
Technological University Dublin Technological University Dublin
ARROW@TU Dublin ARROW@TU Dublin
Articles NanoLab
2015-2
Cell viability assessment using the Alamar blue assay: A Cell viability assessment using the Alamar blue assay: A
comparison of 2D and 3D cell culture models comparison of 2D and 3D cell culture models
Franck Bonnier
Technological University Dublin
Mark Keating
Technological University Dublin
Tomasz P. Wrobel
Jagiellonian University Cracow
See next page for additional authors
Follow this and additional works at: https://arrow.tudublin.ie/nanolart
Part of the Other Cell and Developmental Biology Commons
Recommended Citation Recommended Citation
Bonnier, F. et al. (2015) Cell viability assessment using the Alamar blue assay: A comparison of 2D and 3D
cell culture models,
Toxicology in Vitro,
29, pp. 124-131. doi:10.1016/j.tiv.2014.09.014
This Article is brought to you for free and open access by the NanoLab at ARROW@TU Dublin. It has been accepted
for inclusion in Articles by an authorized administrator of ARROW@TU Dublin. For more information, please contact
Funder: SFI

Cell viability assessment using the Alamar blue assay: A comparison of 2D and 3D cell
culture models
F. Bonnier
1*
, M. Keating
1
, T. Wróbel
2,3
,
K. Majzner
2
, M. Baranska
3
, A. Garcia
1
, A. Blanco
4
H.J. Byrne
1
1. Focas Research Institute, Dublin Institute of Technology (DIT), Camden Row,
Dublin 8, Ireland
2. Jagiellonian Univiversity, Fac Chem, PL-30060 Krakow, Poland
3. Jagiellonian Univiversity, JCET, PL-30348 Krakow, Poland
4. UCD Conway Institute, University College Dublin, Belfield, Dublin 4, Ireland
*Corresponding Author: Franck Bonnier,
Focas Research Institute, Dublin Institute of Technology,
Kevin Street, Dublin 8, Ireland.
E mail address: [email protected]
Ph: +353 1 4027917
Fax: +353 1 4027904
Abstract
Comparisons of 2D and 3D cell culture models in literature have indicated differences in
cellular morphology and metabolism, commonly attributed the better representation of in
vivo conditions of the latter cell culture environment. Thus, interest in the use of 3D
collagen gels for in vitro analysis has been growing. Although comparative studies to date
have indicated an enhanced resistance of cells on collagen matrices against different
toxicants, in the present study it is demonstrated that non-adapted protocols can lead to
misinterpretation of results obtained from classical colorometric dye-based cytotoxic
assays. Using the well established Alamar Blue assay, the study demonstrates how the
transfer from 2D substrates to 3D collagen matrices can affect the uptake of the resazurin
itself, affecting the outcome of the assay. Using flow cytometry, it is demonstrated that
the cell viability is unaffected when cells are grown on collagen matrices, thus the
difference seen in the fluorescence is a result of a dilution of the resazurin dye in the
collagen matrix, and an increased uptake rate due to the larger cell surface exposed to the
surrounding environment, facilitating more effective diffusion through the cellular
membrane. The results are supported by a rate equation based simulation, verifying that
differing uptake kinetics can result in apparently different cell viability. Finally, this work
highlights the feasibility to apply classical dye-based assays on collagen based 3D cell
culture models. However, the diffusion and bioavailbility of test substances in 3D
matrices used in in vitro toxicological assays must be considered and adaption of the
protocols is necessary for direct comparison with the traditional 2D models. Moreover,
the observations made based on the resazurin dye can be applied to drugs or nanoparticles
which freely diffuse through the collagen matrices, thus affecting the effective
concentration exposed to the cells.
Keywords: Alamar blue assay, 3-D Cell culture, collagen gels, extracellular matrix, cell
viability, flow cytometry,
1. Introduction
Significant efforts have been devoted towards the development of more realistic in vitro
cell culture models, better mirroring in vivo conditions. For example, it is well accepted
that, in an environment mirroring the in vivo conditions encountered by the cells, the
observations made are more representative of the cancerous cell phenotype compared to
those found on conventionally used 2D cultures (Breslin and O'Driscoll, 2012; Elliott and
Yuan, 2011; Kimlin et al., 2011). Moreover, providing a microenvironment with
adequate adhesion and proliferation has been reported to allow a more accurate
investigation of cellular homeostasis, differentiation and migration (Kim, 2005; Kim et
al., 2004). The use of collagen gels and more complex multi-component systems such as
Matrigel has become increasingly popular, as they provide the cells with a matrix which
more accurately represents the extra cellular matrix (ECM) (Petersen et al., 1992;
Prestwich, 2008; Weaver et al., 1995). The impact of such matrices on the cell phenotype
and metabolism has been already documented in the literature. For example, it has been
shown that antibodies against B1-integrins exhibit different behaviour when tested in 2D
compared to 3D models (Wang et al., 1998); induced doxorubicin-resistance by the extra
cellular matrix in human osteosarcoma and HT1080 cells
has been demonstrated (Fourre
et al., 2008; Harisi et al., 2007); reduced radiation induced toxicity when cells are grown
in a 3D environment has also been reported (Sowa et al., 2010). The use of collagen
matrices as 3-D cell culture matrices has greatly increased in the last few years and
numerous other publications can be found in the literature describing modification of the
cell phenotype, metabolism or composition when grown in a 3D matrix, compared to 2-D
environments (Lupanova et al., 2010; Wu et al., 2009).
