
spruson.com
The 2023
Pharmaceutical
Patent Review

spruson.com The 2023 Pharmaceutical Patent Review 1
Contents
Introduction 2
2023 Case Law
No pay day for Commonwealth in Sanofi pharma
damages claim: Commonwealth of Australia v Sanofi 4
ToolGen unsuccessful in landmark CRISPR
patent appeal: Toolgen Inc v Fisher 5
Sanofi’s challenge to Amgen PSK9 antibody patents
heard in Federal Court: Sanofi v Amgen Inc 9
Reading between the lines: Sandoz challenge to
rivaroxaban patents unsuccessful: Sandoz AG v
Bayer Intellectual Property GmbH 12
Current pharmaceutical patent cases
before the Federal Court 14
2023 Hot Topics
Skinny labelling in the Australian context: an overview 16
Pharmaceutical Patent Term Extension in Australia 18
“Best Method” Requirement Increasingly Prominent
in Australian Pharmaceutical Patent Disputes 20
Pharmaceutical Policy update 25
About the Spruson & Ferguson
Pharmaceutical Industry Group 27
spruson.com The 2023 Pharmaceutical Patent Review 1

2 The 2023 Pharmaceutical Patent Review spruson.com
Introduction
Welcome to Spruson & Ferguson’s wrap-up of the most notable
developments in pharmaceutical patent law in Australia in
2023. The past year saw a number of important Federal Court
decisions delivered, with the key question of ‘Commonwealth
Government damages’ now on its way to the High Court. We
continue to see pharmaceutical patent cases making their way
to trial, and legal issues which have seen recent attention, such
as the validity of patent term extensions and the best method
requirement, show no let up with a number of these cases
currently before the Courts. Amongst the highlights:
• In the keenly awaited judgment in Commonwealth of Australia
v Sanofi, the Full Court of the Federal Court of Australia has
upheld the trial judge’s decision that the Commonwealth
is not entitled to damages arising from the grant of a 2007
interlocutory injunction preventing Apotex from launching
generic clopidogrel products, on the basis of a patent which
was later found invalid. The High Court has granted special
leave, meaning that it will now proceed to consider an appeal.
It is expected that the hearing will take place this year.
• The Federal Court of Australia has heard an appeal from
a Patent Office opposition decision forming part of the
global patent litigation concerning Amgen’s PCSK9 antibody
patents. In contrast to outcomes in the United States and
Europe, the Australian Patent Office upheld the patent
applications including against claims of lack of fair basis,
sufficiency and best method (Sanofi v Amgen). It remains to
be seen how the Court will approach these issues.
• The Federal Court of Australia has delivered judgment in
Australia’s landmark CRISPR patent dispute, finding that none
of the claims in ToolGen Inc.’s patent application for platform
CRISPR technologies are valid. ToolGen has subsequently
applied to amend its claims and a hearing is scheduled to
Toolgen v Fisher).
• The Federal Court considered construction and inventive
step issues in Sandoz v Bayer, providing important
comments about the preparation of expert evidence in
patent cases.
• We review recent developments in relation to patent term
extensions. After a focus in recent years on the ‘first regulatory
approval’ requirement, we expect there to be ongoing judicial
consideration of the subject matter requirement that a PTE
cover a ‘pharmaceutical substance per se’.
• We tackle the thorny issue of ‘best method’. Australia is one
of the few major patent jurisdictions which has maintained
in its patent law a discrete requirement that a patentee
disclose the “best method” known to it of performing the
invention. Rather than remain in the background or even
fade away, best method challenges have assumed an
increasingly prominent role in Australian patent disputes and
can significantly affect the conduct and strategy of Federal
Court litigation, in particular. Potential pharmaceutical patent
litigants should be mindful of the substantive and procedural
implications best method issues can have in the Australian
iteration of global disputes.
• We provide an update on key pharmaceutical policy issues
and an update on ongoing pharmaceutical patent litigation
in the Courts.
As we continue into 2024, we hope this review provides a
useful practical resource. Please do not hesitate to take the
opportunity to contact our authors, all subject-matter experts in
their respective fields, for advice on the issues raised by these
important decisions.

spruson.com The 2023 Pharmaceutical Patent Review 3
2023 Case Law
spruson.com The 2023 Pharmaceutical Patent Review 3

4 The 2023 Pharmaceutical Patent Review spruson.com
No pay day for
Commonwealth in Sanofi
pharma damages claim:
Commonwealth of
Australia v Sanofi
Author:
Katrina Crooks | Principal, Head of
Spruson & Ferguson Lawyers
Background
In the keenly awaited judgment in Commonwealth of Australia
v Sanofi (formerly Sanofi-Aventis) , the Full
Court of the Federal Court of Australia upheld the trial judge’s
decision that the Commonwealth is not entitled to damages
arising from the grant of a 2007 interlocutory injunction
preventing Apotex from launching generic clopidogrel products,
on the basis of a patent which was later found invalid.
In the financial year 2008, Sanofi’s PLAVIX clopidogrel products
(also sold in Australia as ISCOVER by Bristol-Myers Squibb),
a medication inhibiting the formation of blood clots, was the
third most heavily Government subsidised prescribed drug
in Australia. Commonwealth costs for that year extended to
approximately $170 million.
In August 2007 Apotex commenced legal action to revoke
Sanofi’s Australian patent 597784 covering the product, and
was quickly met with an interlocutory injunction application.
That injunction was granted, and remained in force until the
patent was ultimately found wholly invalid by the Full Court
and special leave for appeal to the High Court was refused.
Apotex’s clopidogrel products were launched on 1 May 2010.
On grant of the interlocutory injunction, Sanofi was required
to give the ‘usual undertaking as to damages’, by which it
undertook to compensate any person adversely affected by
the operation of the injunction. After the patent was revoked,
both Apotex and the Commonwealth brought claims pursuant
to the undertaking, seeking damages for their losses resulting
from the delayed launch of Apotex’s products. Apotex’s claim
was settled. The Commonwealth continued with its claim
which was based on its lost opportunity for pricing decreases
for clopidogrel products which would have been triggered
by a first generic entry, including an immediate mandatory
price reduction, and further price disclosure related price
reductions which would have occurred in the years following.
The Commonwealth’s claim on this basis exceeded $325 million
plus interest.
Key findings and implications
• The Full Court upheld the decision of the trial judge that
the Commonwealth did not succeed in making out its case.
Ultimately this finding arises from a failure to make out the
‘counterfactual’ that Apotex would have launched in the
circumstances at play at the relevant time, had Sanofi not
obtained an interlocutory injunction.
• The appeal decision highlights once again the complexities
in establishing that ‘counterfactual’ to the required
standard. Combined with this difficulty, the Full Court did
not re-consider the trial judge’s finding that it was more
likely than not that the Commonwealth would have been
prepared to reverse statutory reductions in the reimbursed
price for Sanofi’s products triggered by the generic listing
on the PBS, if sale of the generic product was subsequently
restrained by a permanent injunction. Both of these
matters are likely to continue to be raised in interlocutory
injunction hearings as factors requiring a re-evaluation of
the delicate balance between the interests of both parties
in such a scenario.
• The decision highlights again the need for compelling
evidence (supported by contemporaneous documents)
from the ultimate decision-makers at the generic party and
the Commonwealth to convince the Court that, but for the
grant of the interlocutory injunction, the generic product
would have been launched and listed on the PBS in the face
of the significant damages risk if patent infringement was
later made out.
• In overturning the judge’s conclusion that the
Commonwealth’s losses were not a direct consequence of
the interlocutory injunction (because it did not restrain PBS
listing), the Full Court has released some pressure on the
need to explicitly include such a restraint in interlocutory
injunction orders.
First instance decision against the
Commonwealth
The first instance decision delivered in May 2020 was the
first case dealing with a Commonwealth claim for damages in
these circumstances. We reported on this decision here. The
Commonwealth had previously settled claims for compensation
against Wyeth, relating to extended release formulations of
AstraZeneca relating to the “super statin” rosuvastatin
Court decision relating to venlafaxine, in which the generic
party claims were upheld, including third party generic
companies who were not party to the proceedings: Sigma
.

spruson.com The 2023 Pharmaceutical Patent Review 5
At first instance, Nicholas J confirmed that in principle, a claim
by the Commonwealth on an undertaking as to damages
in these circumstances could be made out. However the
Commonwealth failed in its case in several respects:
• The Court found that the Commonwealth’s losses were
not a direct consequence of the interlocutory injunction
granted. In this case the injunction prohibited commercial
activities such as manufacture and sale, but did not
explicitly restrain listing by Apotex of its products on the
Commonwealth’s Pharmaceutical Benefits Scheme (PBS).
There had been some debate at the original interlocutory
injunction hearing as to whether such listing would be
a patent infringing act and so whether the Court should
restrain such an act by injunction. However Apotex in any
event gave a separate undertaking to refrain from PBS
listing its products, given that practically it would not have
been able to meet the guarantee of supply requirements
of such listing in the face of an injunction on supply.
Crucially the Court found that Apotex’s undertaking was not
supported by any undertaking as to damages from Sanofi.
Accordingly, the Court found that the loss was directly
caused by Apotex’s decision not to list on the PBS, not by
the interlocutory injunction itself;
• Despite Apotex’s Australian Managing Director giving
evidence that Apotex would “almost certainly” have
launched “at risk”, Nicholas J was not satisfied that
Apotex’s CEO and ultimate decision-maker, who did
not give evidence, would have authorised a launch if no
interlocutory injunction had been granted;
• The Court found that it was more likely than not that the
Commonwealth would have been prepared to reverse
statutory reductions in the reimbursed price for Sanofi’s
products triggered by the generic listing on the PBS, if sale
of the generic product was subsequently restrained by a
permanent injunction.
Full Court upholds decision against
Commonwealth
On appeal, the Full Court focussed on two key issues: whether
the trial judge erred in finding that:
(a) Apotex would not have sought to PBS list its clopidogrel
products even if it had not been restrained by the interlocutory
injunction (Apotex Launch and Listing Issue); and
(b) the loss claimed by the Commonwealth did not flow directly
from the interlocutory injunction (Directness Issue).
The Court noted that other issues in the appeal all related to
“further hypothetical causative obstacles sequentially secreted
within each other”, all within the overarching hypothetical
scenario where Apotex did list and launch its products in 2008.
However these issues did not arise if one of the two key issues
above was decided against the Commonwealth. That proved to
be the case.
On the Apotex Launch and Listing Issue, the Court reviewed a
significant body of both documentary and testimonial evidence
relied upon at trial, including a substantial number of emails.
Amongst other asserted errors, the Commonwealth argued
that the trial judge had failed to have regard to various parts
of this material and submissions made at trial, and that he
had erred in drawing a ‘Jones v Dunkel’ inference against the
Commonwealth for failing to call Dr Sherman (the ultimate
Apotex decision maker), that is, an inference that evidence
from Dr Sherman would not have assisted the Commonwealth.
Notably, the Commonwealth also claimed that by reason of
judge had lost the advantage usually afforded to a trial judge
with regard to the assessment of credit of witnesses.
The Full Court rejected all of these arguments, affirming
the trial judge’s approach to the evidence. On the question
of delay it found that the trial judge had clearly set out his
reasons, showing that he was very much alive to the detail of
the evidence and its significance. The judge’s reasons were
described as “a most thorough and searching excavation of the
very complicated factual questions which the case generated”.
The Commonwealth therefore failed in showing that had the
injunction not been granted, Apotex would have launched its
products. Its failure in this crucial respect was determinative of
the case.
However the Court also considered the Directness Issue,
on which it found that the trial judge had erred in applying
such a strict causative test. Notwithstanding an intermediate
the grant of the interlocutory injunction and the loss suffered
by the Commonwealth, such loss did flow directly from the
injunction.
Further implications
In December 2023 the Commonwealth was granted special
leave to appeal the Full Court decision to the High Court. The
High Court appeal is expected to be heard this year.
Regardless, it is likely that the implications of this case will
be felt not only in future damages cases, but also at the
interlocutory injunction stage.
A further Commonwealth claim for damages pursuant to
undertakings given by Otsuka and BMS in relation to the
Author
Katrina Crooks
Principal, Head of Spruson &
Ferguson Lawyers

