing, processing, storage, and
distribution of human blood and blood components and amending Council Directive 89/381/EEC
in May 2001 following extensive consultations with all stakeholders during 2000.
Following negotiations between with European Parliament, the Council and the Commission in
2002, Directive 2002/98/EC of the European Community and of the Council on setting standards
of quality and safety for the collection, testing, processing, storage and distribution of human
blood and blood components and amending Directive 2001/83/EC
17
was finally adopted and
Member States were required to have transposed its provisions into their national law by the 8
th
February 2005.
Three further ‘Implementing Directives’ were adopted in 2004 and 2005. These had been
delegated to the Commission via the ‘comitology procedure’. The first implementing
Commission Directive 2004/33/EC of 22nd March 2004 concerned certain technical
requirements for blood and blood components
18
. The second implementing Commission
Directive 2005/61/EC of 30th September 2005 contained traceability requirements and
requirements for the notification of serious adverse reactions and events
19
and the subject of the
third implementing Commission Directive 2005/62/EC of 30th September 2005 was Community
standards and specifications relating to a quality system for blood establishments
20
.
References for Annex III:
1. Council of Europe Agreement No. 26 on the Exchange of Therapeutic Substances
of Human Origin.
2. Council Decision 86/346/EEC accepting, on behalf of the Community, the
European Agreement on the Exchange of Therapeutic Substances of Human
Origin.

100 | P a g e
3. Council Decision 94/358/EC accepting, on behalf of the European Community,
the Convention on the elaboration of a European Pharmacopoeia.
4. Council Directive 65/65/EEC of 26 January 1965 on the approximation of
provisions laid down by Law, Regulation or Administrative Action relating to
proprietary medicinal products.
5. Council Directive 89/381/EEC laying down special provisions for medicinal
products derived from human blood or plasma. OJ L181 of 28.06.1989, p44.
6. Van Aken, W.G. Collection and use of human blood and plasma in Europe.
Council of Europe Press, 1993, p31.
7. Van Aken, W.G. The collection and use of human blood and plasma in the
European Community in 1991. CEC/LUX/V/F/1/59/94. July 1994, p39.
8. Delaney, F.M. The collection and use of blood and plasma in the European
Community in 1993. CEC/LUX/V/F/1/33/95. November 1995, p43.
9. Communication from the Commission to the Council, the European Parliament
and the Economic and Social Committee on Blood Self-Sufficiency in the
European Community. (COM93) 198 final. Brussels, 25 May 1993. P13.
10. Council conclusions of 13 December 1993 on self- sufficiency in blood in the
European Community OJ C 15, 18.1.1994, p. 6–6.
11. Communication from the Commission on blood safety and self-sufficiency 21
st
December 1994 COM (94) 652 final.
12. Council Resolution of 2 June 1995 on blood safety and self-sufficiency in the
Community Official Journal C 164 , 30/06/1995 P. 0001 – 0001.
13. Council Resolution of 12 November 1996 on a strategy towards blood safety and
self-sufficiency in the European Community Official Journal C 374 , 11/12/1996
P. 0001 – 0001.
14. Commission Proposal for a Council Recommendation on the suitability of blood
and plasma donors and the screening of donated blood in the European
Community. COM (97) final of 17.11.1997.
15. Council Recommendation 98/463/EC, OJ L 203 of 21.07.1998, p14.
16. Proposal for a Directive of the European Parliament and of the Council setting
standards of quality and safety for the collection, testing, processing, storage, and
distribution of human blood and blood components and amending Council
Directive 89/381/EEC (2001/C 154 E/14)
COM (2000) 816 final 2000/0323(COD).
17. DIRECTIVE 2002/98/EC of the European Parliament and of the Council of 27
January 2003 setting standards of quality and safety for the collection, testing,
processing, storage and distribution of human blood and blood components and
amending Directive 2001/83/EC.
18. COMMISSION DIRECTIVE 2004/33/EC of 22 March 2004 implementing
Directive 2002/98/EC of the European Parliament and of the Council as regards
certain technical requirements for blood and blood components.
19. COMMISSION DIRECTIVE 2005/61/EC of 30 September 2005 implementing
Directive 2002/98/EC of the European Parliament and of the Council as regards
traceability requirements and notification of serious adverse reactions and events.


102 | P a g e
Annex IV: The Legal Basis and the Legal Framework adopted
Primary Legislation
Healthcare governance within the EU is predominantly a competence of the individual
Member States. Although the principle of subsidiarity embodies that when possible and
reasonable, decision-making power stays with the Member States, the European Union
does have a mandate and obligation in the policy domain of public health. This mandate
is provided in EU primary legislation through Article 168 TFEU, as amended in 1999 by
the Amsterdam treaty. This article specifically provided a legal basis for legislating in
these sectors in its paragraph 4:
‘By way of derogation from Article 2(5) and Article 6(a) and in accordance
with Article 4(2)(k) the European Parliament and the Council, acting in
accordance with the ordinary legislative procedure and after consulting the
Economic and Social Committee and the Committee of the Regions, shall
contribute to the achievement of the objectives referred to in this Article
through adopting in order to meet common safety concerns:
(a) measures setting high standards of quality and safety of organs
and substances of human origin, blood and blood derivatives;
these measures shall not prevent any Member State from
maintaining or introducing more stringent protective measures;’
Secondary legislation
The EU legislation for blood and tissues and cells was proposed, negotiated and adopted
with two basic Acts, Directive 2002/98/EC
398
for blood and Directive 2004/23/EC
399
for
tissues and cells. Each of these Directives defines a scope that covers a major portion of
the substances of human origin that are used for application to patients.
Scope
Directive 2002/98/EC applies to the collection and testing of human blood and blood
components, whatever their intended purpose, and to their processing, storage and
distribution when intended for transfusion. This scope excludes the processing, storage
398
Directive 2002/98/EC of the European Parliament and of the Council of 27 January 2003 setting standards of
quality and safety for the collection, testing, processing, storage and distribution of human blood and blood
components and amending Directive 2001/83/EC (OJ L 33, 8.2.2003, p.30).
399
Directive 2004/23/EC of the European Parliament and of the Council of 31 March 2004 on setting standards of
quality and safety for the donation, procurement, testing, processing, preservation, storage and distribution of human
tissues and cells. (OJ L 102, 7.4.2004, p. 48).

103 | P a g e
and distribution of blood or blood components used for the manufacturing of medicinal
products or medical devices or for application to humans in procedures other than
transfusion (e.g. serum used as drops in the eye, platelet-rich plasma used in orthopaedic
surgery). Blood stem cells (haematopoietic stem cells), even if collected from whole
blood, are explicitly excluded. In general, the provisions apply to the activities of ‘blood
establishments’, and their oversight, up to the point of distribution to hospital blood
banks or to manufacturers of medicinal products or devices. However, some articles are
listed as being applicable also to hospital blood banks that store and issue units of blood
(components) to patients and may do some compatibility testing; inspection and
authorisation obligations do not extend to hospital blood banks. There are provisions,
however, that require traceability to the recipient and haemovigilance reporting after
transfusion.
Directive 2004/23/EC applies to the donation, procurement and testing of human tissues
and cells intended for human application or for manufacturing of products derived from
human tissues and cells but covered by other directives. Where the tissues or cells are
intended for human application, the Directive also applies to processing, preservation,
storage and distribution. Recital (7) confirms that haematopoietic stem cells from any
source, as well as foetal tissues or cells, gametes and embryonic stem cells are also
within the scope. Recital (10) notes that the scope also covers human tissues or cells used
in the manufacture of cosmetic products, noting however, that such use was prohibited
by other current legislation. Human application covers procedures of transplantation and
medically assisted reproduction. The scope is further clarified by the definition of human
tissues and cells that, in effect, excludes other substances of human origin, such as breast
milk or faecal microbiota.
The Directive specifically excludes blood and blood components as well as tissues and
cells used as an autologous graft within the same surgical procedure. The latter intended
to avoid the regulation of surgical procedures where a piece of tissue (e.g. bone or
muscle) is moved from one part of the body to another (e.g., during reconstructive
surgery). Organs or parts of organs to be used for the same function as the entire organ
are also excluded. The use of tissues and cells for research that does not include clinical
application, i.e. in vitro or animal research is excluded from the scope. In line with the
blood Directive, the provisions cover the process from donation to distribution for
clinical application, but not the steps carried out in the facility where the tissues or cells
are applied to patients, although here too, traceability to the recipients and adverse
outcome reporting after clinical application are required.
Key provisions
The two basic Acts provide the framework for the oversight of these sectors, defining
obligations for competent authorities in Member States and for the European
Commission. Member State competent authorities are required to inspect and authorise
blood and tissue establishments and to organise programmes of vigilance reporting in
relation to serious adverse reactions (where a donor or patient has been harmed) and
serious adverse events (where an incident posed a risk of harm to a donor or a patient).

104 | P a g e
The authorities must ensure that imported BTC from third countries are equivalent in
terms of quality and safety to those provided under EU legislation. They must submit
annual summary reports of serious adverse reactions and events to the European
Commission and must communicate with each other when adverse occurrences imply the
need for corrective or preventive action in more than one Member State. There are also
obligation on Member States to submit reports to the European Commission on the
implementation of the legislation and of the measures taken to encourage VUD, at
intervals defined in the Directives. Further generic provisions oblige the Member States
to put penalties in place for non-compliance and to ensure appropriate data protection.
The European Commission has obligations under these basic Acts to submit the Member
State reports on implementation to the Parliament, the Council, the Economic and Social
Committee and the Committee of the Regions and to inform the European Parliament
and the Council of any necessary further measure it intends to take at Community level
in relation to VUD. The European Commission is also required to hold regular meetings
with the competent authorities of the Member States.
Apart from these general oversight provisions, both of the basic Acts also include a
number of technical requirements for ensuring quality and safety at the blood
establishment and tissue establishment level. For example, the blood Directive includes
annexes defining the information to be provided to prospective donors, the elements to be
included in annual activity reports, the labelling requirements for blood components and
the minimum infectious disease and blood group testing requirements for donors.
Tertiary legislation
Most of the technical requirements for blood and tissue establishments are described in
implementing Directives. The basic Act on blood was complemented by three
implementing Directives
400
.
Commission Directive 2004/33/EC as regards technical requirements for blood and
blood components includes detailed provisions on donor eligibility, storage and transport
conditions and specifications for the quality control criteria to be applied for each type of
blood component that was in use at the time. Some limited technical amendments have
since been made to Directive 2004/33/EC
401
.
Commission Directive 2005/61/EC as regards traceability requirements and notification
of serious adverse reactions and events focuses on providing more detailed requirements
for traceability, vigilance reporting and import from third countries.
400
Directives 2004/33/EC as amended, 2005/61/EC, 2005/62/EC, 2006/17/EC as amended, 2006/86/EC as amended
and Directive (EU) 2015/566.
401
Directive 2009/135/EC, Directive 2011/38/EU, Directive 2014/110/EU.

105 | P a g e
Commission Directive 2005/62/EC as regards Community standards and specifications
relating to a quality system for blood establishments focuses on the requirements for
quality management and was amended by Directive 2016/1214
402
to oblige Member
States to take into account the Good Practice Guidelines jointly developed by the
Commission and the European Directorate for the Quality of Medicines and Healthcare
of the Council of Europe and published by the Council of Europe
403
when providing the
required good practice guidelines to their blood establishments.
The basic act on tissues and cells was also complemented by three implementing
technical Directives.
Commission Directive 2006/17/EC as regards technical requirements for donation,
procurement and testing focuses on donor eligibility and testing. Some limited technical
amendments have since been made to this Directive
404
.
Commission Directive 2006/86/EC as regards traceability requirements, notification of
serious adverse reactions and events and technical requirements for coding, processing,
preservation, storage and distribution provides the criteria for the authorisation of tissue
establishments and tissue and cell preparation processes. This directive was amended to
include provisions for the Single European Code, to be applied to tissues and cells
405
.
A third Commission Directive, (EU) 2015/566 regarding the procedures for verifying the
equivalent standards of quality and safety of imported tissues and cells was adopted
more recently. A Commission Decision on conditions of inspections was also adopted
406
,
along with a Commission Decision on agreements with tissue and cell coding
organisations
407
.
402
Directive (EU) 2016/1214.
403
Good Practice Guidelines, included in the Guide to the preparation, use and quality assurance of blood components,
Appendix to Recommendation No. R (95) 15 of the Committee of Minsters on the preparation, use and quality
assurance of blood components adopted on 12 October 1995.
404
Directive 2012/39/EU.
405
Commission Directive 2016/565.
406
Commission Decision 2010/453/EU.
407
Commission Decision 2015/4460.

106 | P a g e
Annex V: Baseline
Part A - Blood and Blood components for transfusion
For this evaluation, a number of key documents have provided information on the baseline prior to the
adoption of the BTC legislation. The key documents and the most significant points highlighted in
them are summarised in this Annex
1. Key Document: Communication from the Commission on blood safety and self-sufficiency in
the European Community 1994.
Background: At its meeting of 13 December 1993, the European Council requested the European Commission
to prepare a report related to the legal provisions and current practices in the Member States on the collection,
control, and treatment of blood and the distribution and trade in blood and blood products with a view to
proposing common safety criteria.
The Commission invited Member States to provide their legal regulations and administrative provisions in this
area. While the results of these activities were not directly made available, the Communication from the
Commission in 1994 draws upon the responses provided by the Member States in written submissions and
during meetings of experts on self-sufficiency and blood safety convened by the Commission and provides an
insight into the status of blood safety, quality and sufficiency at this time and makes note of the future activities
required for improvement in this area.
Key Observations:
It was noted that regulations and practices related to donor recruitment and voluntary non-remunerated
donations in the Member States varied widely from explicit legislation, to general health regulations but
with no precise reference to remuneration, or no specific stipulations. A few Member States stated that in
their legislation non-compliance with the principle of voluntary non-remunerated donations could result in
legal action.
In the Member States at this time, donors were generally voluntary and under no pressure to donate. Some
strictly prohibited directed donations by family or friends or where the donor and recipient know each
other. In situations where blood shortages exist, however, friends and family members of patients in a few
Member States may be asked to donate. In those that were not self-sufficient, physicians could make
treatment conditional on the availability of blood, which sometimes put pressure on the patient as well as
on his/her relatives to donate.
It was evident, at the time, that the interpretation of the concept of non-remuneration was not uniform
throughout the Community. Incentives to encourage blood and plasma donations did take place including
time off work beyond that actually required for the donation and flat rate "expense allowances" were
allowed in many MS.
The donor selection process, also differed across the Community. Whether the donation was for whole

107 | P a g e
blood or plasmapheresis plasma, it was noted that variations existed in the frequency of the clinical
examination, ranging from a medical examination at every donation to none routinely required; in the
person who conducted the donor interview who varied from a physician to a trained staff member of the
collection centre; and in the information programmes for both donors and the general public. The process,
however, generally included the administration of a questionnaire, which again differed both within and
across Member States.
It was noted that it would be beneficial if an agreement was reached as regards the rules and practices for
donor selection, including for first time and repeat donors as well as donors of whole blood, cellular
components and plasma which could be applied across the Community.
Directive 89/381/EEC required that testing of blood and plasma when used as starting materials for blood
derived medicinal products shall comply with the recommendations of the Council of Europe, the WHO
and the European Pharmacopoeia. However, as this Directive did not apply to whole blood, to plasma or to
blood cells of human origin, divergences existed within the Community regarding the testing requirements
for blood and plasma donations.
It was noted that some Member States did not differentiate at the collection stage between blood for
transfusion and blood that is destined or may be used for fractionation; others do. As a consequence, testing
requirements that may be compulsory in one Member State for blood and plasma for fractionation may not
be in another. Concern was raised that this was hindering the free movement of blood and blood products
and are therefore an impediment to the goal of Community self-sufficiency.
At this time, there were preliminary indications that the regulatory controls regarding licencing and
accreditation of blood collection establishments differed widely across the Member States. Many had no
licencing requirements for the collection of blood or plasma; no standard requirements for collection
centres across the country; no routine and/or unannounced inspections by national authorities nor peer
inspections, and differing time periods for licence renewals.
In one Member State, the extraction of blood and blood components for transfusion as well as the
extraction of plasma for fractionation constituted the manufacture of medicinal products, for which a
licence was required. In others, this interpretation did not apply.
It was noted that serious consideration needed to be given to harmonising the licensing and accreditation of
blood collection, processing and distribution establishments across the Community that did not fall within
the scope of Directive 89/381/EEC.
While no specific comment was made on the status of haemovigilance practices in the MS at the time of
this report, it can be assumed that these were not yet developed in the MS or in the very early stages of
development. It was noted that the establishment of a Community-wide surveillance system on blood
transmitted diseases and adverse reactions at both national and Community level could help to keep
transfusion specialists informed, in a timely and orderly fashion, of new infectious agents in particular, their
potential danger, and appropriate measures to be taken to avoid their transmission. It also noted that
existing haemovigilance and pharmacovigilance systems, therefore, would need to be examined in order to
assess their contribution to such a system.
Finally, the document identified the main activities that could be undertaken in the development of a blood

108 | P a g e
strategy including:
Development of scientifically sound policies and agreed procedures in the donor selection process
among blood collection establishments within the Community in order to provide the necessary
reassurances of the safety of blood products originating from whatever Community source;
Implementation of efficient validated and reliable screening tests in the Community;
Development of quality assessment criteria and good manufacturing practices regarding collection,
testing, processing, and transfusion of blood and blood products and patient follow-up procedures;
Development of a haemovigilance system for the collection of epidemiological data related to the blood
transfusion chain;
Development of educational programmes directed towards health professionals on the optimal use of
blood and blood products;
Support for the dissemination of information on blood and blood products and the collection,
processing and transfusion procedures through promotional materials, films, campaigns etc.
2. Comparative analysis of national regulations concerning blood safety across Europe.
Mascaretti et al. Transfusion Medicine, 2004, 14,105–111
In October 2001, representatives of 17 European countries (Albania, Bosnia–Herzegovina, Bulgaria, Croatia,
Czech Republic, Federal Republic of Yugoslavia, Finland, France, Germany, Greece, Italy, Macedonia,
Romania, Slovenia, Spain, Turkey and UK) met in Sarajevo at a course organised by the European School of
Transfusion Medicine (ESTM) to discuss their countries’ regulations concerning different aspects of the safety
of blood transfusion.
This publication presented a preliminary analysis of the activities being undertaken in these countries to ensure
the safety of their blood supply directly before the implementation of the European Blood Directive in February
2005.
The following key observations were noted:
Most countries (13/17) had specific transfusion laws. However, it was noted that the majority of these laws
were rather recent (only two published before 1990).
9/17 countries had hospital-based systems as opposed to national organisations and 9/17 had national
systems while Spain had a ‘mixed system’ with a national programme in 11 of 17 regions. Of the seven EU
states, only four had national programmes. Finland, the country with the lowest population density, was
among the countries with a National Transfusion Service. Of the 9 countries with a hospital-based system,
five (Croatia, Czech Republic, Italy, Macedonia and Slovenia) warranted a greater centralisation of
transfusion organisations.
Quality assurance was common among investigated countries (14/17) and in these was stipulated by law.
Voluntary associations were responsible for donor promotion in the majority of countries (13/17).
Exclusively, voluntary non-remunerated donors were found in 5/17 countries, whereas in the remaining

109 | P a g e
ones, incentives, family replacement and remuneration were mechanisms stimulating blood donation.
In all 17 countries, regulations require that donor suitability be checked by, or under the responsibility of
medical doctors; in most cases, these use selection criteria that are established by official regulations
(15/17).
In 16 countries of 17 (data for Turkey were not available), regulations require that blood and components
were screened for anti-HIV1/2, anti-HCV, HBsAg and anti-treponemal antibodies. In a few nations,
additional tests are required for all blood units donated (however, these were not specified).
Regulations on good clinical use of blood and derivatives were present in most countries but applied only in
some.
Haemovigilance was required by law in 11 countries, but, there was some discrepancy between what was
written and what was done in relation to haemovigilance procedures. It was noted that in 13/17 countries,
transfusion centres were always notified in cases of adverse transfusion reactions.
3. Report from the Commission to the Council, the European Parliament, the European Economic and
Social Committee and the Committee of the Regions – First Report on the application of the Blood
Directive. Brussels, 19.6.2006 COM (2006) 313 final.
Article 26 of Directive 2002/98/EC requires Member States to submit to the European Commission, beginning
on 31 December 2003 and every three years thereafter, reports on the activities that they have carried out in
relation to the implementation of its provisions.
This first Commission report provided an overview of the situation in the 15 Member States that belonged to
the European Union as of 31 December 2003.
The following key observations were noted in this report:
As of December 2003, 14 Member States had designated a competent authority in accordance with the
Directive. Four, however, had more than one – Germany, due to its federal system, had 29; Spain, with its
autonomous communities, had 18; and Denmark and Sweden each had two.
The competent authority in 11 of the Member States had designated, authorised, accredited or licensed
blood establishments to carry out collection, testing, preparation, storage and distribution activities.
The competent authority in 7 Member States had organised inspections and control measures in blood
establishments in order to ensure compliance with the Directive’s requirements. The timeliness of
inspections and control measures, however, varied from every six months to every three years. Emergency
inspections were carried out when necessary.
Six Member States had empowered officials representing the competent authority to carry out inspections
and control measures in blood establishments and facilities of third parties in their State that had been
entrusted by the authorised blood establishment to carry out Evaluation and testing procedures.
Six Member States indicated that their blood establishments were aware that serious adverse events and
reactions had to be notified to the competent authority in accordance with the procedure and notification

110 | P a g e
format.
Eight Member States already had procedures in place to enable blood or blood components associated with
serious adverse events and reactions to be accurately, efficiently and verifiably withdrawn from
distribution.
Nine Member States confirmed that personnel directly involved in the collection, testing, processing,
storage, and distribution of human blood and blood components is qualified for their tasks and has been
provided with timely, relevant and regularly updated training.
Eleven Member States had ensured that each blood establishment instituted and maintained a quality
system based on the principles of good practice. Shortcomings, however, were acknowledged in some
Member States.
All Member States had taken measures to ensure that blood and blood components collected, tested,
processed, stored, released and/or distributed on its territory were traceable from donor to recipient and vice
versa. Eleven Member States indicated that blood establishments had implemented an identification system
for each blood donation and each blood unit and its components.
Nine Member States reported that Evaluation procedures and donation deferral criteria were in place in
blood establishments for all donors of blood and blood components.
Fourteen Member States indicated that provisions are in place for assessing the suitability of individuals to
donate blood, including an examination of and an interview with the donor prior to any donation.
Eleven Member States had taken measures to encourage voluntary and unpaid blood donations with a view
to ensuring that blood and blood components are in so far as possible derived from them.
Fourteen Member States reported that their blood establishments test each donation of blood and blood
components in conformity with requirements listed in Annex IV of the Directive.
Although the requirements for the quality and safety for blood and blood components were adopted through
the Comitology procedure (article 29(f)) after the reporting date, seven Member States reported that their
blood establishments had to ensure that the quality and safety requirements for blood and blood
components met high standards.
4. Trends and Observations in the collection, testing and use of blood and blood components in
Europe. EDQM Report 2001-2011
408
In 2015, the EDQM published its cumulative report on the trends and Observations in the collection, testing and
use of blood and blood components in Europe covering the decade from 2001 to 2011.
The basis for this analysis is formed by data provided annually since 2001 to the Council of Europe by its
Member States and reported on the webpage provided, in individual annual reports, on the collection, testing
and use of blood and blood products in Europe.
408
EDQM Report on the Trends and Observations on the Collection, Testing and Use of Blood and Blood Components in Europe..