Although 3D matrices are increasingly routinely employed for cellular analysis, the
impact of the ECM like microenvironment on the cells is still under investigation. The
degree of modification of the cellular behaviour and metabolism remains unclear and the
comparison between conventional 2D substrates and 3D models is still ongoing.
However, the use of a 3D cell culture environment could lead to difficulties in adapting
established cytological protocols as well as in the interpretation of the results. Although
already intensively used in cancer research, collagen gels have not been adapted to other
fields such as cyto- or nano- toxicology. Moreover, although efforts have been devoted to
explaining the difference of cell behavior when grown on 3D substrates such as collagen
gels, no consideration has been given to the differing cell geometry and more specifically
the cell surface exposed to the microenvironment.
In the present study, the Alamar blue (AB) in vitro cytotoxicity assay has been used as a
model to demonstrate the link between the cell substrate used and the cell geometry,
ultimately also influencing the outcome of the colorimetric assay employed, independent
of the cell viability or proliferation. The results are supported by a rate equation based
simulation, verifying that differing uptake kinetics can result in apparently different cell
viability (Supplementary Information).
2. Materials and Methods
2.1. HeLa Cell line

HeLa cells, immortalized human cervical cells, were obtained from the ATTC (Manassas,
VA, USA). Cells were cultured in DMEM supplemented with 1% L-glutamine (200 mM)
and 10% foetal bovine serum and maintained in a humidified atmosphere containing 5%
CO
2
at 37ºC. HeLa cells were cultured until they reached approximately 80% confluency
before preparing the plates for the cytotoxicity assay. Cells were seeded at a density of 2
x 10
4
cells per well in 6-well-plates (Nunc Lab-Tek
®
). Half the plates were prepared with
collagen gels at a concentration of 2.5 mg/mL, and the other half were directly grown on
the plastic base of the plate as controls. The experiments were performed in triplicate and
readings of the cell viability using the AB assay were performed after 24h. Moreover, it
is important to point out that the whole experiment (in triplicate) has been repeated twice
within three weeks interval in order to confirm the reproducibility of the observations
made.
2.2. Collagen gels
Solutions of collagen I from rat tail tendons (Gibco) were used for preparation of the
collagen gels. The 5mg/mL solution was mixed with sterile 10X phosphate buffered
saline (PBS), sterile distilled water (dH
2
O) and 1M NaOH. The appropriate relative
quantities of these components is determined by the final concentration (2.5 mg/mL) and
volume needed. All the steps were carried out on ice to slow the gelation process. After
mixing, 500 μL of the solution were either placed in 6-wells-plates for the AB
cytotoxicity assay or 1 mL of the solution was placed in a 25 flask for the flow
cytometric analysis, before incubation at 37 °C degrees in a 95% humidity incubator until
a solid gel was formed (about 30 minutes).
2.3. Alamar blue assay
The Alamar Blue® assay is designed to quantitatively measure the proliferation of human
and animal cell lines, bacteria and fungi (Kuda et al., 2003; Mosmann, 1983; O'Brien et
al., 2000; Pettit et al., 2005; S.Al-Nasiry et al., 2007). Over the past 50 years, the AB
assay has been widely used in studies of cell viability and cytotoxicity for biological and
environmental applications (Rampersad, 2012; Vega-Avila and Pugsley, 2011; White et
al., 1996). The bioassay can also be used to establish the relative cytotoxicity of agents
within various chemical classes (Bopp and Lettieri, 2008; Borra et al., 2009; Mikus and
Steverding, 2000; Miret et al., 2006). Using the REDOX indicator resazurin (oxidised
form), it is possible to spectrophotometrically measure the cellular proliferation.
Resazurin is blue and non-fluorescent, whereas resorufin (reduced form) is red and highly
fluorescent. Thus, measuring the changes in the fluorescence of the dye in the
intracellular environment, modifications in the number of metabolic active cells can be
detected. Tetrazolium salts can deliver similar information regarding cell growth, but
present incompatibility problems, the most limiting being the high toxicity of the DMSO
or HCl/isopropanol required for reading the results (Mosmann, 1983). Thus, the AB
assay is generally preferred for kinetic studies. The oxidation-reduction potential of
resazurin is +380 mV at pH 7.0, 25 °C, which means it can be reduced by NADPH (Eo =
320 mV), FADH (Eo = 220 mV), FMNH (Eo = 210 mV), NADH (Eo = 320 mV), as well
as cytochromes (Eo = 290 mV to +80 mV), all part of the cellular respiration metabolic
reactions. However, other enzymes such as the diaphorases (dihydrolipoamine
dehydrogenase (Matsumoto et al., 1990)), NAD(P)H:quinoneoxidoreductase (Belinsky
and Jaiswal, 1993) and flavin reductase (Chikuba et al., 1994) located in the cytoplasm
and the mitochondria can also reduce Resazurin. Therefore, AB reduction is the result of
multiple metabolic reactions and does not necessarily specifically indicate a
mitochondrial dysfunction, but remains a suitable indicator of the cellular health and
viability (Ahmed et al., 1994).