6 The 2023 Pharmaceutical Patent Review spruson.com
ToolGen unsuccessful
in landmark CRISPR
patent appeal
Author: Michael Christie | Principal
The Federal Court of Australia has delivered judgment in
Australia’s landmark CRISPR patent dispute, finding that none
of the claims in ToolGen Inc.’s patent application for platform
CRISPR technologies are valid
. The proceeding is an appeal
from a decision of the Commissioner of Patents in which
the first respondent – a “strawman” named Grant Fisher –
successfully opposed the grant of ToolGen’s application.
The patent application
The case concerned ToolGen’s application for CRISPR/Cas
systems and the use of those systems to introduce a site-
specific, double started break at a target nucleic acid sequence
in a eukaryotic cell.
The application was filed on 23 October 2013 and claims
priority from three provisional applications:
•
filed 23 October 2012;
•
filed 20 March 2013; and
•
filed 20 June 2013.
The application includes two independent claims:
Claim 1. A composition comprising a Type II Clustered
Cas system for use in introducing a site-specific, double
stranded break at a target nucleic acid sequence in a
eukaryotic cell, said CRISPR/Cas system comprising (i)
a nucleic acid encoding a Cas9 polypeptide comprising
a nuclear localization sequence, and (ii) a nucleic acid
encoding a guide RNA that hybridizes to a target nucleic
acid, wherein the guide RNA is a chimeric guide RNA
Claim 10. A method of introducing a site-specific, double-
stranded break at a target nucleic acid sequence in a
eukaryotic cell, the method comprising introducing into the
eukaryotic cell a Type II Clustered Regularly Interspaced
the CRISPR/Cas system comprises:
(a) a nucleic acid encoding a Cas9 polypeptide
comprising a nuclear localization signal, wherein the
nucleic acid is codon-optimized for expression in
eukaryotic cells, and
(b) a nucleic acid encoding a guide RNA that hybridizes
to the target nucleic acid, wherein the guide RNA is a
portion, wherein the target nucleic acid sequence
comprises a first strand that binds to the crRNA portion
and a second strand having a trinucleotide protospacer
and wherein the Cas9 polypeptide and the guide RNA form
a Cas9/RNA complex in the eukaryotic cell, whereby a site-
specific, double stranded break at the target nucleic acid
sequence is introduced.
Several grounds of validity turned on the meaning of the words
“nucleic acid encoding a guide RNA” in both independent
claims. ToolGen sought a broad construction of these words,
arguing that they encompass both DNA which is transcribed
to RNA in a eukaryotic cell and RNA which is transcribed in
vitro prior to it being introduced into a eukaryotic cell.
1
ToolGen
argued that the verb “encoding” can mean both providing the
sequence for producing the guide RNA (through the process
sequence that enables the guide RNA to perform its function.
2
ToolGen placed considerable reliance on claim 19 which, when
read with claim 10, requires that the nucleic acid encoding the
guide RNA is in vitro
Claim 19.
the nucleic acid encoding the guide RNA is in vitro
transcribed RNA.
Justice Nicholas rejected these arguments, finding that claim
10, when read in the context of the specification as a whole,
indicates that the claim is limited to a method in which the
nucleic acid encodes the guide RNA, and that the guide RNA is
transcribed from nucleic acid in the eukaryotic cell.
3
His Honour
found that the term “encoding” should be given its ordinary
meaning as understood by those skilled in the art:
In my opinion, the word “encoding” is used in claim 10
in its conventional sense (i.e. as it would be understood
by a molecular biologist) to refer to the production of a
Cas9 polypeptide by transcription and translation and the
production of a guide RNA by transcription in the cell. The
nucleic acid referred to in the claim provides the information
which is used in the cell to produce the guide RNA. Claim
10 does not encompass a system in which an existing guide
RNA is introduced into the cell.
4
As a consequence of this construction, claim 19 could not be
read sensibly with claim 10 and was found to lack clarity.
5
1 ToolGen

spruson.com The 2023 Pharmaceutical Patent Review 7
The priority date
The hearing of the appeal was conducted on the premise that
if the claims were not entitled to priority based on P1, then a
deferred date of 20 June 2013 established by the filing of P3
would apply. P2 was solely concerned with a method of using
RNA-guided endonucleases in restriction fragment length
polymorphism analysis, and was not considered to disclose the
invention of any of the claims in the patent application.
P1 is a relatively short document; it does not include any
claims and resembles a journal article to which an additional
paragraph headed “Summary of the Invention” had been added.
The CRISPR/Cas9 system described in P1 was derived from
Streptococcus pyogenes and used a single chimeric guide RNA
comprising a crRNA portion fused to a tracrRNA produced in
vitro.
is introduced into the cell in order to transcribe the guide RNA
in vivo. P1 also did not describe what other bacterial species
have Type II CRISPR/Cas systems or how to determine the
endogenous crRNA and tracrRNA sequences for such a species.
The question, then, was whether the disclosure of P1 was
sufficient to establish a priority date for any of the claims in
ToolGen’s application.
The priority date test in Australia is the same as the test for
sufficiency of disclosure. That is, each claim is entitled to
claim priority from an earlier application, provided the earlier
application discloses the claimed invention in a manner that
is clear enough, and complete enough, for the invention to be
performed by a person skilled in the relevant art.
6
In relation to the words “nucleic acid encoding a guide RNA”,
his Honour accepted that it would not be a difficult exercise
for a molecular biologist in possession of the information in P1
coupled with the common general knowledge to use a plasmid
encoding a guide RNA to produce the guide RNA in vivo using
standard techniques that were well known at the priority date.
His Honour also accepted that it would be obvious to the skilled
addressee that he or she could use plasmid DNA encoding a
guide RNA as a means of generating the guide RNA in the cell.
7
encoding a guide RNA, as defined in the claims but, rather,
a guide RNA produced in vitro which is then introduced into
the cell. His Honour found that P1 did not disclose the same
invention as that claimed in ToolGen’s application, and as such,
Claims 1 and 10 (and, with the exception of claim 19, the
dependent claims) are directed to an invention in which the
guide RNA of the claims is introduced into the cell in the
the guide RNA in the eukaryotic cell. P1 does not disclose
any such system either explicitly or implicitly. It follows that
those claims are not entitled to priority based on P1.
8
The next question was whether P1 discloses a system for
cleaving DNA using a Cas9 polypeptide derived from a bacterial
species other than S. pyogenes in a manner which is clear
enough and complete enough for the claimed invention to
be performed by a person skilled in the art. It was common
ground among the parties that P1 disclosed a CRISPR/Cas9
system derived from S. pyogenes. Nicholas J accepted that P1
disclosed, in a general sense, the existence of Cas9 proteins
derived from other bacterial species and the possibility that
they may be used to mediate DNA cleavage in eukaryotic cells.
9
However, the possibility of using Cas9 proteins derived from
other bacterial species was described as just that – a mere
possibility. P1 did not include any further discussion of this
possibility, nor did it present any evidence or commentary from
which it may be inferred that all, or even some, Type II Cas9
proteins derived from other bacterial species could reasonably
be expected to work with particular PAMs to mediate DNA
cleavage in eukaryotic cells.
Moreover, there was nothing disclosed in P1 which would
indicate that S. pyogenes was likely to be representative of
other bacterial species with a Type II CRISPR/Cas system
or that the results of the experimentation with S. pyogenes
derived components provided any reasonable scientific basis
for inferring that Cas9 polypeptides derived from other bacterial
species could also be expected to cleave DNA in eukaryotic
cells.
10
In that context, the evidence showed there was
considerable uncertainty as to whether or not a CRISPR/Cas9
system derived from any particular bacterial species other than
S. pyogenes would work in eukaryotic cells, and that significant
experimental work would need to be done to validate the use of
the system in eukaryotic cells.
11
His Honour found that the work that the person (or team)
skilled in the art would need to undertake at the priority date
to perform the invention of claims 1 and 10 using a bacterial
species other than S. pyogenes would involve a significant
research project:
In my opinion the skilled team would be required to carry out
prolonged research and experimentation and would most
likely encounter significant difficulties along the way. Much
of the work would be non-routine and would be carried out
in circumstances where P1 provided no meaningful guidance
or direction and no assurance of success.
I am persuaded that as at the priority date, P1 did not
enable a skilled team including a molecular biologist
specialising in genome editing in eukaryotic cells and
a microbiologist with expertise in CRISPR/Cas systems
in prokaryotes, to make the compositions of claim 1, or
perform the methods of claim 10, using a bacterial species
other than S. pyogenes, without undue burden.
12
6 Patents Act 1990
Patents Regulations 1991

