TOXICOLOGICAL PROFILE FOR
METHYLENE CHLORIDE
U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES
Public Health Service
Agency for Toxic Substances and Disease Registry
September 2000
ii METHYLENE CHLORIDE
DISCLAIMER
The use of company or product name(s) is for identification only and does not imply endorsement by the
Agency for Toxic Substances and Disease Registry.
iii METHYLENE CHLORIDE
UPDATE STATEMENT
Toxicological profiles are revised and republished as necessary, but no less than once every three years.
For information regarding the update status of previously released profiles, contact ATSDR at:
Agency for Toxic Substances and Disease Registry
Division of Toxicology/Toxicology Information Branch
1600 Clifton Road NE, E-29
Atlanta, Georgia 30333



vii METHYLENE CHLORIDE
QUICK REFERENCE FOR HEALTH CARE PROVIDERS
Toxicological Profiles are a unique compilation of toxicological information on a given hazardous
substance. Each profile reflects a comprehensive and extensive evaluation, summary, and interpretation of
available toxicologic and epidemiologic information on a substance. Health care providers treating
patients potentially exposed to hazardous substances will find the following information helpful for fast
answers to often-asked questions.
Primary Chapters/Sections of Interest
Chapter 1: Public Health Statement: The Public Health Statement can be a useful tool for educating
patients about possible exposure to a hazardous substance. It explains a substance’s relevant
toxicologic properties in a nontechnical, question-and-answer format, and it includes a review of
the general health effects observed following exposure.
Chapter 2: Health Effects: Specific health effects of a given hazardous compound are reported by route
of exposure, by type of health effect (death, systemic, immunologic, reproductive), and by length
of exposure (acute, intermediate, and chronic). In addition, both human and animal studies are
reported in this section.
NOTE: Not all health effects reported in this section are necessarily observed in
the clinical setting. Please refer to the Public Health Statement to identify
general health effects observed following exposure.
Pediatrics: Four new sections have been added to each Toxicological Profile to address child health
issues:
Section 1.6 How Can Methylene Chloride Affect Children?
Section 1.7 How Can Families Reduce the Risk of Exposure to Methylene Chloride?
Section 2.7 Children’s Susceptibility
Section 5.6 Exposures of Children
Other Sections of Interest:
Section 2.8 Biomarkers of Exposure and Effect
Section 2.11 Methods for Reducing Toxic Effects
ATSDR Information Center
Phone: 1-888-42-ATSDR or (404) 639-6357 Fax: (404) 639-6359
E-mail: [email protected] Internet: http://www.atsdr.cdc.gov
The following additional material can be ordered through the ATSDR Information Center:
Case Studies in Environmental Medicine: Taking an Exposure History—The importance of taking an
exposure history and how to conduct one are described, and an example of a thorough exposure
history is provided. Other case studies of interest include Reproductive and Developmental
Hazards; Skin Lesions and Environmental Exposures; Cholinesterase-Inhibiting Pesticide
Toxicity; and numerous chemical-specific case studies.

viii METHYLENE CHLORIDE
Managing Hazardous Materials Incidents is a three-volume set of recommendations for on-scene
(prehospital) and hospital medical management of patients exposed during a hazardous materials incident.
Volumes I and II are planning guides to assist first responders and hospital emergency department
personnel in planning for incidents that involve hazardous materials. Volume III—Medical Management
Guidelines for Acute Chemical Exposures—is a guide for health care professionals treating patients
exposed to hazardous materials.
Fact Sheets (ToxFAQs) provide answers to frequently asked questions about toxic substances.
Other Agencies and Organizations
The National Center for Environmental Health (NCEH) focuses on preventing or controlling disease,
injury, and disability related to the interactions between people and their environment outside the
workplace. Contact: NCEH, Mailstop F-29, 4770 Buford Highway, NE, Atlanta, GA 30341-
3724 • Phone: 770-488-7000 • FAX: 770-488-7015.
The National Institute for Occupational Safety and Health (NIOSH) conducts research on occupational
diseases and injuries, responds to requests for assistance by investigating problems of health and
safety in the workplace, recommends standards to the Occupational Safety and Health
Administration (OSHA) and the Mine Safety and Health Administration (MSHA), and trains
professionals in occupational safety and health. Contact: NIOSH, 200 Independence Avenue,
SW, Washington, DC 20201 • Phone: 800-356-4674 or NIOSH Technical Information Branch,
Robert A. Taft Laboratory, Mailstop C-19, 4676 Columbia Parkway, Cincinnati, OH 45226-1998
• Phone: 800-35-NIOSH.
The National Institute of Environmental Health Sciences (NIEHS) is the principal federal agency for
biomedical research on the effects of chemical, physical, and biologic environmental agents on
human health and well-being. Contact: NIEHS, PO Box 12233, 104 T.W. Alexander Drive,
Research Triangle Park, NC 27709 • Phone: 919-541-3212.
Referrals
The Association of Occupational and Environmental Clinics (AOEC) has developed a network of clinics
in the United States to provide expertise in occupational and environmental issues. Contact:
AOEC, 1010 Vermont Avenue, NW, #513, Washington, DC 20005 • Phone: 202-347-4976 •
med.mc.duke.edu/oem/aoec.htm.
The American College of Occupational and Environmental Medicine (ACOEM) is an association of
physicians and other health care providers specializing in the field of occupational and
environmental medicine. Contact: ACOEM, 55 West Seegers Road, Arlington Heights, IL
60005 • Phone: 847-228-6850 • FAX: 847-228-1856.
ix METHYLENE CHLORIDE
CONTRIBUTORS
CHEMICAL MANAGER(S)/AUTHORS(S):
Jewell D. Wilson, Ph.D.
ATSDR, Division of Toxicology, Atlanta, GA
Margaret E. Fransen, Ph.D.
Syracuse Research Corporation, North Syracuse, NY
Fernando Llados, Ph.D.
Syracuse Research Corporation, North Syracuse, NY
Mona Singh, Ph.D.
Syracuse Research Corporation, North Syracuse, NY
Gary L. Diamond, Ph.D.
Syracuse Research Corporation, North Syracuse, NY
THE PROFILE HAS UNDERGONE THE FOLLOWING ATSDR INTERNAL REVIEWS:
1. Health Effects Review. The Health Effects Review Committee examines the health effects
chapter of each profile for consistency and accuracy in interpreting health effects and classifying
end points.
2. Minimal Risk Level Review. The Minimal Risk Level Workgroup considers issues relevant to
substance-specific minimal risk levels (MRLs), reviews the health effects database of each
profile, and makes recommendations for derivation of MRLs.
3. Data Needs Review. The Research Implementation Branch reviews data needs sections to assure
consistency across profiles and adherence to instructions in the Guidance.
xi METHYLENE CHLORIDE
PEER REVIEW
A peer review panel was assembled for methylene chloride. The panel consisted of the following
members:
1. Harvey Clewell III, M.S., ICF Kaiser International, Ruston, LA.
2. William J. George, Ph.D., Professor of Pharmacology, Director of Toxicology, Tulane University
School of Medicine, New Orleans, LA.
3. Lyman K. Skory, Ph.D., Skory Consulting, Inc., Midland, MI.
These experts collectively have knowledge of methylene chloride's physical and chemical properties,
toxicokinetics, key health end points, mechanisms of action, human and animal exposure, and
quantification of risk to humans. All reviewers were selected in conformity with the conditions for peer
review specified in Section 104(I)(13) of the Comprehensive Environmental Response, Compensation,
and Liability Act, as amended.
Scientists from the Agency for Toxic Substances and Disease Registry (ATSDR) have reviewed the peer
reviewers' comments and determined which comments will be included in the profile. A listing of the
peer reviewers' comments not incorporated in the profile, with a brief explanation of the rationale for their
exclusion, exists as part of the administrative record for this compound. A list of databases reviewed and
a list of unpublished documents cited are also included in the administrative record.
The citation of the peer review panel should not be understood to imply its approval of the profile's final
content. The responsibility for the content of this profile lies with the ATSDR.
xiii METHYLENE CHLORIDE
CONTENTS
FOREWORD ....................................................................... v
QUICK REFERENCE FOR HEALTH CARE PROVIDERS ................................. vii
CONTRIBUTORS ...................................................................ix
PEER REVIEW .....................................................................xi
LIST OF FIGURES ................................................................. xx
LIST OF TABLES................................................................. xxii
1. PUBLIC HEALTH STATEMENT.................................................... 1
1.1 WHAT IS METHYLENE CHLORIDE? .......................................... 1
1.2 WHAT HAPPENS TO METHYLENE CHLORIDE WHEN IT ENTERS THE
ENVIRONMENT? ........................................................... 2
1.3 HOW MIGHT I BE EXPOSED TO METHYLENE CHLORIDE?...................... 3
1.4 HOW CAN METHYLENE CHLORIDE ENTER AND LEAVE MY BODY? ............ 4
1.5 HOW CAN METHYLENE CHLORIDE AFFECT MY HEALTH? ..................... 4
1.6 HOW CAN METHYLENE CHLORIDE AFFECT CHILDREN? ...................... 6
1.7 HOW CAN FAMILIES REDUCE THE RISK OF EXPOSURE TO METHYLENE
CHLORIDE? ............................................................... 7
1.8 IS THERE A MEDICAL TEST TO DETERMINE WHETHER I HAVE BEEN EXPOSED
TO METHYLENE CHLORIDE?................................................ 8
1.9 WHAT RECOMMENDATIONS HAS THE FEDERAL GOVERNMENT MADE TO
PROTECT HUMAN HEALTH? ................................................ 9
1.10 WHERE CAN I GET MORE INFORMATION? .................................. 10
2. HEALTH EFFECTS .............................................................. 13
2.1 INTRODUCTION .......................................................... 13
2.2 DISCUSSION OF HEALTH EFFECTS BY ROUTE OF EXPOSURE ................. 13
2.2.1 Inhalation Exposure .................................................. 15
2.2.1.1 Death ..................................................... 15
2.2.1.2 Systemic Effects ............................................ 29
2.2.1.3 Immunological and Lymphoreticular Effects ...................... 34
2.2.1.4 Neurological Effects ......................................... 35
2.2.1.5 Reproductive Effects ......................................... 39
2.2.1.6 Developmental Effects ....................................... 40
2.2.1.7 Genotoxic Effects ........................................... 41
2.2.1.8 Cancer .................................................... 44
2.2.2 Oral Exposure ...................................................... 49
2.2.2.1 Death ..................................................... 49
2.2.2.2 Systemic Effects ............................................ 49
2.2.2.3 Immunological and Lymphoreticular Effects ...................... 58
2.2.2.4 Neurological Effects ......................................... 58
2.2.2.5 Reproductive Effects ......................................... 58
xiv METHYLENE CHLORIDE
2.2.2.6 Developmental Effects ....................................... 58
2.2.2.7 Genotoxic Effects ........................................... 59
2.2.2.8 Cancer .................................................... 59
2.2.3 Dermal Exposure .................................................... 60
2.2.3.1 Death ..................................................... 60
2.2.3.2 Systemic Effects ............................................ 60
2.2.3.3 Immunological and Lymphoreticular Effects ...................... 62
2.2.3.4 Neurological Effects ......................................... 62
2.2.3.5 Reproductive Effects ......................................... 62
2.2.3.6 Developmental Effects ....................................... 63
2.2.3.7 Genotoxic Effects ........................................... 63
2.2.3.8 Cancer .................................................... 63
2.3 TOXICOKINETICS......................................................... 63
2.3.1 Absorption ......................................................... 65
2.3.1.1 Inhalation Exposure .......................................... 65
2.3.1.2 Oral Exposure .............................................. 66
2.3.1.3 Dermal Exposure ............................................ 68
2.3.2 Distribution ........................................................ 68
2.3.2.1 Inhalation Exposure .......................................... 68
2.3.2.2 Oral Exposure .............................................. 69
2.3.2.3 Dermal Exposure ............................................ 69
2.3.3 Metabolism......................................................... 69
2.3.4 Elimination and Excretion ............................................. 78
2.3.4.1 Inhalation Exposure .......................................... 78
2.3.4.2 Oral Exposure .............................................. 79
2.3.4.3 Dermal Exposure ............................................ 79
2.3.5 Physiologically Based Pharmacokinetic (PBPK)/Pharmacodynamic (PD)
Models ............................................................ 80
2.3.5.1 Methylene Chloride PBPK Model Comparison ...................... 81
2.3.5.2 Discussion of Methylene Chloride PBPK Models .................... 85
2.4 MECHANISMS OF ACTION ................................................ 107
2.4.1 Pharmacokinetic Mechanisms ......................................... 107
2.4.2 Mechanisms of Toxicity.............................................. 109
2.4.3 Animal-to-Human Extrapolations ......................................
115
2.5 RELEVANCE TO PUBLIC HEALTH ......................................... 117
2.6 ENDOCRINE DISRUPTION ................................................ 133
2.7 CHILDREN’S SUSCEPTIBILITY ............................................ 133
2.8 BIOMARKERS OF EXPOSURE AND EFFECT ................................. 137
2.8.1 Biomarkers Used to Identify or Quantify Exposure to Methylene Chloride ...... 138
2.8.2 Biomarkers Used to Characterize Effects Caused by Methylene Chloride ....... 140
2.9 INTERACTIONS WITH OTHER CHEMICALS ................................. 141
2.10 POPULATIONS THAT ARE UNUSUALLY SUSCEPTIBLE ...................... 142
2.11 METHODS FOR REDUCING TOXIC EFFECTS ................................ 144
2.11.1 Reducing Peak Absorption Following Exposure ........................... 145
2.11.2 Reducing Body Burden .............................................. 145
2.11.3 Interfering with the Mechanism of Action for Toxic Effects.................. 147
2.12 ADEQUACY OF THE DATABASE ........................................... 147
2.12.1 Existing Information on Health Effects of Methylene Chloride ............... 148
2.12.2 Identification of Data Needs .......................................... 150
2.12.3 Ongoing Studies .................................................... 162
xv METHYLENE CHLORIDE
3. CHEMICAL AND PHYSICAL INFORMATION ...................................... 163
3.1 CHEMICALIDENTITY .................................................... 163
3.2 PHYSICAL AND CHEMICAL PROPERTIES ................................... 163
4. PRODUCTION, IMPORT/EXPORT, USE, AND DISPOSAL ............................ 167
4.1 PRODUCTION ........................................................... 167
4.2 IMPORT/EXPORT ........................................................ 170
4.3 USE..................................................................... 170
4.4 DISPOSAL ............................................................... 171
5. POTENTIAL FOR HUMAN EXPOSURE ........................................... 173
5.1 OVERVIEW.............................................................. 173
5.2 RELEASES TO THE ENVIRONMENT ........................................ 173
5.2.1 Air .............................................................. 178
5.2.2 Water ............................................................ 179
5.2.3 Soil .............................................................. 180
5.3 ENVIRONMENTAL FATE.................................................. 180
5.3.1 Transport and Partitioning ............................................ 180
5.3.2 Transformation and Degradation ....................................... 181
5.3.2.1 Air ...................................................... 181
5.3.2.2 Water .................................................... 182
5.3.2.3 Sediment and Soil .......................................... 182
5.4 LEVELS MONITORED OR ESTIMATED IN THE ENVIRONMENT ............... 183
5.4.1 Air .............................................................. 183
5.4.2 Water ............................................................ 183
5.4.3 Sediment and Soil .................................................. 185
5.4.4 Other Environmental Media........................................... 185
5.5 GENERAL POPULATION AND OCCUPATIONAL EXPOSURE .................. 186
5.6 EXPOSURES OF CHILDREN ............................................... 187
5.7 POPULATIONS WITH POTENTIALLY HIGH EXPOSURES...................... 189
5.8 ADEQUACY OF THE DATABASE ........................................... 190
5.8.1 Identification of Data Needs .......................................... 191
5.8.2 Ongoing Studies .................................................... 194
6. ANALYTICAL METHODS ....................................................... 197
6.1 BIOLOGICAL SAMPLES ...................................................
197
6.2 ENVIRONMENTAL SAMPLES .............................................. 197
6.3 ADEQUACY OF THE DATABASE ........................................... 203
6.3.1 Identification of Data Needs .......................................... 204
6.3.2 Ongoing Studies .................................................... 205
7. REGULATIONS AND ADVISORIES .............................................. 207
8. REFERENCES ................................................................. 213
9. GLOSSARY ................................................................... 265
METHYLENE CHLORIDE xvi
APPENDICES
A. ATSDR MINIMAL RISK LEVELS AND WORKSHEETS ......................... A-1
B. USER'S GUIDE ........................................................... B-1
C. ACRONYMS, ABBREVIATIONS, AND SYMBOLS ............................. C-1
xvii METHYLENE CHLORIDE
LIST OF FIGURES
2-1 Levels of Significant Exposure to Methylene Chloride - Inhalation ....................... 26
2-2 Levels of Significant Exposure to Methylene Chloride - Oral ............................ 53
2-3 Proposed Pathways for Methylene Chloride Metabolism ............................... 71
2-4 Conceptual Representation of a Physiologically Based Pharmacokinetic (PBPK) Model
for a Hypothetical Chemical Substance ............................................. 82
2-5 Existing Information on Health Effects of Methylene Chloride.......................... 149
5-1 Frequency of NPL Sites with Methylene Chloride Contamination ....................... 177
xix METHYLENE CHLORIDE
LIST OF TABLES
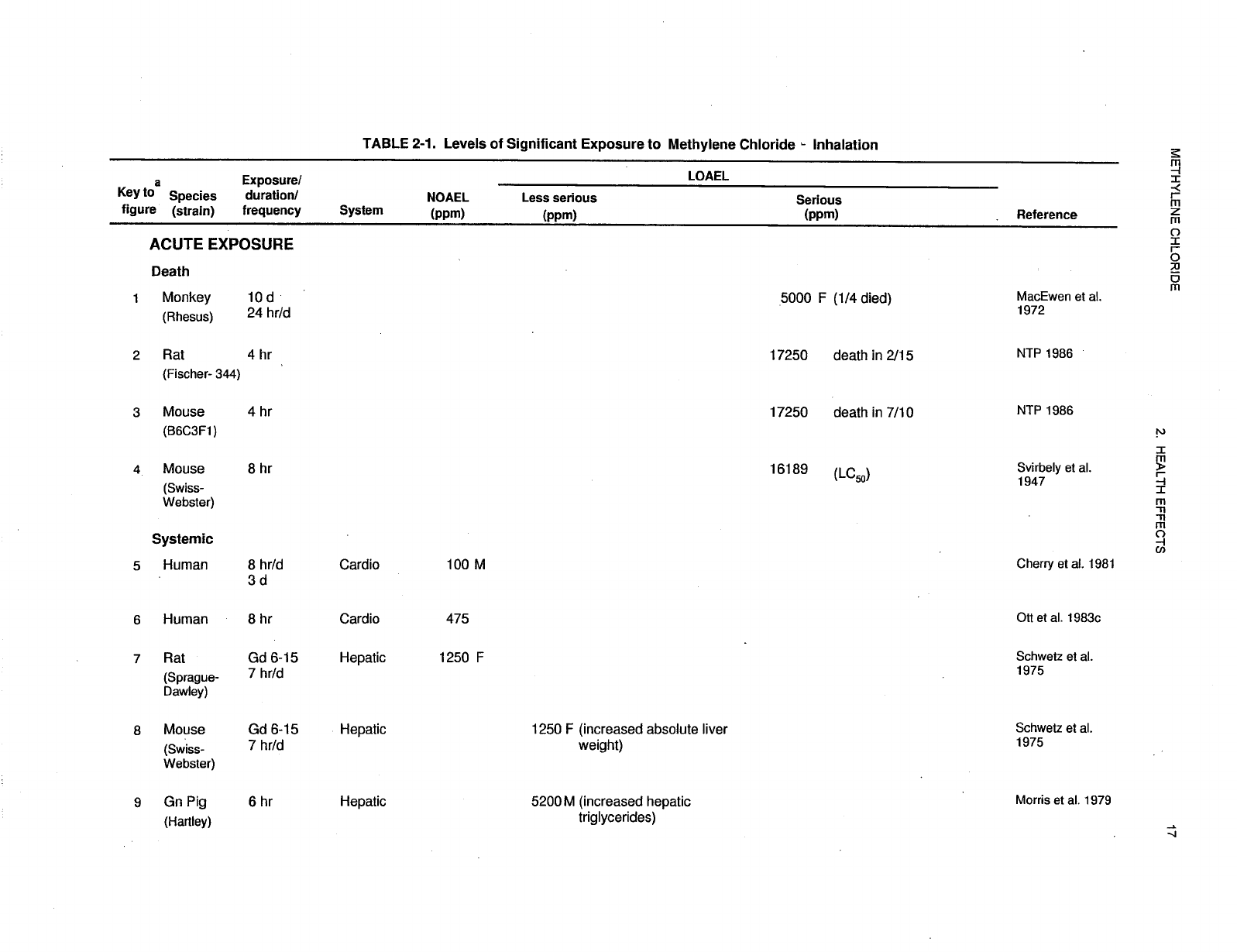
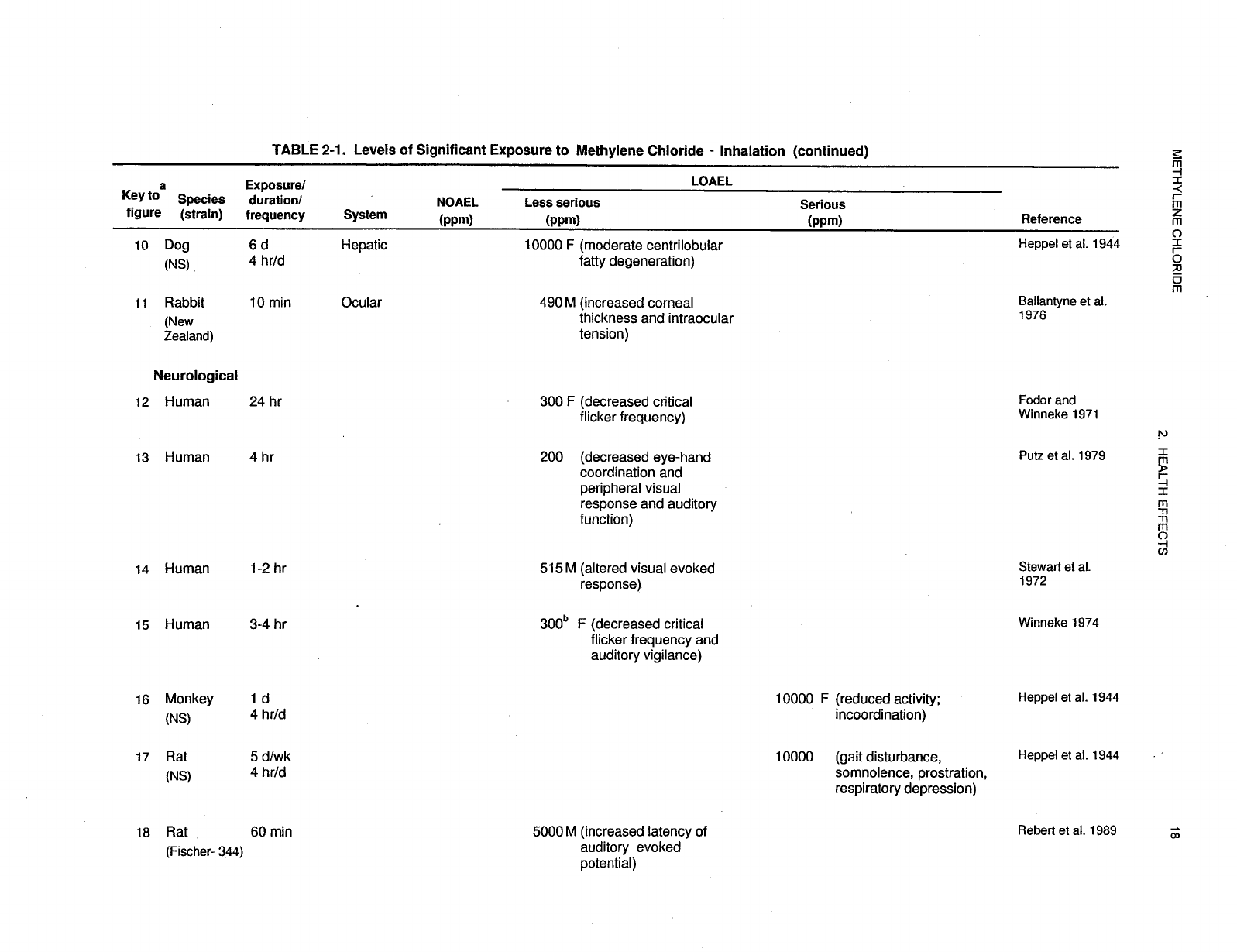
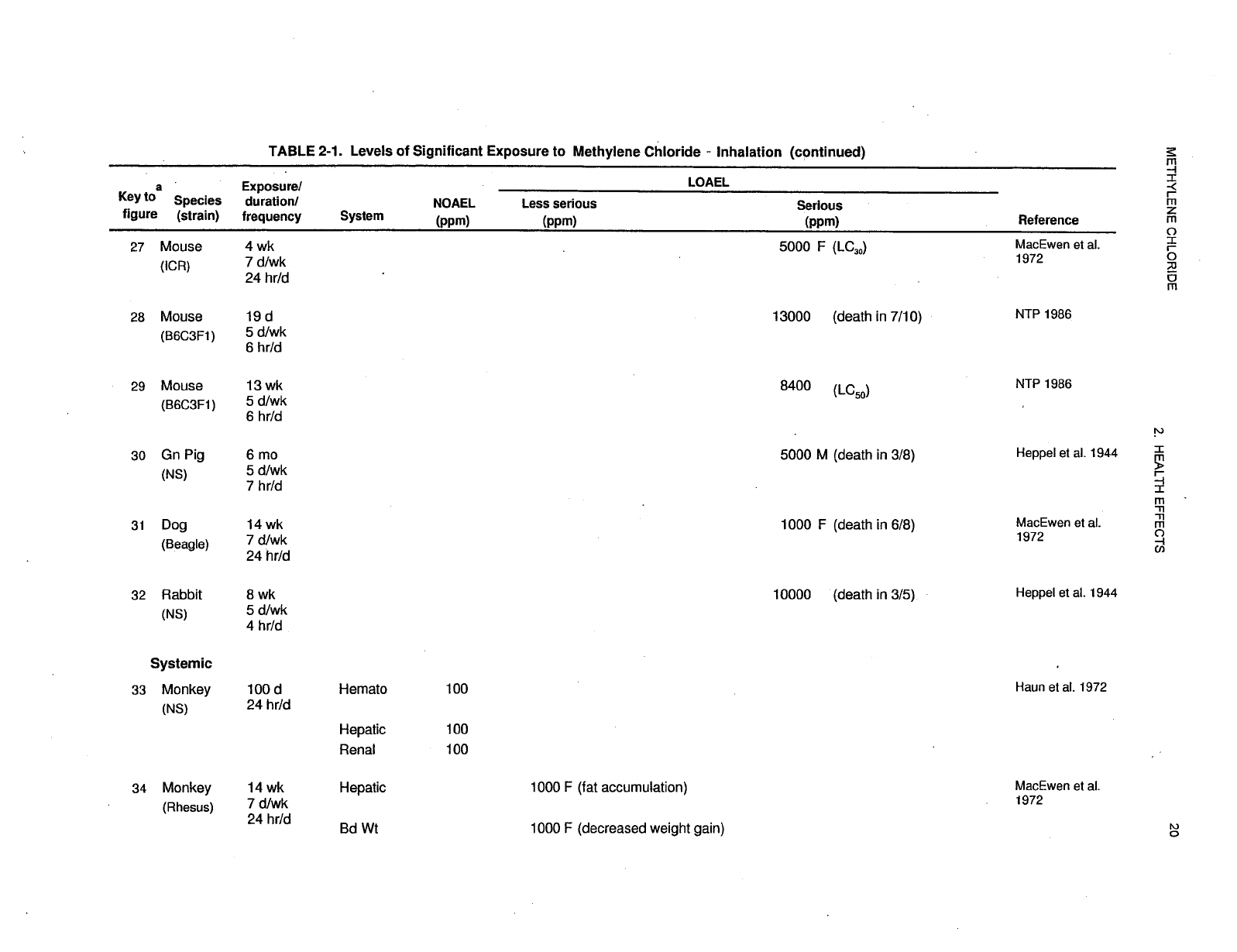
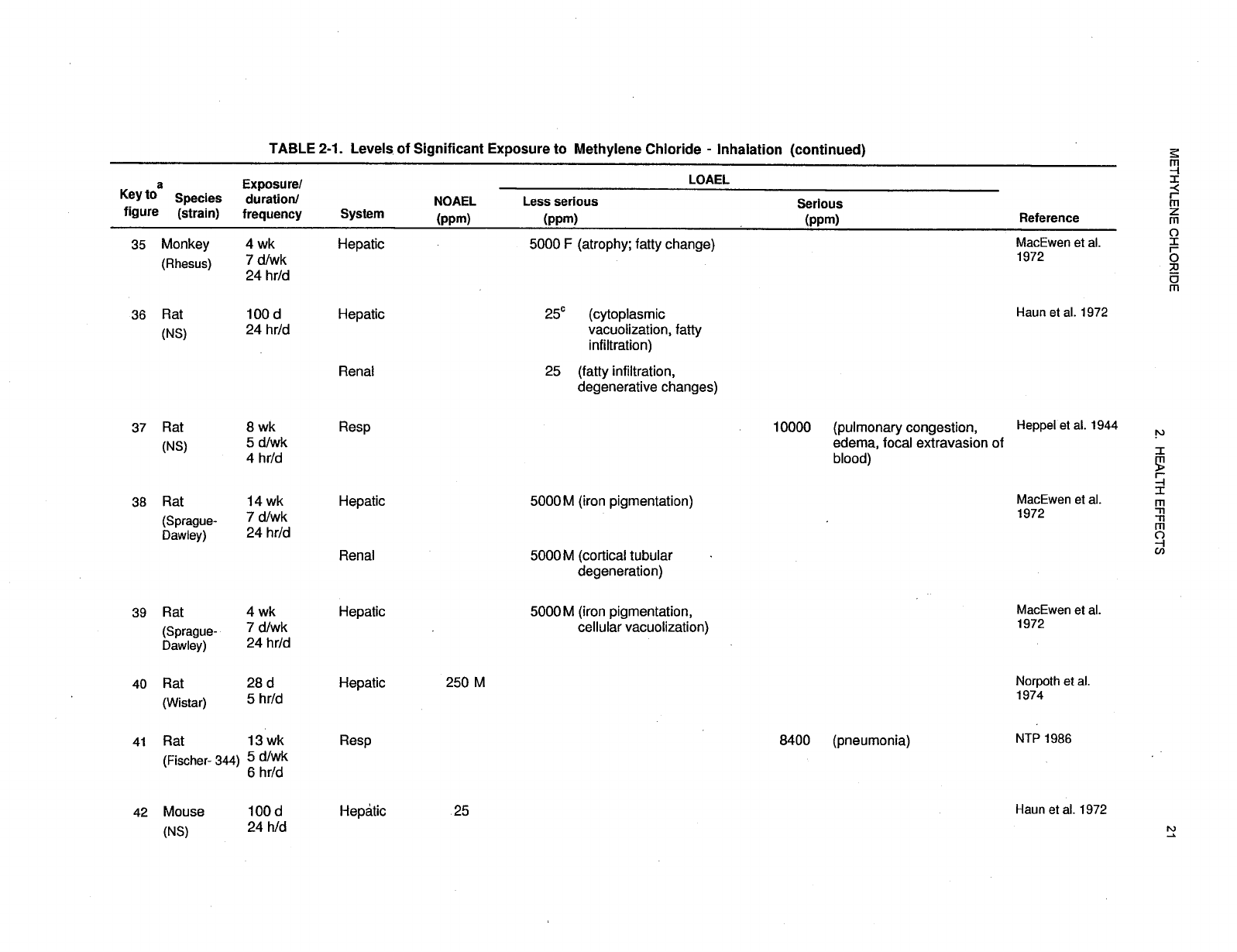
2-1 Levels of Significant Exposure to Methylene Chloride - Inhalation ....................... 17
2-2 Levels of Significant Exposure to Methylene Chloride - Oral ............................ 50
2-3 Genotoxicity of Methylene Chloride In Vitro ........................................ 130
2-4 Genotoxicity of Methylene Chloride In Vivo ........................................ 131
3-1 Chemical Identity of Methylene Chloride .......................................... 164
3-2 Physical and Chemical Properties of Methylene Chloride .............................. 165
4-1 Facilities that Manufacture or Process Methylene Chloride ............................. 168
5-1 Releases to the Environment from Facilities that Manufacture or Process Methylene
Chloride .................................................................... 174
5-2 Summary of Methylene Chloride Levels in Air ...................................... 184
5-3 Ongoing Studies on the Potential for Human Exposure to Methylene Chloride ............. 195
6-1 Analytical Methods for Determining Methylene Chloride in Biological Materials ........... 198
6-2 Analytical Methods for Determining Methylene Chloride in Environmental Samples ........ 199
7-1 Regulations and Guidelines Applicable to Methylene Chloride.......................... 209
1 METHYLENE CHLORIDE
1. PUBLIC HEALTH STATEMENT
This public health statement tells you about methylene chloride and the effects of exposure.
The Environmental Protection Agency (EPA) identifies the most serious hazardous waste sites in
the nation. These sites make up the National Priorities List (NPL) and are the sites targeted for
long-term federal cleanup activities. Methylene chloride has been found in at least 882 of the
1,569 current or former NPL sites. However, the total number of NPL sites evaluated for this
substance is not known. As more sites are evaluated, the sites at which methylene chloride is
found may increase. This information is important because exposure to this substance may harm
you and because these sites may be sources of exposure.
When a substance is released from a large area, such as an industrial plant, or from a container,
such as a drum or bottle, it enters the environment. This release does not always lead to
exposure. You are exposed to a substance only when you come in contact with it. You may be
exposed by breathing, eating, or drinking the substance, or by skin contact.
If you are exposed to methylene chloride, many factors determine whether you’ll be harmed.
These factors include the dose (how much), the duration (how long), and how you come in
contact with it. You must also consider the other chemicals you’re exposed to and your age, sex,
diet, family traits, lifestyle, and state of health.
1.1 WHAT IS METHYLENE CHLORIDE?
Methylene chloride, also known as dichloromethane, is a colorless liquid that has a mild sweet
odor, evaporates easily, and does not burn easily. It is widely used as an industrial solvent and
as a paint stripper. It can be found in certain aerosol and pesticide products and is used in the
manufacture of photographic film. The chemical may be found in some spray paints, automotive
cleaners, and other household products. Methylene chloride does not appear to occur naturally
in the environment. It is made from methane gas or wood alcohol. Most of the methylene
2 METHYLENE CHLORIDE
1. PUBLIC HEALTH STATEMENT
chloride released to the environment results from its use as an end product by various industries
and the use of aerosol products and paint removers in the home.
More information on the properties and uses of methylene chloride may be found in Chapters 3
and 4.
1.2 WHAT HAPPENS TO METHYLENE CHLORIDE WHEN IT ENTERS THE
ENVIRONMENT?
Methylene chloride is mainly released to the environment in air, and to a lesser extent in water
and soil, due to industrial and consumer uses. Many chemical waste sites, including NPL sites,
contain methylene chloride and these might act as additional sources of environmental
contamination through spills, leaks, or evaporation. Because methylene chloride evaporates
readily, most of it is released into the air. In the air, it is broken down by sunlight and by
reaction with other chemicals present in the air. About half of the methylene chloride disappears
from air in 53 to 127 days. Although methylene chloride does not dissolve easily in water, small
amounts may be found in some drinking water. Methylene chloride that is present in water is
broken down slowly by reactions with other chemicals or by bacteria. Over 90% of the
methylene chloride in the environment changes to carbon dioxide (CO
2
), which is already
present in air. It takes about 1 to 6 days for half the methylene chloride to break down in water.
When methylene chloride is spilled on land, it attaches loosely to nearby surface soil particles. It
moves from the soil into the air. Some may also move into groundwater. We do not know how
long it remains in soil. We do not expect methylene chloride to build up in plants or animals.
More information on what happens to methylene chloride in the environment may be found in
Chapters 4 and 5.
3 METHYLENE CHLORIDE
1. PUBLIC HEALTH STATEMENT
1.3 HOW MIGHT I BE EXPOSED TO METHYLENE CHLORIDE?
You may be exposed to methylene chloride in air, water, food, or from consumer products.
Because methylene chloride evaporates easily, the greatest potential for exposure is when you
breathe vapors of contaminated air. Background levels in air are usually at less than one part
methylene chloride per billion parts (ppb) of air. Methylene chloride has been found in some
urban air and at some hazardous waste sites at average concentrations of 11 ppb of air. The
average daily intake of methylene chloride from outdoor air in three U.S. cities ranges from 33 to
309 micrograms per day (1 milligram is equivalent to 1,000 micrograms, 1 mg = 1,000 µg.)