The AB assay was carried out according to manufacturer’s instructions. Briefly, control
medium was removed; the cells were rinsed with PBS and 2 mL of an AB solution (5%
[v/v] solution of AB dye) prepared in fresh medium (without FBS or supplements) were
added to each well. Following 3 hours incubation, AB fluorescence was quantified at the
respective excitation and emission wavelength of 540 and 595nm using a Tecan Genios
microplate reader. The results were averaged over 3 different independent experiments
(n=3, each conducted with one week interval) with 3 replicates per experiment (3 x 6 well
plates), each replicate being prepared from different T75 flasks in order to take into
account the biological variability. Finally, for each plate the reading was also done in
triplicate (values obtained from 3 different wells averaged) in order include the technical
variability due to the efficiency of AB assay, sensitivity of the plate reader or simply
related to the sample preparation. For each experiment, wells containing only the AB
solution without cells were also prepared and incubated for 3h. The fluorescence
measured in those was used as a background and subtracted. However, although this
protocol is routinely used in the literature, some variations in the AB solution
concentration and exposure time have been made purposely throughout this study. These
modifications will be clearly highlighted and discussed at the appropriate points in the
manuscript, for clarity. Again, each experiment was conducted in triplicates (3 x 6 well
plates) which have been prepared from different flasks in order to have independent
replicate based on different cell populations.
2.4 Flow cytometry assays
2.4.1. Apoptosis assay
This study was performed using a Partec CyFlow® Space Flow Cytometer (Partec UK
Limited, Germany). The default Partec FloMax® flow cytometry software has been used
for the analysis of the samples, but the Beckman Coulter Summit software and the
FCSExpress Research Edition have been used for the reanalysis of the samples. The QC
control of the instrument was performed using Spherotech 6 and 8 peak beads. HeLa cells
(1x10
6
cells//flask) were seeded in T-25 flasks and incubated in a 5% CO
2
incubator at
37°C for 24 hours. These conditions were selected to avoid cells becoming over
confluent, which might lead to misinterpretation of the apoptosis assay due to cellular
death caused by the stress of the culture conditions and medium depletion causing cell
starvation. The experiments have been conducted in triplicate. However, in order to keep
replicates as independent from each other as possible, 3 T75 flasks were initially seeded
and from each one of them two T25 were prepared for flow cytometry, with only one
containing a collagen gel as described above. Following incubation, the cells were
washed twice with pre-warmed PBS and were collected by trypsinization, after which the
trypsin was removed by centrifugation. The cells were then washed twice with pre-
warmed PBS and stained with the YOPRO1/Propidium iodide (PI) dyes (Life
Technologies), whereby 1 μL of YOPRO1 dye (100 µm) and 1 µL of PI (1mg/mL) were
used to stain 1x10
6
cell/ml. After staining of a cell population, apoptotic cells show a
green fluorescence, whereas necrotic cells show green and red fluorescence. After
incubation on ice for 30 min, the cells were analysed by flow cytometry within 30
minutes, using 488 nm excitation and reading the fluorescence of YoPro with a IBP
527/30G filter and PI with a IBP 682 B filter. Unstained and single control samples have
been used in order to set up the instrument and gates in order to visualize 3 groups: live
cells, apoptotic cells and necrotic cells. Samples with over-confluent cell populations
leading to medium depletion which is ultimately associated with a high cellular death
have been used as biological controls. 15,000 single cells per sample have been analysed.
3. Results
3.1. Cell viability monitoring using the AB assay
Conventional in vitro cytotoxicity assays such as AB, Neutral Red and MTT (3-(4,5-
Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) have been developed for rapid
screening of cellular proliferation, viability, metabolic activity, lysosomal and
mitochondrial activity (Davoren et al., 2007; Herzog et al., 2007; Mukherjee et al., 2010;
Naha et al., 2010). AB is widely used for cytotoxicity assay and has been selected for the
low induced cytotoxicity from the redox indicator giving more freedom in the
concentration of reazurin used.

Figure 1: Comparison of Alamar blue assay results obtained after compensation of the AB solution
concentration for cells grown on collagen gels (blue) and plastic (red) for 24hrs using both 5% and 6.7%
AB solutions. The experiments have been repeated twice in triplicates and the error bars indicate the
overall variability observed
The viabilities of the HeLa cells grown on a 2D plastic surface and a 3D collagen matrix
have been evaluated using the AB assay according to the standard protocol described in
Section 2.3. Figure 1 displays the AB fluorescence measured after 24h for cells cultured
on both plastic substrates and collagen gels. The Y axis is the fluorescence intensity after
subtraction of background signal of the AB with no cells but incubated for 3 hr.
After 24h incubation, the cells grown on collagen gels exhibit significantly higher
(~21%) fluorescence intensity than those grown on the 2-D substrate (p<0.001). The
statistical variations represented by the error bars show small variance in the fluorescence
read at 24h. The difference in the fluorescence measured indicates a higher degree of
reduction of the AB dye by the cells grown on collagen gels. In absence of exogenous
agents with toxic properties for the cells, the difference in the readings indicates an
increased viability compared to the cells seeded directly on plastic. Therefore, in order to
validate this observation, further approaches have been used.
3.2 Flow cytometry
As collagen gels are purported to better represent in vivo conditions, a possible cause for
the strong increase of the cell viability obtained using the AB assay is a better
biocompatibility of the cells with the 3D matrix. In order to validate the observations
made using the AB assay, additional assays were needed to give more information
regarding the cellular behaviour when grown on plastic compared to the collagen gels.
Flow cytometry is a powerful tool for the analysis of large cell population based on
fluorescent dyes. Moreover, the main advantage for this study was the possibility to label
the cells while in suspension rather than still attached and thus the labeling process
should be independent of the growth substrate.
For the purpose of this work, the apoptosis assay has been selected to visualize the
amount of live / dead cells in the suspension to get an indication of the possible higher
toxicity/better biocompatibility of the substrates.