111 | P a g e
While the reporting of data is open to the 46 Member States of the Council of Europe, the majority of consistent
data is provided by the EU MS and is therefore relevant as a data source to identify certain improvements that
have been made in the decade which saw the introduction and implementation of the European Blood
Directives.
The data presented in this report refers mainly to the usage of blood and blood components in the Member
States and to the occurrence of infectious markers in donors and trends in the presentation of donors for
donation, however, three key points were also noted which demonstrate improvements from the baseline in
2001 prior to the implementation of the European Blood Directives.
The following key observations were noted in this report:
While there were no changes in the proportion of Member States which test all donations with the
following serological tests: Anti-HIV 1+2 / HBsAg, Anti-HCV, Anti-HTLV and Syphilis. Only anti-HBc
testing increased by 1.1 absolute percentage points per year (p=0.4 %). A substantial increase was
observed, however, in the implementation of HIV- and HBV-NAT for testing (respectively 3.8 and 4.5
absolute percentage points per year) of all donations, leading to 56 % (HIV-NAT), 47 % (HBV-NAT) and
53 % (HCV-NAT) of Member States testing all donations in 2011.
There was no indication of a consistent trend in the proportion of countries with a national or expert
committee in place for blood transfusion (while this is not a legislative requirement, it is good practice and
is recommended by the Council of Europe Guide to the Quality and Safety of Blood and Blood
Components. Also no significant change was observed in the proportion of countries that had implemented
labelling of at least 50 % of donations or have at least 50 % of donations covered by standards. However,
the proportion of Member States which labelled component codes (allowing for more complete traceability
of donations) significantly increased over the years, up to 100 % since 2009 (p=3 %).
There was an overall increase of 2.7 percentage points per year in the proportion of Member States, which
have established a quality assurance system. Since 2008, all reporting Member States had either had
established QS in place or planned to establish one.
5. Peer Review of Blood Services in Romania 26-28 July 2004 (Pre-Accession) Mission Report
Reference 10255 (EC Reference)
In advance of the accession process of Romania to the European Union in 2005, a Peer Review mission was
carried out by the European Commission accompanied by relevant experts between 26 and 28 July 2004 in
order to assess the situation with regard to the country’s blood services.
The aim of the mission was to consider the proposed Romanian legislation related to blood, to identify
difficulties and weaknesses, and to propose, if appropriate, suitable and sustainable solutions in order to
facilitate the transposition of Community legislation related to blood (Directive 2002/98/EC, Commission
Directive 2004/33/EC) and the implementation of its requirements in the lead up to accession.
During this peer review mission, the review team highlighted a number of key issues to be addressed by the
Romanian Government and existing Blood Service in order to meet the requirements of the European Blood
Legislation.
It was recommended that the State designate a Competent Authority, without operational functions related

112 | P a g e
to the blood services, as well as an independent body authorised to carry out inspections.
It was noted that the modernisation of blood establishments and hospital blood banks in terms of facilities
and equipment was of high priority.
It was also noted that the establishment of both national and local programmes on donor recruitment and
retention based on voluntary, non-remunerated blood donations, with a clearly identified and separate
budget, was urgently required. The use of family and replacement donors at this time was noted.
It was also recommended that procedures for the notification of adverse events and reactions needed to be
introduced and implemented.
There was a need for training programmes, particularly those related to Quality management, to be
introduced – at national, regional and local levels - to upgrade staff awareness and knowledge in order to
comply with the requirements of the Blood Legislation.
Part B- Tissues and cells
Key document 1
Communication from the Commission to the Council, the European Parliament, the European
Economic and Social Committee and the Committee of the Regions on the application of Directive
2004/23/EC on setting standards of quality and safety for the donation, procurement, testing,
processing, preservation, storage and distribution of human tissues and cells. Brussels, 2010
COM(2009)708 final
Background 1
Article 26 of Directive 2004/23/EC required Member States to submit to the European
Commission, before 7 April 2009 and every three years thereafter, a report on the activities
undertaken in relation to the provisions of the Directive, including an account of the
measures taken in relation to inspection and control.
The Commission is required to transmit these reports to the European Parliament, the
Council, the European Economic and Social Committee and the Committee of the Regions,
and to provide them with a report on the implementation of the requirements of the
Directive, in particular as regards inspections and monitoring.
This report was based on the replies to questionnaires on transposition and implementation
that Member States sent to the Commission on a yearly basis and is particularly focussed
on the year 2008.
This first Commission report provides an overview of the situation in the 27 Member States
at the time.
Key observations 1
The following key observations were noted in this report:
All Member States indicated they had designated a competent authority in accordance with
the requirements of the Directive. In 21 Member States, the designated competent authority
was responsible for all types of tissues and cells. France, Greece, Portugal, Finland and the
United Kingdom had a specific competent authority for reproductive tissues and cells.
Under Article 5, the competent authority or authorities must ensure that tissue and cell
procurement complies with the stipulated requirements. The procurement organisations do

113 | P a g e
not have to be accredited/designated/authorised/licensed by the competent authority or
authorities but the conditions of procurement need to be verified. These could be checked via
an inspection of the procurement organisation or via an inspection of the tissue establishment
receiving tissues and cells from a particular procurement organisation. In this respect, six
Member States (Bulgaria, Germany, Denmark, France, Ireland and the United Kingdom)
carried out 53 inspections of procurement organisations during 2008.
An accreditation/designation/authorisation/licensing system of tissue establishments was in
place in 23 Member States. The system was decentralised in five Member States where the
process was channelled through federal states, regions or autonomous communities. Three
Member States did not have an accreditation/designation/authorisation/licensing system in
place at the end of 2008. Although the accreditation/designation/authorisation/licensing
system was for the most part established in the Member States, around half of them indicated
that they had yet to finalise the accreditation/designation/ authorisation/licensing of each
individual tissue establishment in their territory.
14 Member States had specific systems for authorising tissue and cell preparation processes.
In the other Member States, in the absence of specific authorisation systems, tissue
and cell preparation processes were normally verified and authorised during a general
inspection for the purpose of accreditation/designation/authorisation/licensing of a
tissue establishment. In some Member States, a different institution, independent from the
competent authority or authorities, is responsible for validating and authorising of the
preparation process. This is the case in Romania, where the
Medical College of Physicians is responsible for the approval of the preparation processes.
Only three Member States conducted inspections solely for the purpose of authorising
preparation processes in 2008.
As of 31 December 2008, a total of 1 716 tissue establishments were
accredited/designated/authorised/licensed: 42 skin establishments, 172 musculo-skeletal
establishments, 63 ophthalmic establishments (cornea, sclera, etc.), 49 vascular
establishments (heart valves, vessels, etc.), 193 haematopoietic stem cell establishments
(other than cord blood), 91 cord blood banks, 769 reproductive tissue and cells
establishments, 270 multi-tissue establishments and 67 other types of tissue and cells
establishments (chondrocyte cells, genetically modified cells, keratinocyte cells, myeloblast
cells, etc.).
Comprehensive inspection systems were in place in 23 Member States. Only 15 Member
States had conducted initial or regular inspections of tissue establishments in 2008. 4
Member States did not yet have inspection systems in place.
11 Member States had clearly identified tissue establishments explicitly authorised to import
tissues and cells. 8 Member States had a register of tissue establishments in third countries
from which imports were performed. Sixteen Member States reported that they imported
tissues and cells from third countries during 2008. Slightly less than 50% of Member States
importing tissues and cells use bilateral agreements to verify the equivalence of standards for
quality and safety of the tissues and cells. International standards like EATB, AATB, JACIE,
WMDA and NETCORD are also used, depending on the tissue and/or cell involved.

114 | P a g e
In many cases data concerning volumes of imports were not available; Member States
indicated that 1 122 units of haematopoietic stem cells (HSC), 2 281 units of musculo-
skeletal tissue, 4 units of skin and 7 units of reproductive tissues and cells were imported
during 2008.
Only nine Member States had a register of tissue establishments authorised to export tissues
and cells to third countries. 14 Member States exported tissues and cells during 2008. In
many cases data concerning volumes of exports are not available, but Member States
indicated that 269 units of HSC, 489 units of ophthalmic tissue, 6 225 units of musculo-
skeletal tissue and 10 units of amniotic membrane were exported.
19 Member States had created an annual report on the activities of tissue establishments that
made the reporting of the yearly activities by tissue establishment easier. 16 Member States
received annual reports from their tissue establishments corresponding to 2008 activities.
Only 12 Member States had made the tissue establishments' reports publicly available during
2008. 20 Member States indicated that they have a public register available (Belgium,
Bulgaria, Czech Republic, Denmark, Germany, Estonia, Ireland, Spain, France, Italy,
Cyprus, Lithuania, the Netherlands, Austria, Poland, Portugal, Slovenia, Romania, Finland
and the United Kingdom).
All Member States except for Greece and Latvia had a vigilance system in place to report,
investigate, register and transmit information about serious adverse events and reactions,
which may influence the quality and safety of tissues and cells. Criteria for the reporting of
adverse events to the competent authority had been laid down by 22 Member States. Criteria
for the reporting of adverse reactions to the competent authority had been laid down by 21
Member States. In accordance with Article 7(1) of Directive 2006/86/EC, Member States
must submit to the Commission an annual report on the serious adverse reactions and events
notified to the competent authority. The first annual report on this subject was submitted to
the Commission by only 13 Member States.
All the reporting Member States comply with the minimum testing requirements of Directive
2006/17/EC. Some Member States applied other tests in addition to those established as
minimum requirements in the Directive, in particular NAT testing for HIV/HBV/HCV.
Under Article 24(1), tissue establishments have to have a written agreement with a third
party each time an external activity takes place which may influence the quality and safety of
tissues and cells. 22 Member States indicated that tissue establishments in their territory had
notified third-party agreements.
By July 2009, 26 Member States had notified to the Commission their national transposition
measures in relation to Directive 2004/23/EC. National transposition measures in relation to
Directives 2006/17/EC and 2006/86/EC had been communicated to the Commission by 25
Member States. In July 2009, there were five infringement procedures open for failure to
achieve full transposition of the Directives in two Member States.
Comment: This report demonstrated that implementation of the legislative requirements was

115 | P a g e
progressing since the finalisation and publication of the Tissues and Cells Directive in 2004,
however, there was still some variance in practices with a number of MS needing to
undertake significant work to meet the legislative requirements.
Concerns regarding the development of specific systems for authorising the tissue and cell
preparation process; finalisation of the accreditation/designation/authorisation/licensing
process in respect of each individual establishment; the carrying out of inspections in all
Member States; monitoring of imports/exports; fulfilment of the reporting requirements
(tissue establishments' annual reports on activities, register of
accredited/designated/authorised/licensed tissue establishments at the level of the Member
States and at EU level) and preparation of annual reports on adverse events and reactions for
the Commission.
Key document 2
Peer Review of Tissues and Cell Banking and Transplantation in Romania. Mission Report July
2004. Mission Reference: 10257. (EU Reference)
Background 2
This Peer Review Mission was undertaken in order to perform and analysis of the tissue
and cell retrieval, processing and grafting system in Romania. It was commissioned by DG
SANCO and DG Enlargement/TAIEX of the European Commission and took place over 3
days in July 2004.
The main focus of this mission was to determine whether adequate administrative rules,
infrastructure and capacity were in place within the tissue and cells transplant system in
order to ensure compliance with EU Directive 2004/23/EC.
Key Observations 2
During this peer review mission, it was noted that traditional tissue banking services were
poorly developed in Romania, though there was some banking and transplantation of
corneas, skin, bone and tendons.
The banks that existed were located in specialist hospital units and focused their activity on
trying to supply tissue for the clinical departments in which they were based. There were
no centralised services with multi-tissue retrieval or banking and there appeared to be no
involvement of the blood services in the development of tissue banking. It was noted that
gamete and haemopoietic progenitor cell services were better resourced and more
developed.
Tissue retrieval from deceased donors was organised almost exclusively from the very
small number of brain dead (heart-beating) organ donors, despite the acknowledgement
that a far greater source of potential donors would be non-heart-beating cadavers, and the
absence of any legal barrier. Successful programmes for non-heart beating tissue donation
existed in the past, notably for corneas, but were abandoned or stopped once the legal
requirement for donor family consent was introduced.
There has never been a large scale programme of public education regarding tissue

116 | P a g e
donation and Romanian colleagues described a reluctance among hospital staff to approach
bereaved families for consent. They also made reference to logistical challenges, given
that the majority orthodox Christian community prefers to bring the deceased home before
burial, sometimes for a number of days. It was agreed that these barriers could be
overcome with adequate public and professional support and education.
Importation of tissue grafts must be authorised by the Ministry and was generally
considered to be a prohibitively expensive option, with some exceptions such as heart
valves.
The delegation did not see reports or evidence of tissue exportation. At least two
approaches had been made by US owned ‘for profit’ tissue banks, proposing that bone be
retrieved from cadavers and sent to their facilities for processing. A proportion of the
processed bone (20%) would have been returned as payment. Given the scarcity of
cadaveric bone donors in Romania, these proposals could not be taken further.
The delegation noted that the recently adopted European Directive on tissues and cells
included a number of requirements that needed to be addressed in Romania, to ensure
eventual compliance. These included:
o Designation of a Competent Authority/ies;
o Establishment of a system for inspection and accreditation, designation, licensing or
authorisation of tissue establishments;
o Establishment of a system for inspection and accreditation, designation,
authorisation or licensing for all implied processes and activities;
o Maintenance of a registry of accredited establishments and activities;
o Assurance of traceability from donor to recipient;
o Protection of donors (privacy, health, consent etc.);
o Establishment of a system for the notification of serious adverse reactions or events
following tissue or cell transplantation;
o Establishment of a system to control Import / Export of tissues and cells;
o Establish a comprehensive Quality System in all tissue establishments;
o Notification of adverse or serious events following tissue or cell transplantation;
o Assessment of whether imported tissues comply with equivalent safety and quality
standards to those required by the directive
o Ensuring availability of appropriate staff and facilities as detailed in the directive
and its annexes.
Key document 3
Peer Review of Tissue and Cell Banking and Transplantation in Bulgaria. Mission Report July
2004.

117 | P a g e
Background 3
This Peer Review Mission was undertaken in order to perform and analysis of the tissue
and cell retrieval, processing and grafting system in Romania. It was commissioned by DG
SANCO of the European Commission and took place over 3 days in July 2004.
The main focus of this mission was to determine whether adequate administrative rules,
infrastructure and capacity were in place within the tissue and cells transplant system in
order to ensure compliance with EU Directive 2004/23/EC.
Key Observations 3
The delegation noted that the situation with regard to the programme for transplantation of
tissues and cells in Bulgaria included key elements which would facilitate future
improvement; the legal framework was well advanced, there existed a National
Transplantation Agency, there were many competent practitioners ready and willing to
improve the system and international co-operation was already developed and should
facilitate further improvement in time.
The new Transplantation Act (Effective January 1
st
2004) was quite comparable to the new
EU Directive. However, it was not clear if technical specifications for authorisation and
inspection activities had been clearly or adequately defined.
Furthermore, the inspection system proposed needed to be clarified; inspection guidelines
and work in the education and training of inspections was required.
The public tissue banks visited during the mission required significant upgrade to facilities
and equipment in order to meet the requirements of the new EU Directive.
Considerable work and education was required regarding the implementation of quality
systems required to meet the specifications to be defined in the new EU Directive.
It was noted that the co-existence of private-for-profit and public tissue and cell banking
activities contributes in upgrading the activity level of transplants of tissues and cells but
could lead to some undesirable distortion and must be regularly re-evaluated and remain
under the strict control of the national authorities and the representatives of national
society.
The quality and safety of reproductive tissue and cells needed to be addressed in
legislation, as it was not covered under the newly effective ‘Transplant Act.

118 | P a g e
Annex VI: Cases sent by national courts to the European Court of
Justice for a preliminary ruling on questions of Union law relating to
the BTC sectors.
ECJ Cases
1. C-421/09 – Humanplasma: Case about the implementation of rules on VUD to
imports of blood for transfusion;
Description of the case
This reference for a preliminary ruling concerns the interpretation of Articles 28 EC and 30
EC. The reference to the Court was made in proceedings between Humanplasma GmbH
(‘Humanplasma’), a company established under Austrian law, and the Republik Österreich
(Republic of Austria) concerning the legislative prohibition on the importation of erythrocyte
concentrates provided from blood donations, which were not entirely unpaid.
The case having been brought before it, the Landesgericht für Zivilrechtssachen Wien
(Regional Civil Court, Vienna) decided to stay the proceedings and to refer the following
question to the Court for a preliminary ruling:
‘Does Article 28 EC (in conjunction with Article 30 EC) preclude the application of a national
provision under which the importation of erythrocyte concentrates from Germany is permitted
only where the blood was donated without any payment having been made (with not even
expenses being covered), that being a condition which is also applicable to the obtaining of
erythrocyte concentrates within Austria?’
Summary of the Judgment
Article 28 EC, read in conjunction with Article 30 EC, must be interpreted as precluding
national legislation that provides that the importation of blood or blood components from
another Member State is permitted only on the condition, which is also applicable to national
products, that the donations of blood on which those products are based were made not only
without any payment being made to the donors but also without any reimbursement of the
costs incurred by them in connection with those donations.
Such legislation which has the aim, first, of ensuring that blood and blood components
marketed in the Member State at issue satisfy the criteria of high quality and safety and,
second, of attaining the objective enshrined in Article 20(1) of Directive 2002/98 setting
standards of quality and safety for the collection, testing, processing, storage and distribution
of human blood and blood components, that is, encouraging voluntary and unpaid blood
donations, addresses human health concerns such as those acknowledged in Article 30 EC.

119 | P a g e
Therefore, those objectives are, in principle, capable of justifying a restriction of the free
movement of goods.
However, considered in isolation, the obligation that the blood donation should have been
made without any of the costs incurred by the donor having been reimbursed is not necessary
in order to ensure the quality and safety of the blood and the blood components. That
conclusion is supported by the fact that neither Directive 2002/98 nor Recommendation No
R (95) 14 of the Committee of Ministers to the Member States of the Council of Europe, to
which that directive refers, requires donations to be completely unpaid but provide that small
tokens, refreshments and reimbursements of travel costs connected with the donation are
compatible with voluntary, non-remunerated donation, with the result that those elements
cannot be considered as liable to compromise the quality and safety of those donations or the
protection of human health.
Such legislation therefore goes beyond what is necessary to attain the objective pursued, that
is, to ensure the quality and safety of the blood and of the blood components.
2. C-512/12 - Octapharma France: Case about the extent to which MS can apply
more stringent requirements in line with Directive 2002/98/EC
Description of the case
This request for a preliminary ruling concerns the interpretation of Article 168 TFEU, of
Article 2(2) of Directive 2001/83/EC of the European Parliament and of the Council of
6 November 2001 on the Community code relating to medicinal products for human use, and
of Article 4(2) of Directive 2002/98/EC of the European Parliament and of the Council of
27 January 2003 setting standards of quality and safety for the collection, testing, processing,
storage and distribution of human blood and blood components and amending Directive
2001/83.
The request was made in proceedings between Octapharma France SAS (‘Octapharma’) and
the Agence nationale de sécurité du médicament et des produits de santé (ANSM) (National
Agency for Medicinal Product and Health Product Safety), and the Ministère des Affaires
sociales et de la Santé (Ministry of Social Affairs and Health) concerning the Agency’s
decision of 20 October 2010 setting out the list and fixing the characteristics of labile blood
products (‘the decision of 20 October 2010’), on the ground that the Agency placed on that
list plasma prepared by means of an industrial process such as, inter alia, fresh frozen plasma,
leucocyte-reduced, virus-inactivated by solvent-detergent (‘plasma SD’).
Taking the view that the resolution of the dispute before it depends on the interpretation of
European Union law, the Conseil d’État decided to stay the proceedings and to refer the
following questions to the Court of Justice for a preliminary ruling:

120 | P a g e
(1) Is plasma from whole blood which is prepared by a method involving an industrial
process and which is intended for transfusions capable of having the provisions of Directive
[2001/83, as amended by Directive 2004/27] and those of [Directive 2002/98] applied to it
simultaneously, as regards not only its collection and testing, but also its processing, storage
and distribution; for that purpose may the rule laid down [in Article 2(2) of Directive 2001/83,
as amended by Directive 2004/27] be interpreted as meaning that the Community legislation
on medicinal products alone applies to a product which falls simultaneously within the scope
of another piece of Community legislation only where that latter is less strict than the
legislation on medicinal products?
(2) Must the provisions [of Article 4(2) of Directive 2002/98] be interpreted, where
necessary in the light of Article 168 TFEU, as allowing the maintenance or introduction of
national provisions which, because they submit plasma which is prepared by a method
involving an industrial process to a stricter regime than that to which medicinal products are
subject, provide justification for setting aside the application of all or part of the provisions of
Directive [2001/83, as amended by Directive 2004/27], in particular those which make the
marketing of medicinal products subject to the sole condition of the prior grant of a marketing
authorisation and, in the affirmative, under what conditions and to what extent?’
Summary of the Judgment
1. Directive 2001/83 on the Community code relating to medicinal products for human
use, as amended by Directive 2004/27, and Directive 2002/98 setting standards of quality and
safety for the collection, testing, processing, storage and distribution of human blood and
blood components and amending Directive 2001/83 must be interpreted as meaning that
plasma from whole blood which is prepared by a method involving an industrial process and
which is intended for transfusions comes, in accordance with Article 109 of Directive
2001/83, within the scope of Directive 2002/98 with respect to its collection and testing, and
within the scope of Directive 2001/83, as amended by Directive 2004/27, with respect to its
processing, storage and distribution, on condition that it satisfies the definition of a medicinal
product under Article 1(2) of the latter directive.
2. Article 4(2) of Directive 2002/98 setting standards of quality and safety for the
collection, testing, processing, storage and distribution of human blood and blood
components, read in the light of Article 168 TFEU, must be interpreted as meaning that it
allows the maintenance or introduction of national provisions which make plasma which is
prepared by a method involving an industrial process subject to a more rigorous regime than
that to which medicinal products are subject solely with respect to its collection and testing.
3. C-528/13 – Léger: Case about whether MS can permanently defer men having
sex with men from donating blood on safety grounds

121 | P a g e
Description of the case
This request for a preliminary ruling concerns the interpretation of point 2.1 of Annex III to
Commission Directive 2004/33/EC of 22 March 2004 implementing Directive 2002/98/EC of
the European Parliament and of the Council as regards certain technical requirements for
blood and blood components.
The request was made in proceedings between Mr Léger and the Ministre des Affaires
sociales, de la Santé et des Droits des femme (Minister for Social Affairs, Health and
Women’s Rights) and the Établissement français du sang (French Blood Agency) concerning
the refusal to accept Mr Léger’s blood donation on the ground that he had had sexual relations
with another man.
The Tribunal administrative de Strasbourg decided to stay the proceedings and to refer the
following question to the Court for a preliminary ruling:
‘In the light of Annex III to Directive [2004/33], does the fact that a man has sexual relations
with another man constitute in itself sexual conduct placing him at a risk of acquiring severe
infectious diseases that can be transmitted by blood and justifying a permanent deferral from
blood donation for persons having engaged in that sexual behaviour, or is it merely capable of
constituting, in the light of the circumstances of the individual case, sexual behaviour placing
him at a risk of acquiring infectious diseases that may be transmitted by blood and justifying a
temporary deferral from blood donation for a period determined after cessation of the risk
behaviour?’
Summary of the Judgment
Point 2.1 of Annex III to Commission Directive 2004/33 implementing Directive 2002/98 as
regards certain technical requirements for blood and blood components must be interpreted as
meaning that the criterion for permanent deferral from blood donation in that provision
relating to sexual behaviour covers the situation in which a Member State, having regard to
the prevailing situation there, provides for a permanent contraindication to blood donation for
men who have had sexual relations with other men where it is established, on the basis of
current medical, scientific and epidemiological knowledge and data, that such sexual
behaviour puts those persons at a high risk of acquiring severe infectious diseases and that,
with due regard to the principle of proportionality, there are no effective techniques for
detecting those infectious diseases or, in the absence of such techniques, any less onerous
methods than such a counter indication for ensuring a high level of health protection of the
recipients. It is for the referring court to determine whether, in the Member State concerned,
those conditions are met.
In that connection, it is for the referring court to determine in particular whether the
questionnaire and individual interview with a medical professional, provided for in Annex II
B(2) to Directive 2004/33, are able to identify more precisely the type of behaviour presenting

122 | P a g e
a risk for the health of recipients, in order to impose a less onerous contraindication than a
permanent contraindication for the entire group of men who have had sexual relations with a
man.
4. C-296/15 – Medisanus: Case about whether a MS can envoke rules on VUD
and national self-sufficiency as a restriction on the free movement of plasma
derived medicinal products.
Description of the case
This request for a preliminary ruling concerns the interpretation of Article 2 and Article 23(2)
and (8) of Directive 2004/18/EC of the European Parliament and of the Council of 31 March
2004 on the coordination of procedures for the award of public works contracts, public supply
contracts and public service contracts, read in conjunction with Article 83 of Directive
2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the
Community code relating to medicinal products for human use, with Article 4(2) of Directive
2002/98 and with Article 18 TFEU.
The request was made in proceedings between Medisanus d.o.o. and Splošna Bolnišnica
Murska Sobota (Murska Sobota General Hospital, Slovenia) (‘the hospital’) concerning the
legality of a clause in the tender specifications relating to a public procurement procedure
launched by that hospital for the supply of medicinal products.
The Državna revizijska komisija za revizijo postopkov oddaje javnih naročil (State Public
Procurement Tribunal) decided to stay the proceedings and to refer the following question to
the Court for a preliminary ruling:
‘Must Directive [2004/18], in particular Article 23(2), Article 23(8) and Article 2 thereof,
read in conjunction with
– Directive [2001/83], in particular Article 83 thereof;
– Directive [2002/98], in particular Article 4(2) thereof; and
– the TFEU, in particular Article 18 thereof,
be interpreted as precluding a specification that industrially manufactured medicinal products
must be obtained from “Slovenian plasma” (a requirement based on the domestic legislation
...)?’
Summary of the Judgment
Article 2 and Article 23(2) and (8) of Directive 2004/18/EC of the European Parliament and
of the Council of 31 March 2004 on the coordination of procedures for the award of public
works contracts, public supply contracts and public service contracts, and Article 34 TFEU

123 | P a g e
read in conjunction with Article 36 TFEU, must be interpreted as precluding a clause in the
tender specifications for a public contract which, in accordance with the law of the Member
State to which the contracting authority belongs, requires medicinal products derived from
plasma, which are the subject matter of the public procurement at issue, to be obtained from
plasma collected in that Member State.

124 | P a g e
Annex VII: Written Parliamentary Questions concerning the BTC
legislation since 2002
To read the full question and answer go to this webpage and enter the parliamentary term, the
question number and the year:
http://www.europarl.europa.eu/plenary/en/parliamentary-questions.html
SUBJECT
Number of
questions
(N=77)
Number of specific subject related
questions
Blood
53
15 on MSM; 12 on sufficiency of supply; 9
on infection crises; 8 on VUD; 2 on free
market and 7 others
Tissues & Cells
24
7 on cord blood banking; 3 on sufficiency
of supply; 4 on VUD; 3 on
transposition/implementation; 7 others
Number of
questions
Topic
Question number/year
BLOOD
15
Deferral of Men having sex with Men (MSM) from
blood donation:
Gender discrimination or justifiable safety measure
to protect recipients from infectious disease risks.
P-1842/2003
P-3639/2005
E-5739/2006
E-4492/2006
E-3671/2009
E-006484/2011
E-012319/2013
E-006727/2014
E-011959/2015
E-007504/2015
E-006958/2015
E-007052/2015
E-007026/2015
E-007293/2015
E-005284/2016
12
Sufficiency of the blood (and plasma) supply
E-0594/2005
E-5800/2007
E-5473/2010
E-012357/2011
E-012355/2011
E-010598/2011

125 | P a g e
E-010557/2011
E-002285/2014
E-008923/2015
E-006689/2016
E-001017/2018
E-005795/2018
9
Infection/ transmission crises
E-2307/2005
E-1505/2005
E-4937/2006
E-4736/2006
E-9712/2010
E-000324/2011
E-002446/2013
E-008278/2014
E-003401/2016
8
Voluntary unpaid donations
P-2174/2006
P-4259/2007
P-2557/2007
E-4962/2008
P-2933/2008
P-2779/2010
E-005884/2011
P-005818/2011
2
Free market
E-2314/2003
E-4719/2007
1
Epilepsy as an exclusion criteria for donation
E-2796/2006
1
Separation between blood products and plasma
E-4201/2008
1
Blood testing using NAT
E-001921/2011
1
Assistance for individuals with disabilities in
donation
P-001468/2013
1
Blood substitutes
E-005542/2014
1
Ebola risks
E-010202/2014
1
Age limit for donation
E-005013/2015
TISSUES AND CELLS
7
Private Cord blood banking and the measures taken
to harmonise sampling models, maximise clinical
use and ensure traceability.
E-3677/2002
E-4710/2009
E-0037/2010
E-010212/2011
O-000032/2011
E-003479/2014
E-002705/2017
3
Sufficiency and supply
E-008172/2012
E-010175-12
P-004347/2014

126 | P a g e
4
Voluntary unpaid donations
E-1361/2005
P-1755/2007
P-1735/2007
E-1727/2008
1
Illegal trafficking between EU and USA
E-4095/06
1
On consultations concerning technical procurements
for donation and testing of human tissues and cells.
E-3345/2006
1
Financing human embryonic stem cells for research
and care
E-1384/2007
3
Transposition/ compliance assurance
H-0241/2009
E-010578/2010
E-000687/2011
1
Testing requirements
E-007210/2011
1
Preservation of tissues and cells
E-009595/2011
1
Concerning the classification of medical products
(ATMPs) and the application of the hospital
exemption.
E-013101-13
1
Alert systems
E-007415/2013

127 | P a g e
Annex VIII: Interpretation Questions arising in the National Competent Authority (NCA) Meetings with the
Commission
409
Part 1: Blood
No.
Meeting of
Competent
Authorities
(blood)
Reference to
relevant text of
Directive(s):
Issue:
Outcome:
1.
September 2005
Point 2.4 of
Annex V of
Directive
2004/33/EC
(Quality Control
Parameters)
Quality Control Specification requires that the
protein content of fresh frozen plasma should be not
less than 50g/l.
Makes quality control testing of FFP unnecessarily
cumbersome and expensive without any measurable
increase in safety and quality.
Small expert meeting (May 2006) to come up with a
recommendation that can be presented to the
Regulatory Committee for an opinion and possible
amendment of the Directive.
NCA meeting blood (Sept 2006) agreed
with expert recommendation that 50g/l
threshold for total protein content in
fresh frozen plasma is not unnecessarily
cumbersome.
2.
September 2005,
September 2006
and September
Point 1.2 of
Annex III of
Directive
Required Haemoglobin Levels in Donors prior to
Donation: threshold at 125g/l for women and 135g/l
for men.
April 2010 NCA meeting concluded
there was currently no conclusive
evidence to justify a reduction in Hb
409
On each occasion it was highlighted by the Commission that “working interpretations” developed by the Commission and/or the NCA’s only represent the views of the Commission services
and/or the NCAs; it is for the Court of Justice to decide on the correct interpretation of Union Law.