Contact with consumer products such as paint strippers or aerosol cans that contain methylene
chloride is another frequent source of exposure. Exposure occurs as a result of breathing the
vapors given off by the product or from direct contact of the liquid material with the skin. The
highest and most frequent exposures to methylene chloride usually occur in workplaces where
the chemical is used; exposure can be dangerously high if methylene chloride is used in an
enclosed space without adequate ventilation. People who work with it can breathe in the
chemical or it may come in contact with their skin. In the past, concentrations ranging from 1 to
1,000 parts of methylene chloride per million parts of air (ppm; 1 ppm is 1,000 times more than
1 ppb) have been detected in general work areas, while higher concentrations (1,400 ppm) have
been detected in samples in the breathing zone of some workers. These exposure levels exceed
the current recommended federal limits. The National Institute for Occupational Safety and
Health (NIOSH) estimated that 1 million workers may be exposed to methylene chloride.
Averages of 68 ppb of methylene chloride in surface water and 98 ppb methylene chloride in
groundwater have been found at some hazardous waste sites. Less than 1 ppb has been found in
most drinking water analyzed. We expect exposure from water and food to be low because very
little methylene chloride has been detected in these sources.
More information on how you might be exposed to methylene chloride is given in Chapter 5.
4 METHYLENE CHLORIDE
1. PUBLIC HEALTH STATEMENT
1.4 HOW CAN METHYLENE CHLORIDE ENTER AND LEAVE MY BODY?
Methylene chloride may enter your body when you breathe vapors of contaminated air. It may
also enter your body if you drink water from contaminated wells, or it may enter if your skin
comes in contact with it. Since methylene chloride evaporates into air rapidly, exposure by
breathing is the most likely source of exposure at hazardous waste sites, in the home, and in the
workplace. When you breathe in methylene chloride, over 70% of it enters your bloodstream
and quickly spreads throughout your body, with most of it going to the liver, kidney, brain,
lungs, and fatty tissue. Increased physical activity or an increased amount of body fat tends to
increase the amount of methylene chloride that remains or accumulates in your body tissue.
About half of the methylene chloride in the blood leaves within 40 minutes. Some of the
methylene chloride is broken down into other chemicals, including carbon monoxide (CO), a
natural substance in the body occurring from the breakdown of hemoglobin. Unchanged
methylene chloride and its breakdown products are removed from your body mainly in the air
you breathe out. Small amounts leave in your urine. This usually occurs within 48 hours after
exposure. Although the rate of uptake through the stomach has not been measured, uptake is
likely to be fast. Skin absorption is usually small. Trapping the chemical against the skin with
clothing or gloves can lead to greater absorption and possible chemical burns.
More information on how methylene chloride enters and leaves the body is given in Chapter 2.
1.5 HOW CAN METHYLENE CHLORIDE AFFECT MY HEALTH?
To protect the public from the harmful effects of toxic chemicals and to find ways to treat people
who have been harmed, scientists use many tests.
One way to see if a chemical will hurt people is to learn how the chemical is absorbed, used, and
released by the body; for some chemicals, animal testing may be necessary. Animal testing may
also be used to identify health effects such as cancer or birth defects. Without laboratory
animals, scientists would lose a basic method to get information needed to make wise decisions
5 METHYLENE CHLORIDE
1. PUBLIC HEALTH STATEMENT
to protect public health. Scientists have the responsibility to treat research animals with care and
compassion. Laws today protect the welfare of research animals, and scientists must comply
with strict animal care guidelines.
If you breathe large amounts (800 ppm) of methylene chloride you may not be able to react fast,
remain steady, or perform tasks requiring precise hand movements. You may experience
dizziness, nausea, tingling or numbness of the fingers and toes, and drunkenness if you breathe
methylene chloride for a sufficiently long period of time. In most cases, effects disappear
shortly after the exposure ends. Studies in animals suggest that exposure to higher
concentrations (8,000–20,000 ppm) can lead to unconsciousness and death. There have been
reports of some people becoming unconscious and some people dying after breathing high
concentrations of methylene chloride; accidents of this kind happen more often when methylene
chloride is used without adequate ventilation.
Breathing methylene chloride may cause changes in the liver and kidney in animals, but similar
effects have not been observed in humans. Animal studies indicate that should you be exposed
to high levels of vapors of methylene chloride in air, the vapors may irritate your eyes and affect
your cornea. One study reported these effects at concentrations of 490 ppm; however, the effects
usually disappeared within a few days.
In humans, direct skin contact with large amounts of methylene chloride causes intense burning
and mild redness of the skin. In a workplace accident in which a person was found to have lost
consciousness and partly fallen into an open vat of methylene chloride, extended direct contact
with the liquid caused severe burns of the skin and eyes (cornea); these conditions were
treatable. In rabbits, effects were observed on the eyes (e.g., cornea), but they were reversible
within a few days.
People can smell methylene chloride at about 200 ppm in air. After about 3 hours of exposure at
this level, a person will become less attentive and less accurate in tasks that require hand-eye
6 METHYLENE CHLORIDE
1. PUBLIC HEALTH STATEMENT
coordination. Because people differ in their ability to smell various chemicals, odors may not be
helpful in avoiding over-exposure to methylene chloride.
There is not clear evidence that methylene chloride causes cancer in humans exposed to vapors
in the workplace. However, breathing high concentrations of methylene chloride for long
periods of time did increase the incidence of cancer in mice. No information was found
regarding the cancer-causing effects of methylene chloride in humans after oral exposure. The
Department of Health and Human Services (DHHS) has determined that methylene chloride may
reasonably be anticipated to be a cancer-causing chemical. The International Agency for
Research on Cancer (IARC) has classified methylene chloride in Group 2B, possibly causing
cancer in humans. The EPA has determined that methylene chloride is a probable cancer-
causing agent in humans.
More information on how methylene chloride can affect your health is given in Chapter 2.
1.6 HOW CAN METHYLENE CHLORIDE AFFECT CHILDREN?
This section discusses potential health effects from exposures during the period from conception
to maturity at 18 years of age in humans.
Children and adults may be exposed to low levels of methylene chloride in drinking water.
Small children who live near factories that produce or use methylene chloride could accidently
eat some of the chemical by putting dirty hands in their mouths, but the amount of methylene
chloride in the soil is thought to be too low to be harmful. Children could breathe in methylene
chloride that is used in a number of household products, since it evaporates easily. Also, since
the vapor of methylene chloride is heavier than air, it will tend to stay close to the ground; as a
result, children, being shorter, would breathe in larger amounts than adults during accidental
exposure.
The effects of methylene chloride have not been studied in children, but they would likely
experience the same health effects seen in adults exposed to the chemical. It is also not known if
7 METHYLENE CHLORIDE
1. PUBLIC HEALTH STATEMENT
the way in which methylene chloride is absorbed, metabolized, and eliminated from the body is
different in children than it is in adults. Therefore, adverse effects noted in animals and adult
humans (as discussed in Section 1.5) might also occur in children.
There have not been any reports of a connection between methylene chloride exposure during
pregnancy and birth defects in humans. If a pregnant woman is exposed to methylene chloride, a
small amount may cross the placenta, but not enough to harm the fetus. Studies in animals show
that breathing methylene chloride at relatively high levels during pregnancy may lead to bone
variations, none of which are serious and some of which may be outgrown, in newborn pups.
Methylene chloride has been shown to cross the placenta in rats. Methylene chloride has not
been accurately measured in human milk and there are no animal studies testing to what extent it
can pass into milk.
Sections 2.7 and 5.6 contain specific information about the effects of methylene chloride in
children.
1.7 HOW CAN FAMILIES REDUCE THE RISK OF EXPOSURE TO METHYLENE
CHLORIDE?
If your doctor finds that you have been exposed to significant amounts of methylene chloride,
ask whether your children might also be exposed. Your doctor might need to ask your state
health department to investigate.
Children may be exposed to methylene chloride in consumer household products, such as paint
removers, which contain a large percentage of methylene chloride. In general, the amounts of
methylene chloride in consumer products are low and children are not likely to be harmed unless
large amounts contact the skin or are accidentally swallowed. Using paint removers, especially
in unventilated or poorly ventilated areas, may cause the amount of methylene chloride in the air
to reach potentially dangerous levels. Caution should be used when using paint removers inside
your house; you should follow instructions on the package label for the proper ventilation
8 METHYLENE CHLORIDE
1. PUBLIC HEALTH STATEMENT
conditions when using these products. It is also advisable to make certain that children do not
remain near indoor paint removal activities.
Household chemicals should be stored out of reach of young children to prevent accidental
poisonings or skin irritation. Always store household chemicals in their original labeled
containers. Never store household chemicals in containers that children would find attractive to
eat or drink from, such as old soda bottles. Keep your Poison Control Center’s number next to
the phone.
Sometimes older children sniff household chemicals in an attempt to get high. Your children
may be exposed to methylene chloride by inhaling products containing it. Talk with your
children about the dangers of sniffing chemicals.
1.8 IS THERE A MEDICAL TEST TO DETERMINE WHETHER I HAVE BEEN
EXPOSED TO METHYLENE CHLORIDE?
Several tests exist for determining whether you have had measurable exposure to methylene
chloride. The most direct method measures methylene chloride in the air you breathe out. Your
blood can also be analyzed to determine if methylene chloride is present. However, these tests
are only useful for detecting exposures which have occurred within a few days because
methylene chloride remains in the blood for a very short time. Some absorbed methylene
chloride is stored in fat and slowly returns to the bloodstream. A test to measure
carboxyhemoglobin (COHb), a chemical formed in blood as methylene chloride breaks down in
the body, can also be used as an indicator of exposure. However, this test is not specific, since
smoking and exposure to other chemicals may also increase COHb levels. Your urine can also
be tested for methylene chloride itself or for other chemicals (such as formic acid) that are
produced as methylene chloride breaks down in the body. These tests are not routinely available
in a doctor's office, and they require special equipment. Also, the test for formic acid is not
specific for methylene chloride, since other chemicals, such as formaldehyde, are broken down

9 METHYLENE CHLORIDE
1. PUBLIC HEALTH STATEMENT
to formic acid. The tests may be useful to determine exposure to methylene chloride but do not
by themselves measure or predict health effects.
More information on how methylene chloride can be measured in exposed humans is presented
in Chapters 2 and 6.
1.9 WHAT RECOMMENDATIONS HAS THE FEDERAL GOVERNMENT MADE TO
PROTECT HUMAN HEALTH?
The federal government develops regulations and recommendations to protect public health.
Regulations can be enforced by law. Federal agencies that develop regulations for toxic
substances include the Environmental Protection Agency (EPA), the Occupational Safety and
Health Administration (OSHA), and the Food and Drug Administration (FDA).
Recommendations provide valuable guidelines to protect public health but cannot be enforced by
law. Federal organizations that develop recommendations for toxic substances include the
Agency for Toxic Substances and Disease Registry (ATSDR) and the National Institute for
Occupational Safety and Health (NIOSH).
Regulations and recommendations can be expressed in not-to-exceed levels in air, water, soil, or
food that are usually based on levels that affect animals; then they are adjusted to help protect
people. Sometimes these not-to-exceed levels differ among federal organizations because of
different exposure times (an 8-hour workday or a 24-hour day), the use of different animal
studies, or other factors.
Recommendations and regulations are also periodically updated as more information becomes
available. For the most current information, check with the federal agency or organization that
provides it. Some regulations and recommendations for methylene chloride include the
following:
10 METHYLENE CHLORIDE
1. PUBLIC HEALTH STATEMENT
The EPA requires that releases of methylene chloride of 1,000 pounds or more be reported to the
federal government. The EPA has provided guidelines on how much methylene chloride you
may be exposed to for certain amounts of time without causing risk to human health. It
recommends that exposure of children to methylene chloride in drinking water should not exceed
10 milligrams/liter (mg/L) for 1 day or 2 mg/L for 10 days.
Because methylene chloride is used in processing spices, hops extract, and decaffeinated coffee,
the FDA has established limits on the amounts of methylene chloride that can remain in these
food products.
The OSHA currently has a “permissible exposure limit” (PEL) of 25 ppm for an 8-hour workday
with 125 ppm as a “short-term exposure limit” (STEL) for 15 minute durations for persons who
work with methylene chloride.
NIOSH no longer has a “recommended exposure limit” (REL) for methylene chloride. Because
methylene chloride causes tumors in some animals, NIOSH currently considers it a possible
cancer-causing substance in the workplace and recommends that exposure be lowered to the
lowest feasible limit.
More information on government recommendations regarding methylene chloride can be found
in Chapter 7.
1.10 WHERE CAN I GET MORE INFORMATION?
If you have any more questions or concerns, please contact your community or state health or
environmental quality department or
Agency for Toxic Substances and Disease Registry
Division of Toxicology
1600 Clifton Road NE, Mailstop E-29
Atlanta, GA 30333

11 METHYLENE CHLORIDE
1. PUBLIC HEALTH STATEMENT
* Information line and technical assistance
Phone: 1-888-42-ATSDR (1-888-422-8737)
Fax: (404) 639-6359
ATSDR can also tell you the location of occupational and environmental health clinics. These
clinics specialize in recognizing, evaluating, and treating illnesses resulting from exposure to
hazardous substances.
* To order toxicological profiles, contact
National Technical Information Service
5285 Port Royal Road
Springfield, VA 22161
Phone: (800) 553-6847 or (703) 605-6000
13 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2.1 INTRODUCTION
The primary purpose of this chapter is to provide public health officials, physicians, toxicologists, and
other interested individuals and groups with an overall perspective on the toxicology of methylene
chloride. It contains descriptions and evaluations of toxicological studies and epidemiological
investigations and provides conclusions, where possible, on the relevance of toxicity and toxicokinetic
data to public health.
A glossary and list of acronyms, abbreviations, and symbols can be found at the end of this profile.
2.2 DISCUSSION OF HEALTH EFFECTS BY ROUTE OF EXPOSURE
To help public health professionals and others address the needs of persons living or working near
hazardous waste sites, the information in this section is organized first by route of exposure (inhalation,
oral, and dermal) and then by health effect (death, systemic, immunological, neurological, reproductive,
developmental, genotoxic, and carcinogenic effects). These data are discussed in terms of three exposure
periods: acute (14 days or less), intermediate (15–364 days), and chronic (365 days or more).
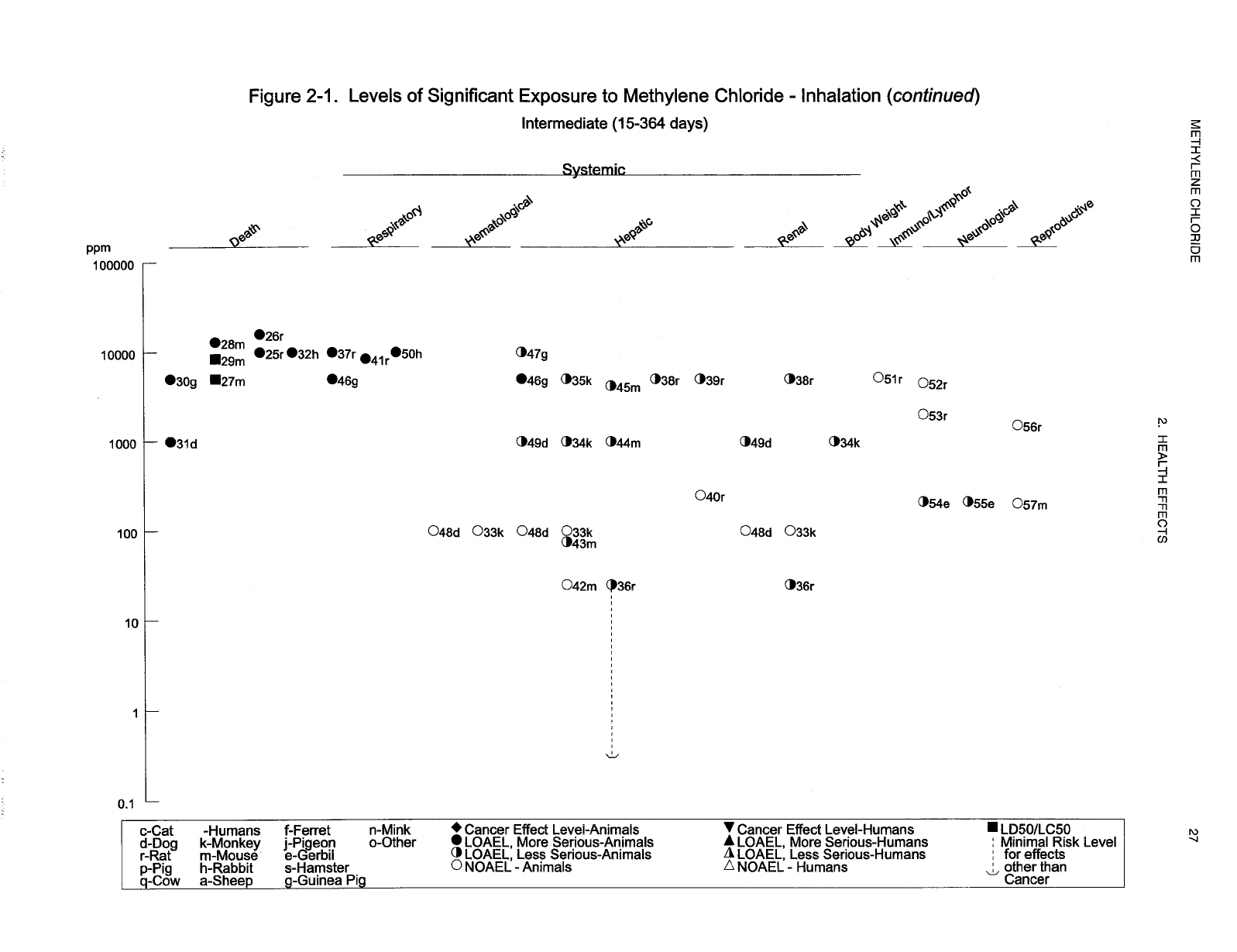
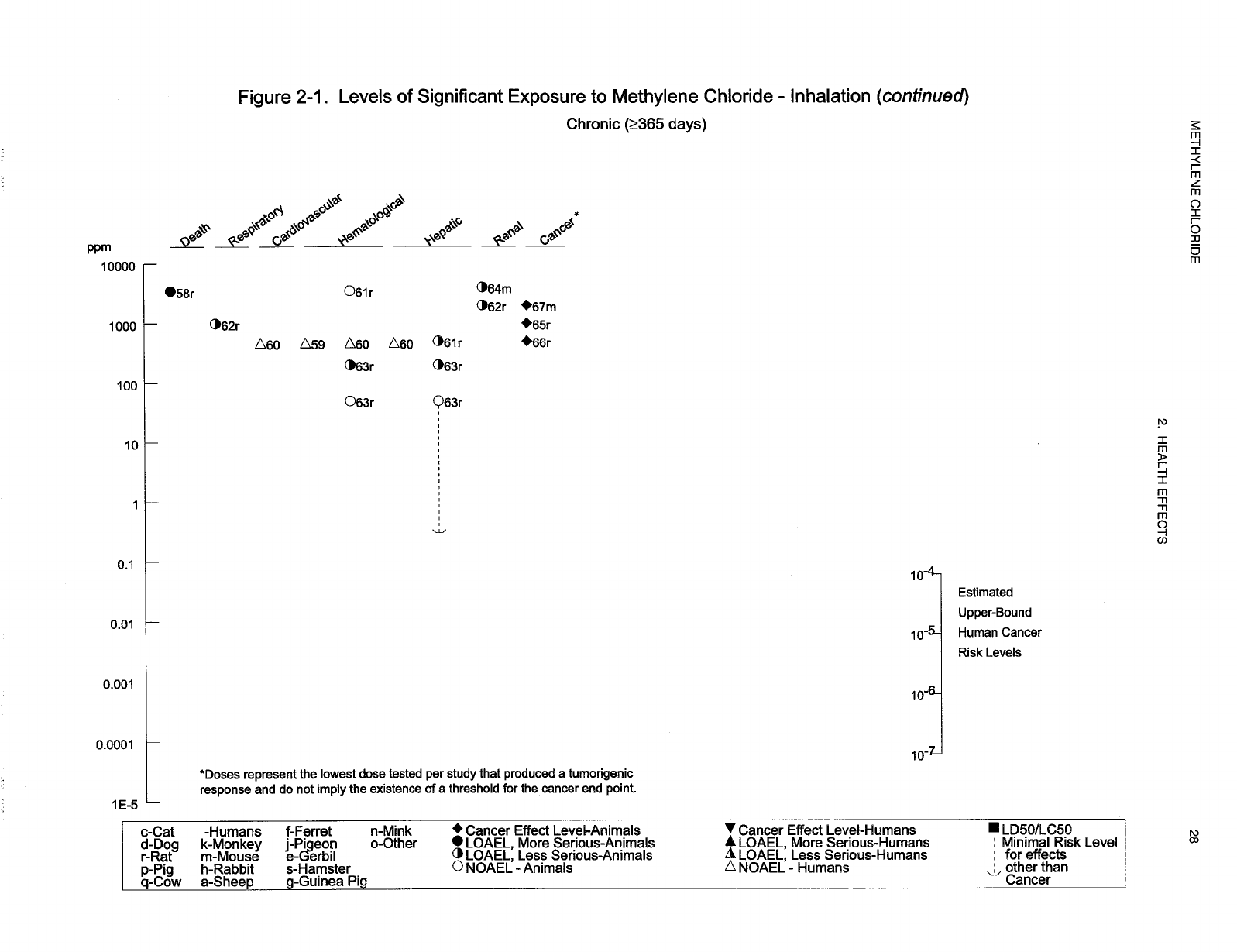
Levels of significant exposure for each route and duration are presented in tables and illustrated in
figures. The points in the figures showing no-observed-adverse-effect levels (NOAELs) or
lowest-observed-adverse-effect levels (LOAELs) reflect the actual doses (levels of exposure) used in the
studies. LOAELS have been classified into "less serious" or "serious" effects. "Serious" effects are those
that evoke failure in a biological system and can lead to morbidity or mortality (e.g., acute respiratory
distress or death). "Less serious" effects are those that are not expected to cause significant dysfunction
or death, or those whose significance to the organism is not entirely clear. ATSDR acknowledges that a
considerable amount of judgment may be required in establishing whether an end point should be
classified as a NOAEL, "less serious" LOAEL, or "serious" LOAEL, and that in some cases, there will be
insufficient data to decide whether the effect is indicative of significant dysfunction. However, the
Agency has established guidelines and policies that are used to classify these end points. ATSDR
believes that there is sufficient merit in this approach to warrant an attempt at distinguishing between
"less serious" and "serious" effects. The distinction between "less serious" effects and "serious" effects is
14 METHYLENE CHLORIDE
2. HEALTH EFFECTS
considered to be important because it helps the users of the profiles to identify levels of exposure at which
major health effects start to appear. LOAELs or NOAELs should also help in determining whether or not
the effects vary with dose and/or duration, and place into perspective the possible significance of these
effects to human health.
The significance of the exposure levels shown in the Levels of Significant Exposure (LSE) tables and
figures may differ depending on the user's perspective. Public health officials and others concerned with
appropriate actions to take at hazardous waste sites may want information on levels of exposure
associated with more subtle effects in humans or animals (LOAEL) or exposure levels below which no
adverse effects (NOAELs) have been observed. Estimates of levels posing minimal risk to humans
(Minimal Risk Levels or MRLs) may be of interest to health professionals and citizens alike.
Levels of exposure associated with carcinogenic effects (Cancer Effect Levels, CELs) of methylene
chloride are indicated in Tables 2-1 and 2-2 and Figures 2-1 and 2-2. Because cancer effects could occur
at lower exposure levels, Figures 2-1 and 2-2 also show a range for the upper bound of estimated excess
risks, ranging from a risk of 1 in 10,000 to 1 in 10,000,000 (10
-4
to 10
-7
), as developed by EPA.
Estimates of exposure levels posing minimal risk to humans (Minimal Risk Levels or MRLs) have been
made for methylene chloride. An MRL is defined as an estimate of daily human exposure to a substance
that is likely to be without an appreciable risk of adverse effects (noncarcinogenic) over a specified
duration of exposure. MRLs are derived when reliable and sufficient data exist to identify the target
organ(s) of effect or the most sensitive health effect(s) for a specific duration within a given route of
exposure. MRLs are based on noncancerous health effects only and do not consider carcinogenic effects.
MRLs can be derived for acute, intermediate, and chronic duration exposures for inhalation and oral
routes. Appropriate methodology does not exist to develop MRLs for dermal exposure.
Although methods have been established to derive these levels (Barnes and Dourson 1988; EPA 1990d),
uncertainties are associated with these techniques. Furthermore, ATSDR acknowledges additional
uncertainties inherent in the application of the procedures to derive less than lifetime MRLs. As an
example, acute inhalation MRLs may not be protective for health effects that are delayed in development
or are acquired following repeated acute insults, such as hypersensitivity reactions, asthma, or chronic
bronchitis. As these kinds of health effects data become available and methods to assess levels of
significant human exposure improve, these MRLs will be revised.
15 METHYLENE CHLORIDE
2. HEALTH EFFECTS
A User's Guide has been provided at the end of this profile (see Appendix B). This guide should aid in
the interpretation of the tables and figures for Levels of Significant Exposure and the MRLs.
2.2.1 Inhalation Exposure
2.2.1.1 Death
Case studies of methylene chloride poisoning during paint stripping operations have demonstrated that
inhalation exposure can be fatal to humans (Bonventre et al. 1977; Hall and Rumack 1990; Stewart and
Hake 1976). Although quantitative estimates of exposure levels were not reported for these cases, levels
of methylene chloride in various tissues were reported: liver (14.4 mg/dL), blood (51 mg/dL), serum
(29 µg/mL), and brain (24.8 mg/100 g) (Bonventre et al. 1977; Hall and Rumack 1990). The cause of
death in these cases was uncertain; however, myocardial infarction was reported in one case (Stewart and
Hake 1976). Death also occurred in two workers involved in oleoresin extraction processes and liquid
cleaning operations (Moskowitz and Shapiro 1952; Winek et al. 1981). Exposure reportedly occurred
from less than 1 hour up to 3 hours, but the concentration of methylene chloride was not reported. The
compound was detected in the lung (0.1 mL/500 g wet tissue), brain (0.27 g/L), and blood (29.8 mg%)
(Moskowitz and Shapiro 1952; Winek et al. 1981). Two cases of lethal poisoning following acute
inhalation of extremely high concentrations of methylene chloride in air (estimated as up to 168,000 ppm)
occurred in two workers burying barrels containing mixed solvents and solid chemical waste in a well
about 2 meters below ground level (Manno et al. 1992). Methylene chloride concentrations in blood of
the two workers were 572 and 601 mg/L, respectively. Blood carboxyhemoglobin (COHb) concen-
trations were about 30% higher than normal. Death appears to have been caused by narcosis and
respiratory depression due to the acute effects of high concentration methylene chloride on the central
nervous system. One death in the United Kingdom resulting from acute inhalation occupational exposure
to methylene chloride (concentration not provided) was attributed to acute narcosis resulting in
respiratory depression (Bakinson and Jones 1985); necropsy revealed evidence of liver and spleen
congestion. One case of fatal gassing occurred during paint-stripping of a chemical tank (Tay et al.
1995). The paint stripper contained 74% w/w (weight by weight) of methylene chloride; the
concentration of methylene chloride vapor within the tank was later estimated to have been well above
100,000 ppm. The worker did not wear a respirator and did not engage a forced ventilation system into
the confined space of the tank. He was found unconscious and died 4 days later. His methylene chloride
blood concentration was 281 mg/L.
16 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Another case of fatal poisoning was reported in Korea: the body of the chief executive of a painting and
coating factory was discovered in an underground trichloroethylene tank in which methylene chloride was
being used to removed rust from iron sheets (Kim et al. 1996a). Autopsy revealed pronounced organ
congestion in the brain, kidneys, liver, and lungs; blood tissue samples contained 3% COHb and
252 mg/L of methylene chloride. This methylene chloride blood concentration is similar to that reported
for other fatalities (Manno et al. 1992; Tay et al. 1995). Concentrations of methylene chloride in other
tissues ranged from 26 mg/kg in the lungs to 75 mg/kg in the brain (Kim et al. 1996a). The data were
sufficient to confirm that the death was due to methylene chloride poisoning.
In another study, no increase in deaths in methylene chloride workers, assessed by life table analysis, was
found after exposure to 30–120 ppm (time weighted averages) for over 30 years (Friedlander et al. 1978).
Fiber production workers exposed to methylene chloride (140–475 ppm) for at least 3 months did not
have a significant increase in mortality (Lanes et al. 1993; Ott et al. 1983b).
Studies in animals confirm that methylene chloride may be lethal after inhalation exposure at high
concentrations. Acute exposure to 16,000–19,000 ppm of methylene chloride for 4–8 hours caused death
in rats and mice (NTP 1986; Svirbely et al. 1947). Also, one of four female monkeys died after 10 days
of continuous exposure to 5,000 ppm methylene chloride (MacEwen et al. 1972). Data suggest there is a
narrow margin between concentrations causing anesthesia and death. An LC
50
of 16,189 ppm was
reported in mice acutely exposed to methylene chloride (Svirbely et al. 1947). No deaths were found in
mice exposed for 4 hours to 16,800 ppm, but 70% of the mice exposed to 17,250 ppm died (NTP 1986).
Repeated exposure from intermediate to lifetime duration at levels ranging from 1,000 to 16,000 ppm can
cause increased deaths in rats, mice, guinea pigs, rabbits, and dogs (Burek et al. 1984; Heppel et al. 1944;
NTP 1986). Results of the different inhalation studies described in the NTP (1986) report illustrate that
with increasing duration, the lethal exposure level decreases. A 19-day intermittent exposure to
6,500 ppm of methylene chloride or a 13-week exposure to 4,200 ppm were not lethal to rats or mice, but
exposure for 2 years at 4,000 ppm reduced survival in female rats and in mice of both sexes (NTP 1986).
Although the same target organs (central nervous system, lungs, liver) are affected in mammals, the
available mortality data suggest differences in sensitivity among species, with dogs being more sensitive
than mice and rats.
An LC
50
value and all reliable LOAEL values for lethality in each species and duration category are
recorded in Table 2-1 and plotted in Figure 2-1.