Figure 2: Flow cytometry apoptosis assay using the Yo-pro/PI dyes. A: Debris have been excluded by
gating them out based on the scatted characteristics (FSC vs SSC), gate 1, B: Aggregated events have been
excluded with gate 2 in FSC-Area vs FSC-Height; C and D: Cells characterized as alive respectively from
plastic (2C) and collagen gels substrates (2D). The experiments have been repeated in triplicate from 3
different T25 flasks.
The results of the apoptosis assay performed after 24h incubation are presented in figure
2. The plot presents the fluorescence measured for both the YOPRO1 (figure 2C and D -
X axis) and the PI (figure 2C and D - Y axis) dyes. The distribution of the data indicates
the proportion of live versus dead cells present in the suspensions harvested from the
collagen gels (figure 2C) and plastic (figure 2D). Apoptotic and necrotic cells incorporate
different levels of the two fluorescent dyes, as well as scatter changes as can be observed
in the scatter bivariate histogram. In the present study, and for comparison with the AB
assay, only the viable cells are of interest. As shown in figure 2, in the case of the cells
grown both on collagen and plastic, the cluster is mainly located in the bottom left area
(with no uptake of any of the dyes), indicating a high proportion of live cells. After
performing the experiments in triplicate from 3 different sets of T25 flasks, the
percentage of live cells has been evaluated to be 84.6% (+/- 5.2%) for those harvested
from the collagen gels whereas for the plastic substrate it was found to be 87% (+/-
2.9%). Therefore, neither of the substrates used has a statistically significant toxicity that
could possibly affect the outcome of the AB assay after 24h incubation, and notably,
according to the standard deviation calculate from the triplicates, there is no significant
difference between the cell populations grown on 2D substrates and 3D matrices.
3.3 Effect of the collagen gels on the AB solution concentration
The observations made using flow cytometry clearly demonstrate that the apparent
difference in the cell viability indicated by the AB assay is not linked to differences in
cell viability. Thus, the question remains as to how to understand and interpret the origin
of the differences in the fluorescence readings of the AB assay.
The collagen matrices are prepared from a solution of collagen initially at 5 mg/mL
which is diluted for the needs of the experiments to 2.5 mg/mL. All the reagents used are
water based and, after gelation and formation of the collagen fibers, the matrices are
similar to wet sponges. Thus, the first concern is about any reduction of the effective
concentration of the AB solution, as presented to the cells. Wells free of cells and only
containing the collagen gels covered with 2 mL of AB solution (5%) were prepared and
incubated for 1h, 2h and 3h. After each time point, the AB was pipetted out of the wells
and the absorbance at 570 nm was measured. The results are expressed as a percentage of
the absorbance obtained from the AB solution placed in the well without any collagen.
After 1h, the absorbance is equal to 76% of the control, after 2h 74% and after 3h 76%.
This demonstrates how quickly the dye from the AB solution can diffuse through the
collagen gels and therefore how the concentration of the AB solution used can be affected
by dilution of the dye solution by the water contained in the matrix. As the collagen gel is
mostly water, it is important to take into account the volume of collagen used to form the
matrix into the calculation of the solution used for the assay.
The effect of the AB solution concentration on the assay can be easily visualised. Based
on the absorbance measurements, it can be estimated that the effective concentration of
the nominally 5% AB solution is reduced by ~25% in the collagen gel, resulting in the
cells being exposed to only 3.75% AB solution. To compensate the dilution of the dyes
into the collagen gels, the assays have been run in parallel using 6.7% AB solutions.
Figure 1 presents the results obtained after 24h for both AB solutions tested on cells
grown directly on plastic and collagen gels. According to the observations made on the
dilution of the dyes in the collagen gels, the results obtained from the cells grown on
plastic with a 5% AB solution should be compared to those obtained from the cells grown
on collagen gels exposed to a solution at 6.7%. Under these conditions, the difference in
the cell viability found is even greater, with a fluorescence intensity 49% more intense
after 24h incubation.
3.4 Understanding the Alamar blue assay

Figure 3: Alamar blue performed on cells grown on collagen gels with high concentration of AB solution
ranging from 25% to 50%. Cells have been exposed to the AB solution for 3h (top) and 5h (bottom). The
error bars indicate the variability observed over 3 different replicates.
The cell viability as measured using flow cytometry highlighted that there is no increased
cell death on the plastic substrates compared to the collagen gels, which rules out
potential toxicity due to the environment of the cells. Thus, two hypotheses could explain
the difference in the fluorescence measured using the AB assay. Firstly, an increased
metabolic activity of the cells when grown on the collagen gels leading to an increased
conversion rate of the resazurin to its fluorescent form. Secondly and more simply,
different rates of uptake of the AB dye by the cells in the two culture environments. The
organization of the collagen matrix can be compared to a network formed by the
polymerized fibers. In this work, the cells have been seeded onto the collagen gels, but,
as discussed above, the dyes from the AB solution freely diffuse through the gels. Uptake
of the dye by the cells occurs by diffusion through the cytoplasmic membrane. Whereas
the cells seeded on plastic have only one side exposed to the surrounding medium, when
seeded on the collagen matrix, the underside of the cells also have direct contact with the
AB solution and thus present a significantly larger surface (membrane) area through
which the dye can diffuse. A higher uptake rate of the AB solution by the cells would
result in a higher fluorescence reading, indicative of a higher metabolic activity or larger
cell population.