8 The 2023 Pharmaceutical Patent Review spruson.com
With regard to the guide RNA itself, P1 disclosed a single
chimeric guide RNA comprising a crRNA portion fused to a
tracrRNA portion without providing any information as to how
it was designed or how its length might be altered.
13
Nicholas
J considered that it would be an undue burden for the skilled
person to redesign the single guide RNA disclosed in P1 or
to design and construct a single guide RNA using a bacterial
species other than S. pyogenes.
14
His Honour found that P1 failed to disclose the claimed
invention in a manner that was clear enough, and complete
enough, for the invention to be performed by a person skilled
in the relevant art. None of the claims were entitled to claim
priority from P1.
Enablement
Patents Act 1990 (Cth) states that a
complete specification must disclose the invention in a manner
which is clear enough and complete enough for the invention
to be performed by a person skilled in the relevant art. The
requirement for enablement is similar to that applicable in other
jurisdictions, particularly Europe and the UK.
The respondents accepted that the patent application, unlike P1,
discloses an invention that comprises “a nucleic acid encoding
a guide RNA”. They did not contend that the invention of the
claims is, in this particular respect, not sufficiently enabled.
However, none of the examples disclosed in the patent
application used CRISPR/Cas9 components from any species
other than S. pyogenes. The respondents submitted that the
patent application did not enable an invention comprising a
system derived from a bacterial species other than S. pyogenes
without undue burden. His Honour accepted that submission
essentially for the reasons given in relation to P1.
15
Similarly, in relation to the guide RNA, there was considered to
be no material difference between the disclosures of P1 and the
patent application regarding the design of the sgRNA including
its tracrRNA component. Accordingly, his Honour found that
the patent application did not provide an enabling disclosure
of a sgRNA having a length different from that disclosed in the
patent application.
16
Support
Patents Act 1990 (Cth) states that the claims
must be supported by the matter disclosed in the specification.
This provision requires that the technical contribution to the art
disclosed by the specification justify the breadth of the claim.
17
In considering the overlapping requirements of enablement and
support, his Honour noted that there may be instances where
providing an enabling disclosure, but not meet the requirement
a claim to an invention for which there was no enabling
disclosure could meet the support requirement because, in
such circumstances, the scope of the monopoly defined by
the claim could not be justified by the technical contribution to
the art.
18
Having found that the invention was not sufficiently
Novelty and inventive step
Three journal articles published after the filing date of P1 but
before the filing date of P3 were relevant to the issues of
novelty and inventive step:
• Cong et al, “Multiplex Genome Engineering Using
Supplementary Materials;
•
Materials; and
• Wang et al, “One-Step Generation of Mice Carrying
Mutations in Multiple Genes by CRISPR/Cas-Mediated
Supplementary Information.
Having decided that ToolGen’s application was not entitled to
the priority date established by P1, his Honour found that claims
1 to 20 lacked novelty and an inventive step, and that claim 21
lacked an inventive step in light of the prior art.
ToolGen has subsequently applied to amend its claims and a
hearing is scheduled to hear that application in May 2024.
17 Merck Sharp & Dohme Corporation v Wyeth LLC
Author
Michael Christie
Principal

spruson.com The 2023 Pharmaceutical Patent Review 9
Sanofi’s challenge to Amgen
PSK9 antibody patents
heard in Federal Court
Author: Michael Christie | Principal
The global patent litigation concerning Amgen’s PCSK9
antibody patents has highlighted the divergent approach taken
by major jurisdictions in assessing the validity of functionally
defined antibody claims. In the US, Amgen’s patent claims were
ruled invalid by the Federal Circuit for lack of enablement.
1
In
corresponding European opposition proceedings, the claims of
Amgen’s patent were found to be enabled, but were subsequently
invalidated by the Board of Appeal for lacking an inventive step.
2
In 2022, a Delegate of the Commissioner of Patents ruled in
Amgen’s favour, finding that five of its patent applications
are valid and should proceed to grant.
3
Sanofi appealed the
decision to the Federal Court. This article summarises the
Delegate’s 2022 decision and provides an update on the
Federal Court appeal, which was heard in November 2023.
4
Patent Office Opposition
The opposed applications
The opposed applications stem from international patent
application no. PCT/US2008/074097. The Australian national
phase application has granted, and its term was extended under
Australia’s pharmaceutical patent term extension provisions.
Amgen filed several divisional applications, five of which were
accepted and subsequently opposed by Sanofi in 2016. The
opposed applications cover Amgen’s cholesterol-lowering
The applicable law
The opposed applications are all subject to Patents Act 1990
(Cth) as it existed prior to the introduction of the Intellectual
Property Laws Amendment (Raising the Bar) Act 2012, which
came into effect in 2013.
The so called “Raising the Bar” amendments were introduced by
the Australian Parliament with the express intention of aligning
Australia’s written description requirements with those of its major
trading partners, particularly Europe and the US. Under the current
Act, the requirements of support and sufficiency apply, meaning
that, as in Europe, the claims must be commensurate with the
technical contribution to the art, and the specification must enable
a skilled person to perform the invention across the full scope of
the claims without undue burden or further invention.
Under the “old” Act, however, the requirements of “full description”
and “fair basis” apply. The standards set by full description and fair
basis are much lower than those set by support and sufficiency,
and challenging a patent on these grounds has been notoriously
difficult (and more often than not, unsuccessful).
The claims
The Delegate broadly grouped the disputed claims into three
classes, namely:
i) epitope claims, which define an isolated monoclonal
antibody by its ability to bind an epitope of PCSK9, the
epitope comprising nominated residues;
ii) residue claims, which define an isolated monoclonal
antibody by its ability to bind one or more specific residues
of PCSK9;
iii) competition claims, which define an isolated monoclonal
antibody by its ability to compete for binding with a
structurally-defined antibody.
5
The claims also include functional language referring to the
ability of the antibody to block or reduce binding of PCSK9 to
construction of certain terms in the claims that were critical to
Sanofi’s opposition.
Clarity
Sanofi opposed the claims of the applications for lack of clarity.
The issues raised in their submissions essentially related to the
use of inexact language in the claims, for instance terms such as
binds, blocks, reduces, neutralizing and competes. The Delegate
rejected these submissions, finding that each term could be given
meaning and that the claims provide a workable standard.
6
Fair basis
Sanofi asserted that the claims of each application were not
fairly based on the matter described in the specification.
The question of fair basis has been expressed by Australia’s
High Court as whether there is a real and reasonably clear
disclosure in the body of the specification of what is claimed,
so that the alleged invention is broadly, that is in a general
sense, described in the body of the specification.
7
Sanofi argued that only two antibodies disclosed in the
applications were actually made, tested and shown to block the
binding of PCSK9 to LDLR, and thereby lower plasma LDL levels.
Sanofi asserted that it is those two antibodies for which the
specification provides a real and reasonably clear disclosure.
8
The Delegate rejected those arguments and pointed to
statements in the specification which described the invention
in broader terms. Although generic and not tied to specific
examples, those paragraphs indicated to the Delegate that the
invention extends beyond the specific antibodies isolated and
characterised in the applications.
9
The invention as described
by the specification was considered to include antigen binding
proteins that bind to the same or an overlapping region of
PCSK9 as bound by the EGFa domain of LDLR or the two
exemplified antibodies.
27
1
2 T 0845/19.
3 Sanofi v Amgen Inc. ‘Sanofi v Amgen’).
4 Sanofi v Amgen Inc NSD876/2022.
5 Sanofi v Amgen
6 Sanofi v Amgen
7 Lockwood Security Products Pty Ltd v Doric Products Pty Ltd
8 Sanofi v Amgen
9 Sanofi v Amgen

10 The 2023 Pharmaceutical Patent Review spruson.com
With regard to the epitope and residue claims, Sanofi asserted
that there is no proof that the antibodies of the invention
bind to one or more of the identified residues. However, the
Delegate emphasised that fair basis is a consideration of the
disclosure provided in the specification, namely what the body
of the specification read as a whole describes as the invention.
It was not necessary for there to be scientific proof of non-
covalent binding between the claimed antibodies and the
identified amino acid residues.
10
The Delegate accepted that the specification does not
demonstrate that the amino acid residues identified as part of
the interaction interface are directly involved in non-covalent
interactions that effect binding between PCSK9 and LDLR or
the two characterised antibodies.
11
Instead, the specification
described X-ray crystallography experiments identifying those
residues on the antigen that are located closest to the antibody
when the two molecules are bound. The Delegate considered
it a reasonable extrapolation to infer that amino acid residues
within the identified region are involved in the non-covalent
interactions that effect binding between PCSK9 and antibody.
12
Accordingly, the Delegate was satisfied that the specification
provides a real and reasonably clear disclosure of the
antibodies encompassed by the epitope and residue claims.
As for the competition claims, the Delegate again pointed to
statements in the specification which described the invention in
broad and general terms, and to paragraphs disclosing means for
identifying competitively binding antibodies. Having construed
the invention in these broad terms, the Delegate was satisfied
that the specification also provides a real and reasonably clear
disclosure of the antibodies encompassed by the competition
claims. Consequently, this ground of opposition failed.
Full description (sufficiency)
The test for sufficiency of the description (under the pre-
Raising the Bar law) has been articulated by Australia’s High
Court as whether the disclosure of the specification will enable
the addressee to produce something within each claim without
new inventions or additions or prolonged study of matters
presenting initial difficulty.
13
Sanofi submitted that the opposed applications do not disclose
an antibody that falls within the scope of any of the claims,
and that a skilled person could not reproduce the antibodies
disclosed in the applications. With regard to the epitope claims,
Sanofi argued that the applications do not disclose a single
antibody that binds an epitope comprising the residues recited
in the claims.
The Delegate re-framed the test for sufficiency by asking
whether, based on the disclosure provided, the addressee will
be able to produce an antibody that binds:
a) an epitope that comprises stated amino acid residues; or
b) to the specific amino acid residues specified;
or whether to do so would require new inventions or additions
or prolonged study of matters presenting initial difficulty.
With regard to the epitope claims, the Delegate was satisfied
that the specification described the binding site or interaction
interface between PCSK9 and LDLR, and that the specification
showed how two exemplary antibodies interact with this region
to block binding between PCSK9 and LDLR. The Delegate
again noted that the epitope, as construed earlier, will include
specific amino acids that directly contact the antibody and also
amino acids that are covered by the antibody. In that context,
the Delegate considered that the residues of PCSK9 that the
specification demonstrates with crystallographic experiments
to be within the region covered by the antibody, can be
considered the epitope, and therefore the epitope claims are
fully described.
14
With regard to the residue claims, the experts for both parties
agreed that the term “binds to” means that the claimed
antibody forms a non-covalent interaction with at least one
of the nominated residues of PCSK9. But the parties’ experts
presented opposing views as to whether the residues specified
in the claims are in fact directly involved in binding. Sanofi did
not present any evidence that the two exemplified antibodies
would not bind at least one of the residues set out in the
claims.
15
Amgen’s expert, however, performed an analysis using
crystal data in the specification and concluded that specific
residues identified in the claims very likely form non-covalent
interactions with the exemplified antibodies or with LDLR.
16
The Delegate accepted Amgen’s submission that the
specification discloses the interaction interface or epitope,
and that the residues identified form non-covalent interactions
between PCSK9 and the antibodies.
17
The question then
became whether the addressee could take this information,
along with the other information provided in the specification,
to generate antibodies that fall within the scope of the claims.
Sanofi submitted that, to make antibodies that will bind to the
relevant residues (or epitope comprising the relevant residues),
the skilled person would have to undertake one of two research
projects. The first was said to require making a biosimilar of
the antibodies disclosed in the application using transgenic
techniques. The second approach would be to seek to obtain
antibodies either by hyperimmunization of transgenic mice
or by other means such as phage display and then carry out
experiments to characterise the antibodies.
Amgen, on the other hand, submitted that the state of antibody
arts was advanced and mature at the priority date and that
armed with the teachings of the application it would be routine
for the addressee to make antibodies of the claimed invention.
The Delegate favoured the proposition presented by Amgen,
and in particular, that antibodies within the scope of the
claims could be produced using well understood mammalian
expression vector methodologies such as cloning the CDRs
of the exemplified antibodies into the framework region of
a known antibody, and that this approach would not require
new inventions or additions or prolonged study of matters
presenting difficulty.
18
10 Sanofi v Amgen
11 Sanofi v Amgen
12 Sanofi v Amgen
13 Kimberly-Clark Australia Pty Ltd v Arico Trading International Pty Limited
14 Sanofi v Amgen
15 Sanofi v Amgen
16 Sanofi v Amgen
17 Sanofi v Amgen
18 Sanofi v Amgen