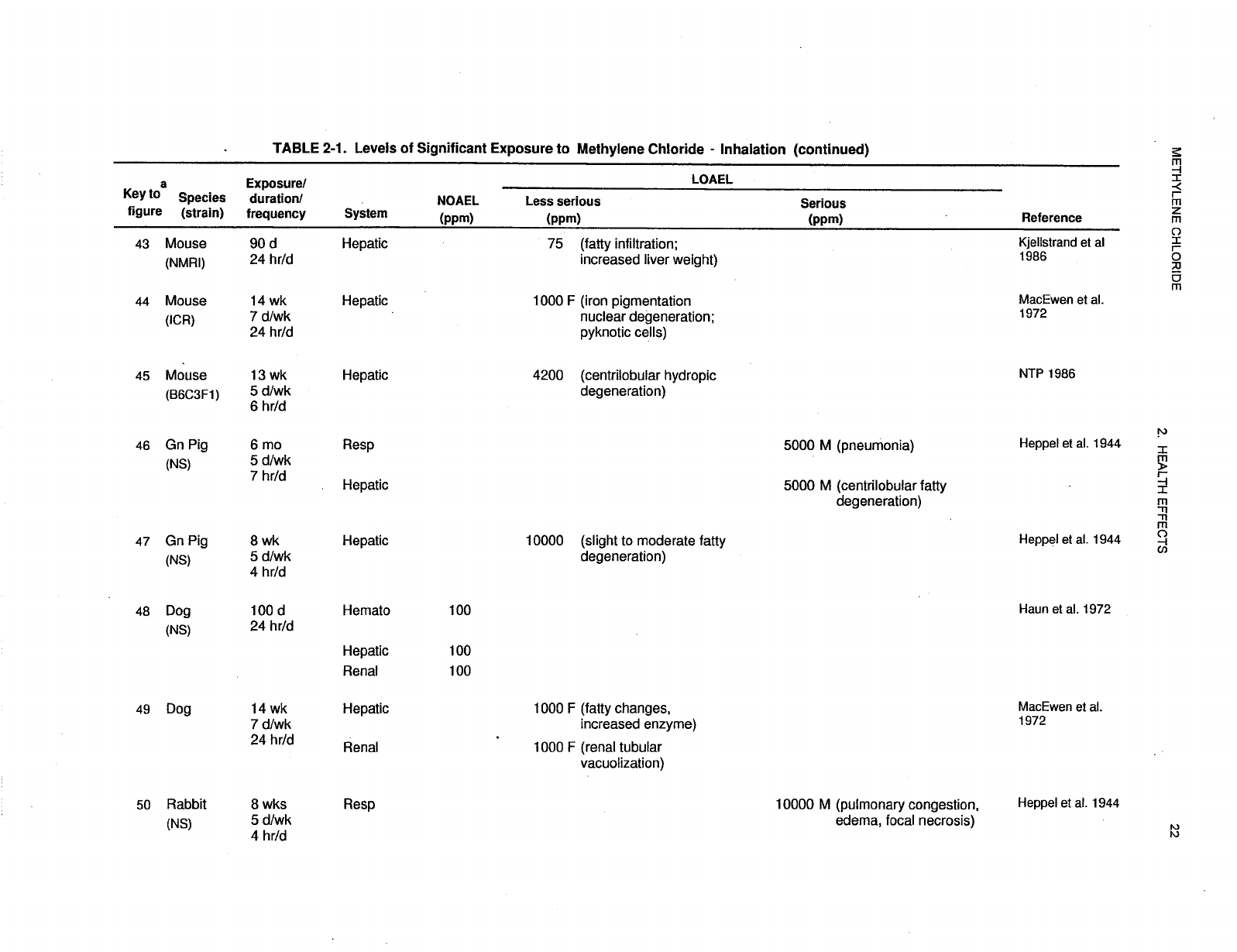
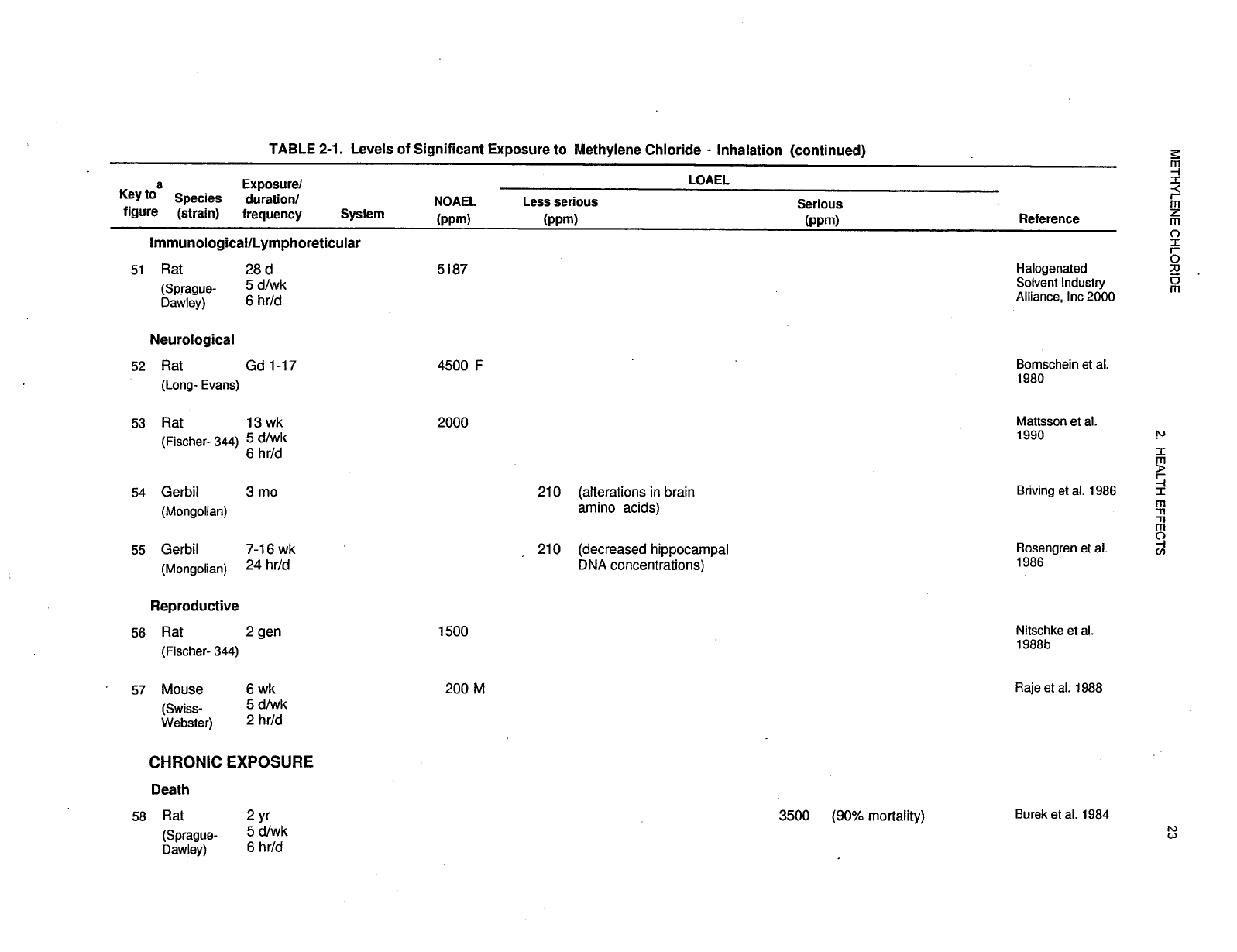
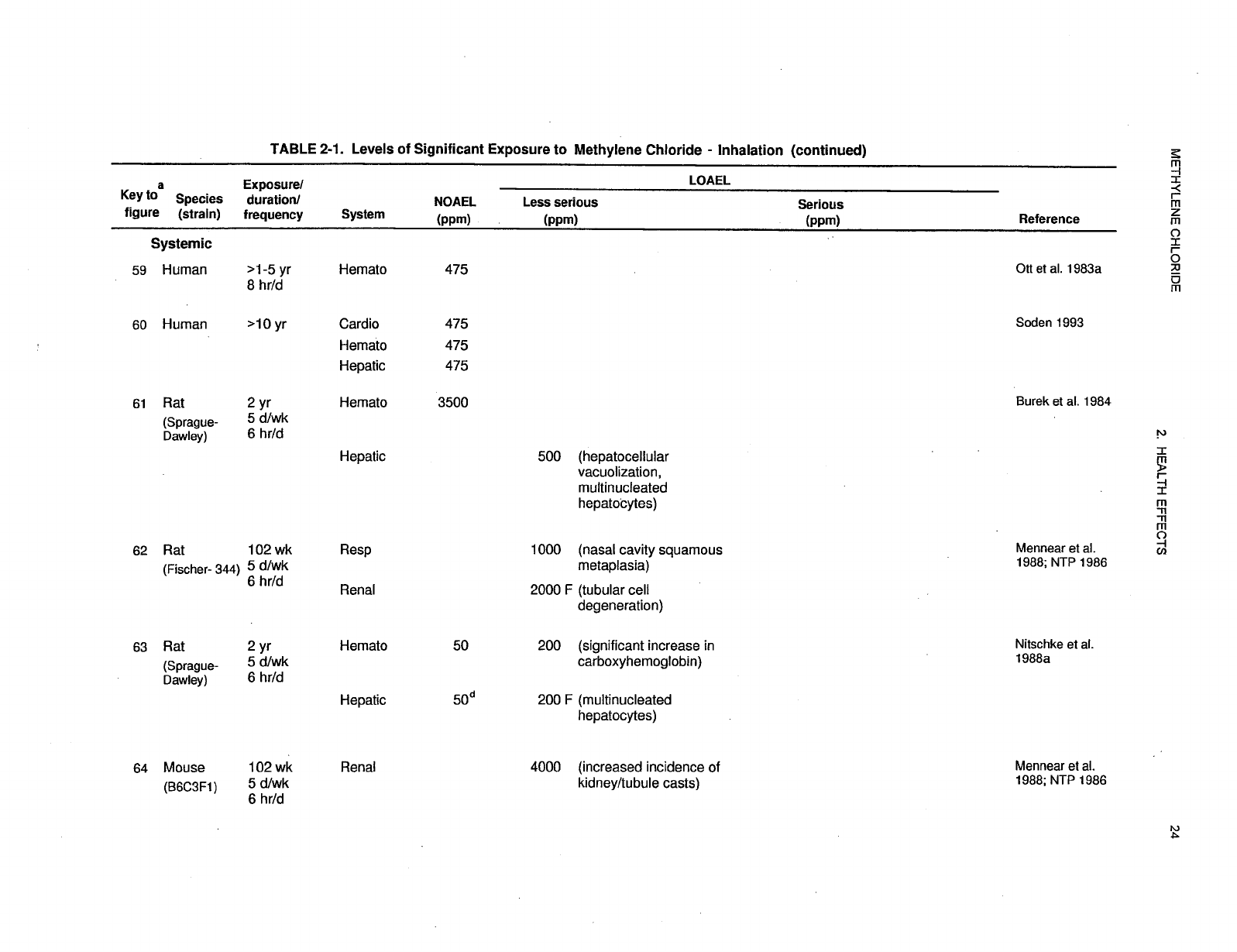
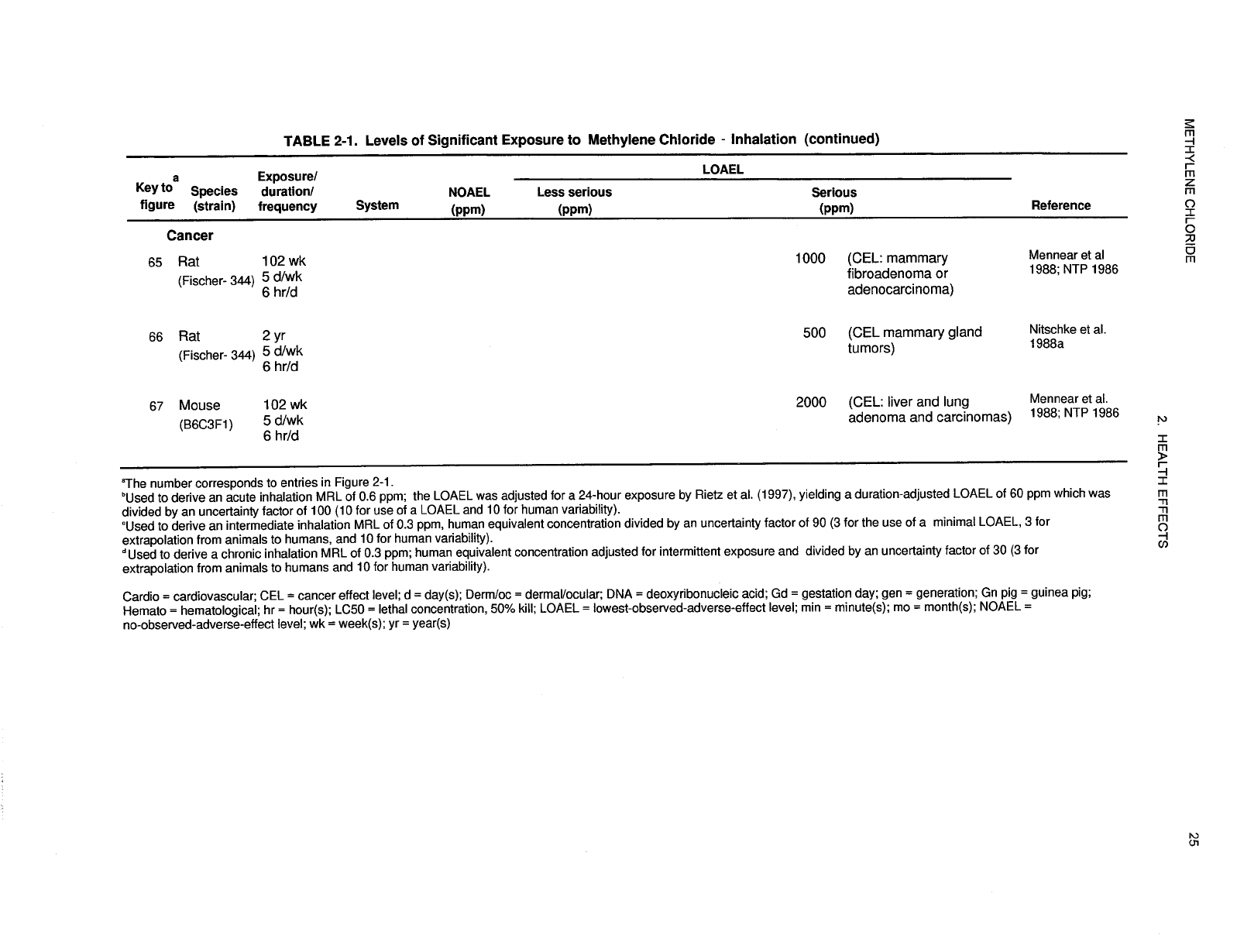

29 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2.2.1.2 Systemic Effects
No studies were located regarding musculoskeletal or dermal effects in humans or animals after inhalation
exposure to methylene chloride. Effects of methylene chloride on the respiratory, cardiovascular,
gastrointestinal, hepatic, renal, and ocular systems are discussed below.
The highest NOAEL values and all reliable LOAEL values for systemic effects in each species and
duration category are recorded in Table 2-1 and plotted in Figure 2-1.
Respiratory Effects. Asphyxia was determined to be the cause of death in the case of a male worker
who was subjected to acute inhalation exposure (concentration unknown) for 1 hour (Winek et al. 1981);
the autopsy revealed bilateral pulmonary congestion with focal hemorrhage. Respiratory symptoms
(cough, breathlessness, chest tightness) were reported in only 4 of 33 cases of acute inhalation exposure
to methylene chloride that were reported to occupational health authorities in the United Kingdom
between 1961 and 1980 (Bakinson and Jones 1985); no exposure levels were provided in this study. No
pulmonary function abnormalities were found in humans exposed to methylene chloride vapors
(50–500 ppm) for 6 weeks (NIOSH 1974). Irritative symptoms of the respiratory tract were more
prevalent among 12 Swedish male graffiti removers, employed to clean underground stations by using
methylene chloride-based solvent, than those of the general population (Anundi et al. 1993). The 8-hour
time-weighted average (TWA) to which these workers were exposed ranged from 18–1,200 mg/m
3
.
Two clinical case studies (Snyder et al. 1992a, 1992b) were reported in which two men who had been
working in confined spaces with a nationally advertised brand of paint remover (consisting of >80% w/w
methylene chloride) presented to the hospital emergency department complaining of dyspnea, cough, and
discomfort in the midchest. In chest x-rays, each of the patients showed alveolar and interstitial
infiltrates. One patient was treated with oxygen and albuterol and his symptoms improved over 48 hours;
a repeat chest x-ray showed complete clearing of the infiltrates. During the next year, the patient
continued to have episodic cough with wheeze and breathlessness which improved with albuterol therapy.
The patient had no prior history of asthma or cough. A methacholine challenge test verified that he had
hyperactive airways. The second patient was treated with oxygen and his symptoms improved during the
next 48 to 72 hours; a repeat chest x-ray taken 3 days later revealed marked, but not complete, resolution
of previously-noted lung infiltrates. Ten days later he was asymptomatic and his chest x-ray was normal.
30 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Pulmonary effects were observed in animals that died following exposure to high concentrations of
methylene chloride (Heppel et al. 1944). Extreme pneumonia was found in 3/14 guinea pigs exposed to
5,000 ppm for up to 6 months, and pulmonary congestion and edema with focal necrosis was found in
3/5 rabbits and 2/16 rats exposed to 10,000 ppm for up to 8 weeks (Heppel et al. 1944). A high incidence
of foreign body pneumonia, involving focal accumulation of mononuclear and multinucleate
inflammatory cells, was observed in 10/20 rats exposed to methylene chloride at 8,400 ppm for 13 weeks
(NTP 1986). The significance of this finding is uncertain since the effect was observed only at the
highest concentration tested. Male B6C3F
1
mice exposed to 4,000 ppm methylene chloride for
6 hours/day, 5 days/week for 13 weeks showed acute Clara cell damage in the lung after a 1-day exposure
to methylene chloride, which appeared to resolve after 5 consecutive daily exposures (Foster et al. 1992).
The appearance and disappearance of the lesion in Clara cells correlated well with the activity of
cytochrome P-450 monooxygenase in Clara cells, as assessed immunocytochemically in the whole lung,
and biochemically in freshly isolated Clara cells. Nasal cavity squamous metaplasia was observed in rats
exposed intermittently to 1,000 ppm methylene chloride in the NTP (1986) bioassay.
Cardiovascular Effects. Studies in humans exposed to methylene chloride vapors between 50 and
500 ppm have not reported significant electrocardiographic abnormalities (Cherry et al. 1981; Ott et al.
1983c; NIOSH 1974). In cohort studies of methylene chloride workers, no increased ischemic heart
disease mortality was observed with chronic time-weighted average exposures from 26 to 1,700 ppm
(Hearne et al. 1990; Lanes et al. 1993; Ott et al. 1983b). There were no differences in cardiac effects, as
measured by a health history questionnaire relating to heart problems (e.g., chest discomfort with
exercise; racing, skipping, or irregular heartbeat), between 150 workers occupationally exposed for more
than 10 years to relatively high levels of methylene chloride (8-hour TWA of 475 ppm), and a similar,
nonexposed group of employees at a polyester staple plant (Soden 1993). The exposed cohort were also
exposed to mean 8-hour TWA concentrations of 900 and 100 ppm of acetone and methanol, respectively.
Data in animals are limited to one study evaluating cardiac arrhythmia in the mouse (Aviado and Belej
1974). Atrioventricular block was observed following acute exposure to methylene chloride at
concentrations greater than 200,000 ppm. Exposure to high concentrations of this sort is not likely to
occur in the environment under normal conditions.
31 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Gastrointestinal Effects. Nausea and vomiting were reported in 13 out of 33 cases of acute
inhalation exposure to methylene chloride that were registered with occupational health authorities in the
United Kingdom between 1961 and 1980 (Bakinson and Jones 1985); concentration levels were not
provided in this study. Dilatation of the stomach was reported in mice after inhalation exposure to
4,000 ppm methylene chloride for 2 years (NTP 1986).
Hematological Effects. In humans, average blood COHb levels measure less than 1% in an
atmosphere free of carbon monoxide, and less than 4% in a normal atmosphere. Blood COHb
concentrations were about 30% higher than normal in two cases of lethal poisoning following acute
inhalation of extremely high concentrations of methylene chloride in air (estimated ~168,000 ppm) in
workers who were burying barrels containing mixed solvents and solid chemical waste in a well about
2 meters below ground level (Manno et al. 1992). Employees monitored at the end of 1 work day
following exposure to methylene chloride at 7–90 ppm (8-hour TWA) had average COHb concentrations
between 1.7 and 4.0% for nonsmokers, and between 4.95 and 6.35% for smokers (Soden et al. 1996).
Additional daily cumulative exposure to methylene chloride did not produce increased levels of COHb.
In volunteers who were exposed to methylene chloride at 200 ppm for 4 hours, blood COHb levels rose to
approximately 5% (Putz et al. 1979); this was equivalent to the levels seen in volunteers after inhaling
70 ppm of carbon monoxide for 4 hours. In nonsmoking volunteers exposed to 50, 100, 150, or 200 ppm
of methylene chloride for 7.5 hours, blood COHb levels rose to 1.9, 3.4, 5.3, and 6.8%, respectively, and
blood COHb levels declined immediately following exposure (DiVincenzo and Kaplan 1981).
Other studies in humans reported increases in the red cell count, hemoglobin, and hematocrit in women
occupationally exposed to concentrations up to 475 ppm during an 8-hour workday, but no effects were
found in men. These effects were judged by the authors to be suggestive of compensatory hematopoiesis
(Ott et al. 1983d). It may be anticipated that stress polycythemia will occur in the majority of individuals,
especially cigarette smokers, who are chronically exposed to methylene chloride vapor concentrations in
the 500 ppm range.
In animals, no significant hematologic or clinical chemistry alterations were reported in dogs and
monkeys exposed continuously to up to 100 ppm methylene chloride for 100 days (Haun et al. 1972). In
the dogs, COHb increased from 0.5 to about 2% during exposure to 100 ppm methylene chloride, but no
significant increase was seen at 25 ppm. In the monkeys, COHb levels were approximately 0.5, 1.7, and
4.5% in controls, 25 ppm, and 100 ppm exposed groups, respectively. No treatment-related effects on
32 METHYLENE CHLORIDE
2. HEALTH EFFECTS
common hematologic parameters (cell counts, hemoglobin concentration differentials, white cell counts,
etc.) were observed among rats chronically exposed to methylene chloride at concentrations up to
3,500 ppm (Burek et al. 1984; Nitschke et al. 1988a).
Musculoskeletal Effects. No musculoskeletal effects have been reported in either animals or humans
after inhalation exposure to methylene chloride.
Hepatic Effects. There is very little published information on the hepatic effects of methylene chloride
in humans. One autoworker, who was exposed to methylene chloride by inhalation and dermally for
1.5 years, was reported to have an enlarged liver in addition to adverse neurological and reproductive
effects (Kelly 1988). NIOSH found workplace levels of methylene chloride to average 68 ppm (range of
3.3–154.4 ppm), which may be an underestimate given evaporation of the volatile liquid from the
applicator pads and cotton gloves that were used; the worker was also exposed to low levels of styrene
(7.2 ppm, range of 1.5–10.4 ppm). Exposure to methylene chloride was verified by a blood COHb level
of 6.4% in a sample taken more than 24 hours after work. The relative contributions of the inhalation and
dermal exposures to the hepatic effect was not determined. There were no alterations in serum enzyme
activity (alkaline phosphatase, alanine aminotransferase [ALT], or lactic dehydrogenase) or in serum
bilirubin, calcium, and phosphorus in humans exposed to methylene chloride vapors (50–500 ppm) for
6 weeks (NIOSH 1974). In a clinical epidemiologic assessment of methylene chloride workers, an
exposure-related increase (not clinically significant) in serum bilirubin was observed in workers exposed
to methylene chloride (up to 475 ppm) and methanol, but there were no concentration-related changes in
serum enzyme levels that could indicate liver injury (Ott et al. 1983a). In Swedish graffiti removers
employed to clean underground stations using methylene chloride solvent, no exposure-related deviations
in serum concentrations of creatinine, aspartate transaminase (AST), ALT, or gamma-glutamyl
transpeptidase were observed (Anundi et al. 1993). Based on these data, the liver appears to be a less
sensitive target organ in humans than it is in rodents (see below).
In animals, the effects of methylene chloride have been studied more extensively. For the most part,
exposure to methylene chloride has resulted in fatty changes in the liver and elevated plasma enzymes.
These effects were reversible when exposure ceased. No histopathological changes were observed in
guinea pigs following acute exposure to 5,200 ppm; however, there was a 2.5-fold increase in hepatic
triglycerides (Morris et al. 1979). When male guinea pigs were exposed to 5,000 ppm of methylene
chloride for up to 6 months, 3/8 died and exhibited moderate centrilobular fatty degeneration of the liver
33 METHYLENE CHLORIDE
2. HEALTH EFFECTS
(Heppel et al. 1944); no deaths, but similar liver histopathology was observed after exposure to
10,000 ppm for 8 weeks (guinea pigs) or 1 week (dogs). Fatty changes in the liver were noted in
monkeys, mice, and dogs continuously exposed to 5,000 ppm for 4 weeks (MacEwen et al. 1972). In
addition, mice exposed to 1,000 ppm exhibited iron pigmentation, nuclear degeneration, and pyknotic
cells (MacEwen et al. 1972). Hepatic microsomal enzymes were elevated at 500 ppm (p<0.01) following
10 days of exposure, but were not increased significantly over control levels in rats exposed to methylene
chloride at 250 ppm for 28 days (Norpoth et al. 1974). Continuous exposure of mice and rats for
100 days to 25 or 100 ppm caused fatty changes in the liver (Haun et al. 1972; Kjellstrand et al. 1986;
Weinstein and Diamond 1972). No effects were seen in mice continuously exposed at 25 ppm, but
cytoplasmic vacuolization was reported in rats at this exposure level (Haun et al. 1972). No adverse liver
effects were reported in dogs or monkeys exposed to up to 100 ppm methylene chloride in the Haun et al.
(1972) study. Using results from the Haun et al. (1972) study, an intermediate inhalation MRL of
0.3 ppm was derived based on the LOAEL of 25 ppm for liver effects in rats. In 13-week studies,
centrilobular hydropic degeneration was observed in female mice exposed to 4,200 ppm, and in both
sexes at 8,400 ppm (NTP 1986). Repeated exposure of rats to 200–500 ppm or greater for 2 years
resulted in increased incidences of hepatocellular vacuolization and multinucleate hepatocytes (Burek et
al. 1984; Nitschke et al. 1988a; NTP 1986), but not at 50 ppm (Nitschke et al. 1988a). In the 2-year NTP
(1986) study, other liver effects in rats included hemosiderosis, focal necrosis of hepatocytes, basophilic
change (females only), hepatocytomegaly, bile duct fibrosis in males, and granulomatous inflammation in
females. The NOAEL of 50 ppm identified in the Nitschke et al. (1988a) study was used as the basis for
derivation of a chronic inhalation MRL of 0.3 ppm.
Renal Effects. Daily exposures to concentrations up to 500 ppm methylene chloride for 6 weeks did
not alter blood urea nitrogen or urine urobilinogen levels in humans (NIOSH 1974). In Swedish graffiti
removers employed to clean underground stations using methylene chloride solvent, no exposure-related
deviations in urinary concentrations of microglobulins or N-acetyl-beta-glucosaminidase were observed
(Anundi et al. 1993).
Renal tubular vacuolization was observed in dogs following continuous inhalation exposure to 1,000 ppm
for 4 weeks and in rats following exposure at 5,000 ppm for 14 weeks (MacEwen et al. 1972).
Nonspecific renal tubular degenerative and regenerative changes were observed after continuous exposure
in rats at 25 and 100 ppm for 100 days (Haun et al. 1972). No significant gross or histopathologic
alterations in the kidneys were reported in dogs or monkeys exposed continuously to up to 100 ppm
34 METHYLENE CHLORIDE
2. HEALTH EFFECTS
methylene chloride for 100 days (Haun et al. 1972). Inhalation exposure to 2,000 ppm for 2 years
resulted in statistically significant increases in the incidences of kidney degeneration in female rats and of
kidney/tubule casts in mice of both sexes (NTP 1986).
Ocular Effects. One human study reported mild eye irritation in males exposed to 500 ppm methylene
chloride vapors after 1 hour (NIOSH 1974). This was most likely due to direct contact of methylene
chloride vapor with the eyes. Irritative symptoms of the eyes were more prevalent among 12 Swedish
male graffiti removers (employed to clean underground stations using a methylene chloride-based
solvent) compared to the general population (Anundi et al. 1993). The 8-hour TWA to which these
workers were exposed ranged from 5 to 340 ppm.
One study reported transient increases in the thickness of the cornea in rabbits that were acutely exposed
to vapors of methylene chloride at 490 ppm (Ballantyne et al. 1976). It is likely that the effects observed
were due to direct effect of vapors on the cornea (see Section 2.2.3.2).
2.2.1.3 Immunological and Lymphoreticular Effects
No studies were located regarding immunological effects in humans after inhalation exposure to
methylene chloride.
Some animal studies have reported alterations in secondary lymphoid organs following exposure to
methylene chloride. Splenic fibrosis was observed in a chronic rat study (Mennear et al. 1988) and
splenic atrophy in an intermediate dog study (MacEwen et al. 1972) at exposure concentrations of
1,000 ppm or greater. The significance of these effects on the spleen as an indicator of chemical insult to
the immune system is not clear since no information was obtained on alterations in germinal cell
proliferation, the lymphocyte subpopulation, or other immunological parameters. For these reasons, these
studies are not presented in Table 2-1. However, a recent study by the Halogenated Solvent Industry
Alliance, Inc. (2000) in which male and female rats were exposed whole body to 5,187 ppm methylene
chloride 6 hours/day, 5 days/week, for 28 days found no evidence of immunotoxicity as judged by gross
and microscopical examination of lymphoid tissues, hematology, or IgM antibody response to sheep red
blood cells (SRBC). This NOAEL is listed in Table 2-1 and plotted in Figure 2-1.
35 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2.2.1.4 Neurological Effects
A number of human studies reveal that the nervous system is perhaps the most important target of acute
methylene chloride toxicity. All 33 cases of acute inhalation exposure to methylene chloride that were
reported to occupational health authorities in the United Kingdom between 1961 and 1980 involved
depression of the central nervous system (Bakinson and Jones 1985). Unconsciousness occurred in 13 of
these cases and other common effects included headache and dizziness; a few instances of confusion,
intoxication, incoordination, and paresthesia were also reported. Acute inhalation exposure to methylene-
chloride-based paint strippers in rooms with inadequate ventilation led to unconsciousness in four cases
and to generalized seizures in one of these (Hall and Rumack 1990); 10/21 respondents to an occupational
health questionnaire reported experiencing dizziness and headache while working in these conditions, but
the symptoms abated when they moved to fresh air. In volunteers, a single 4-hour exposure to 200 ppm
methylene chloride significantly decreased visual and psychomotor performance and auditory function
(Putz et al. 1979). Auditory monitoring, eye-hand coordination, and high-difficulty peripheral brightness
test performances were not degraded until the final hour of exposure, by which time, the level of carbon
monoxide in exhaled breath had risen to 50 ppm and the level of COHb in blood had risen to 5%. A
single 3- to 4-hour exposure to methylene chloride at 300 ppm caused decreased visual and auditory
functions in volunteers, but the adverse effects were reversible once exposure ceased (Fodor and Winneke
1971; Winneke 1974). Winneke (1974) attributed these effects to methylene chloride rather than its
metabolite COHb, since exposure to carbon monoxide at concentrations up to 100 ppm did not cause
similar effects. At the lowest exposure level (300 ppm of methylene chloride), critical flicker fusion
frequency (visual) and auditory vigilance tasks were impaired. These higher-order functions involved
complex visual and central nervous system processes that are assumed to be influenced by the degree of
“cortical alertness” mediated by subcortical structures, especially the reticular formation (Fodor and
Winneke 1971). Similarly, psychomotor performance (reaction time, hand precision, steadiness) was
impaired, but this occurred at higher exposure levels (800 ppm for 4 hours) (Winneke 1974). Since these
parameters are sensitive indicators of overt central nervous system-related depression, drowsiness, or
narcosis, the Winneke (1974) study was selected as an appropriate basis for deriving an MRL for acute
inhalation effects of methylene chloride. Alterations in visual evoked response were observed in humans
exposed to methylene chloride at 515–986 ppm for 1–2 hours (Stewart et al. 1972). In another study,
there were no effects on spontaneous electroencephalogram, visual evoked response, or a battery of
cognitive effects in humans exposed to concentrations of methylene chloride up to 500 ppm (NIOSH
36 METHYLENE CHLORIDE
2. HEALTH EFFECTS
1974). While some changes in tests related to mood have been reported in humans after acute combined
exposure to methylene chloride (28–173 ppm) and methanol (Cherry et al. 1983), no evidence of
neurological or behavioral impairment was observed at exposure levels of 75–100 ppm (Cherry et al.
1981). Dementia and gait impairment were reported in one case of a person exposed to methylene
chloride (500–1,000 ppm) for 3 years (Barrowcliff and Knell 1979). Based on a LOAEL of 300 ppm for
neurological effects (Winneke 1974), an acute inhalation MRL of 0.6 ppm was calculated as described in
Table 2-1 and Section 2.5.
No acute central nervous system effects were observed among 12 Swedish male graffiti removers
employed to clean underground stations using methylene-chloride-based solvent compared to the general
population (Anundi et al. 1993). The 8-hour TWA to which these workers were exposed ranged from
5 to 340 ppm. Two cases of men using a paint remover (>80% methylene chloride by weight) in small
confined spaces were studied by Snyder et al. (1992a, 1992b) in a hospital emergency room. One
reported symptom was severe headache, which disappeared within 24 hours after cessation of exposure.
The authors considered this symptom to be associated with methylene chloride neurotoxicity. No
neurologic effects, as measured by responses to questions relating to neurotoxicity (e.g., recurring severe
headaches, numbness/tingling in hands of feet, loss of memory, dizziness) were reported in a group of
150 employees in a fiber plant occupationally exposed to methylene chloride (mean 8-hour
TWA=475 ppm) for more than 10 years, when compared to a similar, nonexposed cohort (Soden 1993).
In a retrospective epidemiology study, there were no significant associations between potential solvent
exposure and self-reported neurological symptoms (based on a standard battery of medical surveillance
questions) among workers exposed to a variety of solvents, including methylene chloride, at a
pharmaceutical company (Bukowski et al. 1992). However, Bukowski et al. (1992) concluded that
questionnaires were not the most appropriate tool to investigate potential neurobehavioral changes caused
by low-level exposure to solvents, and recommended the use of neurological test batteries. This caveat
would also apply to the study of Soden (1993).
In a group of 34 autoworkers, which included a subgroup of 26 ‘bonders’, complaints of central nervous
system dysfunction were common following occupational exposure to methylene chloride for up to
3 years (Kelly 1988); the precise number of individuals with neurological complaints was not provided,
since the report focused on reproductive effects. The bonding job involved soaking pads from open
buckets of methylene chloride using ungloved hands, so exposures were both by inhalation and by the
dermal route; exposure to methylene chloride was confirmed by analysis of blood COHb levels. In
workplace air samples, NIOSH measured 3.3–154.4 ppm (average=68 ppm) of methylene chloride, but
37 METHYLENE CHLORIDE
2. HEALTH EFFECTS
worker exposure is likely to have been sometimes higher from the evaporation of liquid spilled onto
clothing; bonders were also exposed to styrene at 1.5–10.4 ppm (average=7.2 ppm), which is less than the
threshold limit value (TLV) for that chemical (ACGIH 1999). The neurological complaints reported in
this group included dizziness, lightheadedness, memory loss, personality changes, and depression.
Affected individuals were recommended for further neurological testing, but no test results were reported.
The neurotoxicity of occupational exposure to methylene chloride was examined in a cohort study of
retired airline mechanics who had been chronically exposed to methylene chloride at concentrations
ranging from a mean 8-hour TWA of 105 to 336 ppm, with short-term high exposures ranging from
395 to 660 ppm (Lash et al. 1991). Five categories of variables were assessed: demographic and some
potential confounders, health symptoms, and physiological, psychophysical, and psychological variables.
There were three tests of physiological characteristics (each measuring olfactory, visual, and auditory
parameters) and four tests of psychophysical variables (finger tapping, simple reaction time, choice
reaction time, and complex choice reaction time). Six psychological variables were assessed, most by
more than one test: short-term visual memory, retention measure of visual memory, short-term verbal
memory, and retention measures of verbal memory, attention, and spatial ability. None of the measured
variables were statistically different between the exposed and control groups. However, trends in the
effect sizes appeared within clusters of some variables. In the group of psychological variables showing
effects on memory and attention, the exposed group scored higher than the unexposed group on the verbal
memory tasks but lower on the attention tasks. The major potential confounder was exposure
misclassification. Lack of precision, sampling biases, and random measurement errors might also have
affected the results. However, the authors concluded that overall no effects on the central nervous system
were attributable to chronic, low-level exposures to methylene chloride, a finding they reported as being
consistent with that of Cherry et al. (1981).
White et al. (1995) conducted a 2-year prospective study among workers in the screen printing industry to
investigate the association between exposure to mixed solvents with known neurotoxic properties and
neurobehavioral deficits. Thirty subjects participated in the study which involved medical, demographic,
occupational, and neurological screening, completion of a questionnaire on medical history, life style, and
occupational history, and neurobehavioral assessment using a standard battery of neuropsychological
tests. Subjects were evaluated and tested twice. Air monitoring studies identified workplaces within the
plant that were most exposed to the following chemicals: toluene, methyl ethyl ketone (MEK), mineral
38 METHYLENE CHLORIDE
2. HEALTH EFFECTS
spirits, β-ether, methylene chloride, diacetone alcohol, acetic acid, and lead. A measure of total
hydrocarbon content (THC) was also assessed to account for unidentified solvents in the workplaces.
Generally, exposure to each of the major airborne chemical constituents was below the TLV. Among the
12 subjects classified as having high-acute exposure, performance, adjusted for age and education, was
poorer for tasks involving visual memory and manual dexterity. Whether or to what degree exposure to
methylene chloride contributed to these neurological effects cannot be determined.
Acute studies in animals are consistent with findings in humans that methylene chloride affects the central
nervous system. Narcotic effects of methylene chloride (incoordination, reduced activity, somnolence)
were observed in monkeys, rabbits, rats, and guinea pigs exposed to 10,000 ppm for up to 4 hours
(Heppel et al. 1944); reduced activity was measured in rats exposed to 5,000 ppm (Heppel and Neal
1944). Dogs exposed to 10,000 ppm for 4 hours, first became uncoordinated, then excited and
hyperactive to the extent of bruising themselves, but rapidly recovered afterwards (Heppel et al. 1944).
Somatosensory-evoked potentials were altered in rats after 1 hour of exposure to methylene chloride at
concentration levels of 5,000 ppm or greater (Rebert et al. 1989). Decreased levels of succinate
dehydrogenase were measured in the cerebellum of rats exposed to 500 ppm of methylene chloride for
2 weeks (Savolainen et al. 1981).
Changes in neurotransmitter amino acids and brain enzymes were observed in gerbils after continuous
exposure to 210 ppm for 3 months (Briving et al. 1986; Karlsson et al. 1987; Rosengren et al. 1986). The
DNA concentration decreased in the hippocampus and cerebellum in gerbils exposed to $210 ppm of
methylene chloride, indicating decreased cell density in these brain regions, probably due to cell loss
(Karlsson et al. 1987; Rosengren et al. 1986). Methylene chloride (4,500 ppm) did not affect wheel
running activity and avoidance learning in rats born to dams exposed prior to and/or during gestation
(Bornschein et al. 1980). No treatment-related alterations in sensory evoked potentials, reflexes, posture,
or locomotion were observed in rats exposed at 2,000 ppm (Mattsson et al. 1990).
The highest NOAEL values and all reliable LOAEL values for neurological effects for each species and
duration category are recorded in Table 2-1 and plotted in Figure 2-1.
39 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2.2.1.5 Reproductive Effects
Exposure to methylene chloride has been reported to result in adverse reproductive effects in humans and
animals.
One group of case studies reported reproductive effects in 8 out of 34 men who complained of central
nervous system dysfunction following occupational exposure to methylene chloride for 0.4–2.9 years
(Kelly 1988). The eight men (ages 20–47) were part of a subgroup of 26 ‘bonders’ who applied
methylene chloride to automobile parts during assembly; this involved soaking pads from open buckets of
methylene chloride using ungloved hands, so exposures were both by inhalation and by the dermal route.
In workplace air samples, NIOSH measured 3.3–154.4 ppm (average=68 ppm) of methylene chloride, but
worker exposure is likely to have been sometimes higher from the evaporation of liquid spilled onto
clothing; bonders were also exposed to styrene at 1.5–10.4 ppm (average=7.2 ppm), which is less than the
TLV for that chemical (ACGIH 1999). Exposure to methylene chloride within the group was suggested
by blood COHb levels of 1.2–11% for six nonsmokers and 7.3 and 17.3% for two smokers. All eight men
had recent histories of infertility and complained of genital pain (testicular, epididymal, and/or prostatic);
the testes were atrophied in two workers (ages 27 and 47). Infectious disease was eliminated as a cause of
these reproductive effects. Four men who submitted to testing had reduced sperm counts and the
proportion of sperm with abnormal morphology ranged from 38 to 50%. Uncertainty regarding this study
involves the small number of subjects, the multiple exposure to other organic chemicals, the lack of blood
or urine samples quantifying exposure to methylene chloride, and the lack of a control group. No other
studies were located that reported similar male reproductive effects from exposure to methylene chloride.
Contrary to Kelly’s (1988) findings, Wells et al. (1989) found no evidence of oligospermia in four
workers who had been exposed to levels of methylene chloride that were twice as high as in the Kelly
study, while involved in furniture stripping for at least 3 months. It is not certain whether the longer
duration of exposure (minimum occupational exposure 1.4 years) or exposure to other chemicals in the
Kelly (1988) study contributed to the different outcomes in the two studies.
In a retrospective study of pregnancy outcomes among Finnish pharmaceutical workers during the late
1970s, female workers at eight factories had a higher rate of spontaneous abortions compared to the
general population (Taskinen et al. 1986). It is likely that the women were exposed to multiple solvents,
including methylene chloride, prior to conception, as well as during pregnancy. In the case-control study
40 METHYLENE CHLORIDE
2. HEALTH EFFECTS
of 44 pharmaceutical workers who had spontaneous abortions, exposure to various solvents, including
methylene chloride, was associated with a slightly higher abortion rate (Taskinen et al. 1986). Exposure
to methylene chloride was associated with an odds ratio (OR) for spontaneous abortion of 2.3, but the
increase was of borderline significance (p=0.06; 95% confidence interval [CI]=1.0–5.7). In a logistic
regression model, the odds ratio of spontaneous abortion increased with increasing frequency of exposure
to methylene chloride, but the sample size was too small for statistical significance. In addition, smoking
and alcohol consumption were not considered. The authors mentioned that improved industrial hygiene
procedures that eliminated solvent vapors in the workplace may have contributed to an overall decline in
rates of spontaneous abortion among pharmaceutical workers during the period in question.