Ideally, the measurement of the actual amount of AB dye present in the cells would be
the best approach to investigate differing uptake rates by the cells. However, the dye
diffuses passively through the membrane of the cells and is continuously reduced in the
cytosol. A measurement of the absorbance would register the unreacted, resazurin
(oxidized) form of the dye, whereas fluorescence registers the reacted, resorufin
(reduced) form of the dye.
In order to estimate the effects of different uptake rates of the dye by the two cell culture
models, the protocol for the AB assay has been modified to operate in the regime of
saturated uptake, in which different uptake rates do not affect the quantity of dye uptaken.
One of the advantages of the AB assay is the low cytotoxicity of the dye at low
concentrations which allows the kinetics of the cellular viability over a prolonged time to
be monitored. However, for the purpose of this study, the concentration of the AB
solution was increased up to 50% in order to visualize a point of saturated dye
uptake/conversion, as indicated by the fluorescence intensity reaching a maximum.
Figure 3A presents the results obtained for cells grown on collagen exposed to 25%,
30%, 35%, 40%, 45% and 50% AB solution for 3h. Although, for concentrations up to
35%, the fluorescence slightly increases, it appears that higher concentrations do not
induce a statistically significant increase of the fluorescence. This indicates either
saturation of the dye uptake or of the reduction rate of the resazurin into resorufin.
For varying AB concentrations, measurement of the fluorescence was made every 45
minutes for 7 different time points. After 5h exposure to the AB solution (figure 3B), the
fluorescence still exhibits a maximum intensity at 35% AB solution, but for higher
concentrations, a slight decrease can be seen, likely related to the toxicity of the AB
solution at high concentrations. Similar observations can be made for the cells grown on
plastic substrates (data not shown). A 35% AB concentration was therefore selected in
order to avoid interferences from the cytotoxicity of the solution on the cells which could
make the interpretation of the results more difficult. Figure 4 shows the fluorescence
measured for incubation periods from 3h to 7h15 min (7 time points). Although a small
difference can be seen, with a slightly higher fluorescence for the cells grown on
collagen, it is not comparable to the 21 % difference observed for the standard assay
protocol (3hr, 5% AB). For more precise visualisation, figure 5 displays the difference in
the fluorescence intensities, but also this difference expressed as a percentage of the total
fluorescence measured. The fluorescence is between 1% and 5% higher for the cells
cultured on collagen gels, which is considerably less than the 21% difference found when
working with 5% AB solution. This highlights that, using a higher concentration, the
system has reached saturation. The fact that the fluorescence exhibits similar intensities
also indicates that the cells have reached their maximum reduction rate, which,
importantly, is the same for cells grown on both collagen and plastic. Therefore, the
metabolic activity of the cells as determined by the AB assay is the same for both cell
cultures. This is confirmed in figure 4 as the gap between the two curves remains
constant over time, indicating identical reduction rates of the resazurin into resorufin in
the two cell culture models. If the metabolic activity was higher for cells grown on
collagen, the difference would increase over time. However, the fluorescence difference
between the two substrates remains similar with a maximum difference of about 5% after
4h 15 min. These observations demonstrate that the metabolic activity of the cells in the
two cultures is comparable and therefore the difference in the fluorescence observed
under the standard assay conditions is not related to higher cell viability or proliferation
but to a higher uptake of the dye by the cells. The larger surface area exposed to the
medium from the cells grown on 3D collagen gels increases the passive diffusion into the
cytosol inducing the slightly higher reading obtained which reflects the delay between the
2 models until reaching the saturation point.
The impact of the differences in the effective cell surface areas in 2D vs 3D cell culture
models can be further illustrated using a rate equation model as described in the
Supplementary Information. The model clearly demonstrates how the apparent
differences in cell viability as indicated by the AB assay can be affected by differing
uptake rates of the dye, as a result of different effective cell surface areas (see
supplementary materials). Notably, such a numerical approach could potentially be
employed as a guide to adapting 2D protocols to 3D cultures.

Figure 4: Kinetic of the fluorescence obtained using an AB solution at 35% for cells grown on plastic
(blue) and collagen gels (red) from 3h exposure up to 7h15mins. Reading s have been performed every 45
mins. The values presented are mean fluorescence calculated from experiments conducted in triplicate.
Figure 5: Representation of the difference of the fluorescence obtained for cell grown on plastic and
collagen gels when performing the AB assay using a 35% solution for each time point. The histogram
represents the difference in the fluorescence intensity while the curve is the difference expressed in
percentage. The results presented have been obtained from experiments conducted in triplicate and
averaged.
4. Discussion
According to the observations made throughout this study, there is no evidence that the
transfer from the 2D plastic to the 3D collagen gels has any influence on the cell health
and viability. Clearly, however, the effects of diffusion of the active dyes through the
matrices and their subsequent bioavailability to the cells can lead to misinterpretation of
the results obtained. The concern addressed in the present study relates to the relevancy
of the change of cell behavior, proliferation or resistance against active agents
documented in the literature. The AB assay has been used as a model to highlight the
importance of the cell culture model geometry on the outcome of cytotoxic investigation.
While the cell surface exposed to the surrounding environment will have an impact of the
uptake rates, it is essential to take into consideration the added volume added to the cell
environment associated with the use of 3D protein based substrates. Importantly, the
observations made regarding the resazurin can be applied to other models. Thus, both the
dilution of the solution by the gel and the different uptake routes will also apply to all
solutions/dispersions in vitro such as toxicants, chemotherapeutic agents and
nanoparticles. The 3D matrix acts as a sponge which results in a dilution factor of the
medium and thus of any active agent, drug or nanoparticle in suspension. However, this
effect is often neglected in in vitro studies investigating the cytotoxicity of drugs or
toxicants on cells (Godugu et al., 2013; Lee et al., 2008; Millerot-Serrurot et al., 2010).