128 | P a g e
2007
2004/33/EC
(Haemoglobin
Levels in donors
prior to donation)
At September 2005 NCA Meeting blood, one NCA
highlighted that this threshold had led to an
exclusion of around 15% of female donors in that
country.
At September 2006 NCA meeting blood, suggestion
to allow a capped leeway to lower the haemoglobin
level, in order to decrease the exclusion rate of
donors in some countries or regions due to their
specific populations. Many NCA’s confirmed that
the haemoglobin levels as set in the directive lead to
the exclusion of many potential donors, especially in
young women, specific sub-populations or
geographical areas. The principle of granting more
flexibility to countries concerned by these high
exclusion rates was broadly supported.
The Commission committed to clarifying legal
options available, and requested NCAs to send
written comments and evidence.
levels from those already specified in the
Directive.
(It should be noted that a decision was
taken to allow temporary reduction of
the Hb thresholds during the influenza
pandemic A (H1N1) in order to reduce
the possibility of a shortage of blood and
blood components, Directive
2009/135/EC.)
3.
September 2006
Directive
2002/98/EC
Article 3
‘Definitions’
Donor Leukocytes for Infusion (DLI) explicitly fall
under the Blood Directive, while they are
exclusively used in the frame of bone marrow
transplants, which are regulated by the Tissue and
Cell Directive.
Multiple interpretations among Member States
regarding the legal framework to apply to the DLIs.
This may result in bone marrow transplant centres
NCA meeting T&C (Feb 2007),
considered it recommendable that in
view of the de facto use, Art. 2 of
Directive 2002/98/EC would expressly
exclude from its scope of application
DLI used in the context of
haematopoietic stem cells transplant and
following the same requirements that
these cells.
129 | P a g e
having to be registered and inspected under both
directives.
The European Group for Blood and Marrow
Transplantation (EBMT) regrets this situation and is
calling for having the DLIs regulated only by the
Tissue and Cell Directive.
Several competent authorities confirmed that they
face difficulties indeed in choosing which legal
framework should be applied, leading to differing
national practices.
4.
September 2006
Directive
2002/98/EC
Article 8 –
Inspection and
Control Measures
Inspection frequency of Blood Establishments as
they apply to Mobile Collection Centres:
EMEA and some Member States call for clarification
of Article 8 of Directive 2002/98/EC on inspections
of blood establishments require clarification with
regards to the frequencies of inspections of mobile
collection sites.
NCAs agreed that:
Mobile collection sites are operated and
managed under the quality system of the
reference blood establishment. The
equipment used is supplied by the
reference blood establishment, and its
maintenance and calibration is
performed by the reference blood
establishment. Full information
regarding mobile collection sites’
operation and management, as well as
their equipment, are reviewed during the
inspection of the reference blood
establishment. The inspections of mobile
collection sites’ premises are done as
necessary.

130 | P a g e
5.
Blood Regulatory
Committee
September 2009
and October 2009
Point 1.2 of
Annex III to
Directive
2004/33/EC
& Point 2.2.1 of
Annex III to
Directive
2004/33/EC
Member States confronted with a serious risk of
shortage or an actual shortage in the supply of blood
and blood components directly due to the A(H1N1)
Influenza pandemic, may, on a temporary basis
allow the following derogations:
(a) by way of derogation from point 1.2 of Annex III
to Directive 2004/33/EC, reduce the minimum
haemoglobin levels in donors blood to no less than
120 g/l for females and 130 g/l for males;
and/or
(b) by way of derogation from point 2.2.1 of Annex
III to Directive 2004/33/EC, apply a deferral period
of no less than 7 days after cessation of symptoms of
a flu-like illness.
These derogations were legislated for in
Directive 2009/135/EC published on the
3
rd
November 2009 and applicable until
30
th
June 2010.
6.
April 2010
Annex V of
Directive
2004/33/EC
Quality Control
Parameters
Maximum pH levels for platelets at the end of shelf
life
Member States’ survey supported the findings of the
BEST Study which indicated that pH above 7.4 does
not influence the in vivo recovery and efficiency of
platelets, that the discard of non-compliant platelet
concentrates can be problematic and that it was
likely that there would be an increasing number of
non-compliant units due to the evolution of
collection methods and storage bags.
It was concluded that it was not justified to maintain
the upper pH limit for platelet concentrates as laid
down in Annex V of Directive 2004/33/EC and that
this would require amendment of the Directive
Draft Implementing Directive to amend
Annex V of Directive 2004/33/EC was
presented at this meeting and was voted
in favour of at the Meeting in October
2010.
(Note: At this meeting, the Competent
Authorities called for a review of the
technical requirements specified in
Directive 2004/33/EC given recent
scientific developments in the field)
The Commission adopted the draft,
which was subsequently published as
COMMISSION IMPLEMENTING
DIRECTIVE 2011/38/EU amending
131 | P a g e
through the regulatory procedure.
Annex V to Directive 2004/33/EC with
regards to maximum pH values for
platelets concentrates at the end of the
shelf life.
7.
April 2010
Annex III (Point
1.3) of Directive
2004/33/EC
Protein Levels in Donor’s Blood: to be checked at
least annually.
One NCA considered it appropriate to monitor donor
health when plasmapheresis donors donate regularly.
However, this was considered disproportionate in
some countries where donors are only allowed to
donate plasma between 4 and 6 times per year. It was
suggested that there should be a lower limit set, e.g.
6 donations per year above which an annual protein
test is required.
The Commission noted the concern but
stressed that this issue went beyond the
interpretation of Directive 2004/33/EC
and advised further discussion at the
EDQM level.
8.
Joint Meeting of
the Competent
Authorities and
Regulatory
Committee on
Blood and Blood
Components
October 2010
Annex V of
Directive
2004/33/EC
Quality Control Standards for Fresh Frozen Plasma
requires that Factor VIIIc is measured as a marker of
statistical process control in Fresh Frozen Plasma.
Acceptable results are an average (after freezing and
thawing) of >70% of the freshly collected unit.
One NCA queried if this was in fact the standard
method of quality monitoring in all MS. A
questionnaire was circulated to all CAs and EDQM
was also requested to provide feedback on current
guidance.
9.
Joint Meeting of
the Competent
Authorities and
Regulatory
Directive
2002/98/EC
Article 2 - Scope
PRP (Platelet Rich Plasma) and whether it falls
within scope of Directive 2002/98/EC:
An initial query from MS was put forward at the
Joint Competent Authority/Regulatory Committee
At the Meeting of the Competent
Authorities on Blood and Blood
Components in October 2012, it was
indicated that this procedure could fall

132 | P a g e
Committee on
Blood and Blood
Components
October 2010
NCA meeting May
2011
Meeting in October 2010, as to whether autologous
platelet rich plasma used for e.g. orthopaedic
purposes is within the scope of the SoHO legislation.
The EC committed to reverting on this topic.
At the 2011 meeting, one CA again raised the matter
of PRP (platelet-rich plasma) therapy, which is
among other things used in orthopaedic or cosmetic
therapies. This medical procedure includes the
following steps: blood is collected on site and
thereafter put into a small centrifuge to separate
platelet-rich plasma, which is then re-injected into
the patient, e.g. in the muscles/tendons. PRP therapy
normally takes place in one procedure (thus there is
no storage) in an operating theatre or medical office.
One CA aimed to clarify whether this medical
procedure falls under the scope for collection and
testing under Directive 2002/98/EC, and in particular
the wide reference provided in article 2 stating that
"this Directive shall apply to the collection and
testing of human blood and blood components,
whatever their intended purpose"'.
The feedback provided from eight National
Competent Authorities at this meeting considered
that as PRP is used for autologous purposes within a
single procedure, safety and quality standards laid
down in the legislation are not applicable. Other
NCAs supported this view. It was noted that the
centrifuge equipment falls under the safety and
quality requirements of the Medical Devices
under the scope of the blood directive as
it applies "to the collection and testing of
human blood and blood components,
whatever their intended use …"
The NCA's however expressed that in
practice it would be hard to make this
relatively new procedure comply to the
provisions of the 2002 blood legislation.
It was indicated that this new practice
could be considered in future legal
changes.

133 | P a g e
regulation.
10.
Joint Meeting of
the Competent
Authorities and
Regulatory
Committee on
Blood and Blood
Components
October 2010
Meeting of the
Competent
Authorities on
Blood and Blood
Components May
2011
Directive
2004/33/EC
Annex 3 Point 2.1
Deferral Criteria for CJD and vCJD in Blood Donors
Directive 2004/33/EC Annex 3 Point 2.1:
At the NCAs meeting in October 2010, the European
Medicines Agency indicated its intention to update
the CHMP Position Statement on CJD and plasma
derived and urine derived medicinal products. It was
considering recommending certain exclusion criteria
for blood donors to ensure the safety of plasma-
derived and urine-derived medicinal products.
Possible additional exclusion criteria included
permanent deferral for:
- donors who have spent a cumulative period of 1
year or more in the UK between the beginning of
1980 and the end of 1996,
- recipients of blood transfusions in the UK,
- recipients of transplants and
- donors who have undergone neurosurgery.
Directive 2004/33/EC Annex 3 Point 2.1 describes
the legislative deferral criteria in blood donors for
CJD and vCJD as follows:
Persons who have a family history which places
them at risk of developing a TSE, or persons who
have received a corneal or dura mater graft, or who
have been treated in the past with medicines made
from human pituitary glands. It also states that for
variant Creutzfeldt Jacob Disease, further
precautionary measures may be recommended.
The NCAs expressed serious concern at the
At the Meeting of the Competent
Authorities on Blood and Blood
Components in October 2012 and
following on from previous discussion
on this topic, the Commission services
presented a draft paper for the
preparation of a consensus list of
neurosurgery procedures and transplants
for which blood donors should be
deferred, based on ICD nomenclature.
MS' comments focused on the need to
balance between (1) a too general
deferral, e.g., deferring all patients after
surgery on peripheral nerves, which
would lead to too many unnecessary
deferrals and missed donations and (2) a
too detailed list of deferral criteria which
would be impractical and hard to
implement for staff in blood collection
centres.
It was concluded that it is a MS decision,
taking into consideration the local
epidemiological situation, whether or not
to defer neurosurgeries as well as which
types of neurosurgery. There was a
general preference to keeping deferral
criteria simple.

134 | P a g e
proposals and their impact on the harmonisation of
such criteria in the EU as well as possible impact on
blood donation exclusions and resulting supplies of
blood and blood components.
A questionnaire was sent to NCAs to gather
information on existing national blood safety
measures in relation to Creutzfeldt-Jakob disease and
to estimate the impact of EMA recommendations on
blood supply at national level.
The Commission services presented the replies to the
questionnaire provided by 9 Member States during
the May 2011 meeting.
EMA and ECDC were asked for more
scientific evidence on how to better
define, if possible, the recommended
deferral criteria in relation to
neurosurgery procedures and transplants.
At the meeting of the Competent
Authorities on Blood and Blood
Components April 2013, the
Commission outlined that there is no
deferral for neurosurgery at EU level, as
the final version of the EMA position
paper on plasma derived medicines did
not contain any reference to neurological
deferrals.
Data from one Member State was also
presented showing that exclusion of all
neurosurgery is not expected to
significantly affect blood supply
volumes.
It was concluded that applying deferral
for neurosurgery is a national decision,
but one, which would not significantly
affect blood supply.
11.
Meeting of the
Competent
Authorities on
Blood and Blood
Directive
2004/33/EC
Annex III
HTLVI/II Disease Deferral in donors of plasma
intended for fractionation:
Directive 2004/33/EC sets out permanent deferral

135 | P a g e
Components May
2011
criteria for donors of allogeneic donations, including
HTLV I/II. Some permanent deferral criteria are
excluded for donations used exclusively for plasma
for fractionation e.g. Babesiosis
At the request of the Plasma Protein Therapeutics
Association (PPTA), the EC consulted the NCAs
whether HTLV I/II positive donors should also be
excluded for donations used exclusively for plasma
for fractionation.
It was concluded that the request letter sent by PPTA
should be circulated to all NCAs for analysis and
that feedback should be provided in writing to the
Commission and that feedback would be provided at
the next CA Meeting.
12.
Meeting of the
Competent
Authorities on
Blood and Blood
Components May
2011
Annex III Point
2.2.2 of Directive
2004/23/EC
Definition of “Qualified Practitioner”
The NO Competent Authority requested clarification
on the different interpretations given to the term
"qualified practitioner" in various translations of
Directive 2004/33/EC, Annex III.
Annex III of Directive 2004/33/EC states under point
2.2.2. that acupuncture should be performed by "a
qualified practitioner", otherwise the donor is to be
deferred from donating blood for 6 months, or for 4
months provided a NAT test for hepatitis C is
negative.
NCAs agreed that "qualified
practitioner" as used in Annex III of the
Directive 2004/33/EC should be
interpreted as "qualified healthcare
practitioner". Member States can
implement stricter provisions.
13.
Meeting of the
Competent
Directive
2004/33/EC
Quality Control Requirements for Blood and Blood
Components:
The National Competent Authorities
indicated that the quality requirements
136 | P a g e
Authorities on
Blood and Blood
Components May
2011
Annex V
Quality Control
Parameters
One Competent Authority consulted the other NCAs
on their opinion concerning whether the quality
requirements for residual cells in fresh frozen plasma
are also applicable to plasma for fractionation, or
only to fresh frozen plasma for transfusion.
set out in Directive 2004/33/EC are
appropriate for plasma for transfusion
and not necessarily for blood donations
that are collected with the intention to
only use the plasma for manufacturing of
medicinal products.
14.
Meeting of the
Competent
Authorities on
Blood and Blood
Components
October 2012
Directive
2002/98/EC
Article 2 – Scope
Autologous Serum Eye Drops:
One NCA presented information on a new procedure
to manufacture eye drops from whole blood and
raised the issue of whether this falls within the scope
of European legislation on blood.
Three MS outlined that they regulate these products
as pharmaceuticals.
It was indicated that this procedure could
fall under the scope blood directive as it
applies "to the collection and testing of
human blood and blood components,
whatever their intended use …" which is
defined in Article 2 of Directive
2002/98/EC.
The NCA's however expressed that in
practice it would be hard to make this
relatively new procedure comply to the
provisions of the 2002 blood legislation.
This new practice should be considered
in future legal changes.
15.
Meeting of the
Competent
Authorities on
Blood and Blood
Components
October 2012
Directive
2002/98/EC
Article 2 – Scope
Interleukin Rich Serum:
Two NCAs explained that they do not consider that
this product falls under the EU blood directives.
One NCA stated that they had received an enquiry
about the regulatory status and had determined that it
was not a blood component falling within the scope
of Directive 2002/98/EC; it was a non-industrially
prepared medicinal product that fell outside the
scope of Directive 2001/83/EC.
It was indicated that this procedure could
fall under the scope blood directive as it
applies "to the collection and testing of
human blood and blood components,
whatever their intended use …"
The NCA's however expressed that in
practice it would be hard to make this
relatively new procedure comply to the

137 | P a g e
One NCA compared this technique to eye drops
manufactured from whole blood, and considered that
it did fall under the EU blood directive.
provisions of the 2002 blood legislation.
This new practice should be considered
in future legal changes.
16.
Meeting of the
Competent
Authorities on
Blood and Blood
Components
October 2012
Meeting of the
Competent
Authorities on
Blood and Blood
Components April
2013
Directive
2002/98/EC
Article 3 –
Definitions and
Article 8 –
Inspection and
Control Measures
Harmonisation of terminology: Plasma master files
(PMF) and EU Blood Directives.
Three NCAs had indicated at the last CA meeting
that inconsistencies concerning terminology, in
particular concerning (mobile) collection sites and
the applicable inspection regimes, might lead to
different interpretations, which may in turn result in
confusion and potential safety risks.
This was applicable to both blood and blood
components for transfusion but also to plasma for
fractionation only.
Suggestions were made for terms including: blood
establishment, blood centre, satellite site, and
relocation. The NCAs agreed to establish a group
working on nomenclature and inspection intervals.
At the NCA Meeting in April 2013, One NCA
presented a list of proposed definitions for different
types of establishments. This list is based on a
working document by EMA. The underlying concern
is the practical difficulty of inspecting every blood
establishment every two years (including but not
limited to plasma collection centres).
It was agreed that the definition of a blood
At the NCA Meeting in April 2013, the
Commission services explained that
Article 8(2) of Directive 2002/98/EC
makes no distinction between different
blood establishments. On the other hand,
the definition of inspections in Article
3(m) may be broad enough to also
include off-site inspections. This is
current practice in the tissues and cells
sector based on a guidance document,
although there is no similar document in
blood sector.
The Commission services also explained
that quality and safety cannot be
jeopardised for the sake of longer
inspection intervals.
A group of CAs were requested to
continue to look at both the proposed
definitions of establishments in the PMF
and the definitions of inspections and
that discussions could continue at the
subsequent meetings of the NCAs.

138 | P a g e
Meeting of the
Competent
Authorities on
Blood and Blood
Components
November 2013
Meeting of the
Competent
Authorities on
Blood and Blood
establishment should be linked to its activities and
inspection requirements. One NCA agreed that there
are insufficient resources to inspect all sites at the
same intervals. Four NCAs supported a risk based
inspection model.
During the previous NCA meeting, MS discussed
that inconsistencies concerning terminology, in
particular concerning (mobile) collection sites and
the applicable inspection regimes.
There was a concern that this might lead to different
interpretations, which may in turn result in confusion
and potential safety risks.
It was considered important to further develop
common thinking, given the difficulty of inspecting
all sites on a 2-year basis, as laid down in Art. 8.2. A
small sub-group was therefore asked to look into
nomenclature and inspection intervals.
A new proposal classifying Blood establishments
according to their structure was presented. It was
explained that depending on the activities performed
the different risk levels could be expected and it
could therefore be more suitable with a risk-based
inspection systems, better focusing inspection
resources.
The definition of "inspection" was also discussed in
this context.
Discussions are ongoing on the
definitions of ‘blood establishment’ and
the definition and understanding of the
‘inspection frequency’ in particular as
regards a ‘risk based model’.
139 | P a g e
Components
November 2015
It was noted that the proposal would underline the
need to comply with the 2-year inspection/control
measure requirement in Directive 2002/98/EC but
that the definition of 'inspection' allowed for
measures other than on-site visits to be used (e.g.
desk-based reviews).
A questionnaire was issued to Competent Authorities
in an effort to understand the current practice in
relation to frequency of inspection/control measures
at blood establishments.
17.
Meeting of the
Competent
Authorities on
Blood and Blood
Components April
2013
Directive
2002/98/EC
Article 3
Definitions
Apheresis granulocyte collection:
As this component is new, no parameters for its
quality and safety were laid down in Directive
2002/98/EC and associated Directives.
The procedure requires pre-treatment of donors with
corticosteroids or growth factors. Optimal collection
also requires the use of a sedimenting agent.
Granulocyte collection by apheresis was reported as
occurring in ten MS.
It was requested that CoE should investigate the new
component. CoE will discuss the issue, as the
European Convention on Human Rights requires the
protection of donors as well as the protection of
recipients.
One NCA underlined the necessity of having a
common approach on granulocyte concentrate in the
future, and requested that the efficacy of the
A component monograph for
granulocyte collection by apheresis was
included in the 18
th
Edition of the
Council of Europe Guide to the Quality
and Safety of Blood and Blood
Components (2015). A proviso was
included which stated that the clinical
efficacy, indication and dosage of
granulocyte transfusions have not been
established.
140 | P a g e
component be studied.
18.
Meeting of the
Competent
Authorities on
Blood and Blood
Components April
2013
Annex III of
Directive
2002/98/EC
Blood component labelling:
One NCA requested that the EU remove the
requirement to state the original composition of the
anticoagulant on the base label of the component.
Adding the composition of the anticoagulant does
not provide accurate information to the clinician and
can even be misleading if the label is read as being
an accurate representation of the content of the bag.
Another NCA considered it a good idea to shorten
this list, and only provide essential information to
physicians.
As the requirement is outlined in Annex
III of Directive 2002/98/EC, it can only
be amended by a new Commission
Decision via the comitology procedure.
The Commission took note of this
request for any future revision of EU
blood legislation.
19.
Meeting of the
Competent
Authorities on
Blood and Blood
Components
November 2013
Annex III of
Commission
Directive
2004/33/EC
Acceptance criteria of blood donors aged > 65:
According to Annex III of Directive 2004/33/EC,
donors over the age of 65 can only donate blood and
blood components with the permission of the
physician in the blood establishment, to be given on
an annual basis.
One NCA sought clarification as to whether this
permission should be given on an individual or
group basis. They explained that donors in this
country can safely donate blood until at least the age
of 71, provided that they meet all other relevant
acceptance criteria.
Other countries presented their national approaches.
Six NCAs have all raised the maximum age from 65.
The Commission services recognised
that the wording of the EU legislation is
silent on whether a decision is to be
made on an individual basis or can be
made for a group as a whole, but recalled
that such permission should be granted
by the physician in the blood
establishment.
The organisational structure of blood
establishments within any given country
will therefore determine whether such a
decision could be taken at a de facto
national level, which would appear to be
the case in certain MS.
The Commission services informed the

141 | P a g e
In contrast, three NCAs have an upper age limit of
65. One NCA reminded the group of the need to
check the impact of anti-coagulants. One NCA also
mentioned that they have a list of medicines to verify
in older persons, as these give an indication of
diseases necessitating exclusion.
group that in the implementation survey
seven out of twenty-seven countries
indicated they wanted to review the age
criteria.
The current edition of the Council of
Europe Guide to the Quality and Safety
of Blood and Blood Components states
that the standards set out in the Guide
define age limits for donation (Min 18
and Max 65) and provide discretion to
the responsible physician to accept
donors outside of these limits. It goes on
to state that this medical discretion can
be applied either on an individual basis
for a given donor or else through a
systematic approach based on an
appropriate medical risk assessment.
20.
Meeting of the
Competent
Authorities on
Blood and Blood
Components
November 2014
Point 2.2.1 of
Annex III to
Directive
2004/33/EC
West Nile Virus (WNV) amendment – Directive
2014/110/EU
The Commission presented the amendment of
Commission Directive 2004/33/EC and informed the
group of next steps towards final adoption following
approval of the amendment by the Regulatory
Committee on the quality and safety of blood.
The new Directive would amend the deferral
criterion for West Nile Virus set out in the table
(second column, last row) of point 2.2.1 of Annex III
to Directive 2004/33/EC by replacing it with the
following: ‘28 days after leaving a risk area of
Some participants expressed concern on
that a requirement for individual NAT
testing is too strict, however it was noted
that only few comments were received
during the adoption process.
Two NCAs and EDQM felt that the
requirement for individual NAT testing
was too restrictive and pooled testing
should have been allowed.
See further on this item raised by

142 | P a g e
locally acquired West Nile Virus unless an
individual Nucleic Acid Test (NAT) is negative’.
European Blood Alliance in 2016
21.
Meeting of the
Competent
Authorities on
Blood and Blood
Components
November 2014
Directive
2002/98/EC
Article 2 - Scope
Lymphocyte immunotherapy:
One NCA presented its query on whether the
collection of partner immunocytes to treat women
who have suffered from multiple miscarriages could
be considered to fall under blood or tissues and cells
legislation. Although blood is collected, the
treatment is carried out in the context of assisted
reproduction, a field regulated by the tissues and
cells legislation.
According to this NCA’s evaluation, this therapy
should be regulated pragmatically, as tissues and
cells or as blood, depending on the professional body
that applies the related therapy.
It was highlighted that in future, lymphocytes might
be used for other treatments, and it was therefore
unclear which legal framework would be the most
appropriate.
MS were requested reflect on this issue and send
comments within 2 weeks. Participants pointed to
the general need for a body to decide on such
borderline issues.
At the meeting of the Competent
Authorities on Blood and Blood
Components November 2015, the
Commission services provided feedback
on discussions arising from an
interpretation question on the regulation
of Lymphocyte immunotherapy used to
treat recurrent miscarriage in pregnant
woman.
It was felt that justifiable arguments
could be made for this activity falling
under either the blood or tissues and cells
legislation. This being the case, based on
the specific nature of their national
circumstances (assessment of risks to
human health / desired level of human
health protection / the existence of more
stringent protective measures etc…), and
given the fact that LIT is typically a local
activity not involving cross-border steps,
Member States benefit from a certain
degree of discretion when deciding
whether to classify this activity under
either blood or tissues and cells
legislation.