No adverse effects on reproduction were observed in rats exposed to concentrations up to 1,500 ppm of
methylene chloride for two generations (Nitschke et al. 1988b). In dominant lethal tests involving male
mice exposed to #200 ppm methylene chloride for up to 6 weeks, no microscopic lesions were found in
the testes (Raje et al. 1988). Uterine, ovarian, and testicular atrophy was observed in rats and mice
exposed to vapors of methylene chloride (4,000 ppm) for 2 years (NTP 1986), but the authors considered
this effect to be secondary to malignant hepatic and alveolar neoplasms, as described in Section 2.2.1.8
Cancer. Existing data suggest reproductive toxicity may occur following chronic exposure to relatively
high concentrations of methylene chloride.
The highest NOAEL values and all reliable LOAEL values for each study for reproductive effects are
recorded in Table 2-1 and plotted in Figure 2-1.
2.2.1.6 Developmental Effects
The only information reported on potential developmental effects of methylene chloride in humans was
part of retrospective study of pregnancy outcomes among Finnish pharmaceutical workers (Taskinen et al.
1986). In the case-control study of these workers, exposure to methylene chloride (probably both before
and during pregnancy) was associated through an odds-ratio prediction with a higher risk of spontaneous
abortion (Taskinen et al. 1986). (See Section 2.2.1.5 Reproductive Effects for additional details).
In a study examining the relationship between birth weights and environmental exposures to methylene
chloride from Kodak manufacturing processes in Monroe Country, New York, no significant adverse
41 METHYLENE CHLORIDE
2. HEALTH EFFECTS
effects on birth weight were found among 91,302 single births from 1976 through 1987 (Bell et al. 1991).
The highest predicted environmental concentration of methylene chloride was 0.01 ppm, which is
substantially lower than levels found in occupational settings, but significantly higher than the average
ambient background level of 50 parts per trillion.
A study in rats demonstrated that methylene chloride crosses the placental barrier (see Section 2.3.2.1;
Anders and Sunram 1982). No treatment-related visceral abnormalities were reported in fetuses of mice
and rats exposed to 1,250 ppm of methylene chloride during gestation, but an increase in the incidence of
minor skeletal variants (e.g., delayed ossification of sternebra or extra sternebrae) was observed in both
species; rats also exhibit an increased incidence of dilated renal pelvis. A maternal effect of increased
liver weight was observed (Schwetz et al. 1975). When rats were exposed to 4,500 ppm, maternal liver
weights increased and fetal body weights decreased, but teratogenic effects were not observed and
viability and growth were not affected (Bornschein et al. 1980; Hardin and Manson 1980). Wheel
running activity and avoidance learning were not affected in rats born to dams exposed prior to and/or
during gestation to methylene chloride at 4,500 ppm (Bornschein et al. 1980). Longer-term exposure (for
two generations) to concentrations of 1,500 ppm of methylene chloride did not affect neonatal survival or
neonatal growth in rats (Nitschke et al. 1988b). Although fetal body weights were decreased, the absence
of other fetotoxic effects, major skeletal variants, or significant embryolethality suggests that
developmental toxicity is not a major area of concern following exposure to methylene chloride.
The highest NOAEL values and all reliable LOAEL values for developmental effects for each species and
duration category are recorded in Table 2-1 and plotted in Figure 2-1.
2.2.1.7 Genotoxic Effects
No studies were located regarding genotoxic effects in humans after inhalation exposure to methylene
chloride.
Results of in vivo assays following inhalation exposure in animals were mixed. Inhalation exposure of
mice to methylene chloride for 10 days at concentrations of 4,000 ppm or higher resulted in significant
increases in frequencies of sister chromatid exchanges in lung cells and peripheral blood lymphocytes,
chromosomal aberrations in lung and bone marrow cells, and micronuclei in peripheral blood erythrocytes
(Allen et al. 1990). However, no evidence of chromosomal abnormalities was seen in bone marrow cells
42 METHYLENE CHLORIDE
2. HEALTH EFFECTS
of rats exposed by inhalation to concentrations up to 3,500 ppm for 6 months (Burek et al. 1984). The
difference in responses observed may be due, in part, to species differences or test methodology. The
relevance of these two studies to clastogenic mechanisms in humans is not certain.
More recent in vivo genotoxicity studies have attempted to elucidate the mechanism(s) of genotoxic
action of methylene chloride. Casanova et al. (1992) pre-exposed male B6C3F
1
mice and Syrian Golden
hamsters for 2 days (6 hours/day) to 4,000 ppm of methylene chloride. On the third day, animals were
exposed for 6 hours to a decaying concentration of [
14
C] methylene chloride and then examined for the
presence of DNA-protein crosslinks. DNA-protein crosslinks were detected in mouse liver, but not in
mouse lung, hamster liver, or hamster lung. Similar results were observed in a second experiment
(Casanova et al. 1996). B6C3F
1
mice, exposed to methylene chloride for 6 hours/day for 3 days at
concentrations ranging from approximately 500 to 4,000 ppm formed DNA-protein crosslinks in the liver.
The formation of DNA-protein crosslinks was a nonlinear function of airborne concentrations of
methylene chloride. In addition, mice exposed for 6 hours/day for 3 days to concentrations ranging from
approximately 1,500 to 4,000 ppm showed an increased rate of DNA synthesis in the lung, indicating cell
proliferation, but increased cell turnover was not detected in mouse lung at exposure concentrations of
150 or 500 ppm. Hamsters showed no evidence of cell proliferation in the lung at any concentration, nor
did cell proliferation occur in the livers of either species.
Devereux et al. (1993) analyzed liver and lung tumors induced in female B6C3F
1
female mice by
inhalation of 2,000 ppm of methylene chloride for 6 hours/day, 5 days/week exposure for up to
104 weeks for the presence of activated ras proto-oncogenes. In methylene chloride-induced liver
tumors, mutations, mainly transversions or transitions in base 1 or base 2, were detected, and were similar
to those observed for the H-ras gene in spontaneous liver tumors. Mutations were also identified in the
lung. The K-ras activation profiles in the methylene chloride-induced tumors were not significantly
different from those in spontaneously occurring tumors. No other transforming genes were found in the
nude mouse tumorigenicity assay. The authors were unable to identify any transforming genes other than
ras genes in either mouse liver or lung tumors. Based on liver tumor data, they suggested that methylene
chloride may affect the liver by promoting cells with spontaneous lesions.
Hegi et al. (1994) generated allelotypes of 38 methylene chloride-induced lung carcinomas from female
B6C3F
1
mice exposed 6 hours/day, 5 days/week for 2 years to 2,000 ppm. The allelotypes were
examined for various genotoxic endpoints, and the results compared to genotoxicity findings in two other
43 METHYLENE CHLORIDE
2. HEALTH EFFECTS
reciprocal-cross mouse strains. Throughout the genome, allelic losses occurred infrequently, except for
markers on chromosome 4, which were lost in approximately half of the carcinomas. In lung adenomas,
chromosome 4 losses were associated with malignant conversion. Methylene chloride-induced liver
tumors did not demonstrate chromosome 4 loss, which indicated that this finding was specific for lung
carcinomas. Preferential loss of the maternal chromosome 4 was also observed in carcinomas in B6C3F
1
mice. On chromosome 6, an association between K-ras gene activation and allelic imbalances was also
found in B6C3F
1
mouse lung tumors. When allelotypes of tumors in mice from two reciprocal cross
strains, AC3F
1
and C3AF
1
, were examined and compared with the findings in B6C3F
1
mice, one allele of
the putative chromosome 4 tumor suppressor gene was shown to be inactivated. Whereas the results in
B6C3F
1
mice suggested that nondisjunction events were responsible for the chromosome 4 losses, tumors
from both reciprocal-cross mouse strains appeared to show small interstitial deletions in a chromosomal
region that is homologous with a region in human chromosomes which is often lost in a variety of tumors,
including lung cancers. In human chromosomes, a candidate tumor suppressor gene, MTS1, is located in
this region.
In another genotoxic analysis with the same cohort (Hegi et al. 1994), loss of heterozygosity at markers
near the p53 gene on chromosome 11 and within the retinoblastoma tumor suppressor gene were
examined in methylene chloride-induced liver and lung tumors and compared to spontaneous tumors in
control mice. The authors concluded that inactivations of p53 and the retinoblastoma tumor suppressor
gene were infrequent events in lung and liver tumorogenesis in mice exposed to methylene chloride.
Replicative DNA synthesis was examined by Kanno et al. (1993) to evaluate the potential role of
treatment-induced lung cell proliferation on pulmonary carcinogenicity in female B6C3F
1
mice exposed
to 2,000 or 8,000 ppm of methylene chloride for 6 hours/day, 5 days/week for 2 years. By the end of the
study, there was a statistically significant increase in lung tumors in exposed animals when compared to
controls. Cell proliferation was assessed in the lung after 1, 2, 3, or 4 weeks of inhalation exposure to
2,000 or 8,000 ppm, and after 13- and 26-week exposures to 2,000 ppm, as measured by changes in
labeling indices (LI). The LI of both bronchiolar epithelium and terminal bronchioles were substantially
decreased in mice exposed to 2,000 ppm of methylene chloride for 2–26 weeks. Similar findings, but not
as severe, were observed in mice exposed to 8,000 ppm. The decreases in LI were not accompanied by
cytotoxicity. The authors concluded that high-concentration exposure to methylene chloride for up to
26 weeks reduces cell proliferation in lung epithelial cells in female B6C3F
1
mice.
44 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Maronpot et al. (1995b) assessed replicative DNA synthesis after 13, 26, 52, and 78 weeks of inhalation
exposure of female B6C3F
1
mice to 2,000 ppm of methylene chloride for 6 hours/day, 5 days/week. A
statistically significant decrease in the hepatocyte LI was only observed at 13 weeks. In lung epithelial
cells, the results were similar to those observed by Kanno et al. (1993). No increases in replicative DNA
synthesis were found in liver foci cells or lung parenchymal cells. K-ras gene activation in liver tumors
and H-ras gene activation in lung tumors did not differ among methylene chloride-induced tumors and
those observed in control animals. The authors concluded that these oncogenes were not involved in
mouse tumorigenesis.
Other genotoxicity studies are discussed in Section 2.5.
2.2.1.8 Cancer
No excess risk of death from malignant neoplasms has been detected in workers exposed to methylene
chloride at levels up to 475 ppm (Friedlander et al. 1978; Hearne et al. 1987, 1990; Lanes et al. 1993; Ott
et al. 1983a). Lanes et al. (1990) reported excess mortality associated with cancer of the buccal cavity
and pharynx (combined), and liver and biliary passages (combined) in workers occupationally exposed to
methylene chloride (#1,700 ppm) in the cellulose fiber production industry for more than 20 years.
Although the actual number of cases was small, the excess mortality for the combined liver/biliary cancer
cases was statistically significant (standard mortality rate [SMR]=5.75; 95% CI=1.82–13.78); since three
of the four deaths were biliary cancer, the SMR was 20 (95% CI=5.2–56.0) for that site alone (Lanes et al.
1990). In a follow-up study of the same cohort (Lanes et al. 1993), no new cancer cases were found, but
there was still an excess mortality for the cohort (SMR=2.98; 95% CI=0.81–7.63). Lanes et al. (1993)
concluded that the excess death in this cohort from liver/biliary cancer was “statistically unstable”, but
warranted further monitoring.
Tomenson et al. (1997) studied mortality due to cancer in a group of workers occupationally exposed to
methylene chloride vapors at a mean exposure concentration of 19 ppm (8-hour TWA); the average
length of employment was 9 years. Compared to national and local rates, the occupationally-exposed
group had lower rates for all cancers, including those of the liver, lung, pancreas, and biliary tract. The
authors suggested that the significant reduction in mortality due to lung cancer was likely associated with
restrictions on smoking in the workplace.
45 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Gibbs et al. (1996) examined causes of mortality in an occupationally exposed group of workers similar
to that studied by Lanes et al. (1990, 1993). Exposure was classified categorically, with the airborne
concentrations in the high-exposure group ranging from 350 to 700 ppm, and in the low exposure group
from 50 to 100 ppm. Because there were no jobs in this cohort that did not involve methylene chloride
exposure, a “0” exposure category was created as an internal control. There was no increase in mortality
from cancers of the lung, liver, pancreas, or biliary tract. An unexpected finding was a concentration-
related increase in mortality from prostate cancer among men with 20 or more years of employment.
Another unexpected finding was an increase in mortality from cervical cancer among women with 20 or
more years of employment, although the increase in women was not concentration-related. These results
are not consistent with those from other studies. It should be noted that the confidence intervals were
very large and no potentially confounding variables were measured. The authors concluded that the
results were difficult to interpret biologically and required further investigation.
In a case control study, Heineman et al. (1994) evaluated exposures of men in the petroleum refining and
chemical manufacturing industries to chlorinated aliphatic hydrocarbons (CAHs), including methylene
chloride, as potential risk factors for astrocytic brain tumors. Job-exposure matrices for six individual
CAHs, including methylene chloride, and for total organic solvents, were developed by estimating the
probability of exposure and the frequency and magnitude of exposure to CAH solvents by industry and
by job classification, based on likely solvent usage over 6 decades (1920–1980). An increase in the
incidence of mortality due to astrocytic brain cancer was observed for exposure to four CAHs (carbon
tetrachloride, methylene chloride, tetrachloroethylene, and trichloroethylene); the strongest association
was with methylene chloride. In occupations judged to be associated with methylene chloride exposure,
risk of astrocytic brain cancer increased with increasing exposure, as measured by the job exposure matrix
in conjunction with duration of employment. The authors stated that these trends could not be explained
by exposures to the other solvents.
As first evidence of such an association, these results should be interpreted very cautiously. The principal
limitation of the study was the lack of direct information on exposure to solvents; no quantitative
measurements were made, nor were specific-use records available. Instead, qualitative estimates of
exposure were made by industrial hygienists based on work histories provided by next-of-kin; there were
no workplace records. A lack of quantitative information on the use of specific solvents in various
occupations, the ability of solvents to be used interchangeably for many industrial applications, and the
use of multiple, or mixtures of, solvents also contributed to a high potential for exposure
46 METHYLENE CHLORIDE
2. HEALTH EFFECTS
misclassification. The authors stated that “few individual risks were statistically significant and most
confidence intervals were broad”; interpretation of the results was based on “patterns of trends”
(Heineman et al. 1994). The authors added that “...the trends and consistency of the methylene chloride
and brain cancer association suggest that chance seems unlikely to entirely explain the results.”
In another case-control study, Cocco et al. (1999) examined the occupational risk of central nervous
system cancer among women, using a study design similar to that described for Heineman et al. (1994).
From death certificates in a U.S. 24-state database for the period 1984–1992, the authors identified 12,980
cases of cancers of the brain and other parts of the central nervous system. For each case, four controls
were selected among women who died from nonmalignant diseases (excluding neurological disorders),
frequency-matched by state, race, and 5-year age-group. Job exposure matrices were developed for 11
occupational hazards, including methylene chloride. An estimate of intensity of exposure was developed
for each occupation and industry listed in the U.S. Census code. A final intensity level score and a
probability of exposure score were then developed for each occupation/industry combination appearing in
death certificates of the study subjects. The ORs were calculated with logistic regression for each
workplace exposure, adjusting for marital status, socioeconomic status, and age at death. The authors
found that potential exposure to methylene chloride was associated with a modest, but statistically
significant, 20–30% increase in risk of mortality from central nervous system cancer (OR=1.2; 95%
CI=1.1–1.2). However, the authors characterized the association as “equivocal”, since risk did not show
a clear increase by probability or intensity of exposure. Admitted weaknesses of the study include poor
occupational information in the death certificates, possible diagnostic bias among lower socioeconomic
status cases, and the absence of more detailed information to supplement that provided in the death
certificates.
Cantor et al. (1995) conducted a case control study with 33,509 cases and 117,794 controls, matched for
age, gender, and race, to investigate the association between occupational exposure to workplace
chemicals and the incidence of breast cancer in women. Exposure was indirectly estimated using a job
exposure matrix that ranked the probability and level of 31 workplace exposures, among them methylene
chloride. All workplace chemical exposures were evaluated separately. After adjusting for
socioeconomic status imputed from occupation, the OR in the highest exposure category of methylene
chloride (probability and level of exposure combined) was slightly greater than 1.0, and statistically
significant (OR=1.46, CI=1.2–1.7). The authors caution that this analysis is crude and should be
considered a first-level “hypothesis-generating” evaluation rather than one which is “hypothesis testing.”
47 METHYLENE CHLORIDE
2. HEALTH EFFECTS
They also noted that the study had numerous methodologic limitations, including the use of death
certificates as the primary source of individual information and the use of a job-exposure matrix rather
than quantitative workplace measurements to estimate exposure. Additionally, no adjustments were made
for other risk factors such as smoking, obesity, family history, duration of employment, or other
confounding variables.
In rats exposed to low levels of methylene chloride (100 ppm) for 2 years, there was a nonsignificant
increase in the total incidence of malignant tumors (Maltoni et al. 1988). In mice and rats, inhalation of
very high levels of methylene chloride significantly increased the incidence of liver and lung cancer
(Mennear et al. 1988; NTP 1986) and benign mammary gland tumors (fibroadenomas or adenomas)
(Mennear et al. 1988; Nitschke et al. 1988a; NTP 1986).
In the NTP (1986) study, groups of 50 animals of each sex were exposed to methylene chloride by
inhalation 6 hours/day, 5 days/week for 102 weeks. F344/N rats were exposed to 0, 1,000, 2,000, or
4,000 ppm of methylene chloride and B6C3F
1
mice were exposed to 0, 2,000, or 4,000 ppm. At or above
2,000 ppm, the incidence of liver tumors (mostly hepatocellular adenomas or carcinomas) in mice was
significantly higher than in chamber and historical control groups (NTP 1986); at 4,000 ppm, the
incidence of liver tumors was highly significant (p#0.001). The incidence of combined benign and
malignant liver tumors was high (67–83%) in the treated animals. There was also a statistically
significant increase in the incidence of lung tumors in mice (p<0.001) exposed at 2,000 ppm or above;
these tumors were primarily alveolar/bronchiolar adenomas or carcinomas. The incidence of combined
benign and malignant lung tumors was 54–85% in the treated animals. The NTP (1986) report concluded
that there was “some evidence of carcinogenicity” in male rats and “clear evidence of carcinogenicity” in
female rats, based on the increased incidence of benign mammary neoplasms following 2 years of
inhalation exposure to methylene chloride. The report concluded that there was “clear evidence for
carcinogenicity” for methylene chloride chronic inhalation exposure, based on the increased incidence of
alveolar/bronchiolar neoplasms and of hepatocellular neoplasms.
In two related studies, Kari et al. (1993) and Maronpot et al. (1995b) examined the progressive
development of lung and liver tumors in B6C3F
1
mice exposed via chamber inhalation to 2,000 ppm
methylene chloride for 6 hours/day, 5 days/week, for 104 weeks. In addition, a series of stop exposure
experiments were performed to evaluate the effects of differing exposure durations on tumor
development. Kari et al. (1993) examined histology and histopathology of lung and liver tumors, whereas
48 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Maronpot et al. (1995b) evaluated DNA synthesis and oncogene expression during tumor development.
(mechanistic aspects of Maronpot et al. (1995b) are discussed in Sections 2.2.1.7 and 2.4.2). Chronic
high-concentration exposure to methylene chloride resulted in: (1) an 8-fold increase in the incidence of
animals having lung adenomas or carcinomas as compared to controls; (2) a 13-fold increase in the total
number of lung tumors in each animal at risk; (3) a 2.5-fold increase in the incidence of mice having liver
adenomas or carcinomas compared to controls; and (4) a 3-fold increase in the number of liver tumors in
each animal at risk. The development of the first lung tumors in methylene chloride exposed mice
occurred 1 year earlier than in control animals. In contrast, there was no difference in the latency to first
liver tumor period between exposed and control animals. The incidences of tumors in lungs, but not liver,
continued to increase after cessation of exposure. Maronpot et al. (1995b) found that 26 weeks of
exposure was sufficient to significantly and irreversibly increase the incidence of lung tumors at 2 years,
whereas the incidence of hepatic tumors increased with 78 weeks of exposure, but not with 25 or
52 weeks of exposure. Furthermore, vulnerability to methylene chloride may have been age-related, since
no lung tumor increase was observed in mice that were kept under control conditions for 52 weeks prior
to methylene chloride exposure for 52 weeks. Based on these results, Kari et al. (1993) and Maronpot et
al. (1995b) concluded that methylene chloride is a more potent lung than liver carcinogen in female
B6C3F
1
mice; the differing incidence of lung and liver tumors under various exposure regimes suggests
that the mechanisms of tumorigenesis in these target organs may be different.
The EPA (1985b) reviewed the NTP data on the carcinogenic effects of methylene chloride and
calculated a human potency estimate. The potency factor (q
1
*) which represents a 95% upper confidence
limit of the extra lifetime human risk, is 1.4x10
-2
(mg/kg/day)
-1
. The unit risk estimate (the excess cancer
risk associated with lifetime exposure to 1 µg/m
3
) for inhalation exposures is 4.1x10
-6
. The EPA (1987a,
1987b) lowered this risk estimate to 4.7x10
-7
µg/m
3
on the basis of pharmacokinetics data reported by
Andersen et al. (1987). Based on this value, cancer risk levels of 10
-4
, 10
-5
, 10
-6
, and 10
-7
correspond to
70 years of continuous exposure to 0.06, 0.006, 0.0006, and 0.00006 ppm, respectively. The predicted
cancer risks are considered conservative upper estimates. The actual risk of cancer is unlikely to be
higher and may be substantially lower. These values are recorded in Figure 2-1. EPA is planning to re-
evaluate potential human risks associated with inhalation exposure to methylene chloride based on new
mechanistic data and more recent pharmacokinetic modeling using tissue dosimetry (see Sections 2.3.5
and 2.4).
49 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2.2.2 Oral Exposure
2.2.2.1 Death
Hughes and Tracey (1993) reported a case in which a woman ingested 300 mL of Nitromors, a paint
remover solvent containing 75–80% methylene chloride, and died 25 days later. Ingestion of this paint
remover is known to cause severe corrosion of the gastrointestinal tract, and the autopsy revealed that
death was due to the corrosive effects of the paint remover rather than to the metabolic consequences of
methylene chloride ingestion.
Acute oral LD
50
values of 2,100 (Kimura et al. 1971) and 2,300 mg/kg (Marzotko and Pankow 1987)
were reported for methylene chloride in rats. Ninety-five percent lethality was reported in rats dosed with
4,382 mg/kg of methylene chloride (Ugazio et al. 1973). The cause of death appeared to be respiratory
failure as a result of depression of the central nervous system. Statistically significant increases in
mortality occurred among male rats gavaged with 320 mg/kg/day of methylene chloride and among male
and female mice receiving $64 mg/kg/day for more than 36 weeks (Maltoni et al. 1988).
LD
50
values and LOAEL values for lethality in each species and duration category are recorded in
Table 2-2 and plotted in Figure 2-2.
2.2.2.2 Systemic Effects
No studies were located regarding respiratory, cardiovascular, musculoskeletal, or dermal effects in
humans or animals following oral exposure to methylene chloride. Gastrointestinal, hematological,
hepatic, renal, endocrine, and metabolic effects after oral exposure to methylene chloride are discussed
below.
The highest NOAEL values and all reliable LOAEL values for systemic effects in each species and
duration category are recorded in Table 2-2 and plotted in Figure 2-2.
Gastrointestinal Effects. A fatal single oral dose of Nitromors, a paint solvent containing 75–80%
methylene chloride resulted in severe corrosion of the gastrointestinal tract, perforation, peritonitis,
septicemia, and death (Hughes and Tracey 1993). A man who ingested 1–2 pints of Nitromors in a

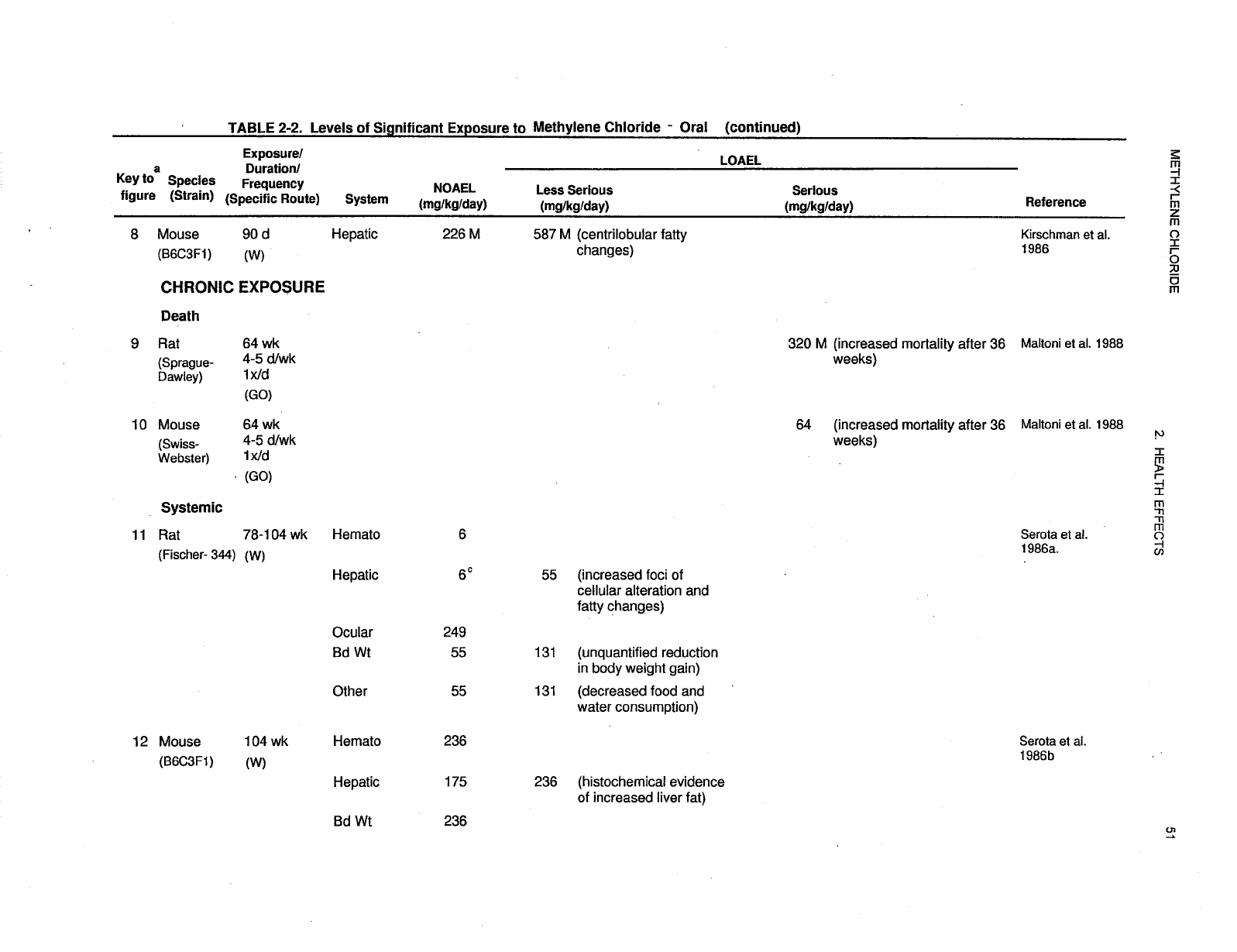
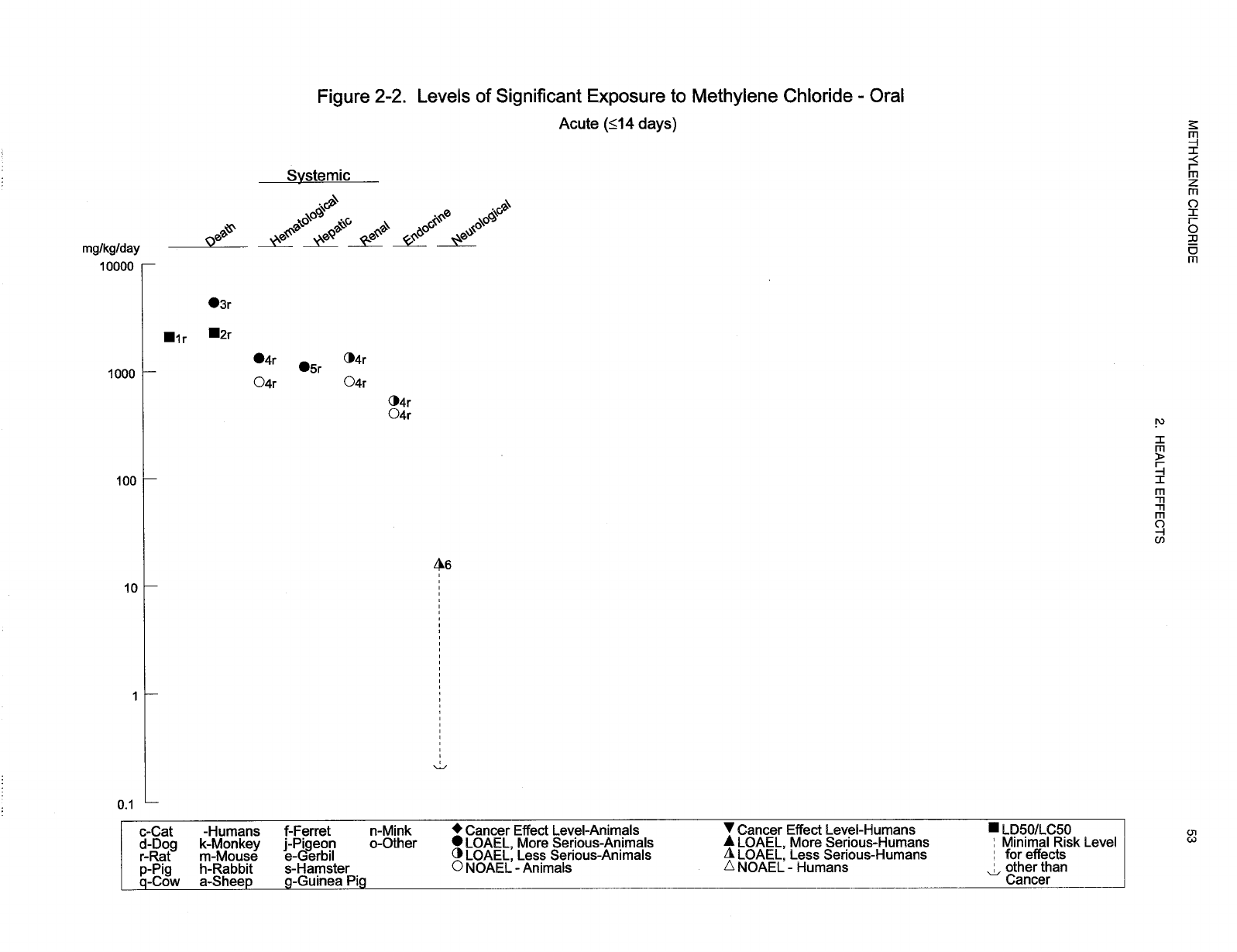

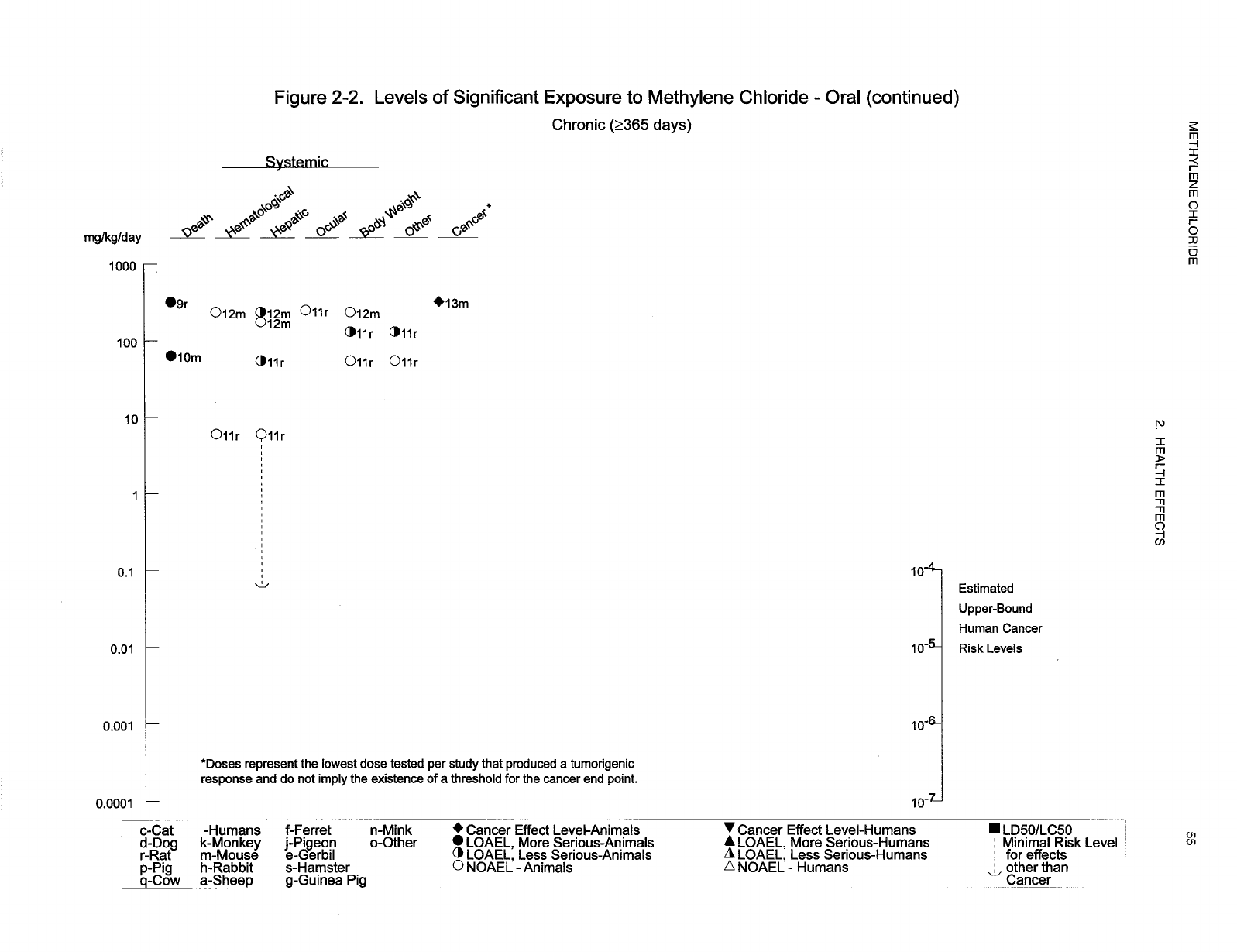
56 METHYLENE CHLORIDE
2. HEALTH EFFECTS
suicide attempt, and started treatment with diuresis and hydrocortisone 1.5 hours later, had episodic
gastrointestinal hemorrhage and duodenojejunal ulceration that developed into diverticula 6 months later
(Roberts and Marshall 1976). No studies were located regarding gastrointestinal effects in animals after
oral exposure to methylene chloride.
Hematological Effects. COHb levels were elevated to 9% in a woman following lethal ingestion of
300 mL of Nitromors, a paint remover solvent containing 75–80% methylene chloride (Hughes and
Tracey 1993). The authors reported that this case study was the first to reveal that ingestion of methylene
chloride results in the formation of COHb, as occurs with methylene chloride inhalation. A man who
ingested 1–2 pints of Nitromors in a suicide attempt exhibited gross hemoglobinuria, a symptom of
intravascular hemolysis (Roberts and Marshall 1976).
In an acute rat study, hemolysis developed within 2 days following a single gavage dose of 1,325 mg/kg
of methylene chloride (Marzotko and Pankow 1987); no such effect was seen at 798 mg/kg. In rats that
were given $420 mg/kg/day of methylene chloride in the drinking water for 3 months, mean hemoglobin
concentrations were elevated in males, and erythrocyte counts were elevated, although mean corpuscular
hemoglobin was reduced, in females (Kirschman et al. 1986); because no quantitative data were
presented, this information is not presented in Table 2-2. In rats of both sexes that were given
55–249 mg/kg/day of methylene chloride in drinking water for 2 years, red blood cell counts and
hematocrit and hemoglobin levels were increased over concurrent control levels (Serota et al. 1986a);
however, Serota et al. (1986a) indicate that half of these increases were statistically significant without
specifying which ones. A dose level of 6 mg/kg/day was a NOAEL. In mice exposed similarly to
60–236 mg/kg/day no significant hematological effects were observed after 104 weeks of treatment
(Serota et al. 1986b).