Although the collagen gels may appear to have a significant impact on the cell behavior
and metabolism, when comparing IC
50
‘s or EC
50
’s calculated from 2D and 3D models,
neglect of the dilution effect in 3D matrices makes the observations questionable.
Apparent systematic variations in viability can be a result of a reduced effective
concentration of the agent or assay used in 3D matrices compared to 2D cultures,
resulting in a dilution factor which could account for the difference in the IC
50
calculated(Godugu et al., 2013; Lee et al., 2008; Millerot-Serrurot et al., 2010) and/or
increased cell resistance to toxic/chemotherapeutic agents
45
.
Conclusion
The use of 3D collagen gels as growth substrates has been reported to affect the cell
phenotype and behavior, exhibited as different degrees of resistance to toxicants or
modified levels of metabolic activity. In the literature, many studies aim to understand
and explain how the use of 3D matrices can induce considerable changes in the results
obtained when testing toxicants, chemotherapeutic agents or nanoparticles compared to
the well known 2D surfaces. Although the interaction of the cells with their surrounding
environment can trigger different signaling pathways in the cells, modifying their
behavior, the present study demonstrates that adaption or comparison of the results
obtained from 2D models with 3D matrix cell culture systems requires consideration of
the geometry and morphology of the cell/substrate interface. Therefore, the diffusion and
bioavailability of test substances in 3D matrices used in in vitro toxicological assays must
be considered and adaption of the protocols is necessary for direct comparison with the
traditional 2D models. The example of the well established AB cytotoxicity assay
highlights that, what can be first interpreted as an increase of the cell viability, is in fact a
result of a reduction of the effective concentration of the assay by dilution in the collagen
matrix, and a difference in the cell area exposed to the surrounding environment of the
cells, resulting in a higher rate of dye uptake by the cells. Thus, in order to clearly
understand how the 3D cell culture model affects the cell resistance and survival, the
protocols used for 2D models need to be carefully improved and adapted to allow direct
comparison of the results obtained.
Acknowledgement
This research was supported by the National Biophotonics and Imaging Platform (NBIP)
Ireland, Higher Education Authority PRTLI (Programme for Research in Third Level
Institutions) Cycle 4, co-funded by the Irish Government and the European Union
Structural Fund and by Science Foundation Ireland Principle Investigator Award
11/PI/1108.A. Blanco would like to acknowledge the ISAC Scholars Program.
References
Ahmed, S.A., Gogal, R.M., Jr., Walsh, J.E., 1994. A new rapid and simple non-
radioactive assay to monitor and determine the proliferation of lymphocytes: an
alternative to [3H]thymidine incorporation assay. J Immunol Methods 170, 211-224.
Belinsky, M., Jaiswal, A.K., 1993. NAD(P)H:quinone oxidoreductase1 (DT-diaphorase)
expression in normal and tumor tissues. Cancer Metastasis Rev 12, 103-117.
Black, J.W., Leff, P., 1983. Operational models of pharmacological agonism. Proc R Soc
Lond B Biol Sci 220, 141-162.
Bopp, S.K., Lettieri, T., 2008. Comparison of four different colorimetric and fluorometric
cytotoxicity assays in a zebrafish liver cell line. BMC Pharmacol 8, 8.
Borra, R.C., Lotufo, M.A., Gagioti, S.M., Barros Fde, M., Andrade, P.M., 2009. A simple
method to measure cell viability in proliferation and cytotoxicity assays. Braz Oral Res
23, 255-262.
Breslin, S., O'Driscoll, L., 2012. Three-dimensional cell culture: the missing link in drug
discovery. Drug Discov Today.
Chikuba, K., Yubisui, T., Shirabe, K., Takeshita, M., 1994. Cloning and nucleotide
sequence of a cDNA of the human erythrocyte NADPH-flavin reductase. Biochem
Biophys Res Commun 198, 1170-1176.
Davoren, M., Herzog, E., Casey, A., Cottineau, B., Chambers, G., Byrne, H.J., Lyng,
F.M., 2007. In vitro toxicity evaluation of single walled carbon nanotubes on human
A549 lung cells. Toxicol In Vitro 21, 438-448.
Elliott, N.T., Yuan, F., 2011. A review of three-dimensional in vitro tissue models for
drug discovery and transport studies. J Pharm Sci 100, 59-74.
Fourre, N., Millerot-Serrurot, E., Garnotel, R., Zahm, J.M., Bonnet, N., Millot, J.M.,
Jeannesson, P., 2008. Extracellular matrix proteins protect human HT1080 cells against
the antimigratory effect of doxorubicin. Cancer Sci 99, 1699-1705.
Godugu, C., Patel, A.R., Desai, U., Andey, T., Sams, A., Singh, M., 2013. AlgiMatrix
based 3D cell culture system as an in-vitro tumor model for anticancer studies. PLoS One
8, e53708.
Harisi, R., Dudas, J., Nagy-Olah, J., Timar, F., Szendroi, M., Jeney, A., 2007.
Extracellular matrix induces doxorubicin-resistance in human osteosarcoma cells by
suppression of p53 function. Cancer Biol Ther 6, 1240-1246.