spruson.com The 2023 Pharmaceutical Patent Review 11
The Delegate was not satisfied that the work required to produce
one antibody embodying each claim using the information
provided in the specification requires anything more than what
is routine in the art, even if such work may be complex, time
consuming and expensive.
19
Similarly, in relation to the competition
claims, the Delegate found that the skilled addressee could use
well-established techniques to produce a library of antibodies
that is then screened using standard techniques to assess for
competition with the reference antibodies.
20
Consequently, this
ground of opposition also failed.
Best method
The Patents Act 1990 (Cth) also requires the complete
specification to describe “the best method known to the
applicant of performing the invention”.
21
Sanofi alleged that the specification failed to disclose the
residues of PCSK9 with which the antibody will form non-
covalent interactions or that form the epitope of the claimed
antibodies. It submitted that by withholding this information,
Amgen concealed the best method by which to achieve the
result which constitutes the invention.
The Delegate rejected this submission, finding that the
specification discloses exemplary antibodies that represent the
invention and provides information that would allow the skilled
addressee to produce antibodies with the same CDRs, and
therefore binding properties, as these antibodies. The Delegate
also noted that there was no evidence that Amgen knew of a
better method than what is disclosed in the specification.
22
Utility
Sanofi also submitted that the invention as claimed failed to
achieve what was “promised” by the specification and therefore
lacked utility.
23
Sanofi argued that none of the claims are limited
to isolated monoclonal antibodies that: a) lower, maintain or
prevent an increase in plasma cholesterol of the subject to which
Promise); or b) have a biological effect of achieving The Promise.
They asserted that only two exemplified antibodies have the
picomolar affinity for PCSK9 and the resultant ability to block
binding of LDLR to PCSK9 to the extent that the antibody is
capable of lowering plasma LDL levels.
The Delegate did not agree with Sanofi’s characterisation of
the “promise” as being limited only to those antigen binding
proteins that will be capable of lowering plasma LDL levels.
Rather, the specification was found to more broadly disclose
antigen binding proteins that bind to particular regions of
PCSK9 to prevent binding to LDLR. The claims, by virtue of
the functional characteristics defined in each of the epitope,
residue or competition claims, were considered to necessarily
encompass those monoclonal antibodies that achieve the more
broadly stated promise of the invention.
24
As such, Sanofi’s
opposition on this ground was unsuccessful.
Federal Court Appeal
Sanofi appealed the Delegate’s decision to the Federal Court. In
the run up to the Federal Court hearing, which commenced in
November 2023, Sanofi filed two interlocutory applications.
Sanofi v Amgen Inc.
interlocutory application seeking orders for discovery and leave
to rely on experimental evidence. In relation to the experimental
evidence, Sanofi sought to rely on experiments conducted
for the purpose of proceedings in other jurisdictions. The
Court refused Sanofi’s application for discovery and permitted
reliance on some but not all of the experimental evidence.
In refusing leave to rely on certain experiments, Nicholas J
considered that the experiments were of little relevance to
Sanofi’s alleged grounds of invalidity. His Honour also observed
that there was significant debate in corresponding European
proceedings about the conclusions that could be drawn from
the experiments, and that introducing those experiments to the
Australian proceedings would likely give rise to a substantial
and undue waste of time and costs.
Experimental evidence for which leave was granted was
considered to be directly relevant to Sanofi’s alleged grounds
of invalidity.
In Sanofi v Amgen Inc.
a further interlocutory application seeking orders to limit the
evidence which Amgen could adduce at the hearing. Amgen
had proposed to rely on declarations made by three experts
in the Patent Office opposition, as well as supplementary
affidavits from those same experts. Sanofi sought to exclude
some of that evidence on the grounds that it was substantially
entirety noting that Sanofi could have raised its concerns at the
first case management hearing but did not do so.
The appeal continues.
19 Sanofi v Amgen
20 Sanofi v Amgen
22 Sanofi v Amgen
23 Patents Act 1990
24 Sanofi v Amgen
Author
Michael Christie
Principal

12 The 2023 Pharmaceutical Patent Review spruson.com
Reading between the
lines: Sandoz challenge
to rivaroxaban patents
unsuccessful
Authors:
James Beckett | Patent Attorney
Katrina Crooks | Principal, Head of
Spruson & Ferguson Lawyers
Summary
In Sandoz AG v Bayer Intellectual Property GmbH
1321, Sandoz AG challenged the validity of two Australian patents
(an anticoagulant) compositions and their uses, for which Bayer
Intellectual Property GmbH is the exclusive licensee. Justice
Rofe found that both patents were valid, and that claims 3 and
4 of patent AU2006208613 were threatened to be infringed
by Sandoz’s intended activities. A key issue at hand was the
construction of the phrase “in hydrophilized form” and the extent
to which the skilled person would refer to materials cited in the
specification to clarify the scope of essential claim features.
Background facts
Bayer Intellectual Property GmbH is the exclusive licensee
of two Australian patents, AU2004305226 (the ‘226 patent)
and AU2006208613 (the ‘613 patent). The ‘226 patent is
directed to compositions comprising rivaroxaban, processes
for preparing said compositions and uses of the composition
for treatment of thromboembolic disorders. The ‘613 patent is
directed to uses of rivaroxaban in rapid-release tablet form for
treatment of thromboembolic disorders. Rivaroxaban is one of
a class of factor Xa inhibitors claimed in a former Bayer patent,
Sandoz is the sponsor of several rivaroxaban therapeutics on
to exploit these products in Australia after November 2023
without Bayer’s approval. Sandoz sought revocation of both the
‘226 and ‘613 patents and Bayer made a cross-claim seeking an
injunction to prevent the threatened infringement of its patents
by Sandoz.
Legal issues and outcomes
Infringement
Bayer alleged that Sandoz’s intended activities threatened to
infringe various claims of both patents.
Subject to the validity of the ‘613 Patent, Sandoz Australia
admitted to threatening infringement of that patent.
The key issue in determining infringement of the claims of
the ‘226 patent was the scope of the phrase “in hydrophilized
form”. This phrase appears in claim 8 (which the remaining
claims ultimately depend on or refer to), which reads:
8. Solid, orally administrable pharmaceutical composition,
morpholinyl)-phenyl]- 1,3-oxazolidin-5-yl} -methyl)-
in hydrophilized form.
(emphasis added)
In the course of the proceedings, both of the primary experts
agreed that they had not encountered the term prior to reading
the patent, and that it was not part of the common general
knowledge in the art. The ‘226 patent does not include a
definition of this term, but does refer to two papers (termed
the “Lerk papers”). Those papers describe a process for
hydrophilization as a means for increasing oral bioavailability
of a drug compound. In short, hydrophilization is described as
a process for coating a hydrophobic drug with a hydrophilic
excipient in order to render it hydrophilic.
The principal dispute was whether it is legitimate for the skilled
person to have regard to the disclosure of the Lerk papers
in order to correctly understand the term used in the claims.
Referring to Justice Greenwood’s decision in Uniline
1
, Justice
Rofe held that “the person skilled in the art may read the prior
art Lerk papers to give context to, and better understand, the
discussion in the ‘226 Patent”.
Consideration then turned to the scope of this phrase. In both
the Lerk papers and the ‘226 patent, the hydrophilization
process involves intensive mixing in the presence of a small
amount of hydrophilic excipient in liquid solution. However,
Bayer asserted that it was not limited to methods which
include a liquid, and could also encompass methods which do
not include any liquid, such as that used to produce Sandoz’s
rivaroxaban products.
Her Honour considered that a skilled person having read the
specification and the Lerk papers, would construe the claims as
referring to the hydrophilization process described in the Lerk
papers, and therefore necessarily including the use of a liquid.
Thus Sandoz’s rivaroxaban products do not infringe the claims
of the ‘226 patent.
Inventive step
An inventive step challenge was brought against both patents
relying on various documents including Bayer’s earlier patent
synthesis, identified it as the most preferred compound of
the class and also noted a number of other beneficial traits,
such as suitability for oral administration and treatment of
thromboembolic disorders.
Under the Patents Act 1990 (Cth) prior to the Raising the Bar
amendments, in order for a document to be prior art under s
1 Uniline Australia Ltd v SBriggs Pty Ltd

spruson.com The 2023 Pharmaceutical Patent Review 13
Evidence of “ascertainment” in patent litigation is commonly
obtained by asking an independent expert witness what search
strategy they would use to research the relevant problem,
undertaking that search and then asking the expert to identify
relevant documents from the results.
In this case, Sandoz had provided WO 919 to its expert before
he reviewed the search results and he had considered it in some
detail. Rofe J noted that it was perhaps unsurprising that WO
919 was then listed by the expert as a high priority document.
In the absence of clear expert evidence that the document
would likely have been ascertained, her Honour considered that
as a patent specification, WO 919 was dissimilar to high impact
journal articles which would be read by all those in the field
to keep up to date. In the circumstances Justice Rofe did not
consider that this threshold had been met.
In obiter, Justice Rofe offered further comments, remarking
that even were the document ascertained, an inventive step
challenge on the basis of WO 919 would still not be successful.
Justice Rofe considered that, in contrast to the decision in
Astrazeneca, where the drug in question was one of a class
of commonly prescribed statins with the same mechanism of
action, the drug in this case (rivaroxaban) was a first in class
compound with a different mechanism of action to existing
Accordingly, while the information in WO 919 may lead the
skilled person to choose rivaroxaban as a starting point, the
hypothetical drug development team would have no guidance
from other compounds in the same class, and there was no
body of knowledge to refer to in order to reliably predict side
effects. Ultimately, Justice Rofe did not consider that the
skilled person would have the requisite expectation of success
that rivaroxaban would pass all the drug development stages
to successful completion of Phase III trials and ultimately be
approved for use in human as a safe and effective once per day
treatment for thromboembolic disorders. A similar finding was
made regarding the ‘613 patent.
The remaining inventive step challenge against the ‘613 patent
was in relation to documents titled the “Blood Abstracts”, which
were a set of three abstracts published in advance of the annual
this instance, her Honour found that the Blood Abstracts would
have been ascertained, understood and regarded as relevant by
In contrast to WO 919, experts for both parties acknowledged
that they would review the ASH abstracts book, in which the
Blood Abstracts made up three of the four abstracts listed under
the index keyword “factor Xa inhibitor”.
7939” had demonstrable effects in various surrogate tests of
thrombosis over a dose range that seemed safe. However,
the Blood Abstracts failed to disclose: the structure, chemical
class, toxicity, formulation chemical form, excipient details or
compound, the skilled team would not (or could not) proceed
any further. Her Honour considered that the skilled person
would not have the requisite expectation of success that
to successful completion of Phase III trials and ultimately be
approved for use in human as a safe and effective once per day
treatment for thromboembolic disorders.
Best method
Sandoz also submitted that the ‘613 patent failed to disclose the
best method of working the invention known to Bayer for making
a rapid release tablet containing rivaroxaban, which Bayer
admitted was that described in PCT/EP2004/012897 (which
is the international phase of the ‘613 patent). While this PCT
application was referred to in the ‘613 patent, a typographical
error had led to the PCT application being mis-numbered.
Sandoz argued this meant that the material contained in the PCT
application was not disclosed in the ‘613 patent.
Justice Rofe accepted that the mistake in the ‘613 patent was
an unintentional typographical error and further considered that
the evidence established that the skilled person would be able
to find the correct reference by searching the patent databases
or engaging a patent searcher. Accordingly, her Honour
considered that the public had been fairly given possession of
the invention described in the ‘613 patent.
Author
James Beckett
Patent Attorney
Author
Katrina Crooks
Principal, Head of Spruson &
Ferguson Lawyers