Hepatic Effects. No studies were located regarding hepatic effects in humans after oral exposure to
methylene chloride.
In rats given two doses (39–1,275 mg/kg each) of methylene chloride by gavage, there were no
significant effects (within 24 hours) in the liver on levels of glutathione, cytochrome P-450, or serum
ALT (Kitchin and Brown 1989). However, at the highest dose level, the activity of ornithine
decarboxylase, an enzyme involved in cell growth, was significantly increased, and DNA damage was
detected in the livers of rats. In another rat study, acute exposure to doses of 1,095 mg/kg/day of
57 METHYLENE CHLORIDE
2. HEALTH EFFECTS
methylene chloride by gavage resulted in liver necrosis (Ugazio et al. 1973). After ingestion of
methylene chloride in drinking water at or above doses of 1,200 mg/kg/day for 3 months, hepatic effects
in rats included centrilobular necrosis, granulomatous foci, accumulation of ceroid or lipofuscin, and
dose-related hepatocytic vacuolation (Kirschman et al. 1986). Liver damage was indicated by increases
in serum ALT in males ($166 mg/kg/day) and females (1,469 mg/kg/day), and by an increase in serum
AST in the latter group. In a parallel 3-month study in mice, there were dose-related hepatic centrilobular
fatty changes over the dose range between 226 and 2,030 mg/kg/day (Kirschman et al. 1986). Chronic
ingestion of methylene chloride in drinking water has been associated with histological alterations of the
liver (cellular foci and areas of cellular alterations) of rats exposed to dose levels of 55 mg/kg/day or
greater (Serota et al. 1986a) and fatty changes in mice exposed to levels of 236 mg/kg/day (Serota et al.
1986b); the NOAELs were 6 and 175 mg/kg/day in rats and mice, respectively. F344 rats and B6C3F
1
mice were exposed for 104 weeks to methylene chloride in deionized drinking water at target doses of 0,
5, 50, 125, or 250 mg/kg/day (for rats) and 0, 60, 125, 185, or 250 mg/kg/day (for mice) (Serota et al.
1986a, 1986b). Based on the calculated intake of 6 mg/kg/day for rats (Serota et al. 1986a), a chronic oral
MRL of 0.06 mg/kg/day was calculated as described in the footnote of Table 2-2.
Renal Effects. Hemoglobinuria as a result of hemolysis was noted in the case of a suicide attempt by
ingestion of the paint remover Nitromors (Roberts and Marshall 1976); the authors indicated that the renal
damage that could have been a consequence of hemolysis was averted by treatment with diuresis and
hydrocortisone. In an acute animal study, administration of a single oral dose of 1,325 mg/kg of
methylene chloride inhibited diuresis in rats (Marzotko and Pankow 1987). In a 3-month study, the pH of
the urine was lowered in all rats that ingested methylene chloride at levels $166 mg/kg/day, and increased
kidney weights were observed in female rats receiving 1,469 mg/kg/day of methylene chloride, but not
607 mg/kg/day (Kirschman et al. 1986).
Endocrine Effects. No studies were located regarding endocrine effects in humans following oral
exposure to methylene chloride. In the only animal study that mentions endocrine effects, administration
of a single oral dose of methylene chloride ($526 mg/kg) to male rats resulted in angiectasis (dilatation of
capillaries) of the adrenal medulla and statistically significant increases in secretion of catecholamines
(epinephrine and norepinephrine) (Marzotko and Pankow 1987). The authors suggested that these
findings appeared to be stress-related.
58 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Ocular Effects. No studies were located regarding ocular effects in humans after oral exposure to
methylene chloride. No treatment-related ophthalmologic findings in rats administered up to 249 mg
methylene chloride/kg/day in the drinking water for 104 weeks (Serota et al.1986a). No further relevant
information was located.
Metabolic Effects. Metabolic acidosis was detected in a man who attempted suicide by drinking
1–2 pints of Nitromors paint remover that contained methylene chloride as the active ingredient (Roberts
and Marshall 1976); he recovered following treatment with diuresis and hydrocortisone.
2.2.2.3 Immunological and Lymphoreticular Effects
No studies were located regarding immunological or lymphoreticular effects in humans or animals after
oral exposure to methylene chloride.
2.2.2.4 Neurological Effects
One and a half hours after drinking 1–2 pints of paint remover (9,000–18,000 mg/kg) that contained
methylene chloride as the active ingredient, a man was deeply unconscious and unresponsive to painful
stimuli; his pupils were reactive, but tendon jerks were depressed and plantar response was absent
(Roberts and Marshall 1976). He was treated by diuresis and hydrocortisone and regained consciousness
by 14 hours after the initial event; at this time, no apparent cerebral damage was detected. The authors
suggested that recovery would have been unlikely without medical intervention.
2.2.2.5 Reproductive Effects
No studies were located regarding reproductive effects in humans or animals following oral exposure to
methylene chloride.
2.2.2.6 Developmental Effects
No studies were located regarding developmental effects in humans or animals following oral exposure to
methylene chloride.
59 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2.2.2.7 Genotoxic Effects
No studies were located regarding genotoxic effects in humans after oral exposure to methylene chloride.
In rats given two doses of 1,275 mg/kg of methylene chloride by gavage (17 hours apart), significant
DNA breakage was detected in the liver 4 hours after the last dose (Kitchin and Brown 1989). Methylene
chloride did not induce a statistically significant increase of micronuclei of polychromatic erythrocytes in
mice when administered at doses up to 4,000 mg/kg (Sheldon et al. 1987). In mice given a single dose of
1,720 mg/kg of methylene chloride, DNA breaks were detected in nuclei from the liver and lung, but not
from stomach, kidney, urinary bladder, brain, or bone marrow; the authors indicated that there was no
evidence of cytotoxicity that might have caused the genetic damage (Sasaki et al. 1998). The variability
in genotoxicity may reflect tissue-specific variation in the metabolism of methylene chloride.
Other genotoxicity studies are discussed in Section 2.5.
2.2.2.8 Cancer
No studies were located regarding carcinogenic effects in humans after oral exposure to methylene
chloride.
Studies in animals provide suggestive evidence that ingestion of methylene chloride may increase the
incidence of liver cancer. In an acute rat study, two doses of 1,275 mg/kg of methylene chloride (given
17 hours apart) caused an increase in the liver activity of ornithine decarboxylase, an enzyme that may
contribute to the promotion of hepatic cancer (Kitchin and Brown 1989). Liver tumors were observed in
female, but not in male rats that ingested methylene chloride (up to 250 mg/kg/day) for 2 years, but the
cancer incidence rates were within historical control ranges (Serota et al. 1986a). Although liver cancer
was observed in male mice, the incidence was not significantly elevated. Female mice did not have
increased liver tumor incidence (Serota et al. 1986b). An increased incidence of mammary tumors was
found in female rats that received methylene chloride by gavage at 500 mg/kg/day for 64 weeks but the
results were not statistically significant (Maltoni et al. 1988). Under the same exposure conditions, an
increased incidence of pulmonary tumors in male mice was statistically significant (p<0.05) when the
increased mortality rate was taken into account (Maltoni et al. 1988). The EPA (1985b, 1987a) reviewed
60 METHYLENE CHLORIDE
2. HEALTH EFFECTS
the available data on the carcinogenic effects of methylene chloride and concluded that there was
borderline evidence for carcinogenicity. The EPA estimated that the upper bound incremental unit
carcinogenic risk for drinking water containing 1 µg/L methylene chloride for a lifetime was
2.1x10
-7
(µg/L)
-1
. This risk estimate derivation was based on the mean of the carcinogenic risk estimates
from the finding of liver tumors in the NTP (1986) inhalation study in female mice; the lung tumor data
from the NTP study were not included in EPA’s analysis. Since the extrapolation model is linear at low
doses, additional lifetime cancer risk is directly proportional to the water concentration of methylene
chloride. Thus, risk levels of 10
-4
, 10
-5
, 10
-6
, and 10
-7
are associated with 0.5, 0.05, 0.005, and
0.0005 mg/L, respectively (0.1, 0.01, 0.001, and 0.0001 mg/kg/day). Because these values are based on
upper bound estimates, the true risk could be lower. These values are shown in Figure 2-2.
2.2.3 Dermal Exposure
2.2.3.1 Death
No studies were located regarding death in humans or animals after dermal exposure to methylene
chloride.
2.2.3.2 Systemic Effects
Respiratory Effects. Shortness of breath was reported among some autoworkers who were exposed to
methylene chloride both dermally and by inhalation for periods of up to 3 years (Kelly 1988). However,
it is likely that inhalation exposure is largely responsible for this effect. No studies were located
regarding respiratory effects in animals following dermal exposure to methylene chloride.
No studies were located regarding the following systemic effects in humans or animals after dermal
exposure to methylene chloride:
Cardiovascular Effects.
Gastrointestinal Effects.
Hematological Effects.
61 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Musculoskeletal Effects.
Renal Effects.
Hepatic Effects. One autoworker who was exposed to methylene chloride by dermal contact and by
inhalation (3.3–154.4 ppm, average of 68 ppm) for 1.5 years was reported to have an enlarged liver in
addition to adverse neurological and reproductive effects (Kelly 1988). This study is described in more
detail under “Inhalation Exposure” in Section 2.1.3.2. The relative contributions of the two exposure
routes to hepatotoxicity were not determined.
No studies were located regarding hepatic effects in animals after dermal exposure to methylene chloride.
Dermal Effects. There are few studies of dermal effects of methylene chloride in humans. In several
cases, workers who were rendered unconscious while stripping furniture using a methylene chloride-
based compound in an open tank became partially immersed in the liquid (Hall and Rumack 1990); first
or second degree chemical burns developed on areas of the body having direct contact with the liquid. In
another case, a worker who was cleaning the interior of a tank with methylene chloride became
unconscious and fell into the solvent when the bucket overturned (Wells and Waldron 1984); during the
30 minutes before he was removed, second and third degree burns developed on the areas of contact. In a
similar workplace accident, in which a man was found dead after 1 hour of exposure to methylene
chloride, chemical burns with excoriation had developed on the areas of contact (Winek et al. 1981).
Ocular Effects. Data are limited regarding ocular effects in humans after dermal exposure to
methylene chloride. Severe corneal burns developed in a worker who was found unconscious and
slumped over with his face partially submerged in an open tank of paint stripper containing methylene
chloride (Hall and Rumack 1990). The duration of exposure was not indicated.
In animals, methylene chloride (0.01–0.1 mL) caused eye irritation and inflammation, increased corneal
thickness, and increased intraocular tension in rabbits following instillation of the liquid solvent into the
conjunctival sac (Ballantyne et al. 1976). Effects were reversible within 3–9 days after treatment. In the
same study, rabbits exposed to vapors of methylene chloride at concentrations of 490 ppm or greater for
10 minutes also showed effects on the eyes. There were small increases in corneal thickness and
intraocular tension. Effects were reversible within 2 days. It is likely that effects observed were due to
62 METHYLENE CHLORIDE
2. HEALTH EFFECTS
direct effect of vapors on the cornea rather than to inhaled methylene chloride or its metabolites acting on
the eyes.
2.2.3.3 Immunological and Lymphoreticular Effects
No studies were located regarding immunological or lymphoreticular effects in humans or animals after
dermal exposure to methylene chloride.
2.2.3.4 Neurological Effects
Neurological effects (loss of memory, loss of concentration, sensory deficit) were reported in a group of
34 male autoworkers, who were exposed to methylene chloride by dermal contact and by inhalation
(3.3–154.4 ppm, average of 68 ppm) for up to 3 years (Kelly 1988). This study is described in more
detail under “Inhalation Exposure” in Section 2.1.3.4. The relative contributions of the two exposure
routes to neurotoxicity were not determined.
No studies were located regarding neurological effects in animals after dermal exposure to methylene
chloride.
2.2.3.5 Reproductive Effects
One group of case studies reported reproductive effects in 8 out of 34 men who complained of central
nervous system dysfunction following occupational exposure to methylene chloride for 0.4–2.9 years
(Kelly 1988). The eight men were part of a subgroup of 26 ‘bonders’ who applied methylene chloride to
automobile parts during assembly; this involved soaking pads from open buckets of methylene chloride
using ungloved hands, so exposures were both by dermal contact and by inhalation (3.3–154.4 ppm,
average of 68 ppm). This study is described in more detail under “Inhalation Exposure” in
Section 2.1.3.5. Infectious disease was eliminated as a cause of the adverse reproductive effects (genital
pain, testicular atrophy, and oligospermia). The relative contributions of the two exposure routes to
reproductive toxicity were not determined. Uncertainty regarding this study involves the small number of
subjects, the multiple exposure to other organic chemicals, the lack of blood or urine samples quantifying
exposure to methylene chloride, and the lack of a control group. No other studies were located that
reported similar male reproductive effects from exposure to methylene chloride. Contrary to Kelly’s
63 METHYLENE CHLORIDE
2. HEALTH EFFECTS
(1988) findings, Wells et al. (1989) found no evidence of oligospermia among four workers who had been
exposed to levels of methylene chloride that were twice as high as in the Kelly study while involved in
furniture stripping for at least 3 months; dermal exposure may have occurred, but this was not specified in
the report. It is not certain whether the longer duration of exposure (minimum occupational exposure 1.4
years) or exposure to other chemicals in the Kelly (1988) study contributed to the different outcomes in
the two studies.
2.2.3.6 Developmental Effects
No studies were located regarding developmental effects in humans or animals after dermal exposure to
methylene chloride.
2.2.3.7 Genotoxic Effects
No studies were located regarding genotoxic effects in humans or animals after dermal exposure to
methylene chloride.
Genotoxicity studies are discussed in Section 2.5.
2.2.3.8 Cancer
No studies were located regarding cancer effects in humans or animals after dermal exposure to
methylene chloride.
2.3 TOXICOKINETICS
Inhalation is the main route of exposure to methylene chloride for humans. Within the first few minutes
of exposure, approximately 70–75% of inhaled vapor is absorbed. However, as the concentration of
methylene chloride in the blood increases, the net uptake is greatly reduced until at steady-state, it is equal
to metabolic clearance, which has a maximum (determined by the fraction of blood flowing to the liver)
of 25% (EPA 1994). Under conditions of continuous exposure to air concentrations of up to
approximately 300 ppm, blood steady state concentrations of methylene chloride are reached in about
4 hours. Pulmonary absorption is influenced by exercise and body fat. In animals, pulmonary absorption
64 METHYLENE CHLORIDE
2. HEALTH EFFECTS
is proportional to magnitude and duration of exposure over a concentration range of 100–8,000 ppm. An
increase of the steady state blood/air concentration ratio at high exposure levels reflects saturation of
metabolic pathways rather than an increased absorption coefficient. There is only qualitative evidence of
oral absorption in humans. In animals, methylene chloride is easily absorbed from the gastrointestinal
tract, particularly from aqueous media. Seventy-five to 98% of an administered dose may be absorbed in
10–20 minutes. There are no quantitative data on dermal absorption of methylene chloride, although it is
known to occur.
Distribution data in humans are lacking, but methylene chloride has been found in human breast milk and
blood. Methylene chloride is widely distributed in animal tissues after inhalation exposure. The highest
concentrations are found in adipose tissue and liver. Methylene chloride has been found in blood from
rats’ fetuses. After acute exposure, methylene chloride disappears rapidly from fat. Distribution of
methylene chloride does not seem to be route-dependent and it does not bioaccumulate in tissues.
There are two main competing metabolic pathways for methylene chloride; one initially catalyzed by
cytochrome P-450 enzymes (CYP2E1) and the other by a theta glutathione-S-transferase (GSSTl-1). The
P-450 pathway (MFO) produces carbon monoxide and carbon dioxide via formyl chloride and the
glutathione pathway (GST) produces carbon dioxide via a postulated glutathione conjugate (S-chloro-
methyl glutathione) and formaldehyde. Both pathways can give rise to toxic metabolites. The oxidative
pathway is preferred at lower exposure concentrations and becomes saturated as exposure levels increase.
Oxidative biotransformation of methylene chloride is similar in rats and humans. In rats, the MFO
pathway is high-affinity low capacity, whereas the GST pathway has lower affinity, but higher capacity.
The GST pathway is more active in mice than in rats and less active in hamsters and humans than in rats.
After inhalation exposure, humans eliminate methylene chloride mainly in expired air, but also in the
urine. In rats, following a single exposure to radioactive methylene chloride, exhaled air had the most
radioactivity, but radioactivity was also found in urine and feces. In exhaled air, the radiolabel was
mostly as carbon monoxide and carbon dioxide. Physiologically based pharmacokinetic (PBPK) models
have been developed to describe disposition of methylene chloride in humans and animals. These models
were designed to distinguish contributions of the two metabolic pathways in lung and liver tissue, to look
for correlations between tumor incidence and various measures of target tissue dose predicted by the
models, and to extrapolate cancer risks from mice to humans.
65 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2.3.1 Absorption
There is no information to determine whether absorption of methylene chloride in children is different
than in adults.
2.3.1.1 Inhalation Exposure
The principal route of human exposure to methylene chloride is inhalation. During absorption through
the lungs, the concentration of methylene chloride in alveolar air in equilibrium with pulmonary venous
blood content approaches the concentration in inspired air until a steady state is achieved. After tissue
and total body steady states are reached through the lungs and other routes, uptake is balanced by
metabolism and elimination. Steady state blood methylene chloride concentrations appear to be reached
after 2–4 hours of exposure (DiVincenzo and Kaplan 1981; McKenna et al. 1980).
Evaluation of pulmonary uptake in humans indicated that 70–75% of inhaled methylene chloride vapor
was absorbed initially (DiVincenzo and Kaplan 1981). Initial absorption of methylene chloride was rapid
as indicated by an uptake of methylene chloride into the blood of approximately 0.6 mg/L in the first hour
of exposure to levels of 100–200 ppm. At a concentration of 50 ppm, the increase in blood methylene
chloride concentration was 0.2 mg/L for the first hour (DiVincenzo and Kaplan 1981). There was a direct
correlation between the steady state blood methylene chloride values and the exposure concentration,
with a proportionality constant of approximately 0.008 ppm in blood per ppm in air (DiVincenzo and
Kaplan 1981). The blood concentrations reached steady state values during the 4th through 8th hour of
continuous exposure to the vapor. Once exposure ceased, methylene chloride was rapidly cleared from
the blood. Six hours after the end of exposure, only traces of methylene chloride were present in the
blood in the highest-concentration group, and pre-exposure baseline blood levels were detected in the
other exposure groups.
Similar to other lipophilic organic vapors, methylene chloride absorption appears to be influenced by
factors other than the vapor concentration. Increased physical activity increases the amount of methylene
chloride absorbed by the body due to an increase in ventilation rate and cardiac output (Astrand et al.
1975; DiVincenzo et al. 1972). Uptake also increases with the percent body fat since methylene chloride
dissolves in fat to a greater extent than it dissolves in aqueous media (Engstrom and Bjurstrom 1977).
Therefore, obese subjects will absorb and retain more methylene chloride than lean subjects exposed to
66 METHYLENE CHLORIDE
2. HEALTH EFFECTS
the same vapor concentration. This does not mean obese subjects are more sensitive to toxicity. Under
controlled conditions, there was a 30% greater absorption and retention of methylene chloride by obese
subjects exposed to 75 ppm for 1 hour as compared to lean subjects (Engstrom and Bjurstrom 1977).
Studies of the relationship of inhalation exposures of animals to their blood methylene chloride
concentrations indicate that absorption is proportional to the magnitude and duration of the exposure over
a methylene chloride concentration range of 100–8,000 ppm. This conclusion is based on the monitoring
of blood methylene chloride concentrations following inhalation exposure in dogs and rats (DiVincenzo
et al. 1972; MacEwen et al. 1972; McKenna et al. 1982). As was the case with humans, blood methylene
chloride levels reached a steady state value as the duration of exposure increased (McKenna et al. 1982).
Studies of blood methylene chloride values during 6-hour exposures of rats to between 50 and 1,500 ppm
of methylene chloride suggest that the steady state blood/air concentration ratio increases as the exposure
concentration increases. The ratio of the steady state methylene chloride concentration in the blood to the
exposure concentration increased from 0.001 to 0.005 and 0.007 as the exposure increased from 50 to 500
to 1,500 ppm, respectively (McKenna et al. 1982). It is postulated that the increased ratio at steady state
results from saturation of metabolic pathways as exposure increases rather than from an increased
absorption coefficient.
2.3.1.2 Oral Exposure
No quantitative studies were located regarding absorption in humans after oral exposure to methylene
chloride. There is qualitative evidence that the compound is absorbed when ingested. A male became
deeply unconscious within 1.5 hours after ingestion of 1–2 pints of a paint remover (9,000–18,000 mg/kg)
(Roberts and Marshall 1976).
In animals, the limited available data suggest that methylene chloride is easily absorbed from the
gastrointestinal tract, particularly if exposed via aqueous media. Ten minutes after treatment, 24% of the
administered dose (50 mg/kg in aqueous solution) was recovered from the upper gastrointestinal tract of
mice when the stomach and small intestinal tissues and contents were analyzed (Angelo et al. 1986a).
Only 2.2% of the methylene chloride was in the stomach and small intestines 20 minutes after compound
administration and less than 1% remained after 40 minutes. At 10 minutes the large intestines and
67 METHYLENE CHLORIDE
2. HEALTH EFFECTS
caecum contained 0.08% of the dose; these values were less than those detected after 20 minutes. Thus,
75% of the dose was absorbed within 10 minutes and about 98% of the dose was absorbed within 20 minutes.
On the other hand, after treatment of mice with 10–1,000 mg methylene chloride in corn oil,
approximately 55% of the administered dose was detected in the stomach and small intestines at
20 minutes and remained there at 2 hours (Angelo et al. 1986a). The large intestines and caecum tissues
and contents contained about 5–8% of the dose at 10 minutes. This value declined to 1–2% at the end of
2 hours. This suggests that the absorption processes are slowed when methylene chloride is presented in
a hydrophobic vehicle. During the 2 hours of observation, approximately 40–45% of the 50 mg dose was
absorbed from the oil vehicle as compared to essentially all of a comparable dose in water. These data are
consistent with the slower emptying of chyme from the stomach when lipids are present, the partitioning
of methylene chloride between the corn oil and the aqueous digestive fluids, the necessity that fats be
emulsified by bile prior to digestion and absorption, and delayed mobilization of lipophilic substances
from the gastrointestinal tract.
Staats et al. (1991) developed a two-compartment model of oral absorption, dependent in part on the
vehicle (aqueous or lipid) and on the lipophilic characteristics of the ingested compound. Absorption
along the gastrointestinal tract was predicted to increase in the first compartment (likely the stomach),
when fat-soluble toxicants are administered in water. When administered in corn oil, the toxicants are
likely to adhere to the lipid vehicle in the first compartment and absorption in the stomach is likely to
decrease. The model also predicts that passage of the compound from the first compartment (stomach) to
the second compartment (small intestine) is enhanced when the vehicle is oil rather than water. However,
existing experimental data suggest that absorption from the second compartment (small intestine) is
generally slow for all lipophilic compounds, irrespective of vehicle. Using previously-determined
absorption and transfer constant values, the model developed by Staats et al. (1991) was able to predict
reasonably experimental data for trichloroethylene. The ability of the model to predict the gastrointestinal
absorption of methylene chloride was not determined. However, in general, the model is consistent with
methylene chloride experimental data (Angelo et al. 1986a).
68 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2.3.1.3 Dermal Exposure
No studies were located regarding absorption in humans following dermal exposure to methylene
chloride.
Dermal permeability constants for rats were obtained for 3 concentrations of methylene chloride (30,000,
60,000, and 100,000 ppm) in air for use in developing a pharmacokinetics model for dermal absorption of
vapors (McDougal et al. 1986). The mean permeability constant was 0.28 cm/hr. The total amount
absorbed was determined to be 34.4, 57.5, and 99.4 mg, respectively, for the three concentrations tested.
2.3.2 Distribution
There is no information to ascertain whether distribution of methylene chloride would be different in
children than in adults.
2.3.2.1 Inhalation Exposure
When methylene chloride is absorbed by the lungs it is expected that it will dissolve in the lipoprotein
components of the blood and enter the systemic circulation after passage through the heart. It is
distributed from the systemic circulation to the body organs. However, no quantitative data were located
which showed the distribution of methylene chloride following human inhalation exposure. Some data
are available which relate to the uptake of methylene chloride by human adipose tissues. These data
indicate that the methylene chloride concentrations in the adipose deposits of lean subjects are greater
than those in obese subjects. However, the total methylene chloride adipose tissue load is greater for the
obese subjects due to their greater adipose mass (Engstrom and Bjurstrom 1977).
Distribution studies in rats demonstrate that methylene chloride and/or its metabolites are present in the
liver, kidney, lungs, brain, muscle, and adipose tissues after inhalation exposures (Carlsson and
Hultengren 1975; McKenna et al. 1982). One hour after exposure, the highest concentration of
radioactive material was found in the white adipose tissue, followed by the liver. The concentration in the
kidney, adrenals, and brain were less than half that in the liver (Carlsson and Hultengren 1975).
Radioactivity in the fat deposits declined rapidly during the first 2 hours after exposure (Carlsson and
Hultengren 1975). Concentrations in the other tissues declined more slowly. On the other hand, after
69 METHYLENE CHLORIDE
2. HEALTH EFFECTS
5 days of exposure to 200 ppm, 6 hours per day, the concentration of methylene chloride in the perirenal
fat was 6- to 7-fold greater than that in the blood and liver (Savolainen et al. 1977).
The animal data are accordingly consistent with the human adipose tissue data discussed above. When
pregnant rats were exposed to 500 ppm of methylene chloride by inhalation for 1 hour, the chemical was
found in fetal blood, but at a lower level than in maternal blood (Anders and Sunram 1982).
2.3.2.2 Oral Exposure
No studies were located regarding distribution in humans following oral exposure to methylene chloride.
In animals, radioactivity from labeled methylene chloride was detected in the liver, kidney, lung, brain,
epididymal fat, muscle, and testes after exposure of rats to a single oral gavage dose of 1 or 50 mg/kg
methylene chloride (McKenna and Zempel 1981). The tissue samples were taken 48 hours after dosing.
At that time, the lowest concentration of radioactivity was found in the fat. The highest concentrations
were in the liver and kidney; this was true for both doses. Radioactivity was found in the blood, liver,
and carcass of mice. Similar results were observed in rats administered doses of 50–1,000 mg/kg
methylene chloride for 14 days (Angelo et al. 1986a, 1986b). At each dose tested, and in each tissue, the
label was rapidly cleared during the 240 minutes after each exposure. These data suggest that methylene
chloride and/or its metabolites do not bioaccumulate in any tissues.
2.3.2.3 Dermal Exposure
In humans, direct dermal contact with pure methylene chloride causes an intense burning, mild erythema
and paresthesia (Stewart and Dodd 1964). Absorption is relatively rapid (McDougal et al. 1986).
2.3.3 Metabolism
Methylene chloride metabolism is not known to be qualitatively different in children than adults.
Information regarding the developmental expression of enzymes that metabolize methylene chloride is
discussed in Section 2.7, Children’s Susceptibility.
70 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Inhalation Exposure
Available data suggest that there are two pathways by which methylene chloride is metabolized. One
pathway utilizes the mixed function oxidase (MFO) enzymes and produces carbon monoxide (CO)
(Figure 2-3, Pathway 1). The other pathway involves the glutathione transferase (GST) and produces
carbon dioxide (CO
2
) (Figure 2-3, Pathway 2). It has been postulated that CO
2
can also be produced by
the MFO pathway if the reactive intermediate in this pathway (postulated to be formyl chloride) reacts
with a nucleophile prior to elimination of the chloride ion and formation of CO (Figure 2-3, Pathway 3)
(Gargas et al. 1986).
The MFO pathway seems to be the preferred pathway for methylene chloride metabolism following
inhalation exposures. Human subjects exposed by inhalation to 500 ppm or greater for 1 or 2 hours
experienced elevated COHb concentrations indicating that methylene chloride was metabolized to CO by
the MFO pathway (Stewart et al. 1972). The COHb concentrations rose to an average of 10.1%
saturation 1 hour after the exposure of 3 subjects to 986 ppm of methylene chloride for 2 hours. The
mean COHb concentration at 17 hours-post exposure remained elevated (3.9% saturation) above the pre-
exposure baseline value (1–1.5% saturation). The exposure of 8 subjects to 515 ppm of methylene
chloride for 1 hour increased the COHb level, which remained elevated above baseline for more than
21 hours.
In human subjects exposed by inhalation to 50–500 ppm of methylene chloride for up to 5 weeks, COHb
concentrations could be predicted from methylene chloride exposure parameters (Peterson 1978).
However, the exhaled breath concentrations of methylene chloride correlated better with exposure
parameters than did COHb concentrations. No differences in methylene chloride metabolism between
male and female subjects were detectable and there was no induction of metabolism to CO during
5-weeks exposure to concentrations ranging from 100 to 500 ppm of methylene chloride (Peterson 1978).
Metabolism of methylene chloride in animals has been shown to be similar to that in humans in an
experiment by Fodor et al. (1973). Albino rats exposed to methylene chloride showed COHb formation,
confirming the observation that methylene chloride is metabolized to CO following inhalation exposures.
Among rats exposed to methylene chloride for 4 hours, concentrations of 500 or 2,500 ppm resulted in
similar maximal blood levels of COHb (Wirkner et al. 1997). A concentration-response relationship
between inhaled methylene chloride and the maximum changes in COHb values was observed when
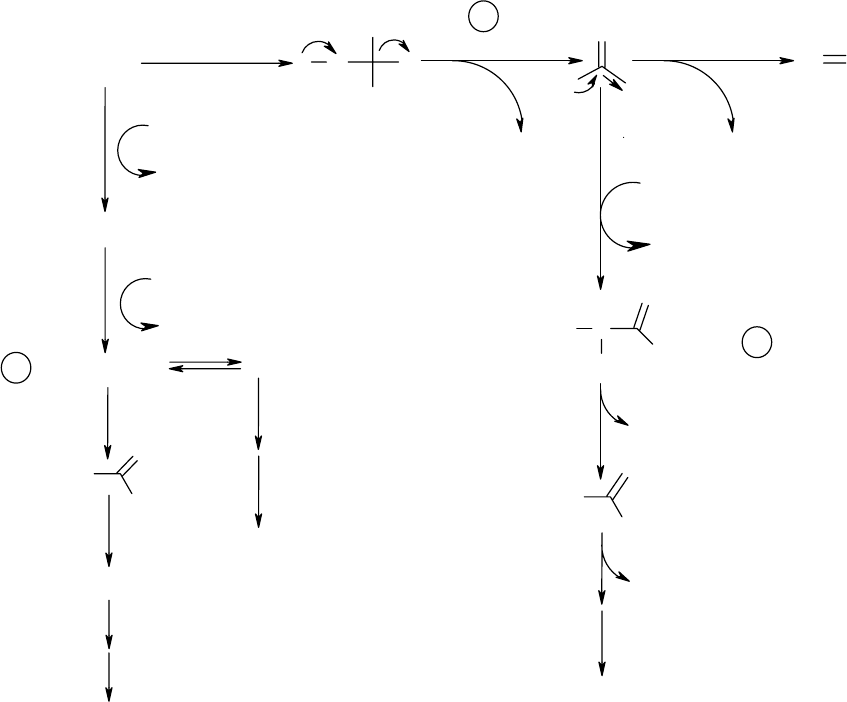
CYT P-450
2
1
H
O
O
2
:C
O
CH
2
X
2
HO X
NADPH
Cl
GSH
CTYOSOL
H
+
+ Cl
-
H
+
+ Cl
GS-CH
2
-X
H
2
O
H
+
+ Cl
GS-CH
2
-OH
CH
2
O + GSH
H
+
O
O
GS
GS
H
H
CO
2
HCOOH + GSH
GSH
H
+
+ Cl
-
NUCLEOPHILE?
(i.e., GSH)
Cl
-
H
Cl
G
S
H
+
3
O
H
CO
2
CO
2
Source: Gargas et al. 1986
1 Mixed Function Oxidase Pathway
2 Glutathione Transferase Pathway
3 Nucleophile Pathway
71
Figure 2-3. Proposed Pathways for Methylene Chloride Metabolism
METHYLENE CHLORIDE
2. HEALTH EFFECTS
72 METHYLENE CHLORIDE
2. HEALTH EFFECTS
4 rabbits were exposed to 1,270–11,520 ppm of methylene chloride for a 4-week period (Roth et al.
1975). The larger the exposure concentration, the longer it took for the changes in blood COHb values to
reach their maximum values. The maximum COHb concentration was observed 1.5 hours after exposure
to 1,270 ppm and 3.5 hours after exposure to 11,520 ppm.
In rats, inhalation of 50 ppm for 6 hours resulted in 26 and 27% of the body burden being recovered as
expired CO and CO
2
, respectively, during the first 48 hours after the exposure period (McKenna et al.
1982). At 1,500 ppm, 14 and 10% of the body burden were recovered as CO and CO
2
, respectively.
These values are consistent with the concept that the enzymes responsible for the metabolic conversion of
methylene chloride to CO and CO
2
become saturated at high-exposure concentrations.
Anders and Sunram (1982) monitored the fetal and maternal blood concentrations of methylene chloride
and carbon monoxide following inhalation exposure of pregnant rats to methylene chloride at 500 ppm
for 1 hour. Whereas the level of methylene chloride in fetal blood was significantly lower than in
maternal blood, the levels of carbon monoxide were about the same. The authors suggested that the
maternal liver has higher biotransforming activity than the fetal liver and that the metabolically generated
carbon monoxide equilibrates between the fetal and maternal circulations.
Thier et al. (1991) investigated whether the human metabolism of methylene chloride was similar to
monohalogenated methanes; these compounds are metabolized via a glutathione-dependent pathway in
human erythrocytes in a subgroup of the human population (called “conjugators”), whereas another
subgroup does not exhibit erythrocyte glutathione-mediated metabolism (called “nonconjugators”).
Blood samples were taken from 10 volunteers who had previously been determined to be either
“conjugators” (subgroup B) or “nonconjugators” (subgroup A) of monohalogenated methanes. The
samples were exposed to radiolabeled methylene chloride, incubated, centrifuged to separate blood
plasma from cellular fraction, and the distribution of radioactivity between the different blood
compartments measured. For individuals from subgroup B, radioactivity in blood plasma increased over
time, reaching 30% in the low-molecular weight fraction and 5% in the high-molecular weight fraction
after 9 hours. For individuals in subgroup A, almost no radioactivity was found in either blood plasma
molecular fraction. In all samples from both groups, no radioactivity was found in either erythrocyte
cytoplasm or membranes. Thus, although dihalogenated methylene chloride does not appear to undergo
metabolism in erythrocytes, some metabolic transformation of methylene chloride appears to have
occurred in the plasma of all individuals classified as “conjugators” (subgroup B), whereas this
73 METHYLENE CHLORIDE
2. HEALTH EFFECTS
metabolism did not occur in “nonconjugators” (subgroup A). The authors concluded that these data
provide evidence for enzyme polymorphism in humans with respect to methylene chloride metabolism.
The contributions of the MFO and GST pathways to the metabolism of methylene chloride were studied
in male rats by Gargas et al. (1986). Based on the data from these studies, the MFO pathway is a high
affinity-low capacity pathway with a metabolic rate of 47 µmol/kg/hour. The GST pathway, on the other
hand, has a lower affinity than the MFO pathway but a higher capacity. As part of this study, rats were
pretreated with an inhibitor of the MFO pathway or a glutathione (GSH) depleting agent. Concentrations
of COHb were measured to assess the relative contribution of each pathway to the total metabolism of
methylene chloride. Pretreatment with the MFO inhibitor essentially abolished CO production by the
high-affinity saturable MFO pathway and limited metabolism to the GSH-transferase pathway.
Concentrations of COHb following treatment with the GSH-depleting agent were increased 20–30%
compared to untreated controls, indicating increased activity of the MFO pathway. Some CO
2
was
produced, indicating that despite some depletion of GSH, some methylene chloride was still metabolized
by the GST pathway or by conversion of CO to CO
2
via the MFO pathway.
There appear to be species differences in pathway preference. The GST pathway is more active in the
mouse than the rat, based on studies of metabolic end products in both species during and immediately
after 6 hours of exposure to 500, 1,000, 2,000, and 4,000 ppm of methylene chloride (Green et al. 1986c).