Herzog, E., Casey, A., Lyng, F.M., Chambers, G., Byrne, H.J., Davoren, M., 2007. A
new approach to the toxicity testing of carbon-based nanomaterials--the clonogenic
assay. Toxicol Lett 174, 49-60.
Kim, J.B., 2005. Three-dimensional tissue culture models in cancer biology. Semin
Cancer Biol 15, 365-377.
Kim, J.B., Stein, R., O'Hare, M.J., 2004. Three-dimensional in vitro tissue culture models
of breast cancer-- a review. Breast Cancer Res Treat 85, 281-291.
Kimlin, L.C., Casagrande, G., Virador, V.M., 2011. In vitro three-dimensional (3D)
models in cancer research: An update. Mol Carcinog.
Kuda, T., Yano, T., , 2003. Colorimetric alamarBlue assay as a bacterial concentration
and spoilage index of marine foods
Food Control 14, pp. 455-461(457).
Lee, M.Y., Kumar, R.A., Sukumaran, S.M., Hogg, M.G., Clark, D.S., Dordick, J.S.,
2008. Three-dimensional cellular microarray for high-throughput toxicology assays. Proc
Natl Acad Sci U S A 105, 59-63.
Lupanova, T., Stefanova, N., Petkova, D., Staneva, G., Jordanova, A., Koumanov, K.,
Pankov, R., Momchilova, A., 2010. Alterations in the content and physiological role of
sphingomyelin in plasma membranes of cells cultured in three-dimensional matrix. Mol
Cell Biochem 340, 215-222.
Matsumoto, K., Yamada, Y., Takahashi, M., Todoroki, T., Mizoguchi, K., Misaki, H.,
Yuki, H., 1990. Fluorometric determination of carnitine in serum with immobilized
carnitine dehydrogenase and diaphorase. Clin Chem 36, 2072-2076.
Mikus, J., Steverding, D., 2000. A simple colorimetric method to screen drug cytotoxicity
against Leishmania using the dye Alamar Blue. Parasitol Int 48, 265-269.
Millerot-Serrurot, E., Guilbert, M., Fourre, N., Witkowski, W., Said, G., Van Gulick, L.,
Terryn, C., Zahm, J.M., Garnotel, R., Jeannesson, P., 2010. 3D collagen type I matrix
inhibits the antimigratory effect of doxorubicin. Cancer Cell Int 10, 26.
Miret, S., De Groene, E.M., Klaffke, W., 2006. Comparison of in vitro assays of cellular
toxicity in the human hepatic cell line HepG2. J Biomol Screen 11, 184-193.
Mosmann, T., 1983. Rapid colorimetric assay for cellular growth and survival:
application to proliferation and cytotoxicity assays. J Immunol Methods 65, 55-63.
Mukherjee, S.P., Davoren, M., Byrne, H.J., 2010. In vitro mammalian cytotoxicological
study of PAMAM dendrimers - towards quantitative structure activity relationships.
Toxicol In Vitro 24, 169-177.
Naha, P.C., Bhattacharya, K., Tenuta, T., Dawson, K.A., Lynch, I., Gracia, A., Lyng,
F.M., Byrne, H.J., 2010. Intracellular localisation, geno- and cytotoxic response of
polyN-isopropylacrylamide (PNIPAM) nanoparticles to human keratinocyte (HaCaT)
and colon cells (SW 480). Toxicol Lett 198, 134-143.
O'Brien, J., Wilson, I., Orton, T., Pognan, F., 2000. Investigation of the Alamar Blue
(resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J
Biochem 267, 5421-5426.
Petersen, O.W., Ronnov-Jessen, L., Howlett, A.R., Bissell, M.J., 1992. Interaction with
basement membrane serves to rapidly distinguish growth and differentiation pattern of
normal and malignant human breast epithelial cells. Proc Natl Acad Sci U S A 89, 9064-
9068.
Pettit, R.K., Weber, C.A., Kean, M.J., Hoffmann, H., Pettit, G.R., Tan, R., Franks, K.S.,
Horton, M.L., 2005. Microplate Alamar blue assay for Staphylococcus epidermidis
biofilm susceptibility testing. Antimicrob Agents Chemother 49, 2612-2617.
Prestwich, G.D., 2008. Evaluating drug efficacy and toxicology in three dimensions:
using synthetic extracellular matrices in drug discovery. Acc Chem Res 41, 139-148.
Rampersad, S.N., 2012. Multiple applications of Alamar Blue as an indicator of
metabolic function and cellular health in cell viability bioassays. Sensors (Basel) 12,
12347-12360.
S.Al-Nasiry, N.Geusens, M.Hanssens, C.Luyten, R.Pijnenborg, 2007. The use of Alamar
Blue assay for quantitative analysis of viability, migration and invasion of
choriocarcinoma cells. Human Reproduction 22, 1304-1309.
Sowa, M.B., Chrisler, W.B., Zens, K.D., Ashjian, E.J., Opresko, L.K., 2010
Three-dimensional culture conditions lead to decreased radiation induced cytotoxicity in
human mammary epithelial cells. Mutat Res 687, 78-83.
Vega-Avila, E., Pugsley, M.K., 2011. An overview of colorimetric assay methods used to
assess survival or proliferation of mammalian cells. Proc West Pharmacol Soc 54, 10-14.