14 The 2023 Pharmaceutical Patent Review spruson.com
Author
Fiona Deng
Lawyer
Author
Lucy Hartland
Special Counsel
Current pharmaceutical
patent cases before the
Federal Court
Authors:
Lucy Hartland | Special Counsel
Fiona Deng | Lawyer
1. Otsuka Pharmaceutical Co. v Generic Health
Pty Ltd NSD121/2012
In similar fashion to The Commonwealth v Sanofi, these
proceedings concern a claim for compensation by the
Commonwealth and the relevant generic company, pursuant
to the usual undertaking as to damages made on grant of
an interlocutory injunction in 2012 in connection with Otsuka
Pharmaceutical Co., Ltd’s (Otsuka) claim that Generic Health
Pty Ltd had infringed certain patents concerning aripiprazole
by the Full Federal Court in Otsuka Pharmaceutical Co., Ltd
matter during 2023, possibly because of the current status of
the Sanofi proceedings.
2. H Lundbeck A/S & Anor v Sandoz Pty Ltd
NSD647/2014
These complex and long running proceedings between H
Lundbeck) and Sandoz Pty Ltd (Sandoz)
Court following the decision in H. Lundbeck A/S v Sandoz
Pty Ltd
contractual patent licence did not cover the extended term of
the patent. The Federal Court proceedings are currently stayed
until the final determination of a review in the Administrative
Appeals Tribunal of the decision H Lundbeck A/S v Sandoz Pty
Ltd
to Sandoz for the extended period.
3. Pfizer Ireland Pharmaceuticals & Anor v
Samsung Bioepis Co. Ltd & Ors NSD331/2022
These patent infringement proceedings which concern
Samsung Bioepis’ biosimilar etanercept product Brenzys, were
commenced by Pfizer Ireland Pharmaceuticals against multiple
with Biogen MC as an interested party) following protracted
preliminary discovery proceedings. There are a number of
interlocutory events on foot including an application to amend
the patent and an application to strike out the proceeding.
4. Novartis AG & Anor v Pharmacor Pty Limited
ACN 121 020 835 NSD506/2023
Novartis AG alleges that Pharmacor has threatened to infringe its
has cross claimed to revoke the relevant claim of the patent.
The proceedings are set down for hearing over several days in
April and May this year. Among other interlocutory applications
dealt with in 2023, an interlocutory application for the hearing
of a separate question (as to relevant time at which a patent
applicant’s knowledge of the best method is to be fixed for
Patents Act 1990 (Cth)) was
dismissed in August 2023, with that matter to be dealt with
together with all other issues at final trial.
5. Cipla Australia Pty Ltd v Bristol-Myers Squibb
Holdings Ireland Unlimited Company & Anor
NSD911/2023
These proceedings concern two patents owned by Bristol
directed at inhibiting Factor Xa in the prothrombinase. These
proceedings are still at an early stage with the parties currently
dealing with discovery matters.
6. Cipla Australia Pty Limited v Bayer Intellectual
Property GMBH VID124/2023
These proceedings concerning two patents covering
of the proceedings concerning the same patents between
Sandoz AG and Bayer Intellectual Property GmbH.
7. Samsung Bioepis Au Pty Ltd v Formycon AG
NSD1167/2023
These patent invalidity proceedings concern two patents
Other cases
The following cases of note are the subject of separate
case notes:
• Commonwealth of Australia v Sanofi (formerly Sanofi-
Aventis)
Court)
• Sandoz AG v Bayer Intellectual Property GmbH
1321 (now on appeal to the Full Court)
• Sanofi v Amgen Inc. NSD876/2022 (judgment reserved)

spruson.com The 2023 Pharmaceutical Patent Review 15
2023 Hot Topics
spruson.com The 2023 Pharmaceutical Patent Review 15

16 The 2023 Pharmaceutical Patent Review spruson.com
Skinny labelling in the
Australian context: an
overview
Author: Andrew Rankine | Principal
Key takeaways
• In Australia, new indications for known pharmaceuticals
may be protected by several patent claim formats, including
method of treatment claims, Swiss type claims and EPC
2000 claims.
• Various forms of “skinny labelling” may be adopted by
suppliers of generic and biosimilar medicines in an attempt
to avoid infringement of second medical use patents when
supplying a known pharmaceutical product for an off-
patent indication in Australia.
• Typically, skinny labelling involves omitting patented
indications from the prescribing information (i.e., label) for
a generic or biosimilar product, with or without an express
statement that the product is not supplied for use in any
patented indications. Such measures require regulatory
approval. Sponsors of generics and biosimilars may also
communicate directly with prescribers and pharmacists
regarding the permissible use of their products.
• In a leading Australian case, although such measures were
effective to avoid infringement of Swiss type claims, they
were not effective to avoid infringement of method of
treatment claims because, on the facts of that case, the
generic sponsor had “reason to believe” its product would
be used for a patented indication despite skinny labelling.
• The more complex regulatory arrangements applicable
to biosimilars and other high-cost medicines may provide
opportunities for more robust forms of skinny labelling (e.g.,
omitting patented indications from the scope of regulatory
approval and/or reimbursement arrangements), although
such measures are yet to be tested before Australian courts.
Patents for second medical uses
Second medical use patents confer exclusive rights relating
to the use of known pharmaceutical substances for new
therapeutic indications. An example is provided by the patents
granted to Warner-Lambert in several jurisdictions relating
to the use of pregabalin (a pharmaceutical previously known
and used in the management of seizures) for the treatment
of certain types of pain (see Warner-Lambert Company LLC v
Apotex Pty LtdPregabalin Case)).
In Australia, several claim formats may be used to protect
a new therapeutic indication for a known pharmaceutical
substance, including the following:
• Method of treatment claims typically have the form “a
• Swiss type claims are purpose-limited process claims
manufacture of a medicament for the treatment of [disease
• So-called EPC 2000 claims are purpose-limited product
While method of treatment claims are prohibited in some
jurisdictions, they are generally permissible in Australia: see
Apotex Pty Ltd v Sanofi-Aventis Australia Pty Ltd
Leflunomide case).
Skinny labelling as a defensive strategy
Skinny labelling refers to strategies that a supplier of generic
or biosimilar medicines may adopt in an attempt to avoid
infringement of a second medical use patent when supplying
a medicine for an “off patent” indication. As discussed below,
skinny labelling may take a number of different forms.
Amendments to pharmaceutical labelling
In common with many other jurisdictions, Australia’s regulatory
regime for therapeutic goods requires the supplier of a
prescription medicine to publish a document (generally known
as “prescribing information” or a pharmaceutical “label”)
providing information necessary for the medicine’s safe and
effective use. Among other things, this prescribing information
or label records the therapeutic indications for which use of the
product has been granted regulatory approval in Australia.
In its simplest form, skinny labelling involves omitting from
prescribing information for a generic or biosimilar product
one or more indications that remain patent-protected, while
retaining those indications that are “off patent”. A more
elaborate version of skinny labelling involves also including
in the generic or biosimilar prescribing information an
express statement that the product is not supplied for use in
accordance with one or more indications that remain patent-
protected. Amendments to prescribing information require the
approval of Australia’s Therapeutic Goods Administration (TGA).
Whether the TGA will approve the inclusion of a “disclaimer” in
generic or biosimilar prescribing information will depend upon
the circumstances of each individual case.
In addition to the strategies mentioned above, the supplier of a
generic or biosimilar product may communicate with Australian
prescribers and pharmacists to inform them that its product
should not be prescribed or dispensed for use in one or more
patented indications.
Such strategies have been considered in a number of Australian
pharmaceutical patent cases:
• In the Leflunomide Case, Australia’s High Court ruled that
a disclaimer included in the prescribing information for
Apotex’s generic leflunomide product was effective to avoid
infringement of Sanofi’s method of treatment claim covering
the use of leflunomide for the treatment of psoriasis,
enabling Apotex’s product to be supplied for the off-patent
rheumatoid arthritis indication.

spruson.com The 2023 Pharmaceutical Patent Review 17
• In the Pregabalin Case, a skinny labelling strategy, coupled
with undertakings to notify prescribers and pharmacists
that Apotex’s generic pregabalin products were only
supplied for use in the treatment of seizure disorders was
ineffective to avoid a preliminary injunction restraining
supply of the generic product, in light of evidence that the
patented pain indication comprised almost the entirety of
the relevant Australian market for pregabalin products.
• In Mylan Health Pty Ltd v Sun Pharma ANZ Pty Ltd
Fenofibrate Case), a skinny
labelling strategy, coupled with undertakings to notify
prescribers and pharmacists that Sun’s generic fenofibrate
products were only supplied for use in the off-patent
hypercholesterolaemia indication, was effective to avoid
infringement of Swiss type claims relating to the use of
fenofibrate in the treatment of diabetic retinopathy, but was
not effective to avoid infringement of method of treatment
claims covering the latter indication.
The Fenofibrate Case serves to highlight an important
distinction between Swiss type claims and method of treatment
claims under Australian law, as it currently stands. In that case,
Australia’s Full Federal Court held that infringement of Swiss
type claims is governed, not by the manufacturer’s intention,
but rather by what the medicament is manufactured “for” as
indicated by (for example) the physical characteristics of the
medicament as it emerges from the manufacturing process,
including its formulation, dosage, packaging and labelling. On
the facts of the Fenofibrate case, Sun’s skinny labelling strategy
was sufficient, in the Full Court’s view, to establish that its
generic fenofibrate products were not “for” use in the treatment
of diabetic retinopathy and thus would not have infringed
Mylan’s Swiss type claims.
By contrast, the Full Court held that Sun would have infringed
Mylan’s method of treatment claims for the diabetic retinopathy
indication (had they been valid). That is because, having
regard to all of the relevant circumstances, Sun had “reason to
believe” their generic fenofibrate products would be used for
the patented indication, despite skinny labelling. Similar findings
are likely to be made where (for example) a patented indication
comprises the overwhelming majority of the Australian market
for the pharmaceutical product in question, as occurred in the
Pregabalin Case.
Additional strategies for suppliers of biosimilars
and other high-cost medicines
Regulatory marketing approval for small-molecule generic
medicines is typically granted on the basis of an appropriate
bioequivalence study, without the need for the generic sponsor to
provide clinical-trial data demonstrating efficacy. In such cases,
regulatory marketing approval granted for the small-molecule
generic product typically encompasses all of the indications for
which the reference (i.e., branded or originator) pharmaceutical
product has been granted marketing approval in Australia.
Australia’s process for regulatory approval of biosimilars is
different. Commonly, data submitted in support of an application
for regulatory approval of a biosimilar includes results of one
or more clinical trials, demonstrating efficacy of the biosimilar
for at least one therapeutic indication, coupled with material
supporting an inference that the biosimilar will also be efficacious
in other therapeutic indications for which the reference product
is approved in Australia (“extrapolation of indications”).
This more complex regulatory pathway may afford the sponsor
of a biosimilar additional strategies to limit the indications for
which its product is granted marketing approval in Australia to
“off patent” indications.
In most cases, supply of biologicals and other high-cost
medicines in Australia is subsidised by the Australian
Government under the Pharmaceutical Benefits Scheme
(PBS). The indications for which supply of a medicine will be
reimbursed under the PBS is determined by the Australian
Government, acting on the advice of an expert committee (the
Pharmaceutical Benefits Advisory Committee; PBAC).
There may be scope for a sponsor of a biosimilar or other high-
cost medicine to limit the PBS listing of its product to exclude
patented indications. In particular, PBS-subsidies are ordinarily
not available where a product is supplied “off label” (i.e.,
supplied for a therapeutic indication for which that product has
Exclusion of patented indications from the scope of regulatory
marketing approval and/or PBS-reimbursement for biosimilars
and other high-cost medicines has the potential to afford the
suppliers of such products with a more robust form of “skinny
labelling”. However, the availability of such strategies will
depend upon the approach adopted by the TGA and PBAC in
each individual case. Whether such strategies will be effective
to avoid infringement of second medical use claims is yet to be
tested before the Australian courts.
Author
Andrew Rankine
Principal