143 | P a g e
22.
Meeting of the
Competent
Authorities on
Blood and Blood
Components
November 2015
Meeting of the CAs
on Blood and
Blood Components
May 2016
Directive
2004/33/EC,
Annex III, Point
2.1
Basocellular Epithelioma as a Donor Exclusion
Criterion:
One NCA raised the topic of the exclusion of donors
with a history of basocellular epithelioma from
donation. It was pointed out that Directive
2004/33/EC, Annex III, 2.1. requires permanent
deferral for malignant diseases except 'in situ cancer
with complete recovery'. It was explained that in a
strict sense a basocellular epithelioma is not a
carcinoma in situ, but an invasive carcinoma with a
very low metastatic potential. Given the relatively
high and increasing frequency of this condition, the
exclusion is having a significant impact on the blood
supply. It was proposed that such donors should be
accepted for donation and arguments to support the
proposal were presented. The discussion on the topic
was postponed to the next meeting.
The NCA presented a discussion on whether donors
with basocellular epithelioma should be excluded
from donation as is currently the case. Basocellular
epithelioma is the most frequent case of skin cancer.
The NCA suggested that these donors should not be
excluded based on the low metastatic potential (only
0.03%) and the lack of documented cases of
transmission and asked for other NCAs views on
this.
One other NCA supported this suggestion.
At the meeting of the Competent
Authorities on Blood and Blood
Components in December 2016, one
NCA gave an update on deferral of
potential donors with a history of
basocellular epithelioma.
The NCA suggested to consider
basocellular epithelioma as a localised
cancer and, based on which, following
complete recovery, such patients would
be free to be blood donors (as stipulated
in point 2.1 of Annex III of Commission
Directive 2004/33/EC). This suggestion
was generally supported in the meeting
and it was noted that it is for each
Member state to decide on the length of
any temporary deferral.
A potential need to consider a change in
the legislation in the future was raised by
the participants, in particular with a view
to alignment with the tissues and cells
legislation. The Commission took note
of this request for any future revision of
EU blood legislation.
23.
Meeting of the
Competent
Directive
2004/33/EC
Potential amendment of Directive 2004/33/EC as
regards temporary deferral criteria for donors of
At the meeting of the Competent
Authorities on Blood and Blood

144 | P a g e
Authorities on
Blood and Blood
Components
December 2016
Annex III Point
2.2.1
allogeneic blood donations regarding West Nile
Virus
Directive 2004/33/EC Annex III Point 2.2.1 (as
updated by Directive 2014/110/EU)
The Commission introduced the request by the
European Blood Alliance (EBA) on the potential
amendment of Directive 2004/33/EC as regards
deferral criteria on WNV. Following amendment in
Directive 2014/110/EU, the deferral criterion now
reads as: ’28 days after leaving a risk area of locally
acquired West Nile Virus unless an individual
Nucleic Acid Test (NAT) is negative.’ EBA
questioned firstly, whether this wording precluded
the use of mini-pool NAT testing and, secondly,
whether ‘a risk area of locally acquired West Nile
Virus’ equates to ECDC risk assessment terminology
for WNV.It was noted that the topic would be
discussed with EBA during the stakeholder meeting
of 2
nd
December organised back-to-back with this
competent authorities meeting. All participants had
been invited to attend that meeting.
Components June 2017, the Commission
services debriefed the group on the
discussion in the December 2016 ad-hoc
stakeholder meeting on the deferral
criterion for West Nile Virus in Directive
2004/33/EC.
During this stakeholder meeting, the
European Blood Alliance (EBA) pointed
out some difficulties for blood
establishments in the implementation of
the criterion as amended in Directive
2014/110/EU.
On the first point (the use of mini-pool
NAT), the group agreed that the cost
implications put forward by EBA
suggested that a broad interpretation of
the text should be favoured. The ECDC
representative confirmed that the
sensitivity level of mini-pool NAT is
sufficiently high so as to not warrant a
credible safety risk if used. One NCA
pointed out that the type of NAT used is
not the decisive factor for them but
rather the sensitivity level of the
individual donor sample, whether this is
tested as part of a mini-pool or not. The
group supported a working
interpretation, which leaves the

145 | P a g e
discretion to MS to decide whether to
use the deferral period or use NAT.
Where NAT testing is permitted, MS
would have the discretion to decide
which type of NAT testing is permitted
and any conditions, which should be
placed on its use i.e. whether a risk
assessment is necessary to justify the use
of a particular type of NAT or to set an
acceptable sensitivity level.
On the second point (harmonisation of
terminology surround ‘WNV at risk
area’), the Commission services
presented the four types of risk area
defined in the ECDC terminology for
WNV risk assessments. The group
agreed that the definition of an ‘affected
area’ in the ECDC risk assessment
terminology is consistent with the term
‘risk area of locally acquired West Nile
Virus’ in Directive 2004/33/EC.
24.
Meeting of the
Competent
Authorities on
Blood and Blood
Components
December 2016
Directive
2002/98/EC
Article 2 - Scope
Platelet Rich Fibrin (PRF) Membrane:
One NCA raised an issue on Regulation of Platelet
Rich Fibrin (PRF)–membrane. It was highlighted
that PRF is one of those that falls on the borderline,
being a blood component that is used for purposes
other than transfusion.
As with previous similar products, the
collection and testing of blood are
covered by the EU blood legislation ‘for
whatever the intended purpose’.
However, for the rest of the process it is
less clear which legal requirements
apply. This issue has been raised in the
past, also in relation to the preparation of
this type of product using bedside

146 | P a g e
devices. The participants stressed that
clarity is needed on the regulatory status
of PRF and that this should be
considered in the ongoing evaluation of
blood, tissues and cells legislation.
Part 2: Tissues and Cells
No.
Meeting:
Reference to
relevant text
of
Directive(s):
Issue:
Outcome:
1.
Meeting of
Competent
Authority
of Tissues
and Cells
February
2007
Article 2 of
Directive
2004/23/EC:
Scope
Regulation of Donor Lymphocytes (DLI):
Donor Lymphocytes fall within the definition of "blood
components" provided by Directive 2002/98/EC, but in practice,
Directive 2002/98/EC would not be de facto applicable when
DLI are used for haematopoietic stem cells transplants. In this
instance, they would fall within the scope of Directive
2004/23/EC.
During discussions at the Blood
Competent Authority Meetings, it was
recommended that Art. 2 of Directive
2002/98/EC would expressly exclude
from its scope of application DLI used
in the context of haematopoietic stem
cells transplant.
2.
Meeting of
Competent
Authoritie
s on
Tissues
and Cells
May 2008
Scope Article
2 of Directive
2004/23/EC
Regulatory Status of Amniotic Membrane used for
homologous procedures:
One NCA raised concern regarding the inclusion of some 'tissues
and cells for non-homologous use' in the scope of the Advanced
Therapy Regulation. In particular the use of amniotic membrane
in the eye could be classified as a medical product, subject to
Meeting of the Competent Authorities
on Tissues and Cells December 2011:
Amniotic membrane is also used as a
wound dressing and/or barrier for
treatment and management of burn
wounds and wounds of various
etiology, its preparation and use being

147 | P a g e
market approval while many EU tissue banks regulated under
Directive 2004/23/EC procure, process, store and supply this
tissue for ocular use. It was felt that if this product was to be
regulated as a medicinal product it would result in it not being
available for many patients in the future.
It was considered that amniotic membrane use can be considered
as homologous because the function it performs is the same
whether on the placenta or on the eye (same essential functions)
and therefore within the scope of Directive 2004/23/EC. This
coincides with the position taken by the FDA.
similar to the one mentioned during
the Meeting of Competent Authorities
in May 2008.
In this regard, one NCA called for a
confirmation that amniotic membrane
used as a wound dressing and/or
barrier for treatment and management
of bum wounds and wounds is
covered by the Directive 2004/23/EC.
It was concluded that amniotic
membrane used as a wound dressing
and/or barrier for treatment and
management of bum wounds is
covered by the Directive 2004/23/EC
and that this view should be also
communicated to CAT.
The NCAs concluded that this issue
highlights the need for a mechanism
to discuss borderline products such as
this and to have a more clear
understanding of ‘homologous’ (i.e.
‘same essential function’) vs. ‘non-
homologous’ use.
3.
Meeting of
the
Competent
Authoritie
s on
Tissues
Definitions of
‘Storage’,
‘Distribution’
and
‘Transport’
within
Storage of Tissues and Cells for end use:
One NCA requested clarification on the requirement to
inspection ‘storage’ of tissues and cells for end-use i.e. at
hospitals/healthcare establishments where the tissues/cells may
be stored before final use. This NCA indicated they had adopted
Meeting of the Competent Authorities
October 2009:
A further discussion was undertaken
at this meeting during which the
Commission services outlined the
various approaches being taken in the
148 | P a g e
and Cells
May 2009
Directives
2004/23/EC,
2006/17/EC
and
2006/86/EC.
a risk-based approach in order to prioritise such establishments
to be inspected and accredited/designated/authorised/licensed.
At the beginning of the year, another NCA performed a survey
among Member States regarding this issues and it seemed that
most MS do not expressly accredit/designate/authorise/license
storage for end-use.
MS in relation to the regulation of
such end-use storage facilities. Some
MS do not authorise these facilities in
any way, other MS authorise the
facilities if storage of tissues and cells
occur for greater than 48 hours and
other MS designate the responsibility
for control of these storage facilities
to the supplying tissue establishment.
Joint meeting of the Competent
Authorities and the Regulatory
Committee on Tissues and Cells May
2010:
The Commission services explained
the provisions relating to the terms of
"transport", "storage" and
"distribution" within the meaning and
scope of Directives 2004/23/EC,
2006/17/EC and 2006/86/EC.
Based on these provisions the
following may be concluded:
Distribution of tissues and cells is an
operation to be carried out by the
tissue establishment before human
application and it is the last point in
the chain regulated by the Tissues and
Cells Directives.
Storage of tissues and cells, being a
step before distribution, is regulated
by the Tissues and Cells Directives

149 | P a g e
only when it is carried out by the
tissue establishments before the
transport and delivery to the
organisations responsible for human
application.
Storage in organisations responsible
for human application is not covered
by the Tissues and Cells Directives.
In any case, traceability of tissues and
cells shall be ensured from the donor
to the recipient (Art. 8 of Directive
2004/23/EC).
The Commission services noted that,
notwithstanding the above and the
exclusion of storage after distribution
from the scope of the Tissues and
Cells Directives, the objective of
Directives 2004/23/EC, 2006/17/EC
and 2006/86/EC is according to recital
13 of Directive 2004/23/EC to:
"...establish standards for each one of
the steps in the human tissues and
cells application process' Therefore, it
is important to ensure safety and
quality of tissues and cells notably in
terms of storage conditions,
traceability and effective recall
procedures, also for this step which is
not covered by Directives
2004/23/EC. 2006/17/EC and

150 | P a g e
2006/86/EC.
4.
Meeting of
the
Competent
Authoritie
s on
Tissues
and Cells
May 2009
Scope Article
2 of Directive
2004/23/EC
Regulatory Status of Pancreatic islets:
The Commission services indicated that it receives recurrent
questions on whether pancreatic islets are covered by the Tissues
and Cells Directive, the Advanced Therapies Regulation or
whether they should be classified as organs.
The NCAs agreed that, without
prejudice to an eventual future
opinion of the CAT (Committee on
Advanced Therapies), pancreatic
islets, as far as they are used for the
same initial function and as far as they
are not substantially manipulated (for
instance cultured), should be
considered under the scope of the
Tissues and Cells Directives.
5.
Meeting of
the
Competent
Authoritie
s on
Tissues
and Cells
May 2009
Point 2.5.b)
of Annex II
of Directive
2006/17/EC
Nucleic Acid Testing for Syphilis and the need to retest
samples:
A query/clarification was raised in relation to the need to retest
after 180 days for syphilis if a NAT was performed at donation.
It was highlighted that Point 2.5.b) of Annex II of Directive
2006/17/EC states that "Where tissues and cells of allogeneic
living donors can be stored for long periods, repeat sampling and
testing is required after an interval of 180 days.".
Point 2.6 of the same Annex specifies that "If in a living donor
(except bone marrow stem-cell and peripheral blood stem-cell
donors) the ‘donation sample’ is additionally tested by the
nucleic acid amplification technique (NAT) for HIV, HBV and
HCV, testing of a repeat blood sample is not required".
It was concluded as reasonable to
assume that no repeat testing for
syphilis after 180 days should be
necessary when point 2.6 of annex II
of Directive 2006/17/EC applies, as
contrarily to HIV, HBV and HCV,
Syphilis is not a donation exclusion
criterion as such.
6.
Meeting of
Annex III of
Testing of Partner Donations (not for direct use) in ART:
Meeting of the Competent Authorities

151 | P a g e
the
Competent
Authoritie
s on
Tissues
and Cells
October
2009
Directive
2006/17/EC
Annex III of Directive 2006/17/EC requires the testing of donors
at the time of each donation of gametes in order to assess the risk
of cross contamination during cryo-preservation. Some NCAs
argued that this is costly and excessive and that testing at a fixed
timeframe would be sufficient. The Commission services
requested MS to submit scientific evidence in support of these
arguments.
Joint meeting of the Competent Authorities and the Regulatory
Committee on Tissues and Cells May 2010:
In the preceding months an expert group had been established as
a response as a response to the concerns raised about the
requirements of testing for each partner donation of reproductive
cells as discussed at the Competent Authority Meeting, 19-20
October 2009. The WG included three NCAs and ESHRE. The
objective of the group is to collect and analyse the available
evidence-base. Key aspects include: potential risk of cross-
contamination during cryopreservation; potential risk of cross-
contamination during other processing and storage steps, and
mix-up of gametes.
In parallel, Commission services requested the assistance of the
ECDC in examining the potential health risks associated with
changing the protocol for testing from each donation to periodic
testing (e.g. every 12th or 24th months).
Meeting of the Competent Authorities on Tissues and Cells
December 2010:
for Tissues and Cells June 2011:
The Working Group on Testing of
Partner Donations (not for direct use)
in ART provided feedback indicating
it had taken into account ECDC's risk
assessment on change of testing
requirements for reproductive cells in
partner donation, as well as the
experience and studies provided by
the participating NCAs.
The Working Group considered that it
was not needed to maintain the
current testing requirements for
partner donation as laid down in
Annex III of Directive 2006/17/EC
and this was agreed by the National
Competent Authorities. This will
require a future amendment of the
Directive, through the regulatory
procedure.
It is the responsibility of the NCAs to
ensure that ART tissue establishments
have in place the appropriate safety
and quality systems, which does not
affect the safety and quality of
reproductive cells and/or human
health when donors are tested at up to
24-month time intervals.
152 | P a g e
The ECDC presented the draft risk assessment on change of
testing requirements for reproductive cells in partner donation to
the Competent Authorities. It underlined that given the limited
data available the findings of the risk assessment should be
treated with caution. The Commission services asked the
Competent Authorities to review and provide comments on the
ECDC risk assessment by 31 January 2011.
Meeting of Competent Authorities for
Tissues and Cells December 2011:
Taking into account the
recommendations in the ECDC's risk
assessment on change of testing
requirements for reproductive cells in
partner donation and following the
discussions with NCAs during the
meeting in June 2011, the
Commission presented a draft
decision amending the current legal
testing requirements for partner
donation as laid down in Annex III of
Directive 2006/17/EC.
All participants agreed with the draft.
Following this outcome, there were
also calls to review the screening
requirements for sperm and oocyte
donors (non-partner) to ensure that the
requirements were up to date with
scientific knowledge and testing
advances.
7.
Meeting of
Competent
Authoritie
s on
Tissues
Annex III of
Directive
2006/17/EC
Starting point for Embryonic Cells Lines:
One NCA queried as to which should be considered the starting
point for embryonic stem cell lines derived from frozen
embryos, the donors of the gametes or the embryonic stem cell
Meeting of Competent Authorities
December 2012:
In one NCA, several universities and
companies are developing new
therapies based on "rest-embryos".

153 | P a g e
and Cells
October
2009
lines themselves? Such embryonic stem cell lines may end up in
clinical trials or in use as advanced therapies.
These tissues and cells are subject to the Tissues and cells
Directives as far as donation, procurement and testing is
concerned; to the Directive 2001/20/EC where used for Clinical
Trials and to Regulation 1394/2007 when used as Advanced
Therapies Medicinal Products.
As these regulations fall under different authorities or bodies
(Commission, National Competent Authorities and EMEA), the
need for a multi-party discussion was expressed in order to
ensure complementarities.
One NCA stated that it consider that the starting point for the
embryonic stem cell lines are the donors and that testing should
be performed in accordance with Directive 2006/17/EC.
The Commission services reaffirmed this indicating that as
embryos used for stem cell lines have been frozen, there is a
legal requirement to test the donors at moment of donation
according to Annex III of Directive 2006/17/EC.
Such embryos were originally created
through IVF for the purpose of
fertility treatment, but are no longer
needed by the partner/couple.
Years after their creation, these
embryos can be donated by the
partner couples to research and
development.
This second donation raises some
specific questions:
(a)Are the original tests of partner
donors, relevant if the embryos are
now donated to third parties,
potentially for allogeneic use?
(b) Are these donated embryos
intended for human application?
(c) Due to the time window between
the 2 donations, additional testing
might be needed to check on potential
contaminations in processing/storage.
(d) As time windows might be around
10 years, national requirements from
before transposition of Directive
2004/23/EC might need to be
considered.
The NCA presented a proposal to
amend donor testing requirements in
the Directive 2006/17/EC, allowing

154 | P a g e
testing to be performed on material
other than donor serum (i.e. cell lines)
under specific circumstances.
Following discussions, it was agreed
to further reflect on the proposed
amendment. Member States were
requested to consult their experts and
provide comments.
8.
Meeting of
Competent
Authoritie
s on
Tissues
and Cells
October
2009
Scope:
Article 2 of
Directive
2004/23/EC
Facial transplant:
A question was raised as to whether composite and facial
transplant should be considered organs or tissues. It was
indicated that in most MS, facial transplants should be
considered as composite tissue transplants, multi-tissues or
tissues with special status. Composite tissue transplants require
processing similar to organs; however, composite tissue
transplants are not defined as organs as they fulfil no
physiological life-saving function. No conclusion was reached at
the meeting.
Meeting of Competent Authorities on Tissues and Cell
December 2011:
The question whether composite tissues, such as facial transplant
should fall under the Organs or T&C Directive was again
introduced by the Swedish Competent Authority.
Since this question was first raised in 2009, Directive
2010/53/EU was adopted and provides for a definition for
"organs" in Art 3 (h). Several NCAs considered that facial
Meeting of Competent Authorities on
Tissues and Cells December 2012:
The issue was brought again to the
attention of the NCAs for organ
transplantation and during their
meeting in September 2012 it was
agreed that the face is a vascularised
composite tissue requiring similar
processing and safety issues to organ
transplantation. The group of the
Tissues and Cells NCAs agreed with
this.

155 | P a g e
transplants require similar processing and have similar safety
issues to organ transplantation. It was suggested that
Commission should have the same interpretation for other multi-
tissue transplantation procedures (e.g. hand transplantation).
Meeting of the Competent Authorities on Tissues and Cells June
2012:
The Commission services indicated that the definition of Organ
should prevail in the instance of composite tissues but that a
thorough analysis of the technical aspects of such procedures is
needed. Several NCAs provided arguments in support of
including the transplantation of composite tissues under the
Organs Directive (e.g. long-term storage – possible for tissues
and cells, but not valid for organs/composite tissues). Other
NCAs stated that they have already included composite tissues
in their national legislation when transposing the Directive
2010/53/EC (ES).
Council of Europe representative mentioned that it was agreed to
include transplantation of composite tissues in the last edition of
the Guide for Organ Transplantation to be published next year.
9.
Meeting of
Competent
Authoritie
s on
Tissues
and Cells
October
2009
Distribution:
Definition
and concept
in Directives
2004/23/EC,
2006/17/EC
and
2006/86/EC.
Commercial Bone Distributors:
One NCA raised a query about the distribution of commercial
bone products in the EU indicating that these were generally
placed on the EU market through one distributor within one EU
MS and how these should/can be managed under the Tissues and
Cells Directive.
The Commission services stated that Directive 2004/23/EC
clearly stipulates that the import/export, storage and distribution
Due to the important safety aspects
related to the issues raised, it was
agreed that they should be raised in
discussion in the Import-Export
Working Group with the aim to
finding a harmonised approach across
EU Member States.

156 | P a g e
activities can only be performed by Tissue Establishments.
Therefore, all distributors of commercial bone substitutes in all
EU MS need to comply with the EU legislation and need to be
authorised by the NCA accordingly.
Meeting of the Competent Authorities on Tissues and Cells
December 2011:
The issue regarding commercial bone products was again
brought up by one CA who had received several applications
from national commercial distributors for authorisation as a
Tissue Establishment for "storage" and "distribution" of
demineralised bone products. Most often, these are processed
outside Europe (USA) and imported through one European
country (i.e. the point of entry), where the donor history file
review and product release takes place. When distributed in
Europe from this point of entry flexibility in relation to
qualifications for the designated Responsible Person should be
considered.
During discussions NCAs shared their experience and problems
encountered with commercial distribution of such products.
Another concern was raised: the possibility that some of the
products may enter into some MS as medicines or medical
devices, and thus avoid the national controls by NCAs for tissues
and cells.
In this regard, the Commission reminded the NCAs that a
Working Group to work on import-export legal requirements is
being created and called for volunteers.
Mutual recognition of the site (authorisation) certificates for
distributors of bone substitutes (DBMs) was also introduced by
one NCA who suggested that the site certificates issued by the
Many of the issues raised here in
relation to the import of commercial
bone products in the EU via
commercial distributors/brokers were
subsequently addressed in Directive
2015/566 as regards the procedures
for verifying the equivalent standards
of quality and safety of imported
tissues and cells, however the issue of
brokering services was not addressed
in this Directive and was addressed at
a national level.

157 | P a g e
National Competent Authorities for distributors of bone
substitutes should be more informative so they are mutually
recognised within EU. When a commercial distributor in one
MS receives DMBs from a tissue establishment in another EU
country, the site certificate of the latter could attest that the
specified activities (e.g. donation, procurement, testing,
processing, storage, distribution) were authorised and therefore
the appropriate regulatory requirements have been fulfilled. The
minimum information of name and site address of the tissue
establishment, the list of the specified activities and the product
descriptions/codes to which it is applicable, should be included
and thus, these types of site certificates can assist the principle of
mutual recognition and transparency in EU.
Several NCAs supported the above proposal. It was also
suggested that the principles of the model certificate in the
"Manual on inspection of tissue and cell procurement and tissue
establishments" published by the Commission in August 2010
should be applicable for all site certificates.
Meeting of the Competent Authorities on Tissues and Cells June
2012:
Brokers selling T&C without storage; online advertising of
demineralised bone products:
One NCA again raised the question about authorising/licensing
"broker companies" which are not involved in tissues/cells
storage, but just import/export of products from third countries
and their subsequent distribution in EU Member States. Criteria
for authorising/inspecting were discussed. In addition,
advertising DBMs over internet with the possibility of ordering
and their direct distribution to EU hospitals/professionals was
also debated.

158 | P a g e
10.
Joint
meeting of
the
Competent
Authoritie
s and the
Regulator
y
Committe
e on
Tissues
and Cells
May 2010
Annex II and
III of
Directive
2006/17/EC
(HTLV
Testing)
Human T-lymphotropic virus (HTLV) I/II testing:
For a number of years the American Association of Tissue
Banks (AATB) had required systematic HTLV testing for
donations of tissues and cells occurring on US territory.
Recently the AATB board had agreed to align with FDA's
approach of not imposing an HTLV test for processed human
tissues.
Directive 2006/17/EC requires that HTLV-I antibody testing is
performed for donors living in, or originating from, high
incidence areas, with sexual partners originating from those
areas or where donor's parents originate from those areas.
The Regulatory Committee concluded that the Commission
should request the assistance of ECDC to review the elements
put forward by the AATB to justify suspending systematic
testing for HTLV and to assess the possible risks of the change
in HTLV testing for human tissues and cells imported from US
into the EU.
One NCA informed the participants that it has decided to
maintain its requirements for HTLV testing for tissues and cells
imported from the US. Commission agreed to set up a small
working group of volunteering Member States to discuss and
elaborate on recommendations on how to best face this situation.
This issue was again discussed at the
Meeting of the Competent Authorities
on Tissues and Cells December 2010
and the Meeting of the Competent
Authorities on Tissues and Cells June
2011 and December 2011.
Following the discussion of the
working group on HTLV testing and
based on ECDC's risk assessment on
HTLV transmission by tissue/cell
transplantation, during the NCAs
meeting in June 2011, it was
recommended to amend Annex 11 and
III of the Directive 2006/17/EC by
changing from high "incidence" to
high "prevalence". This amendment
was subsequently implemented.
11.
Joint
Article 2 of
Pancreatic Islet Cell Transplantation:
Meeting of the Competent Authorities

159 | P a g e
meeting of
the
Competent
Authoritie
s and the
Regulator
y
Committe
e on
Tissues
and Cells
May 2010
Directive
2004/23/EC:
Scope
The Human Tissue Authority in one NCA raised a question in
relation to autologous treatment with pancreatic islets in the
same surgical procedure, where the pancreas is removed,
transported to a separate laboratory for processing and returned
to the operating theatre for application. The patient remains in
theatre while processing takes place.
The question raised was whether this procedure would be
excluded from the scope of Directive 2004/23/EC according to
its Article 2. In addition, the meaning of the term "banking" in
recital 8 was questioned in relation to the above mentioned
surgical procedure.
The NCA explained that the question relates to other similar
treatments e.g. a pelvic sarcoma treatment where part of the
pelvis is removed, transported to a different hospital where it is
irradiated and then returned back to the patient. Another NCA
suggested that the transport of the tissues from the operating
room to the place of processing under certain conditions can be
considered banking as referred to in recital 18 of Directive
2004/23/EC.
on Tissues and Cells 6-7 December
2010:
The Commission services presented
the legal framework that applies to the
situation as raised in the May 2010
meeting:
Tissues and cells used as an
autologous graft within the same
surgical procedure are excluded from
the scope of Directive 2004/23/EC.
However, recital 8 explains that the
exclusion is justified only because the
quality and safety considerations
associated with this process
(autologous same surgical procedure
without any banking process) are
completely different (from ‘normal’
tissues and cells).
The considerations are different
because it was assumed at the time
that in an autologous transplantation
within the same surgical procedure,
the cells would remain during the
whole process in the operation theatre,
hence there would be no risks in this
case for cross-contamination or mix-
up of the cells during transport or
during processing. During the

160 | P a g e
discussion, several NCAs referred to
the next case as a typical example for
exclusion: dissection and use of a
peripheral vena for immediate use as
bypass during cardiovascular surgery.
The term ‘banking process’ is not
defined in Directive 2004/23 but
needs to be interpreted in the light of
COM initial proposal and of the CoE
recommendation R(94)/141.
Based on the above consideration it
was concluded that:
The term ‘banking process’ cannot be
considered only as storage and
preservation.
The processing of autologous grafts in
other establishments with a view to
their application within the same
surgical procedure was considered to
be within the scope of Directive
2004/23/EC, as well as the storage (if
relevant), transport and distribution
operations that are carried out with a
view to this processing.