Wang, F., Weaver, V.M., Petersen, O.W., Larabell, C.A., Dedhar, S., Briand, P., Lupu,
R., Bissell, M.J., 1998. Reciprocal interactions between beta1-integrin and epidermal
growth factor receptor in three-dimensional basement membrane breast cultures: a
different perspective in epithelial biology. Proc Natl Acad Sci U S A 95, 14821-14826.
Weaver, V.M., Howlett, A.R., Langton-Webster, B., Petersen, O.W., Bissell, M.J., 1995.
The development of a functionally relevant cell culture model of progressive human
breast cancer. Semin Cancer Biol 6, 175-184.
White, M.J., DiCaprio, M.J., Greenberg, D.A., 1996. Assessment of neuronal viability
with Alamar blue in cortical and granule cell cultures. J Neurosci Methods 70, 195-200.
Wu, Y.M., Tang, J., Zhao, P., Chen, Z.N., Jiang, J.L., 2009. Morphological changes and
molecular expressions of hepatocellular carcinoma cells in three-dimensional culture
model. Exp Mol Pathol 87, 133-140.

Cell viability assessment using the Alamar blue assay: A comparison of 2D and 3D cell
culture models
F. Bonnier
1
, M. Keating
1
, T. Wróbel
2,3
,
K. Majzner
2
, M. Baranska
3
, A. Garcia
1
, A Blanco
4
H.J. Byrne
1
1. Focas Research Institute, Dublin Institute of Technology (DIT), Camden Row,
Dublin 8, Ireland
2. Jagiellonian Univ, Fac Chem, PL-30060 Krakow, Poland
3. Jagiellonian Univ, JCET, PL-30348 Krakow, Poland
4. UCD Conway Institute, Unversity College Dublin, Belfield, Dublin 4, Ireland
Supplementary Information
S.1 Mathematical model describing the relation between uptake rate and cell surface
area
The system can be simply modeled using a rate equation approach, by which the
differences between low dose and high dose responses, as well as the impact of AB
solution dilution by the collagen matrix and the greater uptake rate of cells in 3D gels
compared to 2D substrates may be more easily visualized. The diffusive uptake and
release of the AB dye by the cell is described by
dN
int
dt
= k
12
.A.B.D – k
21
.A.N
int
(t) Equation 1
where N
int
is the amount of dye internalized by the cells, k
12
is the rate of internalization,
D is the % dose of the dye, and k
21
is the rate of reverse diffusion from the cells. A is a
factor which normalizes for the effective exposure of the cells, depending on the

substrate, and B normalizes for the reduction of the effective concentration of the dye
solution in the 3D gels.
The saturated uptake of the dye is simulated by introducing a concentration of receptors
(Black and Leff, 1983), N
recp
, which are occupied according to
dN
recp
dt
= k
A
.N
recp
(t).N
int
(t) Equation 2
where k
A
is a receptor/dye binding rate. The fluorescence reading, I
AB
, from the AB can
then be represented by
dI
AB
dt
= k
A
.N
recp
(t).N
int
(t)- k
tox
.N
int
(t) Equation 3
The second term allows for a reduction of AB fluorescence due to dye toxicity.
Using such a model, the complete time evolution and concentration dependence of the
AB fluorescence can be simulated. To simulate the differences between the 3D and 2D
cell culture environments, the effective area parameter A, is reduced from 1 for 3D to 0.5
for 2D, mimicking a 50% reduction of the effective area of the cells exposed to the AB
solution. In the 3D environment, the effective concentration of the AB solution is reduced
by 25%, and so a value of B =0.75 is used. For the purpose of demonstrating the
appropriateness of the model for this study, Figure S.1 shows, for example, the dose
dependent %fluorescence predicted for 3hrs and 5 hrs. For collagen gels, values of A=1
and B=0.75 were employed. The figure qualitatively reproduces the behavior observed in
figure 5, whereby an increasing %fluorescence is observed in the range 25-50%
concentration of AB solution after 3hrs, whereas a maximum in %fluorescence is
observed at a concentration of ~30% after 5hrs.
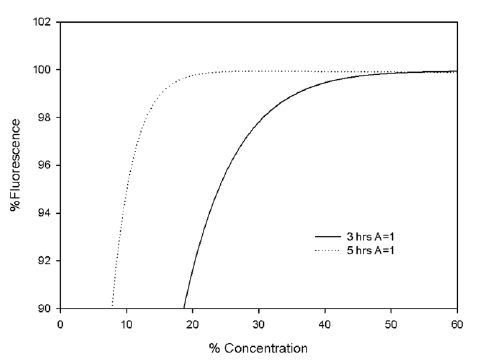
Figure S.1: Comparison of the simulated AB fluorescence after 3 and 5 hrs as a function of
%Concentration of AB solution for the 3D gels (A = 1, B =0.75)
Using the same parameters, the simulated AB fluorescence from the 3D matrices (A = 1,
B = 0.75) after 3hrs can be compared with that of the 2D substrates (A = 0.5, B = 1), as
shown in Figure S.2. It can be seen that, although there is a significant difference of
~25% between the viability values for the two cell culture substrates at an AB
concentration of 5%, this has reduced to ~5% at an AB concentration of 35%. Although it
cannot be considered a precise description of the system, the numerical simulation does
indicate that the observed differences in AB fluorescence can be accounted for simply by
the differences in the cell surface area which is accessible by the dye for diffusion.

Figure S.2: Comparison of the simulated AB fluorescence after 5 hrs as a function of %Concentration of
AB solution for the 3D gels (A = 1, B =0.75) and the 2D substrates (A = 0.5 B =1)