The cytochrome MFO pathway leading to CO and COHb was shown to be saturated at the 500 ppm-
exposure level. After saturation of this pathway had occurred, the blood levels of methylene chloride in
the rat increased almost linearly with concentration, indicating little further metabolism in this species. In
contrast, there was evidence for significant metabolism of methylene chloride in the mouse at high-
concentration levels by the GST pathway, which leads to CO
2
. Comparison of expired CO
2
levels after
4,000-ppm exposure for 6 hours showed almost an order of magnitude more CO
2
produced per kilogram
of body weight in the mouse than in the rat. A marked difference was seen in the rate of clearance of
methylene chloride from tissues, as measured by its elimination in expired air. Although cleared from
blood rapidly in both species, the rate of clearance from rat tissues was markedly slower than in the
mouse. This slow release in the rat sustained metabolism for up to 8 hours after the end of exposure and,
consequently, had a marked effect on the overall body burden of metabolites. Methylene chloride was
cleared from tissues in the mouse in less than 2 hours. Overall, saturation of the cytochrome P-450 MFO
pathway occurred at similar levels in both species, but significantly more methylene chloride was
74 METHYLENE CHLORIDE
2. HEALTH EFFECTS
metabolized by the GST pathway in the mouse when assessed either from the blood levels of methylene
chloride or by CO
2
formation at high-concentration levels.
The isoenzymes involved in the metabolism and biotransformation of methylene chloride have recently
been identified for each major pathway. The MFO pathway involves cytochrome P-450 2E1 and the
glutathione-mediated pathway involves a Θ (theta) class glutathione S-transferase, GSTT1-1 (Blocki et al.
1994; Mainwaring et al. 1996b; Meyer et al. 1991). GSTT1-1 is expressed in a number of human organs
in a tissue-specific manner which is different from the pattern of expression of other glutathione
S-transferases, specifically those from the α, µ, and π classes (Sheratt et al. 1997). A recent in vitro study
by Sheratt et al. (1997) detected very low levels of GSTT1-1 in human lung cells, suggesting that the
human lung is likely to have a low capacity for activating methylene chloride into reactive metabolites.
Although Mainwaring et al. (1996b) found that the overall distribution of mRNA and protein for
GSTT1-1 and GSTT2-2 in the liver and lungs of humans was lower than in mice and rats, they found
localized high concentrations of GSTT2-2 enzyme in human bile-duct epithelial cells. However, since the
enzyme did not localize to the nucleus in these cells, the risk of genotoxic effects from methylene chloride
metabolism would appear to be small. Mainwaring et al . (1996b) also found locally high concentrations
of GSTT1-1 mRNA in a small number of Clara cells and ciliated cells of the alveolar/bronchiolar junction
in one human lung sample out of four. Mainwaring et al. (1996b) also found that rates of metabolism of
methylene chloride were low in human tissue, and lower in the lung than in the liver. Thus, it is possible
that, in some individuals, specific cell types may be vulnerable to genotoxic effects from reactive
intermediates of methylene chloride metabolism, although the overall risk is likely to be low.
The oxidative cytochrome P-450 2E1 is presumed to metabolize methylene chloride to carbon dioxide via
a reactive intermediate, formyl chloride; this metabolic pathway also produces carbon monoxide (Green
1997; Reitz 1990). The glutathione pathway metabolizes methylene chloride to carbon dioxide following
the formation of both formaldehyde and a glutathione conjugate, putatively chloromethyl glutathione. No
carbon monoxide is produced during glutathione-mediated metabolism (Green 1997; Reitz 1990).
Neither the formyl chloride, nor the chloromethyl glutathione, nor any other glutathione conjugate of
methylene chloride, has been isolated and characterized. However, according to Green (1997), the
formation of these reactive intermediates is consistent with the end products formed, and with the
enzymes and cofactors available.
75 METHYLENE CHLORIDE
2. HEALTH EFFECTS
An extremely detailed analysis of these two metabolic pathways was conducted in mice, rats, hamsters,
and humans, both in vivo and in vitro. These studies provide evidence for the concentration-dependent
behaviors of the two pathways and compare the metabolic rates by each pathway in the four species
(Bogaards et al. 1993; Green 1991, 1997; Reitz et al. 1989). In vivo, the cytochrome P-450 MFO
pathway was the major route of metabolism at low-concentration inhalation exposures to methylene
chloride. In both rats and mice, saturation of the MFO pathway occurred at concentration levels of
500 ppm and above, with maximum COHb levels reported to be 12–15%. In vitro studies verified that
this pathway was similar in all four species. In contrast, the glutathione S-transferase pathway was the
major metabolic pathway at exposure concentrations used in the rodent cancer bioassays, and showed a
linear concentration response. Furthermore, metabolic activity in mouse tissue was more than 10-fold
greater than metabolic activity in rat tissue. The glutathione-mediated metabolic rates in hamster and
human tissues were even lower than those observed in the rat (Green 1997). In human tissues, metabolic
rates in the lung were about 10-fold lower than those in the liver (Green 1997). Although the Θ (theta)
class glutathione S-transferase (enzyme 5-5 in rat) has a high specific activity for metabolizing methylene
chloride (Meyer et al. 1991; Sheratt et al.1997), the µ-class GSTs (enzymes 3-3, 3-4, and 4-4) are, as a
group, 800-fold more abundant in rat liver (Blocki et al. 1994); in rat liver cytosol, the Θ-class enzyme
(5-5) and the µ-class group of enzymes (3-3, 3-4, and 4-4) are each responsible for half of the metabolism
of methylene chloride to formaldehyde (Blocki et al. 1994). In addition, another Θ-class enzyme (12-12)
is present in rat liver, but its lability during isolation has prevented analysis of its specific activity (Meyer
et al. 1991). Although Schroder et al. (1996) demonstrated that the human erythrocyte GSTT1-1 is
polymorphic, from N-terminal modification, and differs from liver and lung GSTT1-1, it is not yet known
whether liver and lung human GSTT1-1 are polymorphic. Thus, the enzymatic basis for methylene
chloride metabolism in different tissues in different species is not completely elucidated.
Nelson et al. (1995) examined the distribution of erythrocyte GSTT1-1 polymorphisms among five
different ethnic groups: North American Caucasians, African-Americans, Mexican-Americans, Chinese,
and Koreans. Polymerase chain reaction (PCR)-based genotyping of erythrocyte GSTT1-1 demonstrated
that significant ethnic variations occur. The prevalence of the nonfunctional genotype (i.e., the one
lacking the ability to metabolize methylene chloride) was highest among Chinese (64%), followed by
Koreans (60%), African-Americans (22%), Caucasians (20%), and Mexican-Americans (10%). These
data suggest that there are ethnic differences in metabolizing capacity; additionally, substantial variations
in GSTT1-1 polymorphisms also occur within ethnic groups (Nelson et al. 1995)
76 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Kim et al. (1996b) conducted a series of experiments using the vial equilibration technique on freshly
isolated liver tissue of male Sprague Dawley rats in order to characterize the metabolism of methylene
chloride. Several different hepatic microsomal preparations were used to compare and contrast the MFO-
and GSH-mediated pathways. To produce glutathione depletion in one study, rats were injected
intraperitoneally with 250 mg phorone/kg body weight in corn oil prior to sacrifice and preparation of
liver homogenate for in vitro testing; controls received corn oil only. A glycerol buffer preparation
significantly inhibited methylene chloride metabolism while a sucrose buffer containing EDTA and KCl
did not. The use of substrates for P-450 2E1 (i.e., ethanol, pyrazole) completely inhibited methylene
chloride metabolism, indicating that the methylene chloride was metabolized preferentially by the MFO
pathway under the conditions of this study. Pretreatment with phorone to produce hepatic glutathione
depletion had little effect on the metabolic rate of methylene chloride, demonstrating that the glutathione-
mediated metabolism was not a major pathway under the conditions of the study. The authors concluded
that these results demonstrate that at low exposure levels, little methylene chloride is metabolized by the
GSH pathway, and thus these results confirm the results of other investigators.
Several PBPK models have been developed that can be used to predict tissue-specific exposures to
methylene chloride, taking into account absorption, distribution, and metabolism. A PBPK model was
used to provide quantitative estimates of the levels of methylene chloride in various organs of four
mammalian species (rats, mice, hamsters, and humans) following inhalation exposure (Andersen et al.
1987). The model, which incorporates a variety of variables representing the blood and tissue
concentrations of methylene chloride, exhaled methylene chloride, and instantaneous rates of metabolism
by each pathway, was validated by comparing predictions of concentrations of methylene chloride in
blood with time-course data obtained with Fischer-344 rats, Syrian Golden hamsters, B6C3F
1
mice, and
human volunteers. The predicted values for each of the four species were in agreement with the
experimental data. The model was also shown to predict the appearance and elimination of methylene
chloride metabolites reasonably well.
Other PBPK models are discussed in Section 2.3.5.
Oral Exposure In a lethal poisoning case, a 56 year-old woman ingested approximately 300 mL of
Nitromors, a paint remover solvent whose major ingredient is 75–80% methylene chloride (Hughes and
Tracey 1993). COHb levels in the patient were increased to about 9% in blood samples taken several
hours later. According to the authors, this case demonstrated that conversion of ingested methylene
77 METHYLENE CHLORIDE
2. HEALTH EFFECTS
chloride to COHb occurs in humans; previously, the conversion of ingested methylene chloride to COHb
had only been reported in rats.
No other studies were located regarding metabolism in humans after oral exposure to methylene chloride.
Animal data on metabolism indicate that the process appears to be similar for inhalation and oral
exposures. When rats were gavaged with a single oral dose of 526 mg/kg of methylene chloride, COHb
levels in blood rose to nearly 10% (Wirkner et al. 1997). When methylene chloride was administered to
rats and mice by gavage at daily doses of 50 and 200 mg/kg in the mice and 50 mg/kg in the rats, there
was a dose-dependent biotransformation of methylene chloride to CO
2
and CO (Angelo et al. 1986a,
1986b). Pulmonary excretion of methylene chloride, CO, and CO
2
could be detected within 30 minutes
of administration. Initially, exhaled CO
2
levels from the methylene chloride subjects exceeded CO levels
in the rats. However, the amount of exhalant did increase with time. A similar profile of metabolism was
apparent in mice based on exhaled methylene chloride, CO, and CO
2
values except that the exhaled CO
2
values support studies which indicated that the GST pathway is more important in mice than in rats
(Green et al. 1986c, 1988).
Immediately following administration of doses of 500 or 1,000 mg/kg of methylene chloride in corn oil,
the values for pulmonary excretion of methylene chloride, CO
2
, and CO were all lower than when
methylene chloride was administered in aqueous solution, reflecting the slower absorption of methylene
chloride from the corn oil vehicle (Angelo et al. 1986a). Three hours after administration the corn oil,
pulmonary excretory patterns were similar, but still had lower values than those for the methylene
chloride in aqueous solution.
Pankow and Jagielki (1993) studied the in vivo metabolism of methylene chloride to carbon monoxide as
measured by the COHb level in blood under the following conditions: pretreatment with methanol;
simultaneous administration of methanol; and pretreatment with glutathione-depleting chemicals. Male
Wistar rats were administered 6.2 mmol (=0.4 mL)/kg body mass methylene chloride via gavage and
>148 mmol/kg of methanol. Six hours following methylene chloride administration, blood samples were
taken for COHb determination. The animals were then sacrificed and glutathione levels were measured in
the liver. The authors concluded that the cytochrome P-450 2E1 oxidative pathway is responsible for the
formation of COHb and the metabolism of methylene chloride to carbon monoxide, and suggested that
the two metabolic pathways of methylene chloride (i.e, oxidation cytochrome P-450 2E1 and conjugation
78 METHYLENE CHLORIDE
2. HEALTH EFFECTS
via glutathione/glutathione-S-transferase) may be independent at study doses in rats. However, the
rationale for this suggestion was unclear.
A comparison of the rates of pulmonary elimination of methylene chloride, CO
2
, and CO for oral and
intravenous exposures of rats indicated that biotransformation rather than absorption was the rate-limiting
factor that controlled the CO and CO
2
production (Angelo et al. 1986b). The same situation was true in
mice when the pulmonary elimination patterns of the methylene chloride in water were compared to those
from intravenous injection (Angelo et al. 1986a). This was not the case when the methylene chloride was
given in corn oil.
These data suggest that both the MFO and GST pathways can participate in the metabolism of methylene
chloride. Factors which influence the metabolism of methylene chloride by the oral exposure route are
the rate of absorption and the distribution of the MFO and GST enzyme systems in the various tissues and
the transportation of methylene chloride via the aqueous blood components to the liver or via the
chylomicrons to systemic circulation.
Dermal Exposure No studies were located on the metabolism of methylene chloride after dermal
exposure to humans or animals.
2.3.4 Elimination and Excretion
Elimination and excretion of methylene chloride in children are not expected to be different from adults;
however, this has not been investigated.
2.3.4.1 Inhalation Exposure
Methylene chloride is removed from the body primarily in expired air and urine. In 4 human subjects
exposed to 100 ppm of methylene chloride for 2 hours, an average of 22.6 µg (0.003%) methylene
chloride was excreted in the urine within 24 hours after the exposure; in 7 subjects exposed to 200 ppm of
methylene chloride for 2 hours, the corresponding value was 81.5 µg (0.006%) (DiVincenzo et al. 1972).
The percentage values are based on a respiration rate of 1 mg/m
3
and the assumption that methylene
chloride is completely absorbed. No data were found on amounts recovered in feces. Methylene chloride
79 METHYLENE CHLORIDE
2. HEALTH EFFECTS
excretion in the expired air was most evident in the first 30 minutes after exposure. Initial postexposure
methylene chloride concentrations in expired breath following 2- and 4-hour exposure periods were about
20 ppm and dropped to about 5 ppm at the end of 30 minutes. Small amounts of methylene chloride
remained in the expired air at 2.5 hours.
In rats, methylene chloride was excreted in the expired air, urine, and feces following a single 6-hour
exposure to 50, 500, or 1,500 ppm of methylene chloride (McKenna et al. 1982). Exhaled air accounted
for 58–79% of the radioactive concentration. At the 50 ppm exposure only 5% of the exhaled label was
found as methylene chloride. The remainder was exhaled as CO and CO
2
. As the exposures increased, so
did the amount of unmetabolized methylene chloride exhaled. Methylene chloride accounted for 30% of
the label from the 500 ppm concentration and 55% of the label for the 1,500 ppm concentration. Exhaled
methylene chloride, CO
2
, and CO accounted for 58, 71, and 79% of the inhaled methylene chloride for
the 50, 500, and 1,500 concentrations, respectively. Urinary excretions accounted for 7.2–8.9% of the
absorbed dose and 1.9–2.3% of the absorbed dose was in the feces.
2.3.4.2 Oral Exposure
No studies were located regarding excretion in humans after oral exposure to methylene chloride.
Expired air accounted for 78–90% of the excreted dose in rats in the 48-hour period following a 1 or
50 mg/kg methylene chloride dose in aqueous solution (McKenna and Zemple 1981). The radiolabel was
present in the exhaled air as CO and CO
2
, as well as in expired methylene chloride. The amount of
methylene chloride in the expired air increased from 12 to 72% as the dose was increased from 1 to
50 mg/kg. Radiolabel in the urine accounted for 2–5% of the dose under the above exposure conditions,
while 1% or less of the dose was found in the feces. These data indicate that the lungs are the major
organ of methylene chloride excretion even under oral exposure conditions.
2.3.4.3 Dermal Exposure
No studies were located regarding excretion in humans or animals after dermal exposure to methylene
chloride.
80 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2.3.5 Physiologically Based Pharmacokinetic (PBPK)/Pharmacodynamic (PD) Models
Physiologically based pharmacokinetic (PBPK) models use mathematical descriptions of the uptake and
disposition of chemical substances to quantitatively describe the relationships among critical biological
processes (Krishnan et al. 1994). PBPK models are also called biologically based tissue dosimetry
models. PBPK models are increasingly used in risk assessments, primarily to predict the concentration of
potentially toxic moieties of a chemical that will be delivered to any given target tissue following various
combinations of route, dose level, and test species (Clewell and Andersen 1985). Physiologically based
pharmacodynamic (PBPD) models use mathematical descriptions of the dose-response function to
quantitatively describe the relationship between target tissue dose and toxic end points.
PBPK/PD models refine our understanding of complex quantitative dose behaviors by helping to
delineate and characterize the relationships between: (1) the external/exposure concentration and target
tissue dose of the toxic moiety, and (2) the target tissue dose and observed responses (Andersen et al.
1987; Andersen and Krishnan 1994). These models are biologically and mechanistically based and can
be used to extrapolate the pharmacokinetic behavior of chemical substances from high to low dose, from
route to route, between species, and between subpopulations within a species. The biological basis of
PBPK models results in more meaningful extrapolations than those generated with the more conventional
use of uncertainty factors.
The PBPK model for a chemical substance is developed in four interconnected steps: (1) model
representation, (2) model parametrization, (3) model simulation, and (4) model validation (Krishnan and
Andersen 1994). In the early 1990s, validated PBPK models were developed for a number of
toxicologically important chemical substances, both volatile and nonvolatile (Krishnan and Andersen
1994; Leung 1993). PBPK models for a particular substance require estimates of the chemical substance-
specific physicochemical parameters, and species-specific physiological and biological parameters. The
numerical estimates of these model parameters are incorporated within a set of differential and algebraic
equations that describe the pharmacokinetic processes. Solving these differential and algebraic equations
provides the predictions of tissue dose. Computers then provide process simulations based on these
solutions.
The structure and mathematical expressions used in PBPK models significantly simplify the true
complexities of biological systems. If the uptake and disposition of the chemical substance(s) is
81 METHYLENE CHLORIDE
2. HEALTH EFFECTS
adequately described, however, this simplification is desirable because data are often unavailable for
many biological processes. A simplified scheme reduces the magnitude of cumulative uncertainty. The
adequacy of the model is, therefore, of great importance, and model validation is essential to the use of
PBPK models in risk assessment.
PBPK models improve the pharmacokinetic extrapolations used in risk assessments that identify the
maximal (i.e., the safe) levels for human exposure to chemical substances (Andersen and Krishnan 1994).
PBPK models provide a scientifically sound means to predict the target tissue dose of chemicals in
humans who are exposed to environmental levels (for example, levels that might occur at hazardous waste
sites) based on the results of studies where doses were higher or were administered in different species.
Figure 2-4 shows a conceptualized representation of a PBPK model.
PBPK models for methylene chloride are discussed in this section in terms of their use in risk assessment,
tissue dosimetry, and dose, route, and species extrapolations.
2.3.5.1 Methylene Chloride PBPK Model Comparison
The first methylene chloride-specific pharmacokinetic model was developed by Andersen et al. (1987) for
use in methylene chloride human risk assessment, formulated by integrating information on mouse
physiology, methylene chloride solubility characteristics, and metabolic rate constants to describe the
disposition of methylene chloride in target tissues. This model was actually a modification of earlier
models developed by Ramsey and Andersen (1984) for generic PBPK analysis of volatile chemicals and
by Gargas et al. (1986) for study of the kinetics and metabolism of dihalomethanes. The model could
predict the time-course of the parent chemical and the production of metabolites by both GSH and MFO
pathways. A more comprehensive model developed by Andersen et al. (1991) is also capable of
describing the production of carbon monoxide during oxidative metabolism by the MFO pathway and the
production of COHb by CO binding to blood hemoglobin. This model (Andersen et al. 1991) provided a
coherent description of experimental data from both rodents and humans. In an earlier draft of this
toxicological profile, the Andersen et al. (1991) model was used to conduct route-to-route extrapolation
of the inhalation data in humans from the Putz et al. (1979) study to develop an equivalent oral dose that
was evaluated as the basis for an acute oral MRL. (However, a different critical study has been selected
and has been modeled as described below). The model calculated the peak level (6.37%) of carbon
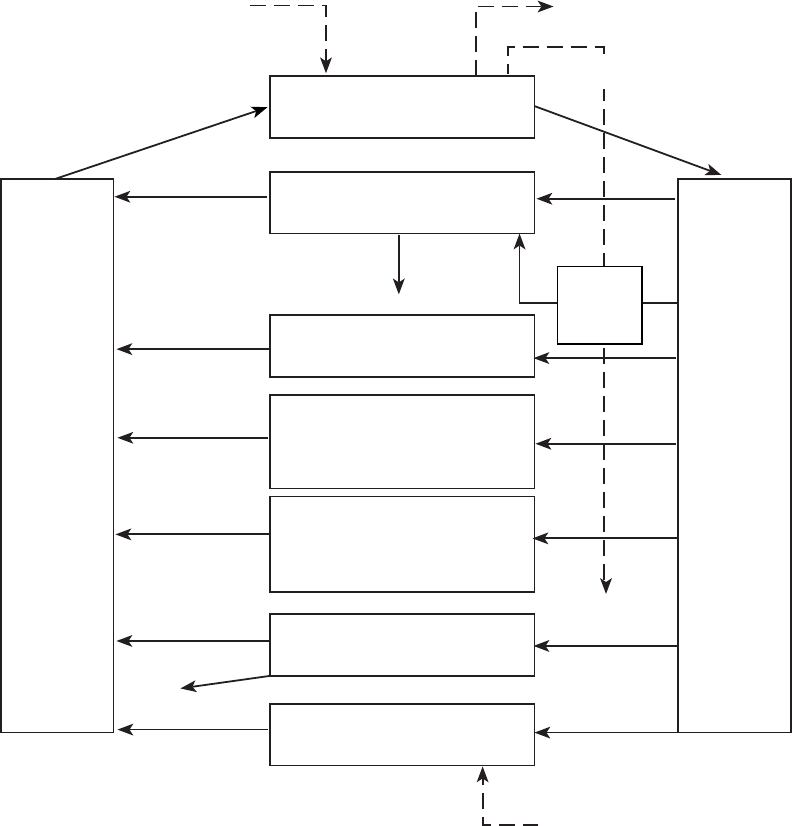
Inhaled chemical Exhaled chemical
Ingestion
Skin
Kidney
Richly
perfused
tissues
Slowly
perfused
tissues
Fat
Liver
Lungs
V
E
N
O
U
S
B
L
O
O
D
GI
Tract
V
max
K
m
Urine
Chemicals in air
contacting skin
Feces
A
R
T
E
R
I
A
L
B
L
O
O
D
82 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Figure 2-4. Conceptual Representation of a Physiologically Based
Pharmacokinetic (PBPK) Model for a Hypothetical Chemical Substance
Source: adapted from Krishnan et al. 1994
Note: This is a conceptual representation of a physiologically based pharmacokinetic (PBPK)
model for a hypothetical chemical substance. The chemical substance is shown to be absorbed
via the skin, by inhalation, or by ingestion, metabolized in the liver, and excreted in the urine or
by exhalation.
83 METHYLENE CHLORIDE
2. HEALTH EFFECTS
monoxide in the blood that produced a neurological effect (decreased visual and psychomotor
performance and auditory function) following exposure to 200 ppm of methylene chloride (Putz et al.
1979). The equivalent concentration of methylene chloride in drinking water that will produce the same
neurological effect was 865 mg/L. Using a daily drinking water consumption value of 2 L and an average
human body weight of 70 kg, the LOAEL was calculated to be 25 mg/kg/day. Reitz et al. (1988, 1989),
Reitz (1990, 1991), and Andersen and Krishnan (1994) used the Andersen et al. (1987) model to make
predictions about GSH-mediated target tissue doses associated with tumorigenesis, or the lack thereof, in
the long-term inhalation and drinking water bioassays. Using target tissue dosimetry as a concentration
surrogate for atmospheric concentrations of methylene chloride, these investigators calculated a unit
inhalation risk for methylene chloride that was two orders of magnitude lower than EPA’s, using the same
linearized multistage methodology.
Dankovic and Bailer (1994) used the models developed by Andersen et al. (1987) and Reitz et al. (1988,
1989; Reitz 1990, 1991) to examine the effects of exercise and intersubject variability on estimation of
human dose levels of methylene chloride. Casanova et al. (1992) developed an extended PBPK model for
DNA-protein crosslink (DPX) formation in mouse liver, based on the model developed by Andersen et al.
(1987).
Reitz et al. (1997) and DeJongh et al. (1998) have expanded earlier models (Andersen et al. 1987,
DeJongh and Blaauboer 1996, Ramsey and Andersen 1984) to include simulations of brain concentrations
of methylene chloride. Reitz et al. (1997) used their model to make route-to-route dose extrapolations
based on brain methylene chloride concentrations. Winneke (1974) had concluded that neurological
effects (decreased visual and auditory functions) of acute exposure to methylene chloride were based
primarily on the properties of the parent compound, and not on the accumulation of COHb, which had
been the conclusion of Putz et al. (1979). Based on the study of Winneke (1974) in humans, an inhalation
exposure to 300 ppm methylene chloride for 4 hours was predicted to result in a similar peak brain
methylene chloride concentration as an exposure to 565 mg methylene chloride/L drinking water, both
exposures correspond to approximately 4 mg/L of brain tissue. Using a daily drinking water consumption
value of 2 L and an average human body weight of 70 kg, the LOAEL was calculated to be 16 mg/kg/day.
This LOAEL was used as the basis for the acute oral MRL (see Section 2.5). DeJongh et al. (1998) used
a rat PBPK model to estimate brain methylene chloride concentrations in rats corresponding to inhalation
exposures to the rat 15-minute or 6-hour LC
50.
84 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Earlier models have also been extended to simulate humans during pregnancy and lactation. The Reitz et
al. (1997) model is based on an earlier model of trichloroethylene pharmacokinetics in the pregnant rat
(Fisher et al. 1989) and includes compartments representing mammary tissue, placenta, and the fetus. The
Reitz et al. (1997) model has been used to simulate fetal doses of methylene chloride in rats that were
exposed to methylene chloride by the inhalation route. Exposure to 1,250 ppm methylene chloride
(4,342 mg/m
3
) for 7 hours/day during gestation days 6–15 was predicted to result in a dose to the fetus of
3.67 mg/L of fetus. The corresponding drinking water intake in the human that would result in the same
fetal dose was estimated from the human PBPK model to be 5,000 mg/L drinking water. Fisher et al.
(1997) described an extension of the PBPK model of Ramsey and Andersen (1984) which simulates
amounts and concentrations of methylene chloride in human breast milk that would result from inhalation
exposures. The human lactation model includes a breast milk compartment and parameters for simulating
breast milk production and intake of breast milk by nursing infants. Simulations of a workday maternal
exposure to methylene chloride at an air concentration of 50 ppm (174 mg/m
3
) yielded a predicted intake
in the infant of 0.213 mg/24 hours.
Advances in modeling methylene chloride pharmacokinetics have also been achieved by linking
methylene chloride models to other PBPK models or to other types of simulation or prediction models.
For example, OSHA (1997) developed a PBPK model that utilizes a Monte Carlo approach to simulate
variability in methylene chloride pharmacokinetics. The model outputs a probability distribution of the
amount of methylene chloride metabolized in the lung through the glutathione-S-transferase pathway.
Poulin and Krishnan (1999) developed a structure-based PBPK model that uses information on chemical
structure to estimate tissue:blood and tissue:air partition coefficients needed to run the model. This
approach can be used to model volatile organic chemicals for which empirically-based estimates are not
available (Poulin and Krishnan 1999). A PBPK model of methylene chloride has been linked to a PBPK
model of toluene (Pelekis and Krishnan 1997). The composite model has been used to simulate the
pharmacokinetic and toxicodynamic outcomes (e.g., COHb concentrations) that might result from
interactions between methylene chloride and toluene at the level of the mixed function oxidase pathway.
85 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2.3.5.2 Discussion of Methylene Chloride PBPK Models
The Reitz (1990) Model (Also the Andersen et al. [1987], the Reitz et al. [1988, 1989, 1997],
and Andersen and Krishnan [1994] Models)
Risk assessment. An inhalation to oral route extrapolation was pharmacokinetically modeled using
data from the Haun et al. (1972) study. Reitz (1990) incorporates PBPK principles into estimating excess
lifetime cancer risk for humans continuously exposed to 1 µg/m
3
. Using the linearized multistage model,
the upper-bound estimate of excess lifetime human cancer risk is 2.8x10
-4
ppm which differs from that of
EPA (4.1x10
-6
) by 2orders of magnitude. It should be noted that EPA used default assumptions of low-
dose linear extrapolation and of surface area adjustment to account for interspecies differences.
Description of the model. Reitz (1990) used the mathematical model developed by Andersen et al.
(1987), which can quantitatively describe the production of metabolites in target tissues by either the
MFO or glutathione (GSH) pathway, to test whether model predictions are consistent with the results
obtained in the rodent inhalation and drinking water cancer bioassays of methylene chloride. The MFO
pathway is oxidative and appears to yield carbon monoxide as well as considerable amounts of carbon
dioxide. The glutathione-dependent pathway produces formaldehyde and carbon dioxide, but no carbon
monoxide. Potentially reactive intermediates are formed in each of the metabolic pathways for methylene
chloride. Distribution of methylene chloride metabolism between these pathways is dose dependent. The
MFO pathway is a high-affinity, limited-capacity pathway which saturates at relatively low airborne
concentrations (about 200–500 ppm). In contrast, the GSH pathway has a lower affinity for methylene
chloride, and does not appear to saturate at experimental concentrations (<5,000 ppm). Thus, at low
concentrations, most of the methylene chloride is metabolized by the MFO pathway. As exposure
concentrations increase and the MFO pathway saturates, metabolism by the secondary GSH pathway is
observed.
Predicted amounts of methylene chloride metabolism produced by each pathway (mg equivalents of
methylene chloride metabolized per L of liver tissue per day) are presented in Reitz’s (1990) analysis.
The predicted amounts of MFO metabolites formed in lung and liver tissue are nearly identical for the 2
concentration levels in the mouse inhalation cancer bioassay (2,000 and 4,000 ppm) because saturation of
the MFO pathway is reached between 200 and 500 ppm of administered methylene chloride. However,
mouse liver and lung tumors are consistently higher in the 4,000 ppm exposure group than in the
86 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2,000 ppm group in this study. Therefore, there is no concentration-response relationship between rodent
tumor incidence and MFO-metabolite levels. The PBPK model also predicts that levels of MFO
metabolites produced by drinking water administration of 250 mg/kg/day methylene chloride to B6C3F
1
mice should be similar to the amounts of MFO metabolites produced in the inhalation study. Because no
statistically significant increases in tumors in the drinking water study were observed, MFO metabolites
would not be predicted to be involved in rodent tumorigenicity. Predicted rates of metabolism of
methylene chloride by the GSH pathway correlate more clearly with the induction of lung and liver
tumors, or lack thereof, in the two chronic studies. In the inhalation study, predicted levels of GSH
metabolites at 4,000 ppm are higher than predicted levels of GSH metabolites at 2,000 ppm. In contrast,
predicted levels of GSH metabolites formed in target tissues during drinking water administration of
methylene chloride are very low. Thus, the pattern of tumor induction, or lack thereof, in both studies
shows a good correlation with the rates of metabolism of methylene chloride by the GSH pathway.
Because the concentration-dependency of GSH-metabolite formation is nonlinear at low concentrations,
the delivered dose arriving at the target sites cannot be directly extrapolated from very high inhalation
concentrations (4,000 ppm) to very low concentrations (<1 ppm) typical of human exposure.
Additionally, with regard to interspecies sensitivity, humans would only be more sensitive than mice
(based on the default surface area interspecies adjustment) if the parent chemical were directly
responsible for observed toxicity. In the case of methylene chloride, metabolism mediated by the GSH
pathway is necessary to activate methylene chloride to reactive intermediates, so this assumption is not
applicable. Reitz et al. (1990) incorporates these PBPK findings and principles into estimating excess
lifetime risk for humans continuously exposed to 1 µg/m
3
, using the linearized multistage model; his
upper-bound estimate of excess lifetime human cancer risk is 2.8x10
-4
ppm, which differs from that of
EPA (4.1x10
-6
) by two orders of magnitude (EPA used default assumptions of low-dose linear
extrapolation and surface area adjustment for interspecies differences).
Validation of the model. The model was validated by comparing predicted responses with
experimental data.
Target tissues. The target tissues were the liver and the lung.
Species extrapolation. Human physiological parameters and assumptions about interspecies
sensitivity were derived from PBPK modeling and use in extrapolation from mice to humans.
87 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Interroute extrapolation. The results and conclusions from this model application are limited to
exposure via inhalation.
The Andersen et al. (1991) Model
Risk assessment. A modification of the Andersen et al. (1991) model by Reitz et al. (1997) was used
to derive an acute oral MRL from inhalation data in humans from Winneke (1974). For acute
neurological effects, the associated dose measure was defined as the peak concentration of methylene
chloride in brain tissue of humans exposed to 300 ppm of methylene chloride for 4 hours by inhalation.
The modified PBPK model calculated that the administered inhalation dose was equivalent to 3.95 mg of
methylene chloride per liter of brain tissue. The equivalent administered human concentration in drinking
water that will produce the same neurological effects was 565 mg of methylene chloride/liter. Using a
daily water consumption of 70 kg, the LOAEL was calculated to be 16 mg/kg/day.
Description of the model. Andersen et al. (1991) previously developed a PBPK model expanding the
original PBPK model of Ramsey and Andersen (1984) for the disposition of inhaled volatiles. This
model expands the previously developed Andersen et al. (1987) model by both describing the kinetics of
carbon monoxide (CO), COHb, and parent compound methylene chloride, and by comparing the
inhalation kinetics of CO and methylene chloride in rats and humans. The description of CO and COHb
kinetics was adapted from the Coburn-Forster-Kane equation, which assumes that most heme is bound
with oxygen and that endogenous CO production is constant. Methylene chloride kinetics and
metabolism were originally described by the generic PBPK model for volatile chemicals (Ramsey and
Andersen (1984). Predictions in humans from the model were compared to several experimental data sets
in the literature from human volunteers exposed to CO or to methylene chloride, and to experimental data
collected by Andersen et al. (1991) from six male volunteers exposed to methylene chloride vapors at
concentrations of 100 or 350 ppm for a period of 6 hours.
Physiological and biochemical constants for CO were first estimated by exposing rats to 200 ppm of CO
for 2 hours and examining the time course of COHb after cessation of CO exposure. The CO inhalation
studies provided estimates of CO diffusing capacity under free breathing and for the Haldane coefficient,
i.e., the relative equilibrium distribution ratio for hemoglobin between CO and oxygen. The CO model
was then coupled with the PBPK model for methylene chloride both to predict COHb time-course
concentrations during and after methylene chloride exposures in rats, and to estimate the yield of CO
88 METHYLENE CHLORIDE
2. HEALTH EFFECTS
produced from oxidation of methylene chloride. In rats, only about 0.7 mol of CO was produced from
1 mol of methylene chloride during oxidation. The combined model predicted COHb and methylene
chloride behavior following 4-hour exposures to 200 or 1,000 ppm of methylene chloride, and COHb
behavior following 30-minutes exposure to 5,160 ppm of methylene chloride. The rat PBPK model was
scaled to predict methylene chloride, COHb, and CO kinetics in humans exposed to either methylene
chloride or CO. Three human data sets from the literature were examined: inhalation of CO at 50, 100,
250, and 500 ppm; seven 30-minute inhalation exposures to 50, 100, 250, and 500 ppm of methylene
chloride; and 2-hour inhalation exposures to 986 ppm of methylene chloride. Additional experimental
data from volunteers exposed to 100 or 350 ppm of methylene chloride were also reported. Endogenous
CO production rates and the initial amount of CO in the blood compartment varied in each study, as
necessary, to provide a baseline value of COHb. The combined PBPK model accurately predicted the
experimental findings in all four human studies.
Validation of the model. In rats, the combined model adequately represented COHb and methylene
chloride behavior following 4-hour exposures to 200 or 1,000 ppm of methylene chloride, and COHb
behavior following ½-hour exposure to 5,160 ppm of methylene chloride. In addition, short-duration
exposures conducted with 5,000 ppm of bromochloromethane, with adjustment of metabolic parameters
and partition coefficients for this different chemical, demonstrated that the PBPK model gave a good
description of COHb levels for up to 6 hours postexposure. In humans, the combined PBPK model
provided a good representation of the experimental data in all four studies examined.
Target tissues. Blood was identified as the target. Both concentrations of COHb and methylene
chloride were considered in the model.
Species extrapolation. The application of the model was validated in both rats and humans. The
authors concluded that this model could be useful for developing biological monitoring strategies for CO
and methylene chloride, based on observed COHb blood concentrations following exposure.
Interroute extrapolation. The model was used to convert inhalation data in humans from Winneke
(1974) into equivalent oral doses that were used as the basis for the acute oral MRL (Reitz 1997).
89 METHYLENE CHLORIDE
2. HEALTH EFFECTS
The Dankovic and Bailer (1994) Model
Risk assessment. Risk assessment was not addressed directly in this model application. However,
the results indicated that some occupationally-exposed individuals may receive glutathione S-transferase-
metabolized methylene chloride doses several-fold greater than human doses previously estimated by
Reitz et al. (1989), based on differing levels of physical activity, and inter-individual variability in
methylene chloride metabolism. Therefore, the assessment of excess lifetime human risk associated with
methylene chloride exposure would be affected.