161 | P a g e
12.
Joint
meeting of
the
Competent
Authoritie
s and the
Regulator
y
Committe
e on
Tissues
and Cells
May 2010
Directive
2004/23/EC
Annex II
Point 2.4
Cornea donation in relation to the 24-hour requirement for
blood
Sampling:
In a letter of 8 March 2010, the University Medical Centre of
Freiburg (Germany) claimed that, due to the requirement for
serological viral testing within 24 hours after the donor's death,
the number of cornea donors in Lions cornea donation bank,
Baden-Württemberg had dropped by 25% over a year. It was
explained that often more than 24 hours are needed to fully
explain a corneal donation to the deceased person's relatives.
Most of the NCAs informed that to date they had not
encountered problems of increased cornea loss because of the
testing requirements in Article 4 and Annex II of Directive
2006/17/EC. In addition, many delegations expressed the view
that serological viral testing within 24 hours after donor's death
is necessary and appropriate.
Meeting of the Competent Authorities
on Tissues and Cells 23-24 June 2011:
One NCA presented the preliminary
results of a national study, which
noted that the availability of cadaver
corneas in this country may be
reduced by over 60% due to the 24-
hour limit for post mortal blood
sampling. A study of the Charité
hospital in Berlin was also quoted.
The Group of Competent Authorities
considered that further validation of
assays and data is required, as well as
an improved statistical analysis.
13.
Meeting of
Competent
Authoritie
s on
Tissues
and Cells
December
2010
Article 2 of
Directive
2004/23/EC:
Scope
Human placenta tissues for human consumption:
One NCA raised a question about the legal status of human
placenta tissues for human consumption. Human placenta is
consumed by some birth mothers either raw (placenta
smoothies) or cooked and dehydrated (capsules) or by the baby
in a form of tincture. The placenta derived products are prepared
by a) the birth mother at her home b) a “placenta expert” at
mother’s home c) third party (commercial service). The
questions raised were 1) whether the placenta processing falls
within Directive 2004/23/EC and its implementing measures and
2) whether placenta derived products are covered by the Food
The Commission services presented
the legal framework. It could be
argued that, although derived from
human tissues and cells, the placenta
derived product under question are
intended to be ingested by humans
hence they are covered by the
definition of ‘food’ in Regulation
178/2002/EC on general food law.
162 | P a g e
Law.
14.
Meeting of
Competent
Authoritie
s tor
Tissues
and Cells
June 2011
Article 2 (a)
of Directive
2004/23/EC:
Scope
Procurement of Stem Cells from Autologous Adipose Tissue
:
The Commission was asked whether the procurement of stem
cells from autologous adipose tissue and re-implantation within
the same surgical procedure is considered in or out of scope of
the Cell & Tissue Directive 2004/23/EC.
The device is currently used for reconstructive surgery (e.g.
reconstruction of the breast following mastectomy).
The Commission services briefly presented the main features of
the CE-marked device, which enables real-time access to
adipose-derived stem cells at the bedside. The device automates
and standardises the extraction, washing, and concentration of a
patient's own adipose-derived regenerative cells (ADRCs).
Several NCAs expressed their
opinions on the procedure.
Conclusively, the group of Competent
Authorities for Tissues and Cells
considered that that procurement of
stem cells from adipose tissue using
the above mentioned procedure, when
applied to the same individual within
the same surgical procedure, in the
same operating room, when cells are
used for the same essential function
(e.g. adipose derived regenerative
cells restoring the adipose mass of the
breast following mastectomy for
breast cancer), should be exempted
from the Cell & Tissue Directive
2004/23/EC, based on Article 2.a.
15.
Meeting of
Competent
Authoritie
s on
Tissues
and Cells
December
2011
Regulatory Status of Apheresis of mononuclear cells for
preparation of ATMPs:
In order to produce some ATMPs (e.g. autologous tumour
vaccines) peripheral blood mononuclear cells are collected by an
apheresis procedure. These procedures can be carried out in
hospitals and in blood establishments. The question is whether
the cell product is in the scope of the Blood Directive or in the
scope of the T&C Directive (or both).
Following discussion, it was agreed
by NCAs that, similar with Donor
Lymphocyte Infusion, mononuclear
cells collected by apheresis for ATMP
manufacture are considered to be in
the scope of the EU tissue and cell
Directives.

163 | P a g e
16.
Meeting of
Competent
Authoritie
s on
Tissues
and Cells
December
2011
VUD
VUD; Compensation of Gamete Donors:
One NCA updated the NCAs about the new decisions taken by
the Human Fertilisation and Embryology Authority's (HFEA)
regarding the financial compensation for gamete donors and the
benefits in kind allowed by its legislation (egg sharing). It was
highlighted that without these approaches, this Member State
would face a major shortage of eggs. 40% of the eggs currently
donated in this Member State are ensured through egg sharing,
the remaining 60% being provided by known donors (friends,
relatives). It was also mentioned that the Member State has a
registry of all gametes donors and that counselling before
donation is compulsory.
During the following discussions, some NCAs provided input on
their national legislation and in particular on the types of
financial compensation for gametes donation or benefits in kind.
Discussions also addressed the issue of financial compensation
vs. incentive. The Commission services reminded the NCAs
about the requirements of the Charter of Fundamental Rights of
the EU, which clearly prohibits on "making the human body and
its parts as such a source of financial gain".
It was agreed by the NCAs that this
topic requires further work at Union
level and that a comprehensive
definitions and understanding of
‘compensation’, ‘incentives’ and
‘benefit in kind’ is required.
17.
Meeting of
Competent
Authoritie
s on
Tissues
and Cells
Article 2 of
Directive
2004/23/EC:
Scope
Regulatory Classification of Autologous Endometrial Cells
(Endocell):
A Competent Authority asked the Commission and the CAs for
Tissues and Cells whether the co-culture of autologous
endometrial cells to support embryo transfer at blastocyst stage
may fall under the provisions of Directive 2004/23/EC and about
NCAs agreed that there is no clear
regulatory framework yet for such a
product (e.g. endometrial tissue)
which is produced in one Member
State for use in processing patient
cells in other Member States. The
164 | P a g e
June 2012
its use in other EU Member States.
One NCA stated that this is considered an ancillary therapeutic
product in this Member State, requiring authorisations.
issue of mutual recognition of
processing authorisations could be
discussed at a future date. Some
NCAs considered that in this case
rather the process in the different sites
of utilisation should be authorised,
and not the single product. It was
stated that authorisation for such
processing falls under Directive
2004/23/EC.
18.
Meeting of
Competent
Authoritie
s on
Tissues
and Cells
June 2012
Article 2&3
of Directive
2004/23/EC:
Scope
Faecal microbiota transplants:
One NCA asked whether faeces donated by partner or close
relative transplanted as treatment for Clostridium difficile
infection may fall under Directive 2004/23/EC and what would
be the safety and quality issues to be considered in this case. The
group of NCAs concluded that bacterial flora does not fall under
the provisions of Directive 2004/23/EC.
Meeting of the Competent Authorities
on Tissues and Cells June 2014:
The issue of faecal
donation/transplant or FMT (Faecal
Microbial Transplant) was raised
again.
Meeting of the Competent Authorities
on Tissues and Cells December 2014:
The Commission services informed
the meeting that, to fall under
Directive 2004/23/EC, such
substances would need to be
considered as a 'tissue' or 'cell' and
secondly, would need to be
considered as intended for 'human
application' as per the definitions of

165 | P a g e
these terms in Article 3 of Directive
2004/23/EC. It was clarified that since
they do not meet these criteria these
substances are not considered as
falling within the scope of Directive
2004/23/EC or any particular quality
and safety legislative framework at
Union level. Thus, Member States are
free to decide on the most suitable
framework either by creating a
specific regulatory framework at
national level or by applying one of
the existing national legislative
frameworks, including the tissues and
cells quality and safety requirements,
to these substances.
The Commission services further
pointed out that Article 168(4) TFEU
is unequivocal in laying down a
mandate for the adoption of measures
setting high standards of quality and
safety with respect to all substances of
human origin. As both human breast
milk and faeces are uncontestably
substances of human origin it was
concluded in the meeting that this
Treaty Article should provide a legal
basis for possible future regulation of
these substances in terms of their
quality and safety.
166 | P a g e
19.
Meeting of
Competent
Authoritie
s on
Tissues
and Cells
December
2012
More
Stringent
Safety
Requirements
NAT HCV testing for tissues imported from third countries
and distributed in EU Member States:
One NCA raised the issue of more stringent safety and quality
requirements than those requested by the EU legislation (e.g.
HVC NAT testing) when importing tissues from third countries
(e.g. USA), particularly via other MS where similar more
stringent requirements are not in place.
The NCAs asked questions, including whether an EU Member
State is allowed to prohibit an import when the quality meets
minimum requirements of the EU Directives but not the more
stringent national requirements. Several NCAs with more
stringent testing requirements stated that they already have in
place methods to check these additional requirements and in
some countries tissues/cells distributed from other EU countries
are treated like tissues/cells imported from third countries.
Since Directive 2004/23/EC lays
down only minimum safety and
quality requirements, it was concluded
that it is the responsibility of the
Member States with more stringent
testing requirements (6-7 EU Member
States) to check whether tissues/cells
distributed from other EU Member
States fulfil their national
requirements.
20.
Meeting of
Competent
Authoritie
s on
Tissues
and Cells
June 2013
Therapeutic application of blood cellular components
separated by cytapheresis:
One NCA asked the Commission and the group of Tissues and
Cells CA whether blood cellular components separated by
cytapheresis are covered by the Blood or Tissues and cells
legislation. This clarification would be needed to decide whether
a blood establishment performing such procedures may also
require an authorisation as a tissue establishment.
Several NCAs considered that such activities are covered by the
Blood legislation. One NCA recalled previous discussions in
which the Tissues and Cells CA group agreed that donor
lymphocytes infusion and mononuclear cells should be covered
by the Tissues and Cells legislation. It was mentioned that both
The Commission agreed that this topic
should be further analysed.

167 | P a g e
pieces of legislation, provide the same level of quality and
safety. However, it was underlined that the legal requirements
should be clear for all stakeholders, and it would be preferable to
have the same approach at EU level.
21.
Meeting of
Competent
Authoritie
s on
Tissues
and Cells
June 2013
Article 17 of
Directive
2004/23/EC
Responsible Person – meeting laws in other Member States:
An NCA queried whether the responsibility for meeting any
supplementary national requirements for distribution falls under
the responsibility of the responsible person (RP) at the receiving
tissue establishment in one Member State or of the RP at the
sending tissue establishment in another Member State.
It was pointed out that Article 17(2)(a) of Directive 2004/23/EC
provides that the RP has the duty and responsibility for
"ensuring that human tissues and cells intended for human
applications in the establishment for which that person is
responsible are procured, tested , processed, stored and
distributed in accordance with this Directive and with the laws in
force in the Member State". It was also recalled that during the
Tissues and Cells CA meeting in December 2012, the group
concluded that it was the responsibility of the Member States
with more stringent testing requirements to check whether
tissues/cells sent from other EU Member States fulfil their
national requirements.
Two NCAs confirmed that in their countries, for tissues/cells
received from other MS, the RP needs to confirm that the more
stringent requirements were met. Two NCAs stated that
collaboration with the RP at the sending tissues/cells do not
fulfil the legal requirements of the receiving country whenever
they are aware that sent tissues/cells do not fulfil the legal
It was indicated that this issue could
possibly be looked at in the context of
a potential revision of Directive
2004/23/EC.

168 | P a g e
requirements of the receiving MS. The Commission highlighted
that this question needs also to take into account cases of "direct
distribution".
22.
Meeting of
Competent
Authoritie
s on
Tissues
and Cells
June 2013
Autologous keratinocytes suspension:
An NCA asked whether Directive 2004/23/EC applies to
autologous keratinocyte suspensions. From the technical point of
view, autologous epithelial tissue is collected from a patient in a
hospital and distributed immediately to a laboratory (outside the
hospital). In the laboratory, the keratinocytes are separated
mechanically and enzymatically from the collected tissue, and
the intermediate product is diluted with a physiological sodium
chloride solution. It was clarified that keratinocytes are not
manipulated or cultured and the entire processing takes about
two hours. After processing, the product is sprayed on the same
patient's burned area or wound. In relation to the above question,
one NCA described a similar case, in which adipose cells are
transported for processing to an outside laboratory in a sealed
container, which is closed/opened only in the operating theatre.
It was noted that with regard to
similar question during the Tissues
and Cells CA meeting in December
2010 (pancreatic islets processed in a
laboratory for use in the same surgical
procedure") the advanced answer was
that the exemption foreseen under
Article 2.2(a) of Directive
2004/23/EC should not apply if the
tissues or cells are taken out of the
operating room to a laboratory for
processing. It was considered that
using a “sealed container" does not
provide any additional safety benefit,
and the risk of mix-up or mislabelling
remains unchanged.
23.
Meeting of
Competent
Authoritie
s on
Tissues
and Cells
June 2013
Partner
Donation in
ART
Definition of Partner Donation:
A question was raised on the use of the term ‘partner donation’
in ART defined solely as an intimate relationship between a
male and female partner. They referred to ‘known donors’ e.g. in
a lesbian relationship where one partner donates oocytes to the
other for IVF following fertilisation with donor sperm or where
It was concluded that the definition of
partner donation could be looked at in
the context of a potential future
revision of the Directive.
169 | P a g e
a female relation or friend may do similar for another female
relation or friend.
Two NCAs confirmed that such ‘known’ donors must be treated
as non-partner donors and tested/processed as such.
24.
Meeting of
Competent
Authoritie
s on
Tissues
and Cells
June 2013
Genetic
Screening of
Egg Donors
Genetic Screening of Gametes:
A question was raised concerning the national practices with
regards to genetic screening of egg donors.
Following some discussion, it was
agreed that further reflection was
required on the development of a list
of genetic screening requirements for
donors of non-partner sperm and
oocytes.
25.
Meeting of
Competent
Authoritie
s for
Tissues
and Cells
December
2013
Article 9 of
Directive
2004/23/EC:
Import
Import and distribution of starting materials for ATMPs:
One NCA introduced the issue of the import and subsequent EU
distribution of starting materials for ATMPs with a view to
clarifying whether such activities fall within the scope of the
tissues and cells legislation.
Article 9 of Directive 2004/23/EC lays down that imports of
tissues and cells from third countries are undertaken by tissue
establishments authorised to import while Article 3 of
Regulation 1394/2007 on ATMPs states that the donation,
procurement and testing of tissues and cells contained in ATMPs
shall be in accordance with the tissues and cells legislation but
makes no mention of their import.
The question concerned circumstances when donation,
procurement and testing takes place in a third country and
whether the subsequent import of starting materials for ATMPs
must be authorised under the tissues and cells legislation.
As shown by the presentation, different approaches were

170 | P a g e
adopted across the MS often depending on the level of
processing the tissues and cells have undergone in the third
country.
26.
Meeting of
Competent
Authoritie
s for
Tissues
and Cells
December
2013
Directive
2006/86/EC:
Annex I
Point C(6)
Use of CE marked medical devices – 'when applicable /
wherever possible / when appropriate'
One NCA requested clarification of the meaning of 'Critical
reagents and materials must meet documented requirements and
specifications and when applicable the requirements of Council
Directive 93/42/EEC of 14 June 1993 concerning medical
devices and Directive 98/79/EC of the European Parliament and
of the Council of 27 October 1998 on in vitro diagnostic medical
devices'.
Directive 2006/17/EC, Annex IV, point 1.3.10 states that
'wherever possible, only CE marked medical devices must be
used…' while Annex II, point 2.1 to the same Directive lays
down that 'tests must be carried out by a qualified laboratory,
authorised as a testing centre by the competent authority in the
Member State, using EC-marked testing kits where appropriate.'
The general opinion during the
discussion which followed this
presentation was that while such
phrasing creates ambiguity it also
leaves some room for some much-
needed flexibility in interpreting the
meaning.
.
27.
Meeting of
Competent
Authoritie
s for
Tissues
and Cells
Article 2 of
Directive
2004/23/EC:
Scope
Breast Milk Banking:
The question of how to regulate the allogeneic 'application' of
breast milk has arisen following the proliferation of breast milk
banks across the EU. As the Council of Europe have produced a
questionnaire for its CD-P-TO members on the issue, it was
decided that DE, rapporteur within the CoE group, would
Meeting of the Competent Authorities
on Tissues and Cells June 2014:
The issue of breast milk banking was
raised again.

171 | P a g e
December
2013
present the questionnaire as a way of initiating a preliminary
discussion on this issue within the CA group.
During the discussion it was suggested that a majority of MS
regulate such activity through food safety authorities although
the emergence of applications of breast milk for therapeutic
purposes may require a reassessment of such regulatory
structures and closer cooperation between food safety and T&C
CAs in order to ensure that disease transmission risks and ethical
issues linked to donation are suitably dealt with.
Meeting of the Competent Authorities
on Tissues and Cells December 2014:
In the meeting the Commission stated
that such substances would need to be
considered as a 'tissue' or 'cell' and
secondly, would need to be
considered as intended for 'human
application' as per the definitions of
these terms in Article 3 of Directive
2004/23/EC to fall under Directive
2004/23/EC. It was clarified that if
these substances are not considered as
falling within the scope of
2004/23/EC or any particular quality
and safety legislative framework at
Union level, Member States are free
to decide on the most suitable
framework either by creating a
specific regulatory framework at
national level or by applying one of
the existing national legislative
frameworks, including the tissues and
cells quality and safety requirements,
to these substances.
The Commission services further
pointed out that Article 168(4) TFEU
is unequivocal in laying down a
mandate for the adoption of measures
setting high standards of quality and

172 | P a g e
safety with respect to all substances of
human origin. As both human breast
milk and faeces are uncontestably
substances of human origin it was
concluded that this Treaty Article
should be able to provide a legal basis
for future regulation of these
substances in terms of their quality
and safety.
28.
Meeting of
Competent
Authoritie
s for
Tissues
and Cells
December
2013
SARE
Reporting
Requirements
Reporting of a serious adverse reaction (SAR) upon the birth
of a child from a sperm or egg donor with a genetic disease:
The issue concerned reporting of SAR following sperm
donations from non-partner donors where a genetic disease is
transmitted to the new-born child from the donor. Technically
speaking this SAR does not occur in the recipient but in the new-
born child and therefore does not need to be reported if the
wording of the legislation is followed strictly. Two NCAs felt
that such cases should be reported as SAR.
29.
Meeting of
Competent
Authoritie
s for
Tissues
and Cells
December
2013
Directive
2006/17/EC
Annex III
Identifiable genetic diseases in non-partner donors:
One NCA called for a discussion on what should or could be
done in terms of guidance or conditions on the use of such
reproductive cells once a genetic disease in a donor has been
identified following sperm donation. Other than the requirement
laid down in Annex III to Commission Directive 2006/17/EC
that complete information on the associated risk of genetic
disease transmission must be communicated and explained to the
Meeting of Competent Authorities on
Tissues and Cells June 2015:
One NCA informed the group about
two changes in national legislation
regarding procedures related to sperm
donors identified as carriers of a
hereditary genetic disorder, effective
from April 2015.

173 | P a g e
recipient where there is an identified history of genetic disease in
the donor's family, there are no rules in the EU legislation on the
use or non-use of such reproductive cells.
One NCA explained that the terms used in its country are
'temporary block' and 'conditional block' however the
conditional block is permanent in its nature except that the
donated cells may continue to be used for siblings of children
already born from that donor only. Another NCA pointed out
that, other than on public health grounds, a complete ban on the
use of such cells may be difficult in light of fundamental /
human rights provisions on the right to start a family and stated
that in the case of a potential use for siblings it would expect a
risk assessment to be carried out and the recipient couple to
receive counselling on the risks of genetic disease transmission.
According to these current
requirements, such donors are no
longer placed in either quarantine or
permanent block, but the sperm bank
must update the donor profile/status
on their website each time such
information becomes available.
Customers (clinics and private
individuals) can purchase sperm
straws from these donors only after
having read and accepted a
declaration (even without reading the
donor information with the case
description and risk assessment, with
reference to Convention on Human
Rights and Biomedicine). The sperm
bank must also inform all customers
that already purchased straws from
such donors.
The NCA also confirmed that, after
reintroducing the "permanent block"
status, it will continue informing CAs
about such cases via the RATC
platform.
30.
Meeting of
Competent
Authoritie
s for
Tissues
and Cells
Distribution:
(Directives
2004/23/EC,
2006/17/EC
and
2006/86/EC)
Direct distribution of reproductive cells to end users:
Following discussions on this subject in previous CA meetings
(June 2013), one NCA gave an update of the latest situation and
informed the group that they had come to the conclusion that,
under its national law, sperm banks could not be prevented from
Meeting of Competent Authorities on
Tissues and Cells June 2015:
Following a request for clarification
from the NCAs, the Commission
informed the group that it has

174 | P a g e
December
2013
distributing directly to end users as they felt that this would
infringe free movement of goods rules.
Several NCAs pointed out that they have more stringent rules in
place on who can purchase /use such reproductive cells or the
permission needed from the NCA prior to cross-border
distribution into their MS. However, this was not the case in all
MS.
This provoked the question of how potential end-users and / or
the distributing sperm bank should be made aware of such
additional national rules. A further question was raised querying
the extent of such situation and whether it would be possible to
get data from the distributing TEs in order to establish the true
extent of this issue. CAs were therefore called on to try to obtain
and provide generic data on this situation to be presented at the
next NCA meeting.
considered whether a requirement to
distribute sperm to an authorised
tissue establishment or authorised
organisation responsible for human
application (i.e. a restriction on direct
distribution to natural persons) is in
line with Union law and if such a
restriction is in line with Union law,
can Member States with such
restrictions in place require the
cooperation of the MS of origin in
enforcing them.
The Commission services indicated
that not only would such a restriction
seem to be admissible in order to
implement EU quality and safety
standards, the lack of such a
restriction may be regarded as not
being in line with Union legislation
and in particular the provisions on
traceability and the obligation to
report (serious) adverse reactions.
Meeting of Competent Authorities on
Tissues and Cells February 2017:
One MS subsequently amended its
legislation to fully comply with EU
requirements for traceability and
vigilance.