18 The 2023 Pharmaceutical Patent Review spruson.com
Pharmaceutical Patent
Term Extension in Australia
Authors: Elizabeth Barrett | Principal
Dan Sieveking | Principal
importance, and in recent years have become of particular interest
in Australian pharmaceutical patent law following a number of
Federal Court decisions clarifying the circumstances in which PTEs
may be available. We provide here a summary of the requirements
and some recent Federal Court decisions in this area.
Under Australian patent law, it is possible to apply for a
PTE of up to 5 years for a standard patent that claims a
pharmaceutical substance, in recognition of the exceptionally
long time and regulatory requirements involved in developing
and commercialising a new pharmaceutical substance.
As set out in section 70 of the Patents Act 1990 (Cth), a patent
i) where the claims of the patent encompass:
a) Pharmaceutical substance(s) per se; or
b) Pharmaceutical substance(s) produced by
recombinant DNA technology; and
ii) where that pharmaceutical substance is included in
goods which have received regulatory approval at least five
years following the effective date of the patent.
The application for PTE must be made in the “prescribed
manner” which includes providing evidence to show that the
goods containing the substance are currently included in the
the application for PTE within six months of the earliest inclusion
in the ARTG of goods containing the pharmaceutical substance,
or grant of the patent, whichever is later.
Eligibility Considerations - Subject Matter
Except for substances produced by a process involving the
use of recombinant DNA technology, an extension of term
is only available in respect of a “pharmaceutical substance
per se” being within the scope of a claim of the patent. The
use of the term “per se” requires the claim to the substance
to be unqualified by process, temporal, or environmental,
components (Boehringer Ingelheim International v
Commissioner of Patents
Patents that claim pharmaceutical substances when produced
by a particular process (product by process claims) will not be
eligible (unless that process involves the use of recombinant
DNA technology). In limited circumstances, a substance could
be new and inventive but can only be defined by reference to
the process in which it was made (for example, compound X
composition is undetermined. In such circumstances, a claim
which defines the substance by reference to such method
steps would be regarded as a claim to the substance per se
(see Zentaris AGPharmacia Italia SpA v
Mayne Pharma Pty Ltd
Additionally, case law has established that pharmaceutical
compositions (formulations) comprising a specified amount
of an active ingredient and other components (excipients),
where the mixture provides a physico-chemical interaction
within the human body, can be eligible for PTE (see iCeutica
Pty LtdSpirit Pharmaceuticals Pty Ltd v
Mundipharma Pty Ltd Mundipharma
case, this extended to a slow release formulation.
Claims which limit the use of a known substance to a
particular environment, for example claims drawn to the
use of pharmaceutical substances when used in a new and
inventive method of treatment, are not considered to be claims
to pharmaceutical substances per se (see Commissioner of
Patents v AbbVie Biotechnology Ltd
In a more recent Federal Court decision, Biogen International
GmbH v Pharmacor Pty Ltd
involved a disputed PTE, Biogen had sought an interlocutory
injunction against Pharmacor. Pharmacor argued that a PTE
granted on the basis of EPC2000 claims in the format “Substance
was not to a pharmaceutical substance per se. The Federal Court
was sympathetic to this position and considered that there was
“a sufficiently strong prospect” that the PTE had been “wrongly
granted”
injunction. This position diverges from the construction of such
claims routinely adopted by the Australian Patent Office (i.e. a
product merely suitable for but not limited to the specified use).
Unfortunately, this issue was not finally determined on account of
settlement of the litigation. However, the decision together with
advice in the Australian Patent Examiner’s Manual suggests that a
claim to “substance X for use ....” may not define a pharmaceutical
substance per se that may support a PTE.
Eligibility Considerations – First Regulatory
Approval Date
Patents Act 1990 (Cth), in order to
be eligible for a PTE the period beginning on the date of the
patent and ending on the first regulatory approval date for the
pharmaceutical substance must be at least 5 years.

spruson.com The 2023 Pharmaceutical Patent Review 19
In two appeal judgements handed down concurrently in March
Commissioner of Patents v Ono Pharmaceutical Co. Ltd
Ono) and Merck Sharp & Dohme Corp. v
Sandoz Pty LtdMSD)), the Full Federal Court
has clarified that a PTE for a patent should be based on the
earliest Australian regulatory approval date of a pharmaceutical
substance which is disclosed and claimed in the patent,
irrespective of whether the substance was developed by the
patentee or a competitor.
In Ono, the claims of the patent encompassed two
pharmaceutical substances, one belonging to the patentee
earlier and requiring an extension of time in order to timely
file the PTE application). In MSD the claims of the patent
encompassed two pharmaceutical substances both developed
registered first and within 5 years of the date of the patent),
registered (later than 5 years from the patent date).
In both decisions, it was held that the goods which were
approved first were those upon which the PTE must be
based. Significantly, in Ono and MSD the Full Federal Court
confirmed that the language of the relevant provisions refers
to the first regulatory approval date in respect of any of the
pharmaceutical substances which may be disclosed and
claimed in the respective patent. Accordingly, it is not open to
the patentee to distinguish between its own goods and those
developed by a third party, nor can a patentee exclude from
consideration or nominate for itself the goods upon which a
PTE is to be based, where more than one product falls within
the scope of its patent. It is also clear from MSD that if the
product the subject of the first regulatory approval is registered
within 5 years of the patent date, no PTE will be available for it
or any subsequent product falling within the patent scope.
Interestingly, Ono’s alternative PTE application filed for AU
confirming the position that it is possible to obtain a PTE based
on a third party’s product.
Practical Implications
Claims defining a pharmaceutical substance “for use” require
careful consideration and may not be eligible to support a PTE
or render any resultant PTE susceptible to challenge.
An application for PTE must be based on the broadest reading
of the claim set as a whole. Where the claims of a patent to be
extended cover more than one active ingredient, they should
also be cross-checked against any goods entered into the
ARTG to determine what registered goods are encompassed by
the claims, including those of unrelated third parties.
It is advisable to consider filing one or more divisional
applications during prosecution so that individual
pharmaceutical substances (that are intended to be ARTG
registered) are quarantined in separate applications.
Proceeding in this way will avoid an earlier registration in
respect of one substance precluding a PTE based on later
registered goods in respect of a different substance.
Where it is not possible to file a divisional application, patentees
may consider pre-emptively filing amendments to exclude
earlier ARTG registered goods if appropriate, in order to
facilitate eligibility and allowance of a PTE.
Author
Dan Sieveking
Principal
Author
Elizabeth Barrett
Principal