Description of the model. Dankovic and Bailer (1994) examined the impact of exercise and inter-
individual variability on human dose estimates to methylene chloride, using the models developed by
Andersen et al. (1987) and used by Reitz et al. (1989; Reitz 1990, 1991). Earlier models used
physiological parameters appropriate for humans at rest and metabolic parameters based on average rates
of methylene chloride metabolism. Dankovic and Bailer (1994) increased model parameters describing
cardiac output, alveolar ventilation, and blood flows to tissues to account for exercise, assuming an
8-hour workday exposed to mean methylene chloride concentrations of 25 ppm. The GSH-mediated
metabolized doses for human liver and lung were increased by a factor of 2.9 and 2.4, respectively, as
compared with the metabolized GSH-mediated dose estimates of Reitz et al. (1989). The model was also
modified to account for inter-individual variability in methylene chloride metabolism. Modeled
metabolized GSH-mediated dose estimates for human liver ranged from 0 to 5.4-fold greater than the
doses estimated by Reitz et al. (1989); for human lung, estimates were 0–3.6-fold greater than those of
Reitz et al. (1989). The authors concluded that their results indicated that some occupationally-exposed
individuals may receive GST-metabolized doses several-fold greater than human doses previously
estimated.
Validation of the model. The model used by Dankovic and Bailer (1994) has been previously
validated (Andersen et al. 1987; Reitz 1990, 1991; Reitz et al. 1988, 1989). The authors merely modified
the model parameters to be consistent with light work conditions, as opposed to the resting condition
parameters used in the earlier models, and to reflect inter-individual variability in methylene chloride
metabolism.
90 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Target tissues. Target tissues were the human liver and the human lung.
Species extrapolation. The model has been previously validated for use with rodents and humans.
Interroute extrapolation. The results and conclusions from this model application are limited to
exposure via inhalation.
The Casanova et al. (1996) Model
Risk assessment. The use of DNA-protein crosslinks as a tissue dosimeter of methylene chloride
exposure markedly reduced the upper-bound estimate and improved the precision of the low-dose excess
lifetime liver cancer risk estimates (as defined by the ratio of the upper-bound estimate to the maximum
likelihood estimate), while having only a minor effect on the maximum likelihood estimate. The
reduction in excess lifetime cancer risk was 2 orders of magnitude less than those estimated by using
airborne methylene chloride vapor concentrations of 10, 30, and 100 ppm.
Description of the model. Casanova et al. (1996) developed an extended PBPK model for DNA-
protein crosslink (DPX) formation in mouse liver associated with chronic methylene chloride exposure by
modifying the model originally developed by Andersen et al. (1987). This extended PBPK model
estimated area under the curve for methylene chloride in mouse liver as the independent variable.
Formaldehyde, one of at least two reactive intermediates formed during glutathione-mediated metabolism,
was considered to be the proximate metabolite associated with tumorigenicity. Parameter estimates for
formaldehyde disposition in methylene chloride-exposed mouse liver were derived from the published
literature. The amount of DPX formed in the mouse liver was estimated for methylene chloride
concentrations used in rodent cancer inhalation bioassay (i.e., 2,000 and 4,000 ppm). Using the linearized
multistage model, the tumor incidence data in mice were fitted to two alternative measures of exposure:
DPX yields and airborne concentration of methylene chloride. The 2 dose measures gave similar
maximum likelihood estimates for the excess lifetime cancer risk at administered concentrations ranging
from 10 to 100 ppm, but the upper 95% confidence limit on this risk estimate was reduced by 2 orders of
magnitude when DPX yield, as compared with airborne concentrations, was used as the measure of
exposure. These results demonstrate that in the case of methylene chloride, the use of DNA-protein
crosslinks as a dose surrogate markedly reduced the upper-bound estimate and improved the precision of
the low-dose risk estimates (as defined by the ratio of the upper-bound estimate to the maximum-
91 METHYLENE CHLORIDE
2. HEALTH EFFECTS
likelihood estimate), while having only a slight effect on the maximum-likelihood estimate. However,
Casanova et al. (1996) point out that DNA-protein crosslinks cannot be used directly as a surrogate for
the internal dose in humans because human hepatocytes, unlike mouse hepatocytes, do not appear to form
DNA-protein crosslinks in measurable amounts (Casanova et al. 1996). A surrogate for the internal dose,
RNA adducts of formaldehyde, has been developed and can be detected in human hepatocytes exposed to
methylene chloride (Casanova et al. 1996, 1997). The utility of this measure is under study.
Validation of the model. The model has been previously validated in other applications and for other
endpoints. For this application, predicted concentrations of DPX were compared with actual
concentrations induced by experimental concentrations.
Target tissues. The target tissue effect was the formation of DNA-protein crosslinks in mouse liver.
Species extrapolation. The results and conclusions from this model application are limited to DPX
formation in mouse liver.
Interroute extrapolation. Casanova et al. (1996) point out that DNA-protein crosslinks cannot be
used directly as a surrogate for the internal dose in humans, because human hepatocytes, unlike mouse
hepatocytes, do not appear to form DNA-protein crosslinks in measurable amounts (Casanova et al.
1996). As a surrogate for the internal dose, RNA adducts of formaldehyde have been developed and can
be detected in human hepatocytes exposed to methylene chloride (Casanova et al. 1996). The formation
of this dose surrogate in the hepatocytes of different species is currently being examined.
The Andersen and Krishnan (1994) Model
Risk assessment. The PBPK model-based risk assessment estimated an excess lifetime cancer risk to
humans of 3.7x10
-8
for a lifetime inhalation exposure of 2.8x10
-4
, which is lower by more than two orders
of magnitude than that calculated by the EPA using the linearized multistage model for low-dose
extrapolation and a default body surface correction factor for interspecies scaling.
92 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Description of the model. The mouse PBPK model developed earlier by Andersen et al. (1987) was
validated by comparing model predictions with observed pharmacokinetic data. To predict the tissue
dosimetry of methylene chloride and its metabolites in humans, the physiological parameters of the model
were scaled and methylene chloride-specific parameters for humans were determined. The metabolic rate
constants for humans were estimated from volunteer human exposure studies (Andersen et al. 1991). The
high-dose to low-dose extrapolation and interspecies extrapolation of methylene chloride induced cancer
risk were conducted with the tissue doses of the glutathione pathway metabolite predicted by the PBPK
model. This was validated by demonstration of a concentration-response association between this dose
metric and the degree of methylene chloride-induced rodent cancers rodent bioassay concentrations
between 2,000 and 4,000 ppm. The cancer risk assessment was conducted using the linearized multistage
model to relate tissue dose of methylene chloride glutathione metabolite to the increases in tumor
incidence in mice exposed to high concentrations of methylene chloride by inhalation. In assessing the
excess lifetime cancer risks associated with human exposure to methylene chloride, the authors assumed
that humans are as sensitive (i.e., not more sensitive) as the most sensitive target species; this assumption
is consistent with what is known about methylene chloride chemistry and metabolism, and about
interspecies variability in carcinogenic response to xenobiotics. The PBPK model-based risk assessment
estimated an excess lifetime human cancer inhalation unit risk 3.7x10
-8
per inhalation exposure of
2.8x10
-4
. This human risk estimate is lower by two orders of magnitude than that calculated by the EPA
using the linearized multistage model for low-dose extrapolation and a default body surface adjustment
factor for interspecies scaling.
Validation of the model. The model has been previously validated by comparing predicted with
actual pharmacokinetic data.
Target tissues. The target tissues were the liver and the lung.
Species extrapolation. The animal PBPK model was used for interspecies extrapolation of
pharmacokinetic behavior of methylene chloride by scaling the physiological parameters and determining
chemical-specific parameters in the species of interest (i.e., humans). Metabolic rates for humans were
developed from experimental literature on human volunteers. It was also assumed that humans are as
sensitive (i.e., not more sensitive than) as the most sensitive target species; this assumption is consistent
with what is known about methylene chloride chemistry and metabolism, and about interspecies
variability in carcinogenic response to xenobiotics.
93 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Interroute extrapolation. The results and conclusions from this model application are limited to
exposure via inhalation.
The Reitz et al. (1997) Model
1. Inhalation Route-to-Oral Route Extrapolation in Volunteers
Risk assessment. In this toxicological profile, ATSDR (2000) establishes a minimal risk level (MRL)
for acute inhalation using the study reported by Winneke (1974). Volunteers were exposed to
300–800 ppm of methylene chloride for approximately 4 hours and tested for neurobehavioral effects.
Concentration-dependent adverse effects included decreased performance in auditory vigilance-
performance tasks; and three small decrements in the visual critical flicker fusion frequency. Because
similar exposures to 50–100 ppm of carbon monoxide alone did not produce these effects, the author
concluded that they were mediated by methylene chloride directly and not by its oxidative metabolite,
carbon monoxide. A LOAEL of 300 ppm from this study was used to determine an acute oral drinking
water equivalent. Using pharmacokinetic modeling, Reitz et al. (1997) was able to estimate that an
inhalation concentration of 300 ppm of methylene chloride would produce a target organ-specific (brain-
specific) dose equivalent to that produced by a drinking water concentration of 565 mg/L of methylene
chloride. Multiplying the drinking water concentration by the default daily water consumption rate (2 L)
and dividing by the default human body weight yields an acute oral dose of 16 mg/kg/day.
Description of the model. Reitz et al. (1997) modified the basic PBPK methylene chloride model
developed by Andersen et al. (1987), Reitz et al. (1988), and Andersen et al. (1991) in the following
manner: (1) liver weights for rodents were based on the actual organ weights of control animals which
were sacrificed during chronic toxicity studies at 6–18 months of age; (2) partition coefficients for
methylene chloride derived from in vitro experiments performed by Gargas et al. (1986) were used for
liver, fat, muscle, and blood, and (3) a brain compartment was added to the methylene chloride model so
that central nervous system effects could be assessed in female and male rodents and humans. Size of
rodent brains, blood flow rates to the brain, and partition coefficients for brain tissue were obtained from
either published literature (e.g., Stott et al. 1983; Thomas 1975) or personal communication by the
authors. The modified methylene chloride model thus contained six tissue compartments: fat, muscle
(slowly perfused tissue), rapidly perfused tissue, liver tissue, mammary tissue, and brain tissue.
94 METHYLENE CHLORIDE
2. HEALTH EFFECTS
For acute neurological effects, the associated dose measure was defined as the peak concentration of
methylene chloride in brain tissue (mg/L of brain tissue) of humans exposed to 300 ppm of methylene
chloride for 4 hours, by inhalation. The modified PBPK model calculated that the administered inhalation
concentration was equivalent to 3.95 mg methylene chloride/L of brain tissue. Human exposure patterns
in the PBPK model simulated realistic human drinking water consumption patterns, (i.e., consisting of
bouts of drinking during the day, with and between meals, and little-to-no drinking during the night).
PBPK modeling predicted that peak concentrations of methylene chloride in the brain would increase
rapidly after each episode of drinking water consumption, and then drop sharply, to near-zero, between
bouts of drinking. Additionally, there would be no cumulative effects of repeated exposure. The
equivalent administered human concentration in drinking water was calculated to be 565 mg methylene
chloride/L. Using a daily drinking water consumption value of 2 L and an average human body weight of
70 kg, the LOAEL was calculated to be 16 mg/kg/day.
Validation of the model. Model predictions of brain methylene chloride concentrations in humans or
rats have not been evaluated for comparability with empirical observations. However, this model has
been previously validated with human and animal data.
Target tissues. The target organ of the model was the central nervous system.
Species extrapolation. There was no species extrapolation because the experiment that was
pharmacokinetically-modeled (Winneke 1974) was conducted in human volunteers.
Interroute extrapolation. The PBPK modeling was conducted to extrapolate from the inhalation route
of exposure to an oral route, specifically drinking water concentration of methylene chloride.
2. Inhalation Route-to-Oral Route Extrapolation and Rodent-to-Human Species
Extrapolation Using PBPK Modeling of Subchronic Toxicity Data
Risk assessment. ATSDR established an intermediate inhalation minimal risk level (MRL) using the
study reported by Haun et al. (1972). Rats were exposed continuously to either 25 or 100 ppm of
methylene chloride for 100 days. Data on liver histopathology of rats exposed to 25 or 100 ppm of
methylene chloride were selected as the critical effect. The authors report that liver cytoplasmic
vacuolization and Oil-Red-O staining associated with fatty deposits were observed in rats at both
exposure concentrations. In mice, fatty deposits (but no liver vacuolization) were reported at the higher
95 METHYLENE CHLORIDE
2. HEALTH EFFECTS
exposure concentration, 100 ppm. Additionally, renal changes were also reported in this study. ATSDR
considered the liver effects at 25 ppm to be adverse and established that concentration as a LOAEL.
Using pharmacokinetic modeling and data on human drinking water consumption patterns, Reitz et al.
(1997) estimated that a drinking water concentration of 6,170 mg/L of methylene chloride produces a
target organ-specific dose equivalent to that produced by an inhalation concentration of 25 ppm.
Multiplying the drinking water concentration by the default daily water consumption rate (2 L) and
dividing by the default human body weight yields an intermediate oral dose of 176 mg/kg/day.
Description of the model. For inhalation-to-oral and rat-to-human extrapolations, Reitz et al. (1997)
modified the basic PBPK methylene chloride model developed by Andersen et al. (1987), Reitz et al.
(1988), and Andersen et al. (1991) in the following manner: (1) liver weights for rodents were based on
the actual organ weights of control animals which were sacrificed during chronic toxicity studies at
6–18 months of age; (2) partition coefficients for methylene chloride derived from in vitro experiments
performed by Gargas et al. (1986), were used for liver, fat, muscle, and blood; and (3) a brain
compartment was added to the methylene chloride model so that central nervous system effects could be
assessed in female and male rodents and humans. Size of rodent brains, blood flow rates to the brain, and
partition coefficients for brain tissue were obtained from either published literature (Stott et al. 1983;
Thomas 1975) or personal communication by the authors. The modified methylene chloride model thus
contained six tissue compartments: fat, muscle (slowly perfused tissue), rapidly perfused tissue, liver
tissue, mammary tissue, and brain tissue.
The PBPK model was utilized to compare the average daily production of methylene chloride metabolites
per L of liver in rats exposed to methylene chloride via inhalation for 24 hours/day with the mean daily
production of methylene chloride metabolites per L of liver in humans drinking water that contained
specific concentrations of methylene chloride. The dose measure was defined as the average daily
concentration of metabolites per L of liver tissue. No distinctions were made between metabolites
produced via the MFO pathway and those produced during glutathione conjugation by the GSH pathway.
Total metabolite production was obtained by integrating the rate of metabolite production during
simulation; the result was then divided by the number of 24-hour exposure periods simulated and the
volume of the liver in each species (rodent and human). At inhalation concentrations by rats of 25 ppm
for 24 hours/day for 100 days, the metabolized tissue-specific dose calculated by the model was 1,259 mg
96 METHYLENE CHLORIDE
2. HEALTH EFFECTS
metabolites methylene chloride/L liver/day. In humans, the drinking water concentration yielding an
equivalent concentration of metabolized tissue-specific dose was found to be approximately 6,170 mg/L.
Multiplying this value by 2 L/day (human drinking water consumption rate) and dividing by the default
human body weight of 70 kg yields a daily dose (NOAEL) of 142 mg/kg/day.
Validation of the model. Model predictions of the concentrations of methylene chloride or its
metabolites in liver of humans or rats have not been evaluated for comparability with empirical
observations. However, this model has been previously validated with human and animal data.
Target tissue. The target tissue was the liver.
Species extrapolation. Rat-to-human extrapolation was conducted using appropriate partition
coefficients, metabolic rates, and other species-specific PBPK variables.
Interroute extrapolation. The PBPK modeling was conducted to extrapolate from the inhalation route
of exposure to an oral route, described in terms of drinking water concentration of methylene chloride.
3. Inhalation Route-to-Oral Route Extrapolation and Rodent-to-Human Species
Extrapolation Using PBPK Modeling of Developmental Toxicity Data
Risk assessment. The developmental toxicity of methylene chloride in rodents was studied by
Schwetz et al. (1975) who exposed rats to 1,250 ppm of methylene chloride vapors for 7 hours/day from
day 6–15 of gestation. There were slight, statistically significant increases in the incidence of minor
skeletal variants in the offspring of females exposed to 1,250 ppm of methylene chloride during gestation.
Therefore, 1,250 ppm was considered to be a LOAEL. The PBPK model was used to extrapolate from
pregnant rodents to pregnant humans and from inhalation route to oral route of exposure. Using
pharmacokinetic modeling and data on human drinking water consumption patterns, Reitz et al. (1997)
estimated that a maternal drinking water concentration of 5,000 mg/L of methylene chloride produces a
fetal dose equivalent to that produced by a maternal inhalation concentration of 1,250 ppm. Multiplying
97 METHYLENE CHLORIDE
2. HEALTH EFFECTS
the drinking water concentration by the default daily water consumption rate (2 L) and dividing by the
default human body weight of 70 kg yields an intermediate oral dose of
142 mg/kg/day.
Description of the model. For determination of developmental effects, modifications to the basic
PBPK methylene chloride model (Andersen et al. 1987) were based on procedures reported by Fisher et
al. (1989). The model consisted of the addition of three compartments: mammary tissue; placental tissue;
and fetal tissue. The fetal dose measure was of the parent compound in fetal tissue and was estimated by
integrating the fetal concentration of methylene chloride (expressed as mg/L of fetal tissue) during the
exposure period in order to calculate the total area under the curve (AUC) for the exposure period. The
AUC was then divided by the total number of hours during which gestational exposure occurred to give
the average concentration of methylene chloride during the exposure period. Because it was assumed that
human fetuses would be exposed continuously throughout gestation, the total AUC for the human fetus
was divided by the total gestation period to calculate the fetal concentration of parent compound
methylene chloride (not metabolites), expressed in mg methylene chloride/L of fetal tissue.
Simulation of the exposure paradigm used by Schwetz et al. (1975) with the PBPK model yielded
3.67 mg methylene chloride/L of fetal tissue in the rodent. PBPK simulation of human drinking water
exposures gave equivalent values of mg methylene chloride/L of fetal tissue at methylene chloride
concentrations approximately 5,000 mg/L of water. Multiplying by 2 L/day and dividing by 70 kg body
weight yielded a human LOAEL of 142 mg/kg/day.
Validation of the model. Model predictions of the concentrations of methylene chloride in rat or
human fetal tissues have not been evaluated for comparability with empirical observations. However, this
model has been previously validated with human and animal data.
Target tissues. The target tissue was the developing fetus.
Species extrapolation. Rat-to-human extrapolation was connected using appropriate partition
coefficients, metabolic rates, and other species-specific PBPK variables.
98 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Interroute extrapolation. The PBPK modeling was conducted to extrapolate from the inhalation route
of exposure to an oral route, described in terms of drinking water concentration of methylene chloride.
The Fisher et al. (1997) Model
Risk assessment. The model provides an approach to estimating rates of intake of methylene
chloride and other volatile chemicals by the nursing infant for a given temporal pattern of maternal
exposure and infant nursing. Simulations of a workday maternal exposure to methylene chloride at an air
concentration of 50 ppm (174 mg/m
3
) yielded a predicted intake in the infant of 0.213 mg/24 hours
(Fisher et al. 1997).
Description of the model. Fisher et al. (1997) described a PBPK model for estimating amounts and
concentrations of methylene chloride in human breast milk that would result from inhalation exposures to
methylene chloride (other air borne volatile chemicals are also simulated in the model). The human
lactation model is an adaptation of the PBPK model of Ramsey and Andersen (1984), with the addition of
a breast milk compartment and parameters for simulating breast milk production and intake of breast milk
by nursing infants. The model simulates seven tissue compartments: blood, lung, fat, liver, richly
perfused tissues, poorly perfused tissues, and breast milk. Two metabolic pathways for methylene
chloride are assumed to occur exclusively in the liver. A glutathione-S-transferase pathway is represented
by an allometrically scaled first order rate constant, and a mixed function oxidase pathway is represented
by a Michaelis-Menton function with constants V
max
and K
m
.
The major innovation in this model is simulation of a breast milk compartment, which allows calculations
of the rates of transfer of chemicals from blood into breast milk and rates of transfer to the infant during
breast feeding. The volume of the breast milk compartment is assumed to decrease from an initial volume
of 0.125 L at the beginning of each nursing session to a residual volume of 0.010 L at the end of each
session. The rate of change in the milk volume is simulated as the difference between a zero order
production rate of 0.06 L/hour and the loss rate from nursing, defined as the product the current milk
volume and a first order loss constant of 20/hour. The amount of methylene chloride in breast milk is
calculated using standard PBPK algorithms for flow-limited transfer from blood, assuming that methylene
chloride partitions from blood directly into breast milk according to empirically derived milk/blood
99 METHYLENE CHLORIDE
2. HEALTH EFFECTS
partition coefficients (Fisher et al. 1997). The resulting concentrations in breast milk are used to calculate
rates of intake by the nursing infant for a given temporal pattern of maternal exposure and infant nursing.
Validation of the model. The human lactation model, as described in Fisher et al. (1997), has not
been calibrated against an empirical data set or validated for any specific use of the model.
Target tissues. Output from the model described are estimates of the amount and concentration of
methylene chloride in breast milk and the rate of intake of methylene chloride by a nursing infant.
Species extrapolation. The model is designed to predict the transfer of inhaled methylene chloride to
breast milk in humans. Extrapolation to other species would require modification of the model to account
for different tissue masses, blood flows, and possibly other kinetic variables.
Interroute extrapolation. The model is designed to simulate the pharmacokinetics of methylene
chloride when exposure is by inhalation to airborne methylene chloride. The pharmacokinetics of
methylene chloride would be expected to be different for other routes of exposure; therefore, the output of
the model cannot be extrapolated to other exposure pathways (e.g., dietary, drinking water) or routes
(e.g., oral, dermal) without modification of the model.
The DeJongh et al. (1998) Model
Risk assessment. The model provides an approach to estimating the brain concentrations of
methylene chloride associated with acute inhalation exposures in rats. DeJongh et al. (1998) reported the
results of simulations of exposures at the 15-minute and 6-hour LC
50
in the rat. The predicted methylene
chloride concentrations in brain were as follows: 15-minute LC
50
exposure, 95,781 ppm (331,770 mg/m
3
,
brain 24.3 mM, brain lipid 97.6 mM; and 6-hour LC
50
exposure, 25,181 ppm (87,469 mg/m
3
), brain
17.3 mM (1,594 mg/L), brain lipid 69.5 mM (6,403 mg/L).
Description of the model. DeJongh et al. (1998) developed a PBPK model for estimating brain
concentrations of methylene chloride and other airborne volatile chemicals in rats. The model is an
adaptation of PBPK models of toluene (DeJongh and Blaauboer 1996) and styrene (Ramsey and
Andersen 1984), with the addition of a brain compartment. The model simulates seven tissue
compartments: blood, lung, fat, liver, richly perfused tissues, slowly perfused tissues, and brain. Two
100 METHYLENE CHLORIDE
2. HEALTH EFFECTS
metabolic pathways for methylene chloride are assumed to occur exclusively in the liver. A glutathione-
S-transferase pathway is represented by an allometrically scaled first order rate constant, and a mixed
function oxidase pathway is represented by a Michaelis-Menten function with constants V
max
and K
m
.
The major innovation in this model is the simulation of a brain compartment, which allows calculations of
the rates of transfer of methylene chloride from blood into water and lipid compartments of brain. The
amount of methylene chloride in the brain is calculated using standard PBPK algorithms for flow-limited
transfer from blood, assuming that methylene chloride partitions from blood directly into the brain
according to an empirically-derived blood-brain partition coefficient (Gargas et al. 1989). Partitioning of
methylene chloride between aqueous and lipid compartments in the brain is calculated from an octanol-
water partition coefficient (Meylan and Howard 1995).
Validation of the model. The model, as described in DeJongh et al. (1998), has not been calibrated
against an empirical data set or validated for predicting brain methylene chloride concentrations.
Target tissues. Outputs from the model described in DeJongh et al. (1998) are estimates of the amount
and concentration of methylene chloride in whole brain or brain lipid.
Species extrapolation. The model is designed to predict the distribution of inhaled methylene
chloride to the brain in rats. Extrapolation to other species would require modification of the model to
account for different tissue masses, blood flows, and possibly other kinetic variables.
Interroute extrapolation. The model is designed to simulate the pharmacokinetics of methylene
chloride when exposure is by inhalation to airborne methylene chloride. The kinetics would be expected
to be different for other routes of exposure; therefore, the output of the model cannot be extrapolated to
other exposure pathways (e.g., dietary, drinking water) or routes (e.g., oral, dermal) without modification
of the model.
The Poulin and Krishnan (1999) Model
Risk assessment. The model provides an approach to estimating the concentrations of methylene
chloride in venous blood, and possibly other tissues, associated with acute inhalation exposures. Poulin
and Krishnan (1998) reported the results of simulations of a 6-hour inhalation exposure of an adult human
101 METHYLENE CHLORIDE
2. HEALTH EFFECTS
to 100 ppm methylene chloride (347 mg/m
3
). The predicted methylene chloride concentrations in venous
blood after 6 hours of exposure were approximately 0.5 and 1.5 mg/L for the bounding assumptions, liver
extraction ratio of one or zero, respectively.
Description of the model. Poulin and Krishnan (1999) described a quantitative structure-toxico-
kinetic relationship (QST
k
R) model for estimating venous blood concentrations, and tissue:air and
tissue:blood partition coefficients of methylene chloride (and other airborne volatile chemicals) in
humans. The QST
k
R model is an adaptation of the PBPK model of Andersen et al. (1991) with the
addition of a model for estimating values of the partition coefficients based on molecular structure
fragment and tissue composition information. The model simulates blood, lung, fat, liver, richly perfused
tissues, and slowly perfused tissues. Two innovations in this model are the approach to the simulation of
methylene chloride metabolism in the liver and the inclusion of the structure-based estimation of partition
coefficients.
The rate of metabolism of methylene chloride in the liver is represented as the products of the liver blood
flow rate, the arterial concentration of methylene chloride, and the liver extraction ratio (ratio of hepatic
clearance to hepatic blood flow). This approach does not require specification of values for the kinetic
constants of metabolism (e.g., V
max
, K
m
), but does require specification of a value, or range of values, for
the liver extraction ratio. In the absence of an empirical basis for any given value for the extraction ratio,
bounding estimates for venous blood concentrations, as affected by hepatic metabolism, can be made by
running simulations in which the extraction ratio is assumed to be either zero or one (Poulin and Krishnan
1999).
The octanol:water partition coefficient, the water:air partition coefficient, and the boiling point are
estimated using molecular structure fragment information (Poulin and Krishnan 1996, 1998). The
estimated values for the above parameters are used with tissue composition information (e.g., water and
lipid content) to estimate values for blood:air and tissue:blood partition coefficients.
Validation of the model. Estimates of blood:air and tissue:blood concentration predicted with the
QST
k
R compared well with empirical determinations for methylene chloride (and a variety of other
volatile organic compounds). Measured venous blood concentrations of methylene chloride in humans
exposed to 100 ppm methylene chloride (347 mg/m
3
) for 6 hours (Andersen et al. 1991) were within the
bounding estimates (liver extraction ratio assumed to be zero or one) predicted with the QST
k
R.
102 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Target tissues. Output from the model described in Poulin and Krishnan (1998) are estimates of the
concentration of methylene chloride in venous blood.
Species extrapolation. The model is designed to predict venous blood concentration of methylene
chloride in humans. Extrapolation to other species would require modification of the model to account
for different tissue masses, blood flows, and possibly other kinetic variables.
Interroute extrapolation. The model is designed to simulate the pharmacokinetics of methylene
chloride when exposure is by inhalation to airborne methylene chloride. The kinetics would be expected
to be different for other routes of exposure; therefore, the output of the model cannot be extrapolated to
other exposure pathways (e.g., dietary, drinking water) or routes (e.g., oral, dermal) without modification
of the model.
The Pelekis and Krishnan (1997) Model
Risk assessment. The model provides an approach to estimating the COHb levels in blood that
would result from oral or inhalation exposures to mixtures of methylene chloride and toluene if certain
assumptions are made regarding the mechanism of interaction between the two chemicals: (1) the
interaction occurs at the level of the mixed function oxidase-mediated metabolism of methylene chloride
to carbon monoxide in the liver; and (2) the mechanism of inhibition can be simulated by Michaelis-
Menten type models of competitive, noncompetitive or uncompetitive inhibition of the pathway. Pelekis
and Krishnan (1997) extrapolated the rat model to humans by replacing the rat values with human values
for physiological variables and by assuming that partition coefficients and the metabolism kinetic
constants are the same in the rat and human. The resulting human model was used to predict the area
under the COHb-time curve for a human exposure to the ACGIH 8-hour TLV for methylene chloride
(50 ppm, 174 mg/m
3
), or to a simultaneous exposure to the methylene chloride and toluene at their
respective TLVs (50 ppm, 188 mg/m
3
). The model predicted a 2–9% decrease in COHb levels in the
mixed exposure compared to the exposure to methylene chloride alone, depending on which mechanism
of inhibition of the mixed function oxidase pathway was assumed.
Description of the model. Pelekis and Krishnan (1997) linked a PBPK model for methylene
chloride (Andersen et al. 1991) with a PBPK model of toluene (Tardif et al. 1993). The composite model
was used to simulate the kinetics of COHb production resulting from exposures to mixtures of the two
103 METHYLENE CHLORIDE
2. HEALTH EFFECTS
chemicals in rats or humans. The model simulates seven tissue compartments: blood, gastrointestinal
tract, lung, fat, liver, richly perfused tissues, and slowly perfused tissues. Metabolic pathways for
methylene chloride are assumed to occur exclusively in the liver. A glutathione-S-transferase pathway is
represented by a first order rate constant, and a mixed function oxidase pathway, which produces carbon
monoxide as a product, is represented by a Michaelis-Menton function with constants V
max
and K
m
. The
metabolism of toluene is assumed to occur in the liver through a mixed function oxidase pathway. An
innovation in this model is the simulation of interactions between methylene chloride and toluene at the
level of the mixed function oxidase pathway. This is achieved with Michaelis-Menton equations that
simulate four possible interaction possibilities: (1) no interaction between methylene chloride and
toluene; (2) competitive inhibition of the mixed function oxidase pathway (increase in apparent K
m
); (3)
noncompetitive inhibition of the pathway (decrease in V
max
); or (4) or uncompetitive inhibition (increase
in K
m
and decrease in V
max
). The relationship between carbon monoxide production (including
endogenous production) and percent COHb in blood is simulated using the Coburn-Foster-Kane model,
as implemented in the model described by Andersen et al. (1991).
Validation of the model. The results of simulations were compared with observations made in
studies of single or mixed exposures to methylene chloride and toluene in rats (Ciuchta et al. 1979;
Pankow et al. 1991a, 1991b). Pankow et al. (1991a, 1991b) reported the levels of COHb in rats exposed
to either a single oral dose of methylene chloride (6.2 mmol/kg, 527 mg/kg), or combined single oral
doses of 6.2 mmol/kg methylene chloride (527 mg/kg) and 18.8 mmol/kg toluene (1,732 mg/kg) (Pankow
et al. 1991a, 1991b). The observed COHb level 6 hours after dosing with methylene chloride was 9.3%
compared to 1.7% after combined dosing with methylene chloride and toluene. The corresponding COHb
levels simulated with the PBPK model were 8.7% for dosing with methylene chloride alone, and 2.1% for
the combined dosing, assuming competitive inhibition of the mixed function oxidase pathway, or 0.6%,
assuming either noncompetitive or uncompetitive inhibition (Pelekis and Krishnan 1997). The PBPK
model also predicted COHb levels 12 hours after dosing with methylene chloride alone and the
corresponding 12-hour area under the COHb-time curve that agreed well with observations from Pankow
et al. (1991). Observed peak COHb levels following exposure of rats for 1 hour to 5,000 ppm
(17,368 mg/m
3
) methylene chloride were 10–12% compared to less than 1% when the methylene chloride
exposure occurred 30 minutes after a single intraperitoneal dose of 0.005 mmol/kg (0.46 mg/kg) of
toluene (Ciuchta et al.1979). Corresponding model simulations agreed well with these observations, only
when uncompetitive or noncompetitive inhibition of the mixed function oxidase pathway was assumed.
The competitive inhibition model severely overestimated the peak COHb concentrations observed in the
104 METHYLENE CHLORIDE
2. HEALTH EFFECTS
combined exposure experiment. These results suggest that, of the three interactions mechanisms
simulated, the noncompetitive and uncompetitive mechanisms more accurately predicted the in vivo
observations.
Target tissues. Output from the model described in Pelekis and Krishnan (1997) are estimates of the
COHb level in arterial blood (expressed in units of percent saturation of hemoglobin).
Species extrapolation. The model was calibrated against observed arterial blood COHb levels in rats.
Although Pelekis and Krishnan (1997) have extrapolated the rat model to simulate human exposures to
methylene chloride and toluene, model outputs have not been compared to empirical observations in
humans; therefore, the accuracy of the extrapolation has not been evaluated. Extrapolation to other
species (other than human) would require additional modifications to the model to account for different
tissue masses, blood flows, and possibly other kinetic variables.
Interroute extrapolation. The model is designed to simulate the pharmacokinetics of methylene
chloride and toluene when exposures are by the inhalation or oral routes. COHb levels predicted by the
model are highly sensitive to the assumed value of the gastrointestinal absorption rate constant of
methylene chloride, and less sensitive to the value of the rate constant for absorption of toluene (Pelekis
and Krishnan 1997). This suggests that potential dose level or exposure medium effects (e.g., diet,
drinking water) or interaction effects on the absorption rate constants should be considered in simulations
of the oral route. The kinetics would be expected to be different for other routes of exposure; therefore,
the output of the models cannot be extrapolated to other exposure pathways (e.g., dermal) without
modification of the model.
The OSHA (1997) Model
Risk assessment. The OSHA model provides an approach to estimating various internal dose
surrogates corresponding to inhalation exposures to methylene chloride. OSHA (1997) used the mouse
PBPK model to translate methylene chloride inhalation exposure levels used in an NTP (1986) mouse
bioassay to equivalent amounts of methylene chloride metabolized by the mouse lung glutathione-
S-transferase pathway. The resulting internal dose estimates were used to establish an internal dose-
response relationship for lung tumors observed in the mouse bioassay. The mouse internal dose-response
relationship was then translated into an equivalent human internal dose-response relationship using the
105 METHYLENE CHLORIDE
2. HEALTH EFFECTS
human PBPK model. The mean and 95th percentile cancer risks (based on the maximum likelihood of
the dose-response parameters) associated with a human exposure to 25 ppm methylene chloride
(87 mg/m
3
), 8 hours/day, 5 days/week for 45 years were 1.24x10
-3
and 3.63x10
-3
, respectively.
Description of the model. OSHA (1997) proposed a PBPK model in support of its Final Rule on
limits for occupational exposure to methylene chloride. The OSHA model is based on several more
recent extensions of the Andersen et al. (1991) model as described by Clewell (unpublished report
available as an exhibit in OSHA (1997) and Reitz et al. (1997). The model simulates eight tissue
compartments: blood, gastrointestinal tract, lung, fat, liver, well perfused tissues, poorly perfused tissues,
and bone marrow.
Innovations in the OSHA model include the following. (1) Bone marrow is simulated as a distinct tissue
rather than including it in the well or poorly perfused tissue compartment. (2) Metabolism of methylene
chloride is assumed to occur in the liver and lung tissues. The liver and lung K
m
values for the mixed
function oxidase pathway are assumed to be identical. The values for the V
max
for the mixed function
oxidase pathway in the lung and the rate constant for the glutathione-S-transferase pathway in the lung
are set as fixed fractions of their respective values in liver. These estimates take into account relative
tissue volumes, in vitro estimates of metabolism rates in the two tissues, and the relative abundance of
microsomal or soluble protein in the two tissues. (3) Alveolar ventilation rates are dependent on cardiac
output. The relationship between the two variables is assumed to be a direct proportionality with a
ventilation-perfusion ratio as the proportionality term. (4) Cardiac output, tissue distribution of cardiac
output, and the ventilation-perfusion ratio are related to work intensity allowing various work-related
exposure scenarios to be simulated. (5) Tissue blood flows, as a fraction of the cardiac output, are
constrained so that either the fractional flow to the well-perfused tissues (mouse model) or poorly
perfused tissues (human model) is set as 1 minus the sum of the fractional flows to all other tissues. This
constraint was imposed as an approach to ensure mass balance of flows when a Monte Carlo sampling
approach was used to select parameter values.
Probability distributions for input parameters in the mouse and human versions of the OSHA model were
developed using a Bayesian analysis of empirical data from gas uptake studies in mice and human open
chamber inhalation studies, respectively. The resulting probability distributions were used in a Monte
Carlo approach to implement the PBPK models. This approach results in probability distributions for
model outputs, rather than single point estimates.
106 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Validation of the model. The mouse and human models were calibrated using data from gas uptake
studies in mice and human open chamber inhalation studies (OSHA 1997). Comparisons of model
outputs with empirical observations other than those used in the Bayesian analysis were not reported in
OSHA (1997).