175 | P a g e
31.
Meeting of
Competent
Authoritie
s for
Tissues
and Cells
December
2014
Article 2 of
Directive
2004/23/EC:
Scope
Regulation of lymphocyte immunotherapy (in treatment of
recurrent miscarriage):
One NCA introduced the topic of lymphocyte immune therapy
(LIT) to the group and raised the question of how the steps
leading to its human application should be regulated across the
Union.
LIT is used as a treatment for recurrent miscarriage and involves
the application of allogeneic human cells (lymphocytes) which
are separated from the whole blood collected from a donor, often
the husband or partner of the recipient. In such a situation both
the EU tissues and cells legislation, the EU blood legislation or a
combination of both could be relevant.
Prior to the NCA meeting a survey was conducted on how
activities leading to LIT were currently regulating. While a
considerable majority of respondents indicated that these
activities were regulated under T&C legislation, a variety of
responses were received with some MS also indicating a
combination of both blood and T&C legislation. Other replies
also stated that other regulatory frameworks were used or that
the therapy was not used within their MS. The NCA itself
indicated that, in the absence of legal clarity on the issue, it was
currently adopting a pragmatic approach allowing for the
procurement to take place by establishments authorised to carry
out procurement under the tissues and cells legislation or to
carry out collection under the blood legislation. The NCA also
pointed out that a cross-sector decision-making process is
needed at EU-level to deal with such classification issues.
Meeting of Competent Authorities on
Tissues and Cells June 2015:
The Commission services considered
the following three issues: whether
withdrawal of whole blood leading to
extraction of lymphocytes falls within
the scope of blood or tissues and cells
legislation, whether Member States
may choose to regulate this activity
under either their national blood or
tissues and cells legislation and finally
whether within a Member State, its
authorities have the discretion to
regulate the activity under both their
national blood and tissues and cells
legislation.
The Commission services put forward
the following view for the group's
consideration: It was felt that
justifiable arguments could be made
for this activity falling under either
the blood or tissues and cells
legislation. This being the case, based
on the specific nature of their national
circumstances (assessment of risks to
human health / desired level of human
health protection / the existence of

176 | P a g e
In the discussion which followed one NCA declared that it
considered the initial steps of donation, procurement (collection)
and testing to fall under the blood legislation and stated its
interest in hearing why a number of NCAs indicated that they
regulate it under the T&C legislation.
One NCA pointed out that both donor lymphocytes for infusion
and haematopoietic stem cells fall entirely under T&C
legislation. In particular, a parallel could be drawn with donor
lymphocytes infusion where identical processing techniques are
applied although the actual procurement method is different.
This NCA also pointed out that a greater number of tissue
establishments were equipped to carry out the procurement and
then processing than blood establishments.
From a practical point of view it thus made more sense to allow
TEs to oversee the donation, procurement (collection) and
testing rather than restrict this to BEs many of which are not
further equipped to carry out the further stages leading to LIT.
While a majority in the group were in favour of allowing
procurement under the tissues and cells legislation, one NCA
asked for further clarity.
more stringent protective measures
etc…), and given the fact that LIT is
typically a local activity not involving
cross-border steps, Member States
benefit from a certain degree of
discretion when deciding whether to
classify this activity under either
blood or tissues and cells legislation.
On the final point the Commission
services reminded the group of the
importance of maintaining legal
certainty within any given national
legislative framework but did not
exclude the possibility that in this
specific case authorities within one
Member State could allow the same
activity to be governed under both
sets of legislation, depending on the
establishment which performs it,
provided that this is justified by an
assessment of the risks to human
health and the desired level of
protection specific to that Member
State focusing in particular on largely
equivalent levels of quality and safety
assured under both sets of national
legislation.
32.
Meeting of
Directive
Testing Requirements for Non-Partner Sperm Donors:
Meeting of the Competent Authorities

177 | P a g e
Competent
Authoritie
s for
Tissues
and Cells
February
2017
2012/39/EU,
Annex III
point 4.2.
On the subject of testing requirements for donations other than
by partners, one NCA explained that its country requires non-
partner sperm donors to be tested at the time of their first sperm
collection and each 3 months subsequently. One NCA referred
to the wording and the history of Directive 2012/39/EU, Annex
III point 4.2. In this context, the Commission services agreed to
request ECDC to assess any risks associated with the donor
testing practices for non-partner donations according with this
practice.
on Tissues and Cells November 2017:
ECDC had assessed the risks
associated with the donor testing
protocol for non-partner donations
currently being applied in one MS.
The preliminary conclusions of the
ECDC technical report suggest that
the national protocol largely ensures
an equivalent level of safety.
However, under certain circumstances
a minor increased risk of missing a
window period infection remains,
which could be mitigated by
restricting release for a specified
period. In that case, the protocol could
be considered as more stringent than
the Directive requirement.
Meeting of the Competent Authorities
on Tissues and Cells June 2018:
The Commission services reported
back that, based on the conclusions of
the ECDC technical report, that the
Danish serological testing protocol
can be considered as safe as, or safer
than, the protocol in Directive
2006/17/EC, therefore, it may be
argued that the protocol can be
considered as a more stringent
national requirement as permitted in
Article 4 of Directive 2004/23/EC and
178 | P a g e
article 168 (4)(a) TFEU. With regard
to the NAT testing protocol, the
Commission services concluded that
this could also be considered as a
more stringent national requirement
provided that the national legislation
was changed in line with the
recommendation of ECDC to defer
release of the donated sperm until the
longest diagnostic window period has
lapsed. This change to the legislation
was subsequently implemented.
33.
Meeting of
Competent
Authoritie
s for
Tissues
and Cells
November
2017
Article 2 of
Directive
2004/23/EC:
Scope
Same Surgical Procedure – A Case Study:
One NCA described a case concerning the use of autologous
tissue/cells that are removed from a patient, immediately
processed in a device at the patient's side and returned to the
same patient. Directive 2004/23 excludes from its scope
tissues/cells that are used in 'the same surgical procedure'.
Some stakeholders now suggesting that such the Same Surgical
Procedure exclusion is no longer appropriate as the 'close to the
patient/bedside' technologies are becoming more and more
important and there should be an authorisation of the process,
not just a CE-marking of the device in which the substance is
processed.
This particular case also involved a 'claim' that fat cells improve
conditions such as chronic cystitis, asthma, stroke, etc. CAs
The NCAs considered that a careful
consideration should be given to this
case in the context of the ongoing
Blood, Tissues and Cells Evaluation.

179 | P a g e
considered that such claims highlight the need for a requirement
for demonstrating 'efficacy'.
34.
Meeting of
Competent
Authoritie
s for
Tissues
and Cells
June 2018
Rapid Alerts relating to Sperm Donors:
One NCA issues rapid alerts in the RATC system on gamete
donors being permanently blocked following the identification
of potentially serious genetic disease or hereditary genes for
such diseases in a donor. The NCA reported regarding a recent
recommendation from the ‘Over-implementation Board’ under
the Ministry of Employment to the Government Committee on
Over-Implementation indicating that the NCA should not issue
alerts to other EU MS when a child with a genetic condition has
been born from a particular sperm donor and sperm from that
donor, potentially or certainly carrying the responsible gene, has
been distributed to other EU MS.
The NCAs were unanimously supportive in their efforts to use
the RATC platform to share information of this kind rapidly.
The representatives considered that transmission of genetic
diseases should indeed be seen as ‘serious adverse reactions’ and
that this kind of communication is important for patient safety
and that the practice should not be changed. The representatives
also noted that the RATC platform is an effective way for such a
rapid communication among the MS authorities.
The Commission services reminded
the participants that in the current
2004 legal framework, genetic
transmissions to children from gamete
donors are not explicitly mentioned,
but that this issue had been raised in
the evaluation exercise as a sector
development on which EU legislation
needs to be aligned for clarity.
180 | P a g e
Annex IX: List of Public Health Programme Actions funded to support the implementation of the BTC legislation
The European Commission provides funding for Actions in the area of SoHO through the EU Health Programme
410
, in the form of projects or joint
actions with national authorities.
Part A: Blood and Blood Components
CATIE
Training sessions for inspectors in the field of
blood and blood Components
https://www.catie-europe.eu/
A project promoting common standards and organising
training workshops to disseminate these standards
Creative Ceutical
Creative Ceutical report
https://ec.europa.eu/health/sites/health/files/blood_t
issues_organs/docs/2017_eupbm_authorities_en.pd
f
An EU-wide mapping exercise of the market for blood,
blood components and plasma derivatives, focusing on
their availability for patients
CORDIS
Rapid SPR for parallel detection of pathogens in
blood
Improving the safety of blood and organ supply by
creating the research infrastructure to monitor
emerging pathogens and develop new screening
tests.
A project on blood testing, for the development of
reliable and comparable testing procedures across the
EU.
410
Website Commission- EU Health Programme.
181 | P a g e
DOMAINE
DOnor MAnagement IN Europe
https://www.sanquin.org/research/donor-studies-
projects/domaine
A project creating a safe and sufficient donor
population in Europe: comparing and recommending
good donor management practice
EU - Q - Blood
SOP
Pan-European standard operating procedure
methodology
http://ec.europa.eu/health/ph_projects/2004/action2
/docs/action2_2004_inter_04_en.pdf
A project developing a pan-European standard
operating procedure (SOP) for best practice in ensuring
the quality and safety of blood
Eurobarometer
Eurobarometer
https://ec.europa.eu/health/blood_tissues_organs/eu
robarometers/eb822_en
A report outlining the European public's attitude to
blood and tissue and cells donation and transfusion
and/or application
EuBIS
Development of pan-European standards and
criteria for the inspection of Blood establishments
(EU-Blood-Inspection)
https://www.eubis-europe.eu/
A project laying down a standard operating procedure
methodology on quality and safety of blood.
EUOBU
EU Optimal Blood Use
http://www.optimalblooduse.eu/
A guide on EU Optimal Blood Use
PBM
Patient Blood Management
https://ec.europa.eu/health/sites/health/files/blood_t
issues_organs/docs/2017_eupbm_authorities_en.pd
f
https://ec.europa.eu/health/sites/health/files/blood_t
A project developing Patient Blood Management
182 | P a g e
issues_organs/docs/2017_eupbm_hospitals_en.pdf
SoHO V&S
Vigilance and Surveillance of Substances of
Human Origin
http://www.notifylibrary.org/content/vigilance-
and-surveillance-substances-human-origin-project-
sohovs
A project supporting the Member States in the
establishment of vigilance and surveillance systems for
tissues and cells in transplantation and assisted
reproduction
VISTART
Vigilance and Inspection for the Safety of
Transfusion, Assisted Reproduction and
Transplantation
https://vistart-ja.eu/
A project promoting and facilitating the harmonisation
of inspection, authorisation and vigilance systems for
blood, tissues and cells.

183 | P a g e
Part B: Tissues and cells
Euro-GTP II
European Good Tissue Practices
http://www.goodtissuepractices.eu/
Projects to develop European Good Tissue Practice Guidance and other tools
for TEs, to improve mutual recognition and support harmonisation of
practices across the EU, and help facilitate the movement of tissues and cells
between Member States
Eurobaromete
r
Eurobarometer
https://ec.europa.eu/health/blood_tissues_
organs/eurobarometers/eb822_en
A report outlining the European public's attitude to blood and tissue and cells
donation and transfusion and/or application
SoHO V&S
Vigilance and Surveillance of Substances
of Human Origin
http://www.notifylibrary.org/content/vigil
ance-and-surveillance-substances-human-
origin-project-sohovs
A project supporting the Member States in the establishment of vigilance
and surveillance systems for tissues and cells in transplantation and assisted
reproduction
EQSTB
European Quality System for Tissue
Banking Project
http://www.eqstb-sanco.org/
A project creating the ‘Guide of recommendations for Tissue Banking’ and a
prototype of an online European multinational musculoskeletal tissue
registry
SANCO/2008/
C6/051
Comparative analysis of medically
assisted reproduction in the EU:
regulation and technologies
A project carrying out a comparative analysis of medically assisted
reproduction in the EU

184 | P a g e
POSEIDON
Promoting optimisation, safety,
experience sharing and quality
implementation for donation organisation
and networking in unrelated HSC
transplantation in Europe
https://webgate.ec.europa.eu/chafea_pdb/h
ealth/projects/2006210
A project promoting the optimisation, safety, experience sharing and quality
implementation for donation organisation and networking in unrelated HSC
transplantation in Europe
EUSTITE
EU standards and training for the
inspection of tissues establishments
A project promoting the standardisation and training for the inspection of
tissues establishments across the Member States, in compliance with the
tissues and cells Directive
EUROCET
European registry for organs, tissues and
cells project
http://www.eurocet.org
A project creating a European registry for the data collection on organ, tissue
and cell donation and transplantation activity
EAHC/2012/H
ealth/19
Economic landscapes of human tissues
and cells for clinical application in the EU
https://ec.europa.eu/health/sites/health/file
s/blood_tissues_organs/docs/economiclan
dscapes_humantissuescells_en.pdf
A project identifying key activities and costs, key players in public and
private sectors, legislative and reimbursement schemes across Member
States, and finally emerging technological trends and associated ethical,
legal, and social issues.
ECCTR
The European Cornea and Cell
Transplantation Registry
http://www.ecctr.org/ecctr-database
A project building a common assessment methodology and establish an EU
web-based registry and network for academics, health professionals and
authorities to assess and verify the safety quality and clinical effectiveness of
(new) human tissue transplantations and in ophthalmic surgery.

185 | P a g e
VISTART
Vigilance and Inspection for the Safety of
Transfusion, Assisted Reproduction and
Transplantation
https://vistart-ja.eu/
A project promoting and facilitating the harmonisation of inspection,
authorisation and vigilance systems for blood, tissues and cells
ARTHIQS
Assisted Reproductive Technologies and
Haematopoietic Stem cells for
transplantations
http://arthiqs.eu/
A project developing guidance for establishing a hematopoietic progenitor
cells donor follow-up registry and inspection guidance in Assisted
Reproductive Technologies (ART)
Jacie
Joint Accreditation Committee ISCT
Europe & EBMT
www.jacie.org/
A project developing and maintaining global standards for the provision of
quality medical and laboratory practice in cellular therapy. JACIE offers
accreditation to transplant programmes in order to encourage health
institutions and facilities to establish and maintain quality management
systems
SANCO/2008/
C6/051
Comparative Analysis of Medically
Assisted Reproduction in the EU:
Regulation and Technologies
https://ec.europa.eu/health/sites/health/file
s/blood_tissues_organs/docs/study_eshre_
en.pdf
A project comparing regulatory frameworks at Member State level for
Medically Assisted Reproduction and comparing reimbursement among
Member States

186 | P a g e
Annex X: ECDC – Support for EU Activities on Substances of
Human Origin 2012 – 2018
The European Centre for Disease Prevention and Control (ECDC) is an EU
agency aimed at strengthening Europe's defences against infectious diseases. ECDC
works in three key strategic areas: it provides evidence for effective and efficient
decision-making, it strengthens public health systems, and it supports the response to
public health threats.
In 2012, ECDC appointed its first Senior Expert dedicated to vigilance of infectious
safety of substances of human origin (SoHO), based today within the Epidemic
Intelligence and Response unit. The work of ECDC on this topic underlines the important
role of transfusion, transplantation and medically assisted reproduction in the secondary
spread of infectious diseases. Since the appointment, there is continuous communication
between DG SANTE and ECDC.
Expert consultation meetings
1. Assessing the risk of communicable diseases transmissible through substances of
human origin, prioritisation exercise, ECDC, Stockholm, 20–21 September 2012;
2. Satellite consultation meeting on blood donation screening for evidence of malaria
infection ECDC, Stockholm, 12-13 February 2013;
3. Consultation on spatial definition of areas affected by malaria, Strasbourg, 04
October 2013
4. Consultation expert meeting on areas with the high prevalence of HTLV – I infection,
ECDC, Stockholm, 22nd September 2014;
5. 2nd Meeting of the Working group on prequalification of new blood donors, ECDC,
Stockholm, 20 October 2014;
6. Priority setting for a risk assessment of bacterial infection transmission through
substances of human origin. Stockholm, 24 – 25 September 2015;
7. Assessing the risk and prevention of hepatitis E transmission through substances of
human origin, 27 May 2015, Lisbon, Portugal;
8. Enhanced epidemiological data collection and analysis by establishments for
substances of human origin, Stockholm, 29 – 30 September 2016;
9. Expert meeting related to the Guide for preparedness activities on the prevention of
Zika virus transmission through SoHO in EU/EEA , ECDC, Stockholm, 11-12 May
2017;
10. WNV and blood donations, Vienna, 15-16 March 2018.

187 | P a g e
External Tenders and Contracts
1. Service contract: Validation and Evaluation of the Risk assessment tool for
contamination of blood donations during outbreaks of infectious diseases (EUFRAT)
ref. no: OJ/10/05/2012-PROC/2012/043.
2. Risk Assessment and Prevention of Infectious Disease Transmission through
Substances of Human Origin (West Nile virus, dengue and malaria) Tender
Reference: OJ/25/04/2013-PROC/2013/011.
3. Systematic Review of Scientific Evidence on the Prevalence of HTLV-I Infection,
ref: SRS-14-0098.
4. Risk Assessment and prevention of Chikungunya, Chagas diseases and Leishmaniasis
transmission through SoHO” Tender Reference: SRS-2015-OUT-0499—DCLiGr/ Id
4942.
5. Framework contract ECDC/2016/012 “Assessing the risk of bacterial disease
transmission by substances of human origin” – ID 5676, Specific contract 1 – 2017,
specific contract 2 – 2018, Specific contract 3 – 2019.
6. Tender for the Framework contract “Assessing the risk of parasitic and fungal
diseases transmission by substances of human origin” in preparation.
Scientific Advice on Microbial Safety of SoHO
Maintaining SoHO safety during communicable disease outbreaks
1. 2012 – 2013 – not recorded
2. 2014 – 2018 – 57 Rapid Risk Assessments
Scientific advice
1. Framework for action plan in prevention and control of Hepatitis B and C in
EU/EEA (EU -HEPFRAME)-2013
2. Ebola and SoHO
3. Screening algorithm – sperm donors
4. Syphilis testing
5. TBE transmission through SOHO
Preparedness plans
1. WNV Blood safety preparedness plan and update
2. Zika virus SoHO safety preparedness plan
3. Zika virus SoHO safety preparedness plan-update

188 | P a g e
Annex XI: COLLABORATION WITH COUNCIL OF EUROPE
The work of the Council of Europe in the blood transfusion area started in the 1950's.
The relevant Committees are the European Committee on Blood Transfusion (Steering
Committee) (CD-P-TS); and the Committee on Quality Assurance in Blood Transfusion
Services (Expert Committee) (GTS) that drafts and updates the Guide to the Preparation,
use and quality assurance of Blood Components (currently in its 19th edition). The
relevant section within the Council of Europe is the European Directorate for Quality
Management (EDQM). EDQM focuses on the ethical, legal and organisational aspects of
blood transfusion with a view to ensuring quality, increasing availability, avoiding
wastage, ensuring optimal use of blood supplies and analysing the possible ethical and
organisational impact of new scientific developments.
The work of the Council of Europe (EDQM) in the area of organ, tissue and cell
transplantation started in 1987, contributing actively to the implementation of high
standards for the protection of public health and for the promotion of human rights and
dignity. The relevant Committee is the European Committee on Organ Transplantation
(Partial Agreement) (CD-P-TO) and its Tissue and Cell Guide Drafting sub-group. The
principles guiding the work of the EDQM in this field are ensuring human dignity,
maintaining and fulfilling human rights and fundamental freedoms, non-
commercialisation of substances of human origin and protecting donors and recipients of
organs, tissues and cells.
A current (2019-2021) grant agreement between the European Commission and EDQM
includes a commitment to collaboration by EDQM to work on the following topics:
Development and regular updating of technical SoHO guidance
A proficiency testing scheme for blood establishments
Quality management, auditing and training for blood establishments
Analysis of EU SARE data for blood, tissues and cells, annually
Standardisation of tissue and cell activity data reporting
Development of strategies for increasing plasma collection in Europe
Training of EU vigilance officers to improve SARE reporting
Support for assessment of BTC standards and practices in EU applicant and
neighbouring countries.

189 | P a g e
Annex XII: Stakeholder Consultation Synopsis
1. Consultation Strategy
The intention of the stakeholder consultation activities conducted as part of this
evaluation was to gather views and opinions on the implementation of the BTC
legislation and factual information on what works well and where there is a room for
improvement. The key elements of the consultation included:
A Public Consultation addressing general questions to the public and specific
questions to targeted stakeholders
A large-scale stakeholder event to present the preliminary findings of consultation
and to identify any remaining information gaps
Targeted Consultation with relevant stakeholders to gather specific inputs, to fill
remaining gaps and to explore certain emerging issues in more depth.
This included: bilateral Meetings with key stakeholders; meetings with relevant
EU agencies and with third country BTC Regulators, multi-lateral topic-specific
meetings with selected stakeholders, including donor and patients associations,
industry and professionals working in the sector and Member State authorities.
Validation of emerging evaluation outcomes through discussions with the
representatives of the Competent Authorities on Substances of Human Origin
Expert Group.
The key stakeholder groups identified:
1. Member State competent authorities for blood, tissues and cells;
2. Member State Ministries of Health and other relevant regulatory bodies;
3. professionals working in blood, tissue and cell donation and transfusion and their
professional associations (see Table below);
4. healthcare professionals using blood, tissues and cells in their clinical practice;
5. blood and tissue establishments and procurement organisations and their
professional associations;
6. upstream / downstream service and equipment suppliers and users;
7. donors and their associations;
8. patients and their associations;
9. manufacturers of medicinal products / medical devices that use blood, tissues and
cells as starting materials;
190 | P a g e
10. other EU and national authorities, including authorities for medicinal products
and medical devices, and agencies such as the European Medicines Agency and
the European Centre for Disease Control;
11. relevant international organisations such as the Council of Europe and the World
Health Organisation;
12. ethics bodies;
13. third country regulators and professionals;
14. research and academia;
15. any interested citizen.
Many of the stakeholders listed above are represented by a number of large associations,
all of which engaged actively in the evaluation process. They are described in the
following Table.
TABLE 1: KEY BTC PROFESSIONAL ORGANISATIONS
411
Consortium of BTC
representative societies (CoRE
SoHO)
CoRe SoHO is a consortium of four Scientific Associations (European
Association of Tissue Banks, The European Eye Bank Association,
The European Society for Blood and Marrow Transplantation and the
European Blood Alliance) formed with the goal of providing expert
opinion and supporting data to European Union decision-makers and
their respective organisations in the field of SoHO. In particular, the
consortium aims to actively contribute to legal and regulatory
discussions affecting the SoHO field.
European Blood Alliance
(EBA)
The European Blood Alliance represents non-profit Blood Services in
Europe. Together these organisations collect the majority of EU blood
donations, around 17 million annually, and supply blood and blood
components for around 470 million EU citizens. More information at:
http://www.europeanbloodalliance.eu/
The European Group for Blood
and Marrow Transplantation
(EBMT)
EBMT represents over 4700 physicians and scientists in 568 centres
in 55 countries in the EU and beyond. Through a structure of
committees and working parties, EBMT promotes research, education,
harmonisation of practices and quality improvement through standards
and accreditation.
www.ebmt.org
The European Haemophilia
Consortium (EHC)
EHC is a non-profit umbrella patient organisation representing around
90,000 patients dependent on clothing factors and other types of
plasma derivatives (PD) supplied by manufacturers. More information
at: www.ehc.eu
The European Plasma Alliance
EPA represents 12 European private sector companies that collected
411
This is not a comprehensive list of BTC stakeholders, others are mentioned in this Synopsis.

191 | P a g e
(EPA)
2.5 million litres of plasma for the manufacturing of plasma derived
medicinal products in 2017. Their companies operate in Germany,
Austria, the Czech Republic and Hungary. Their mission is to promote
safe plasma collection practices in the EU with focus on donor health
and donor safety to ensure patients access to safe products. For more
information: epa@pptaglobal.org
The European Hematology
Association (EHA)
EHA is a non-governmental and not-for-profit membership
organisation that promotes excellence in patient care, research and
education in European haematology.
https://ehaweb.org/
The European Society for
Human Reproduction and
Embryology (ESHRE)
ESHRE is a pan European/ OECD professional organisation of 6,000
members that are clinicians, embryologists, psychologists, nurses,
midwifes and lab technicians. It also supports a European Patients
Association. Its aims are to promote interest in, and understanding of,
reproductive science and medicine by teaching and training,
development and maintenance of data registries and research and
dissemination. The EBMT registry contains information on close to
600.000 transplants, and the JACIE accreditation scheme aimed at
improving quality and safety of HCT.
It also aims to inform policy makers in Europe.
https://www.eshre.eu/
The International Federation of
Blood Donor Organisations
(FIODS)
FIODS represents 18 million blood donors from 81 countries in 5
continents and promotes regular, anonymous, voluntary, non-
remunerated blood donation in all countries of the world. More
information at: http://www.fiods-ifbdo.org/
The International
Haemovigilance Network
(IHN)
IHN is a network of professionals, bringing together 38 national
haemovigilance programmes and many interested individuals. Its aims
are sharing experience and knowledge, benchmarking, the
development of international definitions and international
collaboration on haemovigilance. More information at:
http://www.ihn-org.com/
The International Plasma
Fractionation Association
(IPFA)
IPFA represents not-for-profit plasma fractionators and national blood
services collecting plasma for the manufacture of medicinal products.
It supports the supply of plasma from Voluntary Non-Remunerated
Blood Donors.
http://www.IPFA.org
The International Patient
Organisation for Primary
Immunodeficiencies
(IPOPI/PLUS)
IPOPI/PLUS: The International Patient Organisation for Primary
Immunodeficiencies, is an association of national patient
organisations dedicated to improving awareness, access to early
diagnosis and optimal treatments for primary immunodeficiency
patients worldwide. IPOPI has 61 National Member Organisations, 23
of which are in the EU and it represents 60.000 patients worldwide.
The same participant represented the platform of plasma protein user
(PLUS), an umbrella for patient organisations bringing together
patients dependent on plasma Derived Medicinal Products (PDMP)

192 | P a g e
supplied by manufacturers. More information at: https://ipopi.org/
MedTech Europe
MedTech Europe is the European trade association representing the
medical technology industries. It is an alliance of European medical
technology industry associations representing Diagnostics and
Medical Devices manufacturers operating in Europe. It was founded
jointly by EDMA, representing the European in vitro diagnostic
industry, and Eucomed, representing the European medical devices
industry. More information is available at
https://www.medtecheurope.org/
The Plasma Protein
Therapeutics Association
(PPTA)
PPTA is a global trade and standards setting association representing
commercial manufacturers of plasma-derived and recombinant
biological therapies, collectively known as plasma protein therapies.
PPTA also represents for-profit collectors of Source Plasma. Their
members provide around 70% of the world’s needs for Source Plasma
(600 plasma collection centres based in North America and more than
100 in the EU) and >80% of the world’s plasma protein therapy
products. More information at: http://www.ppta.org/
World Marrow Donor
Association (WMDA)
WMDA provides access to the global database to search for
potentially matched bone marrow or peripheral blood haematopoietic
stem cell donors or cord blood products. Their focus is on unrelated
volunteer donors and they maintain standards and run accreditation
and training programmes. The Bone Marrow Donors Worldwide
registry included 94 organisations listing 30,168,410 donors and cord
blood products for international search on February 17th, 2016
www.bmdw.org
The outcome of the consultation activities and an overview of the stakeholder input is
summarised below.
2. Consultation Activities and Key messages
2.1 Roadmap Feedback
The Evaluation Roadmap was published on 17 January 2017
412
. Stakeholders were
invited to submit comments on the Roadmap during a 4-week period. Feedback was
received from 16 stakeholders and published at the SANTE website
413
.
The respondents focused on shortcomings seen in the BTC legislation rather than on the
planned evaluation methodology. The issues raised in their responses were further
elaborated in the subsequent stakeholder consultation activities.
412
Evaluation and Fitness Check (FC) ROADMAP by the Commission.
413
DG SANTE Website on the Evaluation.