20 The 2023 Pharmaceutical Patent Review spruson.com
“Best Method”
Requirement Increasingly
Prominent in Australian
Pharmaceutical Patent
Disputes
Author: Duncan Longstaff | Principal
Australia is one of the few major patent jurisdictions which
has maintained in its patent law a discrete requirement that a
patentee disclose the “best method” known to it of performing
the invention. As outlined in this article, rather than remain in
the background or even fade away, best method challenges
have assumed an increasingly prominent role in Australian
patent disputes and can significantly affect the conduct and
strategy of Federal Court litigation, in particular. The table at
the end of this article summarises the claims, findings and key
issues of selected Australian pharmaceutical patent cases
where best method has proven a pivotal issue. Potential
pharmaceutical patent litigants should be mindful of the
substantive and procedural implications best method issues
can have in the Australian iteration of global disputes.
Best in Australia (but nowhere else?)
Australia inherited its best method requirement from the United
Kingdom, which itself abolished the corresponding requirement
in its own legislation in 1977, coming into line with European
patent law. The United States abolished its (perhaps less
onerous) “best mode” patent revocation ground as part of the
“America Invents” legislative reforms in 2011, although it can still
play some role during examination before the USPTO. Similarly,
Japan has no formal best method requirement, although similar
objections can be raised during examination it is not a ground of
invalidity. South Africa abolished its best method requirement in
2002. New Zealand retains a narrow best method with respect
to methods which would be entitled to protection in their own
right and but (unlike Australia) does not have strict prohibition in
relation to post-acceptance amendments (meaning there is more
scope to “fix” best method issues).
Best method is a question of fact
The best method requirement under Australian law, now
Patents Act 1990 (Cth), is
a factual question focusing essentially on what the patentee (or
its predecessor in title) actually knew at the time the application
complete or divisional application), rather than at the earliest
priority date, and whether any of that known information about
the best method of performing the invention can be adjudged to
have been withheld from the patent specification. The obligation
is merely to disclose that method known by the patent applicant
(not the inventor(s)), including constructively.
Best method of performing “the invention”
The best method requirement pertains to disclosure of “the
invention”. Therefore, the starting point is to identify the
invention, as claimed and described by the specification as a
whole. Features which are ancillary to the invention do not form
part of the “invention” for the purposes of the best method.
Whether a feature is ancillary to the invention depends on
whether that feature is “necessary and important” to carry the
invention into effect. “Best” means what is best in practice
and not in theory. Commercial value or profitability is not of
itself a relevant criterion for determining what is “best”. What is
required is disclosure of the most effective means of carrying
out the invention known to the patentee at the relevant time.
The best method of performing the invention does not have to
be identified as being the best method in the specification.
Best method is similar, but in addition to,
sufficiency
The best method requirement is therefore similar, but in
addition and in important ways different to, the “sufficiency”
requirement to provide clear enough and complete enough
description to perform the invention, and the “support” and
“clarity” requirements to draw claims that are clear and
supported by matter disclosed in the specification. Patents
satisfying the sufficiency, support and clarity requirements are
increasingly be found invalid for failure to disclose the best
method. A tantalising intersection with the law of “sufficiency”
is that a patentee is not obliged to disclose information that is
already known to the skilled addressee by way of the common
general knowledge. It may also be open to the patent applicant
not to disclose relevant information on the basis that it is
available to the skilled addressee by routine experimentation.
This is to be assessed by reference to the importance of the
information in question, the practicality of disclosing it, and the
extent of the burden imposed on the skilled addressee who is
left to rely upon routine experimentation. This difficult argument
was run successfully by the patentee in the GlaxoSmithKline
case summarised in the table below.
spruson.com The 2023 Pharmaceutical Patent Review 21
Basis for best method challenges
Invalidity for failure to disclose the best method is not confined
to scenarios where the patentee knows two (or more) methods
and discloses the inferior method, although that is perhaps
most common and often proven through discovered or other
contemporaneous documents. Invalidity may also arise
where the comparison is between an “umbrella” or general
methodology that is disclosed in the specification and “a
specific method which was sure to provide the benefits of the
invention” which was not disclosed. In either situation, the party
challenging validity needs to identify some basis for asserting
that the disclosure given in the specification is deficient
because it is apparent that the patentee withheld information
material to the best method of performing the invention.
When best method challenges typically arise
Given the fact-dependent nature of best method challenges,
they arise most often in Federal Court of Australia proceedings
(appeals from pre-grant opposition decisions of the Patent
Office or post-grant revocation cases), where discovery,
subpoenas, witness cross-examination and other compulsive
processes are available to parties challenging patent validity
– see, for example, the Servier decision summarised in the
table below. Best method is also increasingly being run as a
ground of opposition in pre-grant inter partes oppositions in the
Patent Office, typically on the basis of detail apparently absent
on the face of the specification or in view of publications or
statements in a declaration by the inventor (eg, as part of the
patent applicant’s evidence on inventive step or entitlement).
Best method objections are rarely raised during examination,
and are discouraged by the Examiner’s Manual published
and used by IP Australia – although the Kineta Inc. decision
summarised in the table below is an example of a patent
application being refused during examination on the basis of a
best method objection.
Case Claim(s) Finding Notable
Les Laboratoires Servier v
Apotex Pty Ltd
FCR 61
(click for judgment)
1. The arginine salt of perindopril
and its hydrates.
2. Pharmaceutical composition
comprising, as active
ingredient, the arginine salt of
perindopril and its hydrates, in
combination with one or more
pharmaceutically acceptable
excipients.
“…in describing only the
general method of classical
salification rather than a
specific method, such as the
which would have provided
the information to the skilled
reader of a method for
obtaining a form of perindopril
arginine which met the
characteristics of the claimed
invention, Servier failed to
describe the best method
known to it of performing the
invention”.
Evidence (of inventors)
demonstrated actual
patentee knowledge
of better methods not
disclosed
Amendment to introduce
best method (pre-
discretionary grounds
Selected Australian Pharmaceutical Patent Cases Involving Best Method Challenges
Impact best method allegations can have on
conduct of patent disputes
Because best method is a question of fact, it can provide fertile
ground for seeking discovery from the patentee. The Court
has adopting an increasingly strict approach to best method
pleadings, which must not be general and speculative in the hope
of finding a best method case after the patentee gives discovery.
The Court may refuse discovery if there is no apparent best
method case without discovery. However, if a sound basis can
be provided for pleading a specific lack of best method case,
discovery orders are often made in Federal Court proceedings
and can prove onerous for patentees, particularly if the patent in
suit was filed many years ago and the invention was developed
during a major R&D programme. Such discovery exercises can
significantly expand the timetable for evidence and pre-trial steps,
as well as the scope and costs of the proceedings overall. Mindful
of this, sometimes the discovery sought with respect to best
method is targeted at specific documents expected to exist, such
as parts of regulatory dossiers – see the successful application
for discovery in the AUPharma decision in the table below. The
potential complications and case expansion arising from best
method discovery and subsequent evidence has also motivated
parties to actively consider whether best method issues might
be considered as “separate questions” before all other issues of
validity and infringement – see the unsuccessful application made
by the patentee in the Novartis decision in the table below.

22 The 2023 Pharmaceutical Patent Review spruson.com
Case Claim(s) Finding Notable
GlaxoSmithKline
Consumer Healthcare
Investments (Ireland)
Partners Pty Limited
(click for judgment)
1. A pharmaceutical composition
comprising
a bilayer tablet having an
immediate release phase of
paracetamol and a sustained
release phase of paracetamol,
the immediate release phase
being in one layer and
comprising from about 10 to
45% by weight of the total
paracetamol; and
the sustained release phase
being in the other layer and
comprising from about 55%
to 90% by weight of the total
paracetamol in admixture with
a matrix forming polymer or a
mixture thereof;
said composition comprising
from 600 to 700mg of
paracetamol per unit dose and
a pharmaceutically acceptable
carrier,
wherein said composition
has an in vitro paracetamol
dissolution profile (as
determined by the USP type
III apparatus, reciprocating
basket, with 250ml of 0.1M
HCl at 37C set at a cycle
speed of 15 strokes/min) with
the following constraints:
• 30 to 48% released
after 15 minutes
• 56 to 75% released
after 60 minutes
•
after 180 minutes.
“…we are not satisfied that
the respondents discharged
their onus of establishing
that the Patent was invalid
on the ground that the
complete specification failed
to specify the particular grade
and viscosity of HPMC or
granulation end points that
might be used to perform the
invention according to the
best method known to the
patent applicant. We agree
with the primary judge that the
best method was disclosed,
albeit at a level of generality
that did not include the
more detailed but inessential
manufacturing and production
information described in
the MAA applicable to the
commercial embodiment.”
Patent survived best
method challenge
because details not
disclosed were inessential
to performance because
they were common
general knowledge /
routine
Selected Australian Pharmaceutical Patent Cases Involving Best Method Challenges
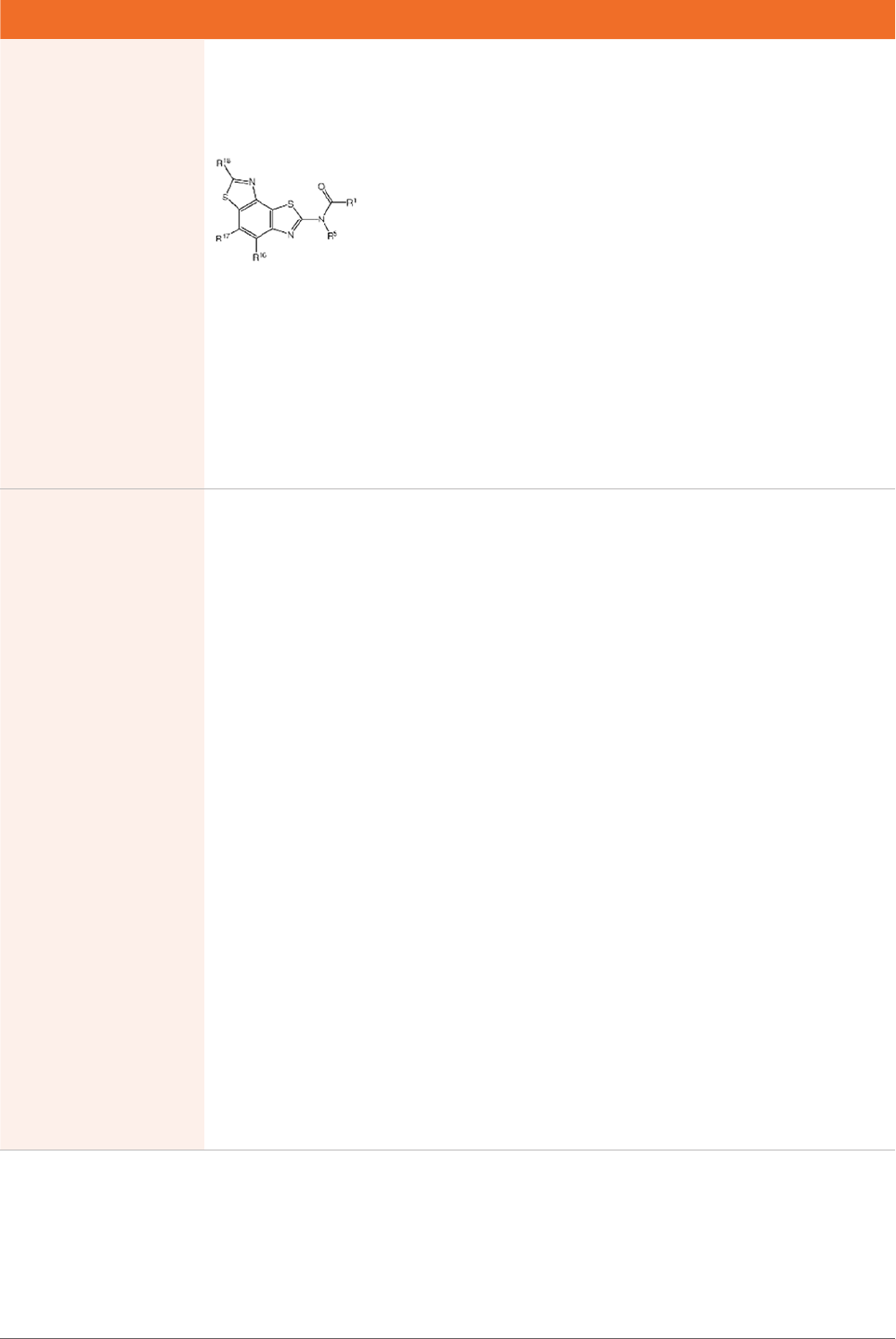
spruson.com The 2023 Pharmaceutical Patent Review 23
Case Claim(s) Finding Notable
Kineta, Inc.
(click for judgment)
Claim 1 as proposed to be
amended is directed to a
compound represented by the
formula:
For the purposes of this decision
it is not necessary to consider the
definition of the variables R1, R5,
R16, R17 and R18. Later claims are
appended to claim 1 and directed
to pharmaceutical compositions
comprising the compound and
methods of treatment comprising
administering the composition.
“The specification does
not set out any method of
preparing the compounds, and
no method is apparent when
the specification is read in the
light of the common general
knowledge. The applicant was
aware that the compounds
could be obtained from a
commercial supplier. I am
satisfied that the specification
does not comply with section
disclose the best method
known to the applicant.”
Patent applicant failed
to disclose the best
method of performing the
invention because it had
failed to disclose that the
only method known to it of
obtaining the compounds
of the invention was
to commission their
synthesis from a particular
supplier
Patent Office decision
upholding objection during
examination
AUPharma Pty Limited v
Mundipharma Pty Limited
(click for judgment)
“Each patent relates to an oral
controlled-release pharmaceutical
composition comprising
oxycodone and naloxone, where
the oxycodone and the naloxone
are present in a ratio within
011 patent), or within the range
the composition releases the
oxycodone and the naloxone”.
The Court orders that:
1. The respondent
produce to the applicant
electronic copies of the
following modules from
the dossier provided to
the Therapeutic Goods
Administration in relation to
each of the respondent’s
a) module 3.2.P.1 titled
“Description and
Composition of the
Drug Product”;
b) section 3.2.P.2.1 titled
“Components of the
Drug Product”;
c) section 3.2.P.2.2 titled
“Drug Product”;
d) section 3.2.P.2.3 titled
“Manufacturing Process
Development”; and
e) module 3.2.P.3.3
titled “Description of
Manufacturing Process and
Process Controls”.
Discovery ordered with
respect to specific
sections of regulatory
dossier where best
method challenge
related to details
of pharmaceutical
formulation, in the
context of a challenge to
a pharmaceutical patent
term extension
Selected Australian Pharmaceutical Patent Cases Involving Best Method Challenges