Target tissues. The mouse and human models have been used to estimate the amounts of methylene
chloride metabolized through the glutathione-S-transferase pathway in liver and lung (OSHA 1997).
Species extrapolation. Separate models for the mouse and human have been developed, enabling
extrapolations of internal dose surrogates between these two species (OSHA 1997). Extrapolation to
other species would require additional modifications to the models to account for different tissue masses,
blood flows, and possibly other kinetic variables.
Interroute extrapolation. The mouse and human models have been used to simulate the
pharmacokinetics of methylene chloride when exposures are by the inhalation route (OSHA 1997). The
models include gastrointestinal tissue compartment and, therefore, could be applied to oral exposures,
provided that validation studies support such uses of the model. The kinetics would be expected to be
different for other routes of exposure; therefore, the output of the models cannot be extrapolated to other
exposure pathways (e.g., dermal) without modification of the models.
2.4 MECHANISMS OF ACTION
2.4.1 Pharmacokinetic Mechanisms
The physical properties of methylene chloride, particularly its lipophilic nature, high vapor pressure, and
high serum/air partition coefficient, suggest that it is likely to be absorbed across the alveolar membranes
of the lung, mucosal membranes of the gastrointestinal tract, and the skin by passive diffusion. Once in
the body, it is widely distributed, with the greatest amounts accumulating in the more lipophilic tissues;
this probably also occurs by passive diffusion.
Andersen et al. (1987), Reitz (1990, 1991), Reitz et al. (1988), and Andersen and Krishnan (1994) have
used a PBPK model to predict amounts of methylene chloride metabolism produced (mg equivalents of
methylene chloride metabolized per L of liver tissue per day) produced by the two major pathways of
107 METHYLENE CHLORIDE
2. HEALTH EFFECTS
methylene chloride metabolism: the MFO pathway and the GSH-mediated pathway. This model
identifies the pathway which appears to activate methylene chloride to reactive intermediates but neither
characterizes the putative metabolite nor describes the mechanisms of action. In essence, the MFO
pathway is oxidative and appears to yield carbon monoxide as well as considerable amounts of carbon
dioxide. The glutathione-dependent pathway produces formaldehyde and carbon dioxide, but no carbon
monoxide. Potentially reactive intermediates are formed in each of the metabolic pathways for methylene
chloride: formyl chloride via the MFO pathway and methylchloroglutathione via the GSH pathway.
Distribution of methylene chloride metabolism between these pathways is dose dependent. The MFO
pathway is a high-affinity, limited-capacity pathway which saturates at relatively low airborne
concentrations (about 200–500 ppm). In contrast, the GSH pathway has a lower affinity for methylene
chloride, and does not appear to saturate at experimental concentrations (<5,000 ppm). Thus, at low
concentrations, most of the methylene chloride is metabolized by the MFO pathway. As exposure
concentrations increase and the MFO pathway saturates, metabolism by the secondary GSH pathway is
observed.
Predicted amounts of methylene chloride metabolism produced by each pathway (mg equivalents of
methylene chloride metabolized per L of liver tissue per day) are presented in Reitz’s (1990, 1991)
analyses. The predicted amounts of MFO metabolites formed in lung and liver tissue are nearly identical
for the 2 concentration levels in the mouse inhalation cancer bioassay (2,000 and 4,000 ppm) because
saturation of the MFO pathway occurs at administered airborne concentrations of approximately
200–500 ppm. However, mouse liver and lung tumors are consistently higher in the 4,000 ppm exposure
group than in the 2,000 ppm group in this bioassay. Therefore, there is no concentration-response
relationship between rodent tumor incidence and MFO metabolite levels. The PBPK model also predicts
that levels of MFO metabolites produced by long-term drinking water administration of 250 mg/kg/day
methylene chloride to B6C3F
1
mice should be similar to the levels of MFO metabolites produced in the
inhalation bioassay. However, no statistically significant increases in tumors in the drinking water
bioassay were observed; therefore, MFO metabolites would not be predicted to be involved in rodent
tumorigenicity by either route of administration. Predicted rates of metabolism of methylene chloride by
the GSH pathway correlate more clearly with the induction of lung and liver tumors, or lack thereof, in
the two chronic studies. In the inhalation study, predicted levels of GSH metabolites at 4,000 ppm are
higher than predicted levels of GSH metabolites at 2,000 ppm. In contrast, predicted levels of GSH
metabolites formed in target tissues during drinking water administration of methylene chloride are very
low. Thus, the pattern of tumor induction, or lack thereof, in both studies shows a good correlation with
108 METHYLENE CHLORIDE
2. HEALTH EFFECTS
the rates of metabolism of methylene chloride by the GSH pathway. Because the dose dependency of
GSH metabolite formation is nonlinear at low concentrations, the delivered dose arriving at the target
sites cannot be directly extrapolated from very high inhalation concentrations (4,000 ppm) to the very low
concentrations (<1 ppm) typical of human exposure.
Casanova et al. (1996) developed an extended PBPK model for DNA-protein crosslink (DPX) formation
in mouse liver, based on the model originally developed by Andersen et al. (1987). The extended PBPK
model estimated area under the curve for methylene chloride in mouse liver as the independent variable.
Tissue-specific yields of DPX were used as the dose surrogate. Estimates were made of the amount of
DPX formed in the mouse liver at methylene chloride inhalation concentrations used in the bioassay (i.e.,
2,000 and 4,000 ppm) and plotted against liver tumor yields in the mouse. DPX thus served as a
concentration surrogate for airborne methylene chloride concentrations. The model assumes that DPX
formation is associated with methylene chloride mouse liver tumorigenicity. Because formaldehyde
produces DPX, and GSH-mediated metabolism of methylene chloride produces formaldehyde as a
reactive intermediate, the authors suggest that formaldehyde is involved in the genotoxic mechanism(s) of
action associated with mouse liver tumorigenicity. When excess lifetime cancer risk was estimated using
the mouse liver tumor data and two alternate dose measures, DPX and airborne vapor concentrations, the
maximum likelihood estimates were similar, but the upper-bound estimate using DPX was two orders of
magnitude lower than that using airborne concentrations. Thus, DPX appears to be a reasonable dose
surrogate for methylene chloride inhalation exposure. However, most other investigators do not consider
DPX formed by the weakly mutagenic activity of formaldehyde to be the putative mechanism of action of
methylene chloride-induced liver tumorigenicity (see Section 2.4.2, Mechanisms of Toxicity).
Furthermore, human hepatocytes do not appear to form DPX in measurable amounts, as do mouse
hepatocytes (Casanova et al. 1996).
2.4.2 Mechanisms of Toxicity
The lung, the blood system, and the nervous system are the major target organs of toxicity associated with
exposure to methylene chloride.
Non-neoplastic Mechanisms. In humans, Snyder et al. (1992a, 1992b) have reported headache, chest
discomfort, cough, and the presence of alveolar and interstitial infiltrates in the lung as a result of short-
term high-concentration vapor exposure to methylene chloride in confined, unventilated rooms or
109 METHYLENE CHLORIDE
2. HEALTH EFFECTS
basements. In B6C3F
1
mice exposed to 4,000 ppm of methylene chloride vapors for 6 hours (Foster et al.
1992), the major initial morphological effect observed in mouse lung was acute Clara cell damage.
However, the damage appeared to resolve after five consecutive daily exposures to methylene chloride.
The appearance and disappearance of the lesion in the Clara cell correlated well with the activity of
cytochrome P-450 monooxygenase in the Clara cell, as assessed immunocytochemically (CYP2B1 and
CYP2B2) in the whole lung and biochemically in the freshly isolated Clara cell (as determined by
ethoxycoumarin O-dealkylation and aldrin epoxidation).
Over 13 weeks (5 days/week) of exposure, the acute Clara cell damage, which developed after a 1-day
exposure but resolved after 5 consecutive exposures, reappeared on reexposure after a 2-day weekly
break. The severity of the lesion diminished as the study progressed. The authors suggest that the reason
for the decrease or disappearance of the lesion was due to an adaptation/tolerance in the Clara cell to
methylene chloride that was linked to a marked decrease of methylene chloride metabolism by
cytochrome P-450 pathways. Glutathione (GST) activity in the Clara cell either remained unchanged or
increased following methylene chloride exposure.
Inhalation and ingestion exposures to methylene chloride result in the production of carbon monoxide
associated mainly with metabolism via the MFO pathway. CO binds to hemoglobin, and can cause
carboxyhemoglobinemia. In two fatal human cases of methylene chloride poisoning, COHb was elevated
to approximately 30% (Manno et al. 1992). Other reports on human and animals show that COHb
increases from baselines of 0–2 to 4–15%, under varying regimes of methylene chloride inhalation
exposure.
Neurotoxicity resulting from exposure to methylene chloride is believed to be associated with the
lipophilic properties of methylene chloride; however, the precise mechanisms of neurotoxicity are not
known. Presumably, the methylene chloride enters cell membranes, which in the case of neurons,
interferes with signal transmission, in a manner similar to general anesthetics (De Jongh et al. 1998;
Sikkema et al. 1995). Neurotoxicity is also assumed to be caused by the hypoxia that results from the
formation of COHb.
Cancer. With regard to tumor induction in the rodent lung and liver, methylene chloride is postulated to
be activated to an unknown reactive intermediate via metabolism. There are two major metabolic
pathways: the MFO pathway, specifically cytochrome P-450 2E1 and glutathione-glutathione
110 METHYLENE CHLORIDE
2. HEALTH EFFECTS
S-transferase-mediated (GSH-GST) pathway. The isoenzyme involved in the GSH-GST pathway has
been identified as a Θ (theta) class glutathione S-transferase, GSTT1-1, which is present in moderate
quantities in the mouse lung, but has been detected only at very low levels in human lung and liver tissue
samples (Mainwaring et al. 1996b; Sheratt et al. 1997). These findings suggest that in humans the lung
and liver are likely to have little capacity to activate methylene chloride into its reactive metabolites.
However, higher than background levels of GSTT1-1 mRNA were detected in a small number of Clara
cells and alveolar/bronchiolar ciliated epithelial cells of one human lung sample (out of four) and of
GSTT2-2 enzyme in the biliary epithelium of the human liver (Mainwaring et al. 1996b). Thus, it is
possible that, in some individuals, these specific cell types may be vulnerable to genotoxic effects from
reactive intermediates of methylene chloride metabolism, although the overall risk is likely to be low.
The MFO pathway is oxidative and appears to yield carbon monoxide as well as considerable amounts of
carbon dioxide. The glutathione-dependent pathway produces formaldehyde and carbon dioxide, but no
carbon monoxide. Potentially reactive intermediates are formed in each of the metabolic pathways for
methylene chloride: formyl chloride in the oxidative pathway, and formaldehyde and chloromethyl
glutathione in the conjugative pathway. Neither formyl chloride nor the glutathione conjugate of
methylene chloride has been isolated or characterized, although Green (1997) reports that their formation
is entirely consistent with available information on glutathione-mediated metabolism. Distribution of
methylene chloride metabolism between these pathways is dose dependent. The MFO pathway is a high-
affinity, limited-capacity pathway which saturates at relatively low atmospheric concentrations
(approximately 200–500 ppm). The GSH pathway, in contrast, has a lower affinity for methylene
chloride, but does not appear to saturate at experimentally produced concentrations (<5,000 ppm). Thus,
the MFO pathway accounts for most of the metabolized methylene chloride at concentrations less than
500 ppm, but as exposure concentrations increase above the MFO saturation level, increases in the
amount of methylene chloride metabolized by the secondary GSH pathway are seen (Reitz 1990). The
concentration dependency of these two metabolic pathways is consistent with the tumor results obtained
in long-term rodent inhalation and drinking water cancer bioassays of methylene chloride and supports
the assertion that GSH-mediated metabolism is responsible for methylene chloride-induced
tumorigenicity in B6CF
1
mice.
There is no evidence to suggest that methylene chloride is a direct acting carcinogen; the marked species
differences in carcinogenicity induced by methylene chloride are not typical behavior of direct-acting
compounds. Methylene chloride also does not exhibit the chemical reactivity towards nucleophiles
111 METHYLENE CHLORIDE
2. HEALTH EFFECTS
normally associated with direct action (Green 1997). Therefore, metabolic activation is required which
interacts in some way with mouse tissues to cause tumors.
A series of bacterial mutagenicity tests has demonstrated that: methylene chloride induction of bacterial
mutagenicity is expressed more strongly in Salmonella typhimurium TA 1535 modified to express a
mammalian GST Θ class enzyme (NM5004 strain) than in the original strain (Oda et al. 1996); meth-
ylene chloride induction of bacterial mutagenicity S. typhimurium strain TA 100 is unaffected by the
presence of GST α or π classes (Simula et al. 1993); methylene chloride is less mutagenic in a S.
typhimurium GSH-deficient strain (TA100/NG11) as compared to TA 100 (Graves et al. 1994a); and
bacterial testing with 3 K12 strains of Escherichia coli showed that methylene chloride (activated by S9
mouse liver fraction) and formaldehyde were mutagenic only in the wild-type E. coli, a characteristic
shared with crosslinking agents; these data initially suggested a mutagenic role for metabolically-derived
formaldehyde in E. coli (Graves et al. 1994a).
These bacterial assays demonstrated that in in vitro tests, methylene chloride was activated by a Θ class
GST enzyme to a bacterial mutagen in S. typhimurium and behaved similarly to formaldehyde in E. coli
tester strains. However, in the Chinese Hamster ovary (CHO) assay involving the hypoxanthine-guanine
phosphoribosyl transferase (HPRT) gene assay, studies of DNA single strand breaks and DNA-protein
crosslinks at mutagenic concentrations of methylene chloride and formaldehyde showed that both these
compounds induced DNA single-strand breaks; only formaldehyde induced significant DNA-protein
crosslinking (Graves et al. 1996). Similar findings were observed in cultured, freshly isolated mouse
hepatocytes (Graves and Green 1996), but not in rat hepatocytes (Graves et al. 1994b, 1995). The authors
concluded that, although formaldehyde might play a role in methylene chloride genotoxicity, its weak
mutagenicity and the absence of methylene chloride-induced DNA-protein crosslinking in the
CHO/HPRT assay suggested that methylene chloride-induced DNA damage and resulting mutations are
likely produced by its glutathione conjugate, putatively chloromethylglutathione. Graves and Green
(1996) also concluded that these results suggested that the mechanism for methylene chloride
tumorigenicity in the mouse liver was likely to be genotoxic and mediated by the GSH pathway.
Observed species differences in liver tumorogenicity between the mouse and the rat might result from
species differences in the amount of GSH-mediated metabolism induced by methylene chloride exposure.
A series of studies have been conducted to elucidate the precise genetic mechanisms of methylene
chloride carcinogenicity. Female B6C3F
1
mice were exposed to vapor concentrations of
112 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2,000–8,000 ppm for 2 years and sacrificed at various intervals to evaluate a number of genotoxic
endpoints.
Replicative DNA synthesis in the bronchiolar epithelium, examined by the use of the labeling index (LI),
indicated that mice exposed to 2,000 ppm of methylene chloride for 2–26 weeks decreased to 40–60% of
controls (Kanno et al. 1993). Mice exposed to 8,000 ppm of methylene chloride have a less dramatic
decrease in LI. No pathological changes were found in the exposed lungs. Thus, high-concentration
exposure to methylene chloride for up to 26 weeks reduces the cell turnover of bronchiolar cells in these
mice; therefore, increased cell proliferation does not appear to be involved in mouse lung tumorogenesis.
DNA single-strand breaks were detected in the livers of B6C3F
1
male mice, but not Syrian Golden male
hamsters, immediately following inhalation exposure to 2,000–8,000 ppm for 6 hours, but not 2 hours
after exposure, suggesting active DNA repair (Graves et al. 1995). The DNA of mouse Clara cells
incubated in vitro with methylene chloride was also damaged at high concentrations. Pretreatment of
mice with a glutathione depletor prior to inhalation exposure caused a decrease in the amount of DNA
damage detected, suggesting a GST-mediated mechanism; similar findings were observed in Clara cells
incubated in vitro with methylene chloride and a glutathione depletor.
Devereux et al. (1993) analyzed liver and lung tumor, induced in female B6C3F
1
female mice by
inhalation of 2,000 ppm of methylene chloride for 6 hours/day, 5 days/week exposure for up to
104 weeks, for the presence of activated ras proto-oncogenes. In methylene chloride-induced liver
tumors, mutations, mainly transversions or transitions in base 1 or base 2, were detected and were similar
to those observed for the H-ras gene in spontaneous liver tumors. Mutations were also identified in the
lung. The K-ras activation profile in the methylene chloride-induced tumors was not significantly
different from the profile in spontaneously-occurring tumors. No other transforming genes were found in
the nude mouse tumorogenicity assay. The authors concluded that at present, no transforming genes other
than ras genes could be identified in either mouse liver or lung tumors. Based on liver tumor data, they
also suggested that methylene chloride may affect the liver by promoting cells with spontaneous lesions.
Hegi et al. (1993) studied allelotypes of 38 methylene chloride-induced lung carcinomas from female
B6C3F
1
mice exposed 6 hours/day, 5 days/week for 2 years to 2,000 ppm. The allelotypes were
examined for various genotoxic endpoints, and the results were compared with genotoxicity findings in
two other reciprocal-cross mouse strains. Throughout the genome, allelic losses occurred infrequently,
113 METHYLENE CHLORIDE
2. HEALTH EFFECTS
except for markers on chromosome 4, which were lost in approximately half of the carcinomas. In lung
adenomas, chromosome 4 losses were associated with malignant conversion. Methylene chloride-induced
liver tumors did not demonstrate chromosome 4 loss, which indicated that this finding was specific for
lung carcinomas. Preferential loss of the maternal chromosome 4 was also observed in carcinomas in
B6C3F
1
mice. On chromosome 6, an association between K-ras gene activation and allelic imbalances
was also found in B6C3F
1
mouse lung tumors. When allelotypes of tumors in mice from two reciprocal
cross strains, AC3F
1
and C3AF
1
, were examined and compared to the findings in B6C3F
1
mice, one allele
of the putative chromosome 4 tumor suppressor gene was shown to be inactivated. Whereas the results in
B6C3F
1
mice suggested that nondisjunction events were responsible for the chromosome 4 losses, tumors
from both reciprocal-cross mouse strains appeared to show small interstitial deletions in a chromosomal
region homologous with a region in human chromosome which is often lost in a variety of tumors,
including lung cancers. In human chromosomes, a candidate tumor suppressor gene, MTS1, is located in
this region.
In another genotoxic analysis with the same cohort (Hegi et al. 1994), loss of heterozygosity at markers
near the p53 gene on chromosome 11 and within the retinoblastoma tumor suppressor gene were
examined in methylene chloride-induced liver and lung tumors and compared to spontaneous tumors in
control mice. The authors concluded that inactivations of p53 and the retinoblastoma tumor suppressor
gene were infrequent events in lung and liver tumorigenesis in mice exposed to methylene chloride.
Replicative DNA synthesis was examined by Kanno et al. (1993) to evaluate the potential role of
treatment-induced lung cell proliferation on pulmonary carcinogenicity in female B6C3F
1
mice exposed
to 2,000 or 8,000 ppm of methylene chloride for 6 hours/day, 5 days/week for 2 years. By the end of the
study, there was a statistically significant increase in lung tumors in exposed animals when compared to
controls. Cell proliferation was assessed in the lung after 1, 2, 3, or 4 weeks of inhalation exposure to
2,000 or 8,000 ppm, and after 13 and 26 weeks exposure to 2,000 ppm, as measured by changes in
labeling indices (LI). The LI of both bronchiolar epithelium and terminal bronchioles were substantially
decreased in mice exposed to 2,000 ppm of methylene chloride for 2–26 weeks. Similar findings, but not
as severe, were observed in mice exposed to 8,000 ppm. The decreases in LI were not accompanied by
cytotoxicity. The authors concluded that high-concentration exposure to methylene chloride for up to
26 weeks reduces cell proliferation in lung epithelial cells in female B6C3F
1
mice.
114 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Maronpot et al. (1995b) assessed replicative DNA synthesis after 13, 26, 52, and 78 weeks of inhalation
exposure by female B6C3F
1
mice to 2,000 ppm of methylene chloride for 6 hours/day, 5 days/week. A
statistically significant decrease in the hepatocyte LI was only observed at 13 weeks. In lung epithelial
cells, the results were similar to those observed by Kanno et al. (1993). No increases in replicative DNA
synthesis were found in liver foci cells or lung parenchymal cells. K-ras gene activation in liver tumors
and H-ras gene activation in lung tumors did not differ between methylene chloride-induced tumors and
those observed in control animals. The authors concluded that these oncogenes were not involved in
mouse tumorogenesis.
DNA-protein crosslinks (DPX) in lung and liver were examined by Casanova et al. (1992, 1996) in male
B6C3F
1
mice exposed to 2,000 and 4,000 ppm of methylene chloride for 6 hours/day for 2 days and in
male Syrian Golden hamsters exposed to 3,500 ppm. The authors suggested that formaldehyde derived
from GSH-mediated methylene chloride metabolism might be forming DPX in mouse liver. Although
DPX were detected in mouse liver, there was no evidence of DPX formation in mouse lung, hamster liver,
or hamster lung. Additionally, DPX are not formed in measurable amounts in human liver tissue
(Casanova et al. 1996). Therefore, the induction of DPX by formaldehyde in mouse liver might be a
species-specific, tissue-specific response. A subsequent in vitro study by this laboratory confirmed the
absence of DPX formation in response to methylene chloride in human, rat, and hamster hepatocytes,
whereas a dose-response was observed in mouse hepatocytes (Casanova et al. 1997). In a different
experiment, a dose-response in the formation of RNA-formaldehyde adducts was observed in hepatocytes
of all four species (mouse>human>rat>hamster). RNA adduct production was related to the expression of
the GSTT1-1 enzyme; the human liver sample lacking GSTT1-1 did not produce RNA adducts.
In vitro tests using two different strains of Salmonella, one with and one without GST expression,
revealed that methylene chloride may be mutagenic by at least two pathways (DeMarini et al. 1997). The
bacterial strains employed were TA100 and RSJ100; the latter is a derivative of the strain TA1535 (not
normally mutagenized by methylene chloride) that contains recombinant rat GSTT1-1. In RSJ100,
methylene chloride was mutagenic at moderate doses, and produced a single class of mutation (GC ÷ AT
transversions) only at the middle C of the target CCC. Although methylene chloride was mutagenic in
TA100, it required a much higher dose to match the mutation frequency in RSJ100. Furthermore, it
produced a variety of lesions and mutations (predominantly GC ÷ TA transversions) at the first and
second positions of the CCC target. The implication of these results is that genotoxic effects of
methylene chloride will be different, depending on the GSTT1 phenotype. Those who lack the functional
115 METHYLENE CHLORIDE
2. HEALTH EFFECTS
gene would only be susceptible to genetic damage from methylene chloride under high exposure levels,
whereas carriers might be vulnerable to genetic damage at much lower exposure levels.
According to Maronpot et al. (1995b), the precise mode of action of methylene chloride-induced mouse
tumorigenicity appears to be elusive and has not yet been confirmed.
2.4.3 Animal-to-Human Extrapolations
The two major difficulties in applying the results of rodent cancer bioassays to humans involve
extrapolation from the high-concentration rodent exposures to the lower concentrations typical of human
exposure conditions; and the interspecies extrapolation. The predominant tumors of interest with regard
to methylene chloride-induced tumorigenicity are mouse lung and mouse liver. However, species
differences among rodent tumorigenic, genotoxic, and morphological responses to methylene chloride, as
well as differences between mice and humans, appear to limit the applicability of mouse tumor data to
humans, according to some authors. Foster et al. (1992) have suggested that the Clara cell may have a
role in mouse lung tumor induction; there is a substantially higher number of Clara cells in the mouse
than in other rodent species or in humans. GSH-mediated activation of the proximate carcinogenic agent
of methylene chloride in mice has been associated with a specific isoenzyme, the Θ (theta) class GSTs;
Sheratt et al. (1997) have shown that this isozyme is expressed at very low levels in human pulmonary
cells in vitro, suggesting that in humans, the lung has little capacity to activate methylene chloride into
biologically reactive intermediates. In vitro studies (e.g., Graves et al. 1995) have shown species
differences in the ability to induce DNA single-strand breaks in mouse, rat, hamster, and human cells that
are compatible with the known rodent carcinogenicity, or lack thereof, in chronic cancer bioassays.
These in vitro studies suggested that humans are unlikely to be more susceptible than rodents to
methylene chloride-induced liver cancer.
In a comparison of rates of methylene chloride metabolism by each of the two major pathways in four
species, the mouse, rat, hamster, and human, Green (1997) presented in vivo and in vitro evidence that the
MFO pathway metabolic rates are similar among all four species and saturated at concentrations of
500 ppm or above. In contrast, the GSH-mediated metabolism is linear over the concentration range
studied; it is also the major metabolic pathway for methylene chloride in mice at concentration levels used
in cancer inhalation bioassays. Furthermore, the activity in mouse tissues is more than an order of
magnitude greater than the activity in rat tissues. Hamster and human tissues show metabolic rates for
this pathway that are even lower than those found in the rat. These findings are corroborated by the
localization study of Mainwaring et al. (1996b), which found higher levels of GSTT1-1 mRNA and
protein in the liver and lung of mice than in rats or humans. In mechanistic terms, Green (1997)
116 METHYLENE CHLORIDE
2. HEALTH EFFECTS
concludes that these studies demonstrate that differences in glutathione-mediated metabolism among
mice, rats, hamsters, and humans are correlated with differences in carcinogenicity of methylene chloride.
This is supported by the levels of glutathione that have been detected in the livers of the different species.
The hepatic concentrations of GSH (in nmol/10
6
cells) were approximately 129 in B6C3F
1
mice (Ruch et
al. 1989), 50 in Sprague-Dawley rats (Jones et al. 1978), 22 in Syrian Golden hamsters, and 21 in humans
(Steinmetz et al. 1988). Thus, the mouse is the most sensitive species to metabolic activation of
methylene chloride by glutathione metabolism, whereas humans appear to be the least sensitive.
However, other evidence suggests that methylene chloride may be potentially carcinogenic in humans.
Mainwaring et al. (1996b) analysis of the distribution of mRNA and protein for GSTT1-1 and GSTT2-2
in the analysis of the liver and lungs of mice, rats, and humans corroborated the finding of Sheratt et al.
(1997) that the overall levels in human tissues are much lower than in those of mice. However, the
immunodetection of localized high concentrations of GSTT2-2 enzyme in human bile-duct epithelial cells
is potentially significant, considering the increased incidence of biliary cancer following chronic exposure
to methylene chloride as reported by Lanes et al. (1990). On the other hand, the GSTT2-2 antibody did
not localize to the nucleus of human biliary epithelial cells, which would tend to reduce the potential
genotoxic effect in humans. Although Mainwaring et al. (1996b) found that rates of metabolism of
methylene chloride were very low in human lung, they also detected higher than background amounts of
GSTT1-1 mRNA in a few Clara cells and ciliated cells of the alveolar/ bronchiolar junction of the lung in
one human sample out of four. Therefore, despite the general low level of GSTT1-1and GSTT2-2 in
human tissue, it is possible that in some individuals, specific cell types within the human liver (bile duct)
and lung might produce genotoxic reactive intermediates as a result of methylene chloride metabolism.
Based on GSTT enzyme distributions and concentrations, the carcinogenic risk from methylene chloride
in humans appears to be low as in rats rather than high as in mice.
The mouse model has been employed in assessment of excess lifetime cancer risks in humans from
inhalation exposure to methylene chloride by EPA and others. Recent data, both in vitro and in vivo,
117 METHYLENE CHLORIDE
2. HEALTH EFFECTS
strongly indicate that the higher sensitivity of the mouse, relative to humans, must be taken into
consideration if mouse data are to be used to estimate potential human health risks.
2.5 RELEVANCE TO PUBLIC HEALTH
Issues relevant to children are explicitly discussed in 2.7, Children’s Susceptibility, and 5.6, Exposures of
Children.
Overview. Methylene chloride has been widely used in industrial processes, food preparation,
agriculture, and consumer products; consequently, there have been numerous studies describing its effects
in a variety of experimental animal species. Humans have not been clinically studied as extensively.
Although methylene chloride uses in agricultural goods and some consumer products have declined in
recent years, there is still potential public health concern due to its continued use in industrial processes,
and continued releases into the environment.
The central nervous system is affected adversely in humans and animals at inhalation exposure levels of
200 ppm or higher (Putz et al. 1979). Effects in animals were also reported on the liver and kidney
following continuous exposure at concentrations of 25 ppm or greater (Haun et al. 1972), and on the
cardiovascular system, but at extremely high exposures (Aviado and Belej 1974). Long-term inhalation
exposure to methylene chloride (500 ppm or greater) increased tumors in some animals (Nitschke et al.
1988a), but did not cause teratogenic or reproductive effects in a two-generation study (Nitschke et al.
1988b). Since inhalation is the principal route of exposure to methylene chloride, most of these effects
have been tested for or observed by this route. Data on effects observed after oral and dermal exposure
are somewhat more limited. Further details are presented below.
Minimal Risk Levels for Methylene Chloride.
Inhalation MRLs.
C An MRL of 0.6 ppm has been derived for acute inhalation exposure (0–14 days) to methylene
chloride. This MRL supersedes the previous MRL of 3 ppm derived in the 1998 draft for public
comment profile. Refer to chapter 7 for additional information.
118
C
METHYLENE CHLORIDE
2. HEALTH EFFECTS
The acute inhalation MRL was derived from the behavioral toxicity study by Winneke (1974) in which a
randomized blind clinical chamber experiment was used to expose 6–20 volunteers to vapors of
methylene chloride (300, 500, or 800 ppm) or filtered air for 3–4 hours. Subjects were tested at
45-minute intervals with standard neurobehavioral tests measuring: (1) critical flicker fusion frequency
(visual); (2) auditory vigilance performance; and (3) performance on psychomotor tasks. The tested
parameters were considered to reflect the status of ‘cortical alertness’ (Fodor and Winneke 1971). A
statistically significant depression in critical flicker fusion (CFF) frequency was observed at all
concentrations. The magnitude of CFF frequency depression was similar at exposure concentrations of
300 and 500 ppm and was larger at 800 ppm. Thus, there was no dose-response at the two lowest
concentrations, and a dose-response was evident at the highest concentration. A decrease in auditory
vigilance performance was observed at 500 ppm and psychomotor task performance was impaired at
800 ppm. Thus, reduced CFF frequency is the most sensitive neurological response to acute inhalation
exposure to methylene chloride. Based on this endpoint, the LOAEL is 300 ppm. A PBPK model for this
experiment was used to adjust the dosage to a 24 hour exposure period, thus resulting in a LOAEL of
60 ppm for the same endpoint (Reitz et al. 1997). The MRL was derived by dividing the LOAEL of
60 ppm by an uncertainty factor of 100 (10 for the use of a LOAEL and 10 for human variability).
An MRL of 0.3 ppm has been derived for intermediate inhalation exposure (15–364 days) to
methylene chloride.
The intermediate inhalation MRL was derived from a study by Haun et al. (1972) in which groups of
mice, rats, dogs, and monkeys were continuously exposed to methylene chloride for 14 weeks at chamber
concentrations of either 0, 25, or 100 ppm. Body weights and clinical signs were monitored throughout
the study. Necropsy was performed and tissues were examined histopathologically and organ-to-body
weight were determined at the end of the exposure. Data on liver histopathology of rats exposed to 25 or
100 ppm of methylene chloride were selected as the critical effect. Liver cytoplasmic vacuolization and
staining associated with fatty deposits were observed in rats at both exposure concentrations; Haun et al.
(1972) did not mention whether there were quantitative differences in the effect observed at the two
exposure levels. The MRL was derived based on a LOAEL of 25 ppm for hepatic effects. Because the
critical effect observed is an extrarespiratory effect (rat liver), a human equivalent concentration (HEC)
was calculated. Since the ratio of the blood:air partition coefficient in the rat to the blood:air partition
coefficient in the human was > 1, the value of 1.0 was used to calculate the LOAEL
[HEC]
(EPA 1994).
This resulted in an MRL of 0.3 ppm by dividing the LOAEL of 25 ppm by an uncertainty factor of 90 (3
for use of a minimal LOAEL, 3 for extrapolation from animals to humans, and 10 for human variability).
119 METHYLENE CHLORIDE
2. HEALTH EFFECTS
C An MRL of 0.3 ppm has been derived for chronic inhalation exposure ($365 days) to methylene
chloride.
The chronic inhalation MRL was derived from a study by Nitschke et al. (1988a) in which groups of
90 male and 108 female Sprague-Dawley rats were exposed to methylene chloride at 0 (controls), 50,
200, or 500 ppm for 6 hours/day, 5 days/week for 2 years. A number of satellite groups were also
exposed to assess the temporal relationship between methylene chloride exposure and evidence of
toxicity. Subgroups of females in the main study were sacrificed after 6, 12, 15, and 18 months of
exposure. The following end points were evaluated: body weight, food consumption rates, organ weights,
hematology, clinical chemistry, urinalysis, pathology, histopathology, and blood COHb levels. Blood
COHb levels were consistently higher than 10% in animals exposed to $200 ppm. No pathologic or
histopathologic nontumor findings were reported except in the liver. Hepatocellular cytoplasmic
vacuolization consistent with fatty changes, and multinucleate hepatocytes were elevated in female rats
exposed to methylene chloride at 200 and 500 ppm; a slight increase in the incidence of hepatocellular
vacuolization was also observed in male rats exposed to 500 ppm.
The NOAEL of 50 ppm was adjusted for continuous exposure (6 hour/day, 5 day/week) resulting in a
NOAEL
[ADJ]
of 8.92 ppm. Whereas the MRL was derived based on hepatic effects (extrarespiratory), a
human equivalent concentration (HEC) was calculated. Since the ratio of the blood:air partition
coefficient in the rat to the blood:air partition coefficient in the human was > 1, the value of 1.0 was used
to calculate the LOAEL
[HEC]
(EPA 1994). This resulted in an MRL of 0.3 ppm by dividing the LOAEL
of 8.92 ppm by an uncertainty factor of 30 (3 for extrapolation from animals to humans, and 10 for human
variability).
Oral MRLs.
CAn MRL of 0.2 mg/kg/day has been derived for acute oral exposure (0–14 days) to methylene
chloride. This MRL supersedes the previous MRL of 0.5 mg/kg/day derived in the 1998 draft for public
comment profile. Refer to chapter 7 for additional information.
The acute oral MRL was derived by route-to-route extrapolation of the data from Winneke (1974) in
which a randomized blind clinical chamber experiment was used to expose 6–12 volunteers to vapors of
methylene chloride (300, 500, or 800 ppm) or filtered air for 3–4 hours. Subjects were tested at
45-minute intervals with standard neurobehavioral tests measuring: (1) critical flicker fusion frequency
120 METHYLENE CHLORIDE
2. HEALTH EFFECTS
(visual); (2) auditory vigilance performance; and (3) performance on psychomotor tasks. A statistically
significant depression in critical flicker fusion (CFF) frequency was observed at all concentrations. The
magnitude of CFF frequency depression was similar at exposure concentrations of 300 and 500 ppm and
was larger at 800 ppm. Thus, there was no dose-response at the two lowest concentrations, and a dose-
response was evident at the highest concentration. A decrease in auditory vigilance performance was
observed at 500 ppm and psychomotor task performance was impaired at 800 ppm. Thus, reduced CFF
frequency is the most sensitive neurological response to acute inhalation exposure to methylene chloride.
Based on this end point, the LOAEL is 300 ppm.
Reitz et al. (1997) modified the basic PBPK model for methylene chloride that was developed by
Andersen et al. (1987), Reitz et al. (1988), and Andersen et al. (1991) as described in Section 2.3.5.2.
The major modification of the model was the inclusion of a brain compartment so that central nervous
system effects could be assessed. Reitz et al. (1997) modeled the Winneke (1974) data to obtain the
target organ (brain) concentrations of methylene chloride associated with administered inhalation
concentrations, and then calculate the human drinking water concentrations (mg/L) that would result in
the equivalent target organ-specific doses. Human exposure patterns in the PBPK model simulated
realistic human drinking water consumption patterns, (i.e., consisting of bouts of drinking during the day,
with and between meals, and little-to-no drinking during the night). PBPK modeling predicted that peak
concentrations of methylene chloride in the brain would increase rapidly after each episode of drinking
water consumption, and then drop sharply, to near-zero, between bouts of drinking. Additionally, there
would be no cumulative effects from repeated exposure.
For acute neurological effects, the associated dose measure was defined as the peak concentration of
methylene chloride in brain tissue (mg/L of brain tissue) of humans exposed to 300 ppm of methylene
chloride for 4 hours by inhalation. The modified PBPK model calculated that the administered inhalation
dose was equivalent to 3.95 mg