193 | P a g e
2.2 Public Consultation
The Online Public Consultation (OPC) was launched on 29 May 2017 and ran for 14
weeks. A summary of the consultation was published in all official EU languages. A
Questionnaire for Citizens, available in all EU official languages and a more detailed
Questionnaire for administrations, associations and other organisations were made
available. The questionnaire for administrations, associations and organisations included
a section with questions on blood and blood components and a section with questions on
tissues and cells, so that respondents could choose to answer for one or both sections.
There were 43 responses from individual citizens and 158 from organisations. The latter
included a broad range of organisations impacted by the legislation, including all of the
key professional societies, donor and patient organisations, national authorities and
industrial associations. Many individual blood and tissue establishments also responded.
Around a third of respondents uploaded additional documents, either position statements
or relevant publications.
A factual summary report of the OPC was published on the DG SANTE web pages along
with the individual submissions
414
.
The respondents came from 23 Member. Stakeholders responding on behalf of
organisations were mostly located in Germany (14%); Italy (11%) and Spain (9%). The
majority of citizen responses came from Austria (21%); Italy (16%); Germany (14%) and
the Netherlands (12%).
Over half of the organisations were national (53%), over a third (35%) had an
International or European reach and the remainder worked at a regional or local level.
Organisations responding to the OPC were mostly Blood and Tissues
Establishments/Registries or their professional associations (51%). Public
Administrations, mainly national competent authorities for blood, tissues or cells, made
up the second largest group (22%). There were also respondents representing
Manufacturers of Medicinal Products or Medical Devices (11%); Healthcare Providers
(9%); Donor Organisations (3%); and Patient Organisations (4%).
A number of key messages emerged from the OPC that are detailed in the published
summary. In particular, the following views and issues were highlighted for each of the
Evaluation Assessment Criteria.
For effectiveness, the majority of respondents expressed the view that the EU legislation
on blood and blood components has increased quality and safety for these substances
(93% of 85 respondents,) and achieved a high level of human health protection for
recipients (92% of 87 respondents) to a great or to some extent, with only one
414
DG SANTE Website on the Evaluation- Public Consultation.
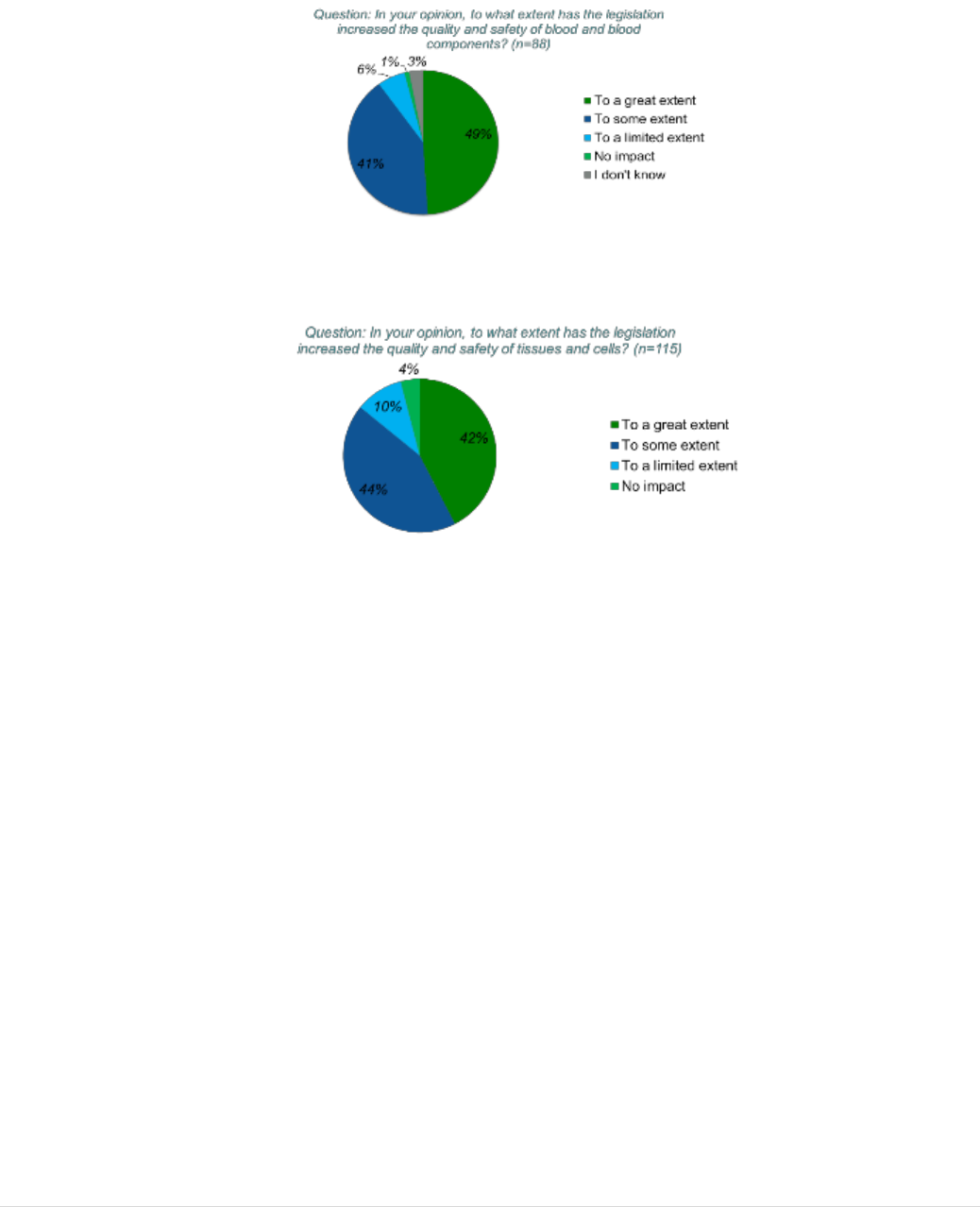
194 | P a g e
stakeholder organisation working in healthcare provision responding that there had been
no impact in either of these two areas (Figures 1 and 2).
FIGURE 1: RESPONSES FOR BLOOD AND BLOOD COMPONENTS
FIGURE 2: RESPONSES FOR TISSUES AND CELLS
However, several provisions are considered to be inadequate or missing. In particular, on
donor protection, out of 86 respondents who answered this question, 32% considered that
the legislation only had a limited or no impact in this area, which is an important view
amongst public authorities and establishments. In summary, the representatives of donor
and patient organisations, public administration, blood and tissue establishments and
other organisations pointed to a number of requirements they considered missing or
inadequate. These included:
inadequate provisions for the protection of the living donor, including donor
evaluation, reporting of adverse reactions and long-term donor follow-up. These are
considered essential for certain types of donation involving unknown health risks;
limited requirements to ensure quality of blood, tissues and cells, as opposed to
safety, including the need to verify the quality criteria of these substances before
release for clinical application; - lack of demonstration of safety and clinical
effectiveness in the recipient, particularly in the context of novel, or even
experimental, preparation processes for blood, tissues and cells;

195 | P a g e
overly limited descriptions of scope, missing a number of substances that are applied
to patients or are donated and used for other purposes but are not currently regulated
at EU level;
inadequate and/or unclear key definitions;
inadequate provisions for ensuring sufficiency of supply, highlighted particularly by
patient groups that see lack of access as a key risk to patients.
Of the 82 respondents representing the blood sector who answered regarding the
challenges to maintaining compliance with blood and blood component legislation, the
majority (61%) stated that the main challenge was inadequate definitions, followed by
limited resources for competent authorities (57%) and blood establishments (44%).
While 35% of respondents considered that requirements were too stringent or detailed,
exactly the same proportion of respondents answered that requirements were not specific
enough.
Over half of 117 respondents (51%) representing the tissues and cells sector identified
barriers preventing the effective implementation of the legislation, including financial
burdens; continuing inconsistencies, in large part due to differing interpretations between
MS; and a lack of coherence with other relevant EU legislation. A majority of
respondents (58% of 115 respondents) also considered that the rules on oversight do not
effectively ensure the full application of the tissues and cells legislation. In particular,
respondents detailed that differing national authorities have varying interpretations of the
tissues and cells legislation, and suggested that oversight activities require further
harmonisation.
For relevance, the key message from most stakeholders representing blood and tissue
and cells fields was that the legislation is not up-to-date with scientific, technological,
epidemiological or societal developments and that the process of updating is not flexible
or quick enough to adapt to them; many examples were provided. It was considered by
many respondents that the more technical aspects of the current legislation should be
moved to guidance that can be rapidly updated, in line with changing risks and
technologies. Particularly, the guidance of the Council of Europe (EDQM) and of ECDC
were identified as suitable references to up-to-date technical standards.
The majority of respondents representing the blood sector considered that the
developments were not significantly addressed, and a notable proportion considered that
the legislation was not suited to the current situation. For instance, the important impact
and potential of pathogen inactivation technologies during processing was considered not
to be addressed and the availability of more sensitive donor testing by nucleic acid
technology was not considered to be adequately reflected. Nearly half of all respondents
(44% of 81 respondents) considered there were gaps in scope of the blood and blood
components legislation, for example: for blood components used for therapeutic purposes
other than transfusion (e.g. serum eye drops, fibrin glue, platelet rich plasma, platelet
lysate, lyophilised plasma); and, for other substances of human origin used

196 | P a g e
therapeutically (e.g. human faeces, breast milk and urine for the manufacture of
medicinal products).
Similarly to above, the majority of respondents representing the tissues and cells sector
considered that the developments are not significantly addressed, or not suited to the
current situation. For example, only 31% of 113 respondents considered that the
legislation is fully adapted to scientific developments relating to processing, and only
29% of 112 respondents considered that the legislation is fully adapted to
epidemiological developments. Similarly to the field of blood and blood components,
those responding on the relevance of the legislation for tissues and cells pointed to the
important developments in donor testing and microbial inactivation technologies that are
not currently addressed and to the risks brought by multiple epidemiological outbreaks
but not mitigated by provisions in the legislation.
For efficiency, the majority considered that the legislation has incurred costs but that
these had been justified by benefits for patients.
Concerning blood and blood components, most respondents (80% of 87 respondents)
considered that the application of the EU blood and blood components legislation
brought regarding costs – which would not have been incurred otherwise – for
themselves, their organisation or stakeholders represented by their organisation. Donors,
manufacturers of downstream products, and public administrators outside the EU
particularly indicated that they had incurred significant costs, while the majority of
respondents considered there were no additional costs. A minority of respondents (11%
of 53 respondents) considered that these costs were not justified by the benefits of the
legislation for patients; most respondents considered they were either partially (58%) or
fully (28%) justified.
For tissues and cells, most respondents, representing a range of fields and organisations,
considered that the application of the EU tissues and cells legislation brought significant
or minor additional costs (59% and 25% from 114 respondents, respectively) – which
would not have been incurred otherwise – for themselves, their organisation or other
stakeholders represented by their organisation. Only a minority of respondents (21% of
94 respondents) considered that these costs were not justified by the benefits of the
legislation for patients; most respondents considered they were either partially (34%) or
fully (43%) justified.
With regard to coherence, inconsistencies between the BTC legal frameworks were
identified, along with inconsistencies related to the borderlines with the EU legal
frameworks on medicinal products and medical devices and with international
frameworks regulating these substances. In general, respondents pointed to the lack of a
common EU-level mechanism to clarify these borders in view of the many innovative
developments in biotechnology.
Concerning blood and blood components, a higher proportion of respondents (49% of 82
respondents) considered that there were also minor (37%) or significant (12%)

197 | P a g e
inconsistencies with medicinal products legislation. In both cases, less than a third of
respondents considered that blood and blood components legislation was fully consistent
and coherent with those legislative frameworks. Similar to medical devices, issues
concerning the consistency of the Directives with medicinal product legislation included
inconsistent requirements (e.g. in the testing of plasma for fractionation compared with
for blood intended for transfusion); borderline issues when blood cells are used as
starting materials for ATMP manufacture; an absence of provisions for international
controls on the quality of certain blood products which limits trade; and inconsistencies
with regards to the regulation of plasma and the definition of 'industrial' processing.
For tissues and cells, 19% of 114 respondents report significant inconsistencies with the
EU legal framework for medicinal products. The main issues highlighted concerned
vigilance and surveillance communication requirements within or between MS, as well
as the role and mandate of EU agencies. Respondents also expressed their view that,
similar than with medical devices, greater clarity is required on borderlines between
tissues and cells and ATMP legislation pointing to heterogeneous implementation of
legislation in/between MS, with implications for safety and quality. Several respondents
pointed to the lack of a common EU-level mechanism to clarify the borders between
different EU legal frameworks, in view of the many innovative developments in
biotechnology.
Regarding EU added value, in general most respondents considered that the positive
impact of the legislation could not have been achieved, or would have been achieved
more slowly, without EU legislation. However, many pointed to the more stringent
national requirements adopted by many Member States as limiting the added value of the
legislation at EU level.
For blood and blood components, the majority of the 86 organisations that responded to
this question, considered that EU legal provisions have added value to regulating the
safety and quality of blood and blood components by greatly (53%) or somewhat (13%)
improving or accelerating what could have otherwise been achieved at a national or
global level.
For tissues and cells, around a third (34%) of 110 respondents considered that EU legal
provisions have added value to regulating the safety and quality of tissues and cells
across all MS in a manner that could not have been achieved by national or global level
measures. Furthermore, 44% of all respondents considered that the provisions greatly
(21%) or somewhat (23%) improved or accelerated what could have otherwise been
achieved at a national or global level, and only 14% considered that the same outcomes
could have been achieved without EU tissue and cells legislation in place.

198 | P a g e
The summary of the OPC, together with the individual submissions, was published by
DG SANTE
415
. The Annex published together with this summary includes a detailed
analysis of replies to the OPC questions covering the BTC evaluation criteria.
2.3 Targeted Stakeholder Consultation
Meetings with CASoHO Expert Group
The Competent Authorities for Substances of Human Origin Expert Group E01718
(CASoHO Expert group) includes the representatives, from competent authorities
responsible for overseeing the implementation of the BTC legislation. The World Health
Organisation, the Council of Europe (EDQM), the European Centre for Disease Control
(ECDC), CHAFEA and European Medicines Agency (EMA) were invited as
observers
416
,
417
.
Aspects of the evaluation were discussed during 10 meetings of the SoHO authorities,
including 4 of the blood competent authorities, at 3 of the tissue and cell competent
authorities, and at expert sub-groups, on vigilance and traceability
418
.
Ad-hoc multi-lateral meetings with EU authorities and stakeholders
Five ad-hoc meetings between invited stakeholders and the BTC competent authorities
were organised to explore specific topics where data or detailed information were
lacking.
Table 2: Topics discussed at meetings with stakeholders and competent authorities
Sub-sector
Date (footnote
reference to
minutes online)
Topics addressed
Stakeholders Present
– see Abbreviations and
descriptions at the beginning of
this document.
Blood
02/12/2017
419
West Nile Virus
testing
Clinical follow-up of
recipients
European Blood Alliance
European Haematologists
Association
Key messages:
- Despite recent revision of Directive 2006/33/EC, stakeholders presented evidence
that the new provisions were too specific and that equivalent safety could be
achieved at a lower cost.
- The EHA argued for the need for systematic follow up of patients following
415
Summary of Responses to the Open Public Consultation for the Evaluation of the Blood, Tissues and Cells
Legislation.
416
DG SANTE Website-Blood, tissues, cells and organs- Events.
417
https://ec.europa.eu/health/blood_tissues_organs/events_en#anchor3.
418
Meeting of the SoHO Vigilance Expert Sub-Group 7 APRIL
2017.
419
Ad-Hoc Meeting between Stakeholders and representatives of members of the Competent Authorities on
Substances of Human Origin Expert Group (CASoHO E01718) 2 December 2017.

199 | P a g e
transfusions and HPC transplantation
Tissues and
cells
22/02/2017
420
Donor safety
Clinical follow-up of
recipients
European Association of Tissue
Banking
European Society for Human
Reproduction and Embryology
European Eye Bank Association
European Society for Blood and
Marrow Transplantation
Key messages:
- Broad consensus on the inadequacy of current tissue and cell donor protection and
follow up provisions
- Many professionals conduct recipient follow up studies.
- General consensus that recipient follow up should be mandated in certain
circumstances, in addition to the follow-up of children born of medically assisted
reproduction.
Blood
22/06/2017
421
Donor safety
Plasma Supply
European Blood Alliance
PPTA
IPFA
FIODS
Key messages:
- Broad consensus on the inadequacy of current blood donor protection and follow
up provisions
- Strong concern from all sectors and patients regarding the EU plasma collection
rates and the reliance on the US for EU supply.
Tissues and
cells
16/11/2017
422
Assisted Reproduction:
Genetics
Defining quality in
ART
Medical devices and
ART
ESHRE
Fertility Europe
Donor conception network
Cryos sperm bank
European Sperm bank
Key messages :
- The possibilities for genetic screening have changed dramatically since the
legislation was adopted and these changes should be reflected in provisions.
- For the field of assisted reproduction, quality should be defined with clear criteria
relating to successful pregnancy and healthy offspring
Greater communication with the regulatory framework for medical devices in
needed due to the reliance for safety and success on those devices.
Blood
10/10/2018
423
Essential medical
Medtech Europe
420
Ad-Hoc Meeting between Stakeholders and representatives members of the Competent Authorities on Substances
of Human Origin Expert Group (CASoHO E01718) 22 February 2017.
421
Ad-Hoc Meeting between Stakeholders and representatives of members of the Competent Authorities on
Substances of Human Origin Expert Group (CASoHO E01718) 22 June 2017.
422
Ad-Hoc Meeting between Stakeholders and representatives of members of the Competent Authorities on Substances
of Human Origin Expert Group (CASoHO E01718) 16 November 2017.
423
Ad-Hoc Meeting between Stakeholders and representatives of members of the Competent Authorities on Substances
of Human Origin Expert Group (CASoHO E01718) 10 October 2018.

200 | P a g e
devices for continuity
of supply
Pathogen reduction
technologies
European Blood Alliance
European Plasma Association
PPTA
IPFA
Key messages :
- High level of dependence of the blood sector on a sustainable supply of medical
devices
- Pathogen inactivation during blood processing can bring increased safety but costs
should be justified and balanced with alternative strategies for achieving
equivalent safety levels.
The evidence (data and references) collected at these meetings were used in the BTC
evaluation.
Bilateral meetings with EU stakeholders
Twenty-one organisations attended bilateral meetings with representatives of the
European Commission in the course of the BTC evaluation.
Overall, the following organisations or companies participated in bilateral meetings with
DG SANTE:
Blood
Tissues and Cells
- European Blood Alliance
- European Hematology
Association
- International Plasma
Fractionation Association Plasma
Protein Therapeutics Association
- Biotest AG
- CSL Behring
- NHS Blood and Transplant
- Establissement Francais du Sang
- Agence nationale de sécurité du
médicament et des produits de
santé
- Medtech Europe
- Cryos International
- European Society for Blood &
Marrow Transplantation
- Nordic Cryobank Group/ European
Sperm Bank
- European Society for Human
Reproduction and Embryology
- Alliance for Regenerative Medicine
- European Eye Bank Association
- Confederation of Danish Entreprise
- International Society for Cell and
Gene Therapy
- Agora Consortium
- European Association of Tissue
Banks
- ICCBBA (ISBT 128 coding
standard)
- Eurocode
The stakeholders representing blood sector, highlighted that there is a lack of adequate
provisions in the EU blood legislation to encourage the Member States to establish or
increase plasma collection for PDMP manufacture, a lack of key definitions of plasma

201 | P a g e
components and the lack of a definition of compensation. The stakeholders also pointed
that the regulation of the collection of human plasma should be consistent with wider
SoHO regulations and a definition for SoHO should be included in the legislation for
greater clarity and the concept of Voluntary Unpaid Donation lacks of clarity in the
legislation.
424
In the meetings, the tissue and cells stakeholders underlined a series of issues that have,
for their members, as described in the following points
425
.
- Some technical requirements to ensure safe tissues of high quality are outdated or
too limited in the Directives. The new Good Practice Guidance and the new
monographs to be published in 2019 by EDQM, as part of the Guide to the Safety
and Quality of Tissues and Cells, are seen as more effective and up to date.
- An absence of legal provisions for the authorisation of clinical users or their
hospitals/clinics to receive and use human tissues for transplant.
- New types of substances of human origin are now used clinically but do not fall
within with the scope of the existing SoHO legislation because of the way the
scope is defined in the BTC directives. These substances remain un-regulated or
regulated differently in Member States.
- Some existing provisions are not evidence based, including those relating to air
quality requirements for processing facilities and some donor selection criteria;
borderlines with ATMPs are not adequately clear.
Bi-lateral meetings with International stakeholders
In meetings with US FDA/CBER
426
with the American Association of Blood Banks
427
and the American Association of Tissue banks
428
participants highlighted the inter-
Member State different approaches to interpretation in the EU, and the challenges that
causes for those wishing to export to the EU, the challenges of keeping legislation up to
date and the ways in which guidance can be recognised by regulators and the
mechanisms for defining regulatory borderlines.
Exchanges were also held with regulatory authorities in in third jurisdictions (e.g. Korea
and Japan)
429
to consult with some key international regulators in the field of BTC to
explore commonalities and differences between the EU regulatory system and those
applies elsewhere for these substances. The discussions focused on approaches to
classification of BTC and BTC-based medicinal products and on the best approaches to
balancing legal requirements with good practice standards.
424
Meeting between PPTA and DG SANTE B4 19 June 2018.
425
Meeting between the Board of the European Association of Tissue Banks and DG SANTE B4 18 September 2018
Meeting between the International Society for Cell Therapy and DG SANTE B4 14 June 2018.
426
Meeting with the U.S. FDA 8 March 2018.
427
Meeting between American Association of Blood Banks (AABB) and the European Commission (DG SANTE B4)
9 March 2018
.
428
Meeting between American Association of Tissue Banks (AATB) and the European Commission (DG SANTE B4)
9 March 2018.
429
Teleconference meeting between the South Korean Ministry of Food and Drug Safety and DG SANTE B4 15
November 2018.
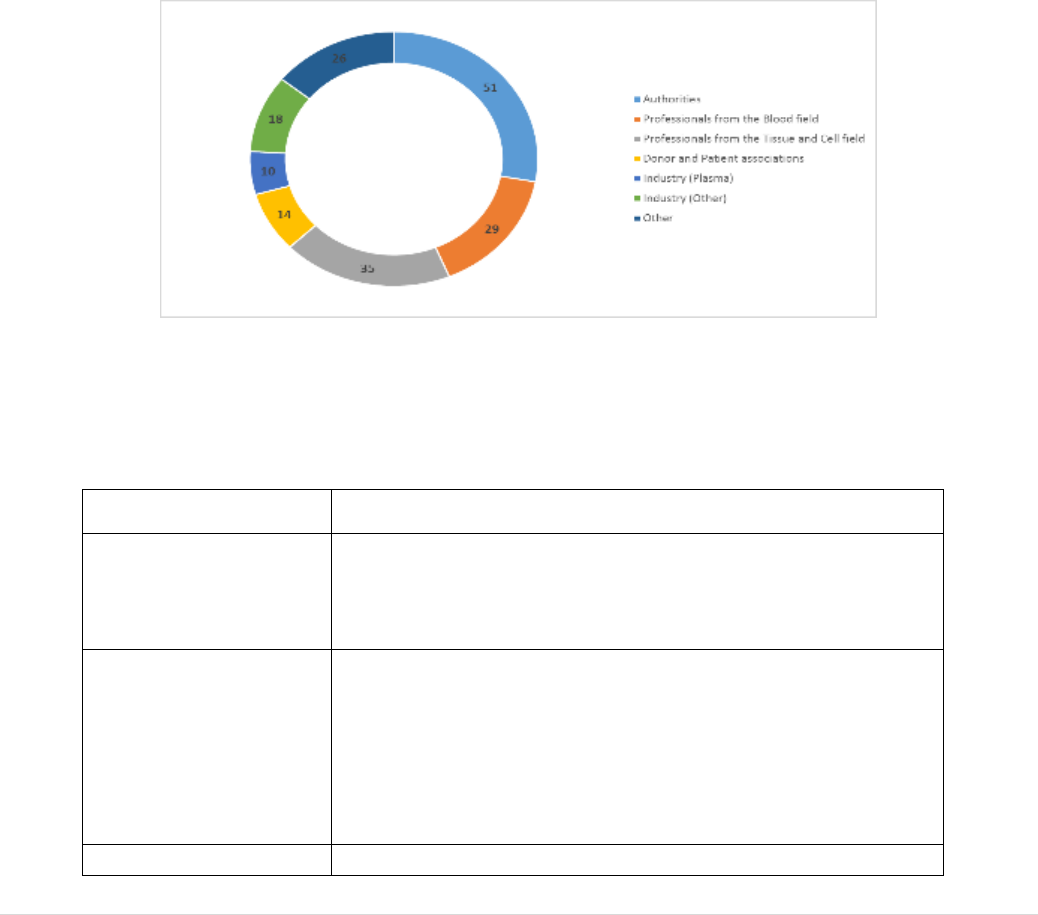
202 | P a g e
2.4 Stakeholder Event
A Stakeholder Event was organised by the European Commission services on 20
September 2017. The main purpose of the event was to provide an opportunity to
validate the main messages that had emerged from open and targeted consultation
activities, and to explore remaining evidence gaps. The meeting was open to all
interested stakeholders. The event was attended by 205 participants bringing together
members of the public, national authorities, patient and donor groups, professionals
working with blood, tissues and cells, industry representatives and other relevant
stakeholders, who had the opportunity to express their views on key topics regarding the
EU Blood, Tissues and Cells legislation. The majority were from EU Member States (21
Member States were represented) and 10 were from non-EU countries (USA, Norway,
Switzerland and the Russian Federation).
The audience represented a variety of sectors and stakeholders, as shown in Figure 3.
Figure 3: Stakeholder groups at the stakeholder event of 20 September 2019.
The meeting was structured around five main themes relating to the BTC legislation. The
key messages that emerged from the presentations and discussions were the following:
THEME
KEY MESSAGES
The key importance of
donors: The gift of life
The current BTC legislation provides inadequate protection
and health monitoring in donors.
Regulatory oversight of
the sectors - How to
ensure safety and
quality?
There are gaps and shortcomings in the oversight provisions
relating to new therapies not included in the current scope, the
lack of mandated recipient follow up, many technical
provisions being outdated, 2-yearly inspection frequency
considered as not cost-efficient and a lack of provisions for
emergency preparedness.
Availability and
Strong concerns were expressed regarding the reliance of the