24 The 2023 Pharmaceutical Patent Review spruson.com
Case Claim(s) Finding Notable
Novartis AG v Pharmacor
FCA 963
(click for judgment)
1. A pharmaceutical composition
comprising:
(i) the AT 1-antagonist
valsartan or a
pharmaceutically acceptable
salt thereof;
and
carboxy-1-oxopropyl)-(4S)-
p-phenylphenylmethyl)-4
amino-2R-methylbutanoic
methyl-pentanoic acid or
pharmaceutically acceptable
salts thereof and a
pharmaceutically acceptable
carrier.
“On balance, I am not
persuaded that the just
resolution of the substantive
question raised by Novartis’s
separate question—namely,
the Act (in its relevant form)
for determining the patent
applicant’s knowledge of the
best method—in accordance
with the overarching purpose,
favours the hearing of that
question separately from and
before any other question in
the proceeding. I am satisfied
that the substantive question
is best determined in the
context of the trial itself.”
Application by patentee,
after giving discovery, for
legal viability of specific
best method challenge to
be heard as a “separate
question” before all other
issues of validity and
infringement refused by
the Court
Author
Duncan Longstaff
Principal
Selected Australian Pharmaceutical Patent Cases Involving Best Method Challenges

spruson.com The 2023 Pharmaceutical Patent Review 25
Pharmaceutical Policy update
Author: Lucy Hartland | Special Counsel
Federal budget
annual expenditure on the Pharmaceutical Benefits Scheme
(PBS) would not increase over the next five years, by contrast
to expenditure on Medical Benefits which are expected to
increase. Such “flat” forecasting for PBS expenditure is not new
and is consistent with previous (pre covid) budget forecasts.
Notwithstanding that expenditure on the PBS has in fact
increased over time, it is clear that efforts to contain the costs
of subsidised medicines will continue.
Another item of note in the current budget is the identification
of a “contingent asset – unquantifiable” being the expectation
of recovery of compensation against pharmaceutical
companies in respect of what proved to be wrongly granted
interlocutory injunction. We have noted key proceedings
involving Sanofi and Otsuka elsewhere in this update. This item
has been noted in past budgets and indicates the government’s
expectation that the compensation to which it claims an
entitlement is significant.
Eighth Community Pharmacy Agreement
of agreements between Minister for Health and Aged Care,
the Pharmacy Guild of Australia and Pharmaceutical Society
of Australia concerning compensation to pharmacists for
dispensing subsidised medicines on the PBS and in connection
with various community pharmacy medication management
programs and services. The CPAs usually last for 5 years but
although the current (seventh) CPA is not due to expire until 30
June 2025, negotiations for the either CPA commenced in mid
2023. On 14 March 2024, the parties announced that they had
reached heads of agreement and that they will work in good
faith to finalise and implement the agreement (to be effective
The New Frontier – Delivering Better Health for all
Australians report
In November 2023, the government delivered its response to
the Standing Committee on Health, Aged Care and Sport report
.
While there are several recommendations and responses of
interest, of note is recommendation 11, which the Government
has accepted. This recommendation is that:
… the Department of Health conduct a comprehensive
consultation process with industry to establish a more
flexible way forward for the repurposing of drugs in
Australia. This should include:
repurposing of drugs for all diseases, not just rare disease.
No detail as to the proposed implementation of this
recommendation is available yet.
While doing no more than “noting” Recommendation 27,
concerning increasing data exclusivity periods to 10 years for
vaccines and orphan drugs, the government stated:
that additional periods of data exclusivity should be made
available for orphan drugs and vaccines, but only where
thorough analysis of the issue and evidence demonstrate
this is necessary to encourage investment in these
important areas. Such analysis would need to consider the
potential impacts on access to medicines and the patent
system, including the flexibilities available in that system,
and the current and ongoing discussions on IP and vaccine
accessibility occurring in international fora
It is therefore possible that this specific issue will be revisited
although the timeframe for that is unknown.

26 The 2023 Pharmaceutical Patent Review spruson.com
Health Technology Assessment Policy and
Methods Review
The long awaited review of Health Technology Assessment policy
has been ongoing during 2023 and is expected to be completed in
April 2024. Affected subsidy/ funding schemes include:
• the Pharmaceutical Benefits Scheme;
• the Medicare Benefits Schedule;
• the National Immunisation Program;
• the Life Saving Drugs Program.
It is hoped that the review will lead to faster and more cost
effective access to new health technologies and that it will help
make Australia a desirable country for first or early launch of
such technologies.
Early notification of generic medicine
applications to the innovator
In 2020, the Therapeutic Goods Administration sought
feedback on a proposed measure for a system of earlier
notification of generic medicine applications to the innovator.
At present, an innovator may be notified of an application
to register a generic medicine via s 26B of the Therapeutic
Goods Act 1989 which requires generic applicants to provide
such notification in certain circumstances. However in many
cases s 26B is not engaged or if it is engaged. The practical
effect is that innovators typically learn of generic medicine
applications upon entry onto the ARTG or through market
intelligence of an impending launch does not result in a
notification to the patentee.
The proposed measure would require first generic sponsors
to notify the patentee when their application is accepted
for evaluation by the TGA, before the TGA commences the
evaluation. In an update in December 2023, the TGA indicated
that feedback on this measure was mixed and the measure
would not be progressed. We note that the 2023 Special 301
Report on Intellectual Property Protection and Enforcement
notes that early notification to patentees remains an area of
concern for the United States. It is therefore possible that further
consideration of notification of generic applications will occur.
Author
Lucy Hartland
Special Counsel
The approval of psychedelics for certain
medical uses
Since 1 July 2023, authorised psychiatrists have been able to
prescribe medicines containing the psychedelic substances
for certain mental health conditions. Under the changes, MDMA
relation to these specific uses, psilocybin and MDMA have
in the Poisons Standard. However, all other uses of psilocybin
Poisons Standard.
As at the date of writing, there were no psilocybin or MDMA
products on the ARTG, however a registered psychiatrist
who has been approved as an authorised prescriber will be
able to access and legally supply an unregistered therapeutic
product containing psilocybin or MDMA to patients under
their care for the specific uses referred to above if all other
clinically appropriate treatment options on the ARTG have
been considered.

spruson.com The 2023 Pharmaceutical Patent Review 27
About the Spruson &
Ferguson Pharmaceutical
Industry Group
At Spruson & Ferguson, our strength lies in our deep expertise in all
aspects of intellectual property law and practice spanning across
the entire IP life cycle. We also recognise that an understanding of
the commercial and regulatory environments in which our clients
operate, is vital to maximise the impact of IP strategy.
Our Pharmaceutical Industry Group includes Principals, lawyers
and attorneys from across our firm working together to meet
the need of our pharmaceutical industry clients. This includes
Andrew Rankine
Principal
Specialisation: litigation
patent and trade mark prosecution experts, and litigators
and commercialisation specialists, allowing us to build a team
with appropriate skill set for any matter. We all share a strong
knowledge of the pharmaceutical industry and ensure that our
advice is always commercially appropriate and relevant.
Our team has many decades of experience in intellectual property,
and represents one of the largest groups of its kind in Australia.
Our patent attorney Principals are PhD qualified, between them
representing a huge breadth of specialist scientific expertise in the
life sciences. Our litigation and commercialisation team are also
highly experienced, our team members having acted in a number
of Australia’s largest pharmaceutical litigation matters.
Annabel Reader
Special Counsel
Specialisation: trade marks
Dr. Catherine Winbanks
Principal
Specialisation: patents
Dr. Charles Tansey
Principal
Specialisation: patents
Dr. Daniel Sieveking
Principal
Specialisation: patents
Dr. Doug Horton
Principal
Specialisation: patents
Duncan Longstaff
Principal
Specialisation: litigation
Dr. Elizabeth Barrett
Principal
Specialisation: patents
Katrina Crooks
Principal, Head of Spruson &
Ferguson Lawyers
Specialisation: litigation
Dr. Michael Christie
Principal
Specialisation: patents
Sylvie Tso
Principal
Specialisation:
commercialisation
28 The 2023 Pharmaceutical Patent Review spruson.com
New Zealand
Pacific Islands
Australia
Singapore
Malaysia
Indonesia
Brunei
Papua New
Guinea
Philippines
Taiwan
Vietnam
Cambodia
China
Macau
SAR
Mongolia
Laos
Myanmar
Thailand
Nepal
Pakistan
India
Sri Lanka
Hong Kong
SAR
10
Offices
25
+
Countries
190
+
IP professionals
480
+
Employees
Jurisdictions with physical offices and/or direct filing
Other jurisdictions serviced
28 The 2023 Pharmaceutical Patent Review spruson.com

30 The 2023 Pharmaceutical Patent Review spruson.com
Our offices
Bangkok
Telephone: +66 2 256 9164
Email: [email protected]
Beijing
Telephone: +86 10 8225 5655
Email: [email protected]
Brisbane
Telephone: +61 7 3011 2200
Email: [email protected]
Hong Kong
Telephone: +852 2161 9999
Email: [email protected]
Jakarta
Telephone: +62 21 252 3853
Email: [email protected]
Kuala Lumpur
Telephone: +60 3 2283 1668
Email: [email protected]
Manila
Telephone: +63 282 467 750
Email: [email protected]
Melbourne
Telephone: +61 3 8637 7131
Email: [email protected]
Singapore
Telephone: +65 6333 7200
Email: [email protected]
Sydney
Telephone: +61 2 9393 0100
Email: [email protected]
© 2024 SPRUSON & FERGUSON. Spruson & Ferguson companies are members of the IPH Ltd group, and part of an ‘ownership group’ for the purposes of the Australian and
New Zealand Code of Conduct for Trans-Tasman Patent and Trade Marks Attorneys 2018 (see https://www.spruson.com/about/ownership-group)
DISCLAIMER
The information contained in this document is provided for general informational and educational purposes only and does not constitute legal or professional advice.
Spruson & Ferguson does not guarantee the accuracy or currency of the information contained in this document, despite making all efforts to ensure it is up-to-date and
free from error at the time of inclusion. The content of this document is not a complete statement of the law on any subject. Professional advice should be sought before
any course of action is pursued. Moreover, transmission of the information in this document is not intended to create, and the receipt does not constitute, an attorney-client
relationship between Spruson & Ferguson and the recipient.