203 | P a g e
sufficiency - Are
patients getting the
blood, tissues and cells
that they need?
EU on the United States for meeting its needs for plasma for
medicinal product manufacture with many advocating for EU
‘Strategic Independence’ for plasma. Other sufficiency
concerns related to wastage of BTC due to outdated or
unjustified donor eligibility provisions.
Legal consistency and
coherence - Regulatory
pathways for Substances
of Human Origin
Concerns were expressed regarding definitions that lack
clarity and result in divergent interpretations and legal
uncertainty, including with regards to the regulatory
classification of BTC substances/products. A case study was
presented that demonstrated how a lack of legal certainty and
harmonisation resulted in an ATMP developer limiting its
area of activity to the UK rather than across several MS.
Challenges of exporting tissues to the EU from the US due to
diverging Member State requirements were highlighted.
A changing world –
Technological, societal,
epidemiological and
international
developments.
There was broad consensus that the changes in epidemiology
and technology that have occurred mean that many provisions
are outdated. Concerns were expressed regarding increasing
commercialisation (or commodification) and there was a call
for cost-effectiveness analysis of any future safety and quality
measures.
A Summary of this Stakeholder Event was published by DG SANTE
430
.
3. Representativeness of Stakeholder Participation
Looking across all stakeholder consultation activities, a total of 288 organisations
participated in the process (see Figure 4) with many taking part in multiple of all
activities.
430
Summary of the Blood, Tissues and Cells Stakeholder Event 20 September 2017.
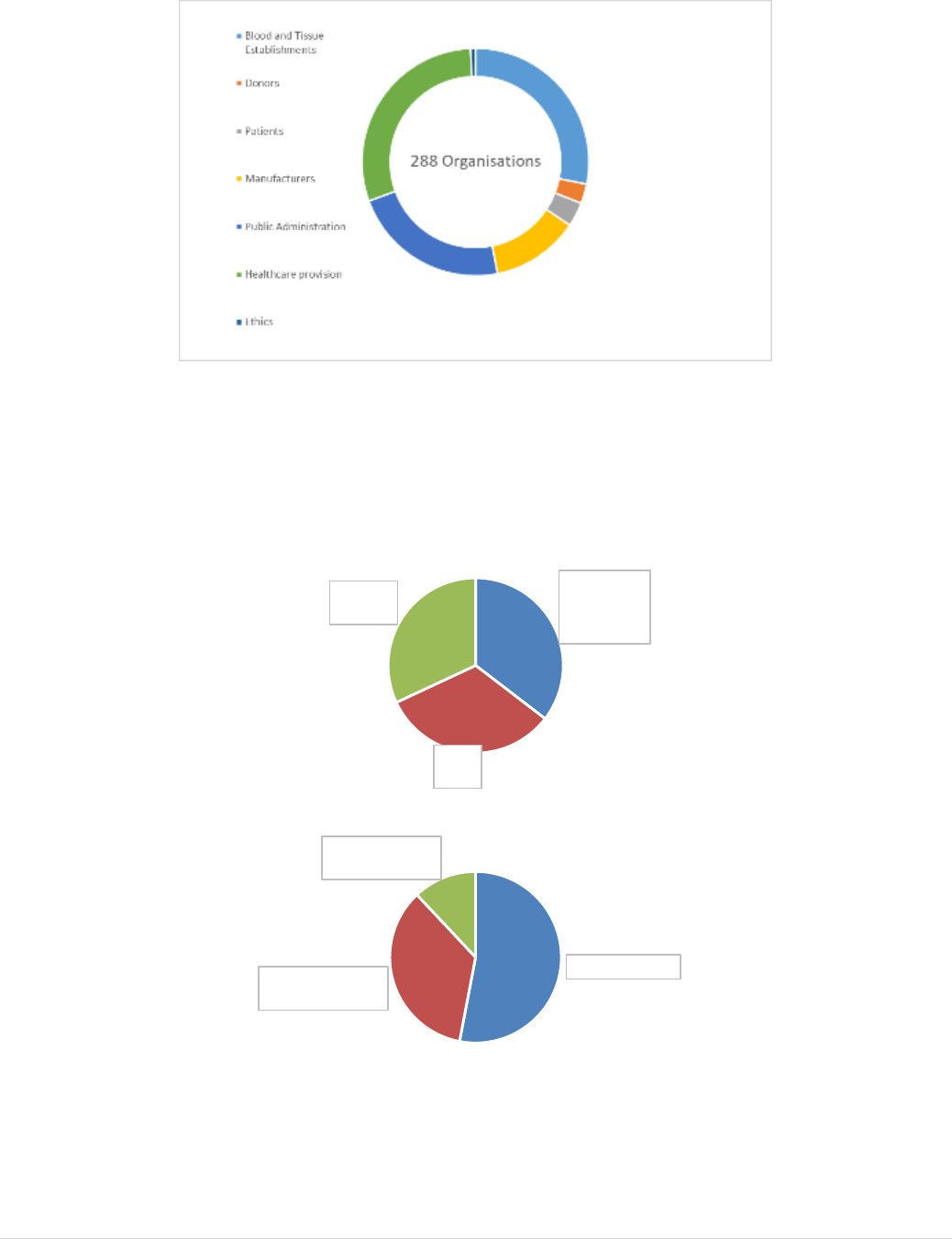
204 | P a g e
FIGURE 4: TYPES OF ORGANISATIONS PARTICIPATING IN STAKEHOLDER CONSULTATION ACTIVITIES.
The organisations that responded to the OPC were evenly distributed between blood,
tissues and cells in terms of experience (Figure 5) and represented a mix of local,
national and international organisations (Figure 6).
FIGURE 5: SUB-SECTOR OF OPC RESPONDERS
FIGURE 6: OPC RESPONDER GEOGRAPHICAL REACH
Blood and
blood
components
35%
Cells
33%
Tissues
32%
National; 53%
European/Intern
ational ; 35%
Regional/local;
12%
205 | P a g e
Target groups of stakeholders identified in the consultation strategy were reached by the
OPC (Figure 7). DG SANTE reached out more actively to some that were less
represented, particularly donors and patients, inviting them to participate in ad-hoc
multilateral stakeholder meetings.
FIGURE 7: OPC RESPONDER CATEGORIES
Of the 43 individual citizens that responded to the dedicated OPC questionnaire, the great
majority (87%) were interested in the consultation because of having direct experience in
one of the sectors concerned. They represented a balanced mix of individuals working in
the public sector (42%), private healthcare industry (33%) and the non-profit sector
(14%).
Only one submission to the OPC was received from an Ethics organisation (The Nuffield
Council of Bioethics). However, a number of the professional associations that
participated, and are categorised under a different heading, also have a mandate for
ethical aspects of the sectors and submitted comments on ethics related topics.
Although a number of organisations classified themselves as Academic/research, it was
agreed with them that, on the basis that their submissions addressed issues for tissue
establishments within the institutions they represent, that they would be grouped with
blood and tissue establishments. However, it should be noted that the European Society
of Blood and Marrow Transplantation and the European Society for Human
Reproduction and Embryology, in particular, also represent the organisations that carry
out the largest amount of research in those fields. The same applies to some extent to a
number of other organisations. It is not, therefore, considered that research and academia
perspectives are missing.
Targeted stakeholder consultation was the tool used to reach out to and engage those
stakeholders that were less well represented in the OPC. The most challenging group of
EU public
administr
ations
22%
Blood/Tis
sue
Establish
ments
51%
Patients
organisati
ons
4%
Donor
organisati
ons
3%
Healthcar
e
providers
9%
Downstre
am
manufact
urers
11%

206 | P a g e
stakeholders to reach were donors and patients and international authorities. Exceptions
to this were blood donors that were represented by the International Federation of Blood
Donors and patients that rely on plasma derived medicinal products, represented by
IPOPI and PLUS. The latter organisations participated actively in the OPC, in the major
stakeholder event and in multi-lateral stakeholder meetings. Patients relying on fertility
treatment or on donor gametes to have children were actively contacted and two
societies, Fertility Europe and the Donor Conception Network, participated in an ad-hoc
multilateral stakeholder meeting. There are no organisations representing replacement
tissue and cell donors. Patients that receive tissue transplants are very diverse, from
cornea transplant recipients to orthopaedic and cardiovascular surgery patients and they
are not organised as tissue recipients.
As international authorities did not submit to the OPC they were actively contacted and
both face-to-face meetings and teleconferences were used to present and discuss the
legislation with them. The United States is the main country involved in exporting
plasma, tissues and cells into the EU, and therefore, efforts were focused there as
described above. A mission by Commission personnel to the United States included an
in-depth workshop with a large number of FDA experts where all of the key issues of
common concern were explored.
The authorities regulating other areas of relevance in the EU, particularly medical
devices and medicinal products, were represented in the OPC submissions but usually in
their capacity of also regulating blood, tissues and cells and usually they commented
from that perspective. To ensure that this sector had its views taken into account
Commission personnel presented the evaluation and invited discussion at a routine
meeting of the Pharmaceuticals Committee and a meeting of the Borderline and
Classification expert group in medical devices. Dedicated meetings were also held with
the European Medicines Agency and the European Centre for Disease Control.
The inputs and contributions received from the stakeholders in the BTC stakeholder
consultation was indispensable to crystallise the key findings of the BTC evaluation
which highlights that the BTC legislation has substantially improved safety and quality
of blood, tissues and cells in the EU. While public confidence in the sectors remains
high, there are important and growing gaps and shortcomings to address.

207 | P a g e
Annex XIII: Examples of technological changes in the processing
of tissues and cells
Sub-sector
Examples of technological advances
Replacement
tissues
Corneas: transplanted whole as standard in the early 2000s are
now routinely laser cut to allow the supply of thin lamellar grafts,
sometimes with more than one patient treated from one cornea
431
.
Skin: the epidermis can be removed or the cells can be removed
from the skin leaving a matrix structure into which the recipient
cells will migrate and grow
432
or the epidermal cells can be
separated and used in suspension (without culturing)
433
.
Bone and tendons: treated in a wide range of often complex ways,
to remove cells, to remove minerals, to reduce or eliminate
contaminants and to prolong preservation times
434
.
Heart valves: increasingly decellularised to improve
recellularisation with the patient’s own cells when implanted
435
.
Haematopoietic
stem cells
Bone marrow, peripheral blood stem cells and cord blood:
preparation often includes sophisticated methods for cell selection,
cell depletion and volume reduction before transplant
436
.
Medically assisted
reproduction
Oocytes: now routinely preserved by vitrification, a process that
was considered highly experimental at the time that Directive
2006/86 was adopted
437
.
Ovarian and testicular tissue: now successfully cryopreserved to
preserve fertility in patients with cancer
438
. Future perspectives
include the use of ovarian tissue to postpone menopause or delay
osteoporosis
439
431
Boynton GE and Woodward MA (2015) Evolving techniques in Corneal Transplantation. Curr Surg Rep. 3(2).
Published online 1 Feb 2015.
432
Hogg P
1
, Rooney P, Ingham E et al. (2012) Development of a decellularised dermis. Cell Tissue Bank. 2013 Sep;
14(3):465-74.
433
Section 18.10 Guide to the Quality and Safety of Tissues and Cells for Human Application. EDQM, 3
rd
Edition
2017.
434
Osborne JC, Kurz A, Trias E et al. (2012) Skeletal Tissue: Specific recovery and processing issues. In: Tissue and
Cell Processing: an Essential Guide Eds: Fehily D, Brubaker, S, Kearney JN and Wolfinbarger L.
435
Chapter 19, Guide to the Quality and Safety of Tissues and Cells for Human Application. EDQM, 3
rd
Edition 2017.
436
Section 21.4 Guide to the Quality and Safety of Tissues and Cells for Human Application. EDQM, 3
rd
Edition 2017.
437
Chian R, Wang Y and Li Y (2013) Oocyte vitrification: advances, progress and future goals J Assist Reprod Genet
(2014) 31:411–420.
438
Chapter 25, Guide to the Quality and Safety of Tissues and Cells for Human Application. EDQM, 3
rd
Edition 2017.
439
Andersen C, Kristensen SG () Novel use of the ovarian follicular pool to postpone menopause and delay
osteoporosis. Reproductive BioMedicine Online (2015) 31, 128–131.

208 | P a g e
where recent developments include mitochondrial replacement
therapy
440
and germline genome editing
441
.
Embryos: Mitochondrial replacement therapy
442
, and germline
genome editing
443
represent two highly innovative developments
that might become important for preventing serious genetic
conditions in the future, although ethical concerns related to these
technologies are considerable
444
,
445
.
440
In mitochondrial replacement therapy, genomic DNA (nuclei, mitotic spindle or polar body(ies)) from an oocyte
with mitochondria affected by a genetic condition is transferred to a donor oocyte. The oocyte is fertilised in vitro,
resulting in an embryo with genomic/mitochondrial DNA from three individuals.
441
Germline genome editing can be applied to correct genetic defects in gametes or embryos.
442
In mitochondrial replacement therapy, genomic DNA (nuclei, mitotic spindle or polar body(ies)) from an oocyte
with mitochondria affected by a genetic condition is transferred to a donor oocyte. The oocyte is fertilised in vitro,
resulting in an embryo with genomic/mitochondrial DNA from three individuals.
443
Germline genome editing can be applied to correct genetic defects in gametes or embryos.
444
Vogel G. (2015) Embryo engineering alarm - Researchers call for restraint in genome editing. Science 347(6228):
130.
445
The Scientist (2017) “Opinion: Ethical Considerations of “Three-Parent” Babies”.

209 | P a g e
Annex XIV: Emerging/Increasing Commercial Activities
Sector
Commercial/non-public sector activity
Haematopoietic
stem Cells
Cord blood banking for future possible family use (‘insurance
against future illness’) – fee basis
Offering treatments for diseases not traditionally treated by cell
transplantation (often without proof of clinical effectiveness
446
)
Other sources of
stem cells
Banking of other sources of stem cells for future family use e.g.
teeth, umbilical cord tissue, adipose tissue
Replacement
tissues
Musculoskeletal processing in subsidiaries of commercial US
banks established in the EU
Import of musculoskeletal tissues from commercial companies
in the US
Import of skin products from commercial companies in the US
Marketing of tissue grafts by surgical instrument companies
Medically assisted
reproduction
Banking of oocytes for supply to IVF clinics (equivalent to
model for sperm banking)
Online distribution of sperm direct to patients
Banking of gametes or reproductive tissue for own future use
(social freezing to allow postponement of family planning)
Corneas
Trend in international cornea banking towards
commercialisation
447
. Suggestions that within 5 years, 50% of
all corneas worldwide could be supplied by a single US
commercial company, if current trends continue
448
.
Plasma collection
Increasing reliance on the commercial sector to collect plasma
in the EU
446
See item 34 in Annex VIII, part 2.
447
Mannis MJ, Sugar J (2018) Is This the Future of Eye Banking? Cornea (editorial) Cornea 37(7): 811-812.
448
M Moshirfar JL Goldberg TW Brown WD Wagner YC Ronquillo (2019) A paradigm shift in eye banking: how
new models are challenging the status quo. Clinical Ophthalmology 2019:13 63–67.

210 | P a g e
Annex XV: A non-exhaustive list of clinical outcome registries in
the BTC sectors
N
o
Data Registry
Geographi
cal scope
Follow-up focus
Source
1.
European
Society for
Blood and
Marrow
Transplantation
(EBMT) patient
registry
Europe
Patients who have
undergone a
haematopoietic stem cell
transplantation (HSCT)
procedure; patients with
bone marrow failures
receiving
immunosuppressive
therapies; and patients
receiving non-
haematopoietic cell
therapies.
https://www.ebmt.org/eb
mt-patient-registry
2.
The IVF
monitoring
consortium
(EIM) hosted
by European
Society of
Human
Reproduction
and Embryology
ESHRE
EU and
Non EU
countries
Children born after ART
https://www.eshre.eu/ei
m
3.
European
Cornea and Cell
Transplantation
Registry
ECCTR
Europe
New human tissue
transplantations and
ophthalmic surgeries
(cornea transplantations)
http://www.ecctr.org/abo
ut-the-ecctr-project
4.
Registry of
Committee of
Nordic Assisted
Reproductive
Technology and
Safety
(CoNARTaS)
Denmark,
Finland,
Norway,
Sweden
Health in mothers and
children born after ART
http://www.conartas.com
/about/
5.
Scandinavian
donations and
transfusions
(SCANDAT)
Sweden,
Denmark
Blood donors, blood
transfusions and transfused
patients
http://www.scandat.se/
6.
International
Surveillance of
Worldwide
Recipients of blood and
blood products that can
https://ihn-org.com/istare

211 | P a g e
Transfusion-
Associated
Reactions and
Events
(ISTARE)
certainly, probably or
possibly be imputed to
blood transfusion. Also,
blood donors

212 | P a g e
Annex XVI: Inconsistencies between EU-legal frameworks
TABLE 1: INCONSISTENCIES BETWEEN THE BLOOD AND TISSUE AND CELL PROVISIONS.
Topic
Inconsistency
Donor testing for
syphilis
This test is mandated for tissue and cells donors (apart from
donors of reproductive tissues and cells) but not for blood
donors, where the risk of transmission is likely to be higher.
Donor testing for
Human T-cell
leukemia virus
(HTLV)
There are specific provisions for performing this test for tissue
and cell donors but none for blood donors, without clear
evidence of a rationale for this difference.
Preparation Process
Authorisation
Directive 2006/86/EC provides for an authorisation for the
processes applied to tissues and cells before they are
distributed for clinical application. There is no such
authorisation process provided for blood and blood
components. In contrast, Annex V of Directive 2006/33/EC
defines a series of blood component specifications that
indicate which preparation processes are permitted.
Traceability/Coding
The tissue and cell legislation provides for a specific Single
European Code that has involved the construction of a public
compendium of authorised tissue establishments, with their
associated codes, and a compendium of tissue and cell product
codes, that must be used to construct the Single European
Code to appear on tissue and cell labels (with some
exemptions and exceptions). There are no specific rules for the
coding of blood and blood components, apart from generic
provisions for traceability and unique identification of donors
and their donations.
Rules on import from
third countries
The tissue and cell legislation has detailed rules for import
from third countries with a requirement for competent
authorities to issue a specific import authorisation to tissue
establishments meeting certain criteria449. There is no such
provision in the blood legislation.
449
Commission Directive 2015/566 implementing Directive 2004/23/EC concerns the procedures for verifying the
equivalent standards of quality and safety of imported tissues and cells.

213 | P a g e
Quality System
There is an obligation on the Commission to provide standards
and specifications for the activities related to a quality system
in blood establishments
450
while no such obligation exists for
tissues and cells.
Immutability
451
of
adverse reactions
This concept is included in the blood legislation where an
imputability grading scale is provided in Directive 2005/61/EC
and reporting requirements are dependent on the imputability
level while no such concept or reporting requirements are
included in the tissue and cell legislation.
Additionally, the requirements for reporting of blood related
serious adverse reactions according to imputability level are
somewhat incoherent within the blood legislation
452
.
TABLE 2: EXAMPLES OF INCONSISTENT OR UNCLEAR CLASSIFICATIONS BETWEEN BTC
AND MEDICINAL PRODUCTS
Substance/therapy
Borderline issue
Reference
Pancreatic islets
Used as an alternative
to pancreas
transplantation in
patients with type 1
diabetes.
The Commission has received recurrent questions on
whether pancreatic islets are covered by the Tissues
and Cells Directive, the Advanced Therapies
Regulation or whether they should be classified as
organs.
Although they are separated by enzymatic digestion,
both CAT
453
and the Tissue and Cell competent
authorities have considered that these fall under the
tissues and cells legislation.
See Part 2, item 4 in
Annex 9.
Isolated hepatocytes
(without expansion)
Used as a ‘bridge’ to
liver transplantation in
patients on the
transplant waiting list.
A Commission survey of EU tissue and cell authorities
indicates the following current situation for cells
separated from tissue by enzymatic digestion without
expansion (including keratinocytes, hepatocytes etc.):
9 Member States regulate as tissues and cells
7 regulate as a medicinal product (ATMP)
2 decide on a case-by-case basis depending on
manipulation and use.
3 do not have this therapy or do not regulate it.
Presentation to the
meeting of T&C
competent
authorities 2019
454
.
450
Directive 2002/98/EC Article 11 paragraph 2.
451
Imputability is defined in Directive 2005/61/EC as ‘the likelihood that a serious adverse reaction in a recipient can
be attributed to the blood or blood component transfused or that a serious adverse reaction in a donor can be attributed
to the donation process’.
452
Article 5 of Directive 2005/62/EC requires reporting of serious adverse reactions to the competent authority if they
are graded as imputability of 2 or 3 while the reporting form included in Annex II part D of the same Directive implies
that reactions of all imputability levels much be reported.
453
The ATMP Regulation introduced a mechanism for requesting scientific recommendations on whether a medicinal
product meets the definition of an ATMP from the Committee of Advanced Therapies (CAT)
453
, based on scientific
criteria.
454
Summary reports of the Tissue and Cell Competent Authority meeting of 13-14 May 2019.

214 | P a g e
Platelet rich plasma
(and platelet rich
fibrin) Produced, in
high volumes, in
hospitals using a
medical device but
unclear if the blood
legislation or tissue and
cell legislation should
also apply. Not used
‘for transfusion’ but in
other procedures such
as orthopaedic or
cosmetic.
Directive 2002/98 would seem to be applicable, in any
case, to collection and testing. Divergent regulatory
systems and quality and safety rules are applied.
A Commission survey of EU tissue and cell authorities
indicates the following current situation:
7 Member States do not regulate
5 apply blood safety and quality requirements
4 apply tissue and cell safety and quality requirements
2 regulate as a medicinal product (non-ATMP)
3 regulate in another way.
See Part 1, items 9
and 25 in Annex 9
Isolated keratinocytes –
sprayed onto patient
skin to promote healing
of burns and other
wounds (without cell
expansion).
A Commission survey of EU tissue and cell authorities
indicates the following current situation for cells
separated from tissue by enzymatic digestion without
expansion (including keratinocytes, hepatocytes etc.):
9 Member States regulate as tissues and cells
7 regulate as a medicinal product (ATMP)
2 decide on a case-by-case basis depending on
manipulation and use.
3 do not have this therapy or do not regulate it.
Presentation to the
meeting of T&C
competent
authorities 2019
455
.
Serum eye drops
Autologous serum
prepared for patients
with dry eye syndrome
– usually stored and
applied at home by the
patient.
Commission services indicated that this procedure
could fall under the scope blood directive as it applies
"to the collection and testing of human blood and blood
components, whatever their intended use …" as
defined in Article 2 of Directive 2002/98/EC. Many
blood and tissue establishments prepare and supply the
product.
A Commission survey of EU tissue and cell authorities
indicates the following current situation:
7 regulate under the blood legislation
3 apply tissue and cell requirements
3 regulate as a medicinal product (non-ATMP)
8 Member States do not regulate
See Part 1, item 15
in Annex 9
Presentation to the
meeting of T&C
competent
authorities 2019
456
.
Amniotic membrane
patches
Transplanted in the eye
to promote corneal
healing or in some cases
as a membrane or skin
replacement therapy.
In some cases,
fragments are
dispersed in autologous
serum
Although the anatomical site of application (the eye) is
different to the original site in the donor (the placenta),
tissue and cell competent authorities considered that
the essential function was the same as in the original
site and therefore that this tissue should be regulated
under Directive 2004/23/EC.
At least one Member State regulates as a medicinal
product when fragments care dispersed in serum
considering that the fragmentation of the membrane
means that it cannot carry out the same essential
function.
See Part 2, item 2 in
Annex 9.
FDA also consider
this to be ‘the same
essential function’.
Autologous adipose
tissue (prepared in
A Commission survey of EU tissue and cell authorities
indicates the following current situation:
Presentation to the
meeting of T&C
455
Summary reports of the Tissue and Cell Competent Authority meeting of 13-14 May 2019.
456
Summary reports of the Tissue and Cell Competent Authority meeting of 13-14 May 2019.

215 | P a g e
hospital)
11 Member States regulate as tissues and cells
2 Member States regulate as medicinal product
(ATMP)
2 do not regulate.
3 decide on a case-by-case basis or regulate in another
way.
competent
authorities 2019
24
.
TABLE 3: EXAMPLES OF SUBSTANCES CONTAINING BTC WHERE THE BORDERLINES
WITH MEDICAL DEVICES HAVE BEEN DISCUSSED
Substance/therapy
Borderline issue
Reference
Platelet rich plasma
(PRP, and platelet
rich fibrin, PRF)
Produced, in high
volumes, in hospitals
using a medical device
but unclear if the
blood legislation or
tissue and cell
legislation should also
apply. Not used ‘for
transfusion’ but in
other procedures such
as orthopaedic or
cosmetic.
Directive 2002/98 is applicable to
collection and testing. Divergent
regulatory systems and quality and
safety rules are applied for the
subsequent steps.
A Commission survey of EU tissue and
cell authorities indicates the following
current situation:
7 Member States do not regulate
5 apply blood safety and quality
requirements
4 apply tissue and cell safety and
quality requirements
2 regulate as a medicinal product (non-
ATMP)
3 regulate in another way.
In all cases, the device to prepare the
PRP and PRF are regulated by the
medical device regulation.
See Part 1, items 9
and 25 in Annex 9
Presentation to the
meeting T&C
competent
authorities 2019
24
.
Decellularised dermis
(skin)
Used for a range of
skin replacement
treatments, including
burns and for
aesthetic surgery. The
removal of donor cells
is considered to
There has been discussion at the MDCG
subgroup Borderline and Classification
on whether tissues from which cells
have been removed (or rendered non-
viable) should be considered as
‘derivatives’ and should therefore fall
under the new Medical Device
Legislation.
A Commission survey of EU tissue and
Presentation to the
meeting of T&C
competent
authorities 2019
457
.
457
Summary reports of the Tissue and Cell Competent Authority meeting of 13-14 May 2019.

216 | P a g e
accelerate the
repopulation of the
tissue with recipient
cells after application.
cell authorities indicates the following
current situation:
13 regulate under the tissues and cells
legislation
7 have no current regulation or do not
have the therapy.
Decellularised heart
valves
Used for heart valve
replacement. The
removal of donor cells
is considered to
accelerate the
repopulation of the
tissue with recipient
cells after application.
There has been discussion at the MDCG
subgroup Borderline and Classification
on whether tissues from which cells
have been removed (or rendered non-
viable) should be considered as
‘derivatives’ and should therefore fall
under the new Medical Device
Legislation.
A Commission survey of EU tissue and
cell authorities indicates the following
current situation:
15 regulate under the tissue and cell
legislation
5 do not regulate or do not have the
therapy.
Presentation to the
meeting T&C
competent
authorities 2019
27
.
Demineralised bone
(with or without the
addition of gel or
putty)
Used in large volumes
for a wide range of
applications where the
stimulation of new
bone growth is
required. The removal
of minerals from bone
makes the naturally
occurring bone-
growth-stimulating
proteins more exposed
and functional.
There has been discussion at the MDCG
subgroup Borderline and Classification
on whether tissues from which cells
have been removed (or rendered non-
viable) should be considered as
‘derivatives’ and should therefore fall
under the new Medical Device
Legislation.
A Commission survey of EU tissue and
cell authorities indicates the following
current situation regarding
demineralised bone combined with
putty or gel:
11 regulate under tissue and cell
legislation
1 regulates as a medical device
1 regulates as a medicinal product (non-
ATMP)
3 do not have the therapy
Presentation to the
meeting T&C
competent
authorities 2019
24
.

217 | P a g e

{kind=link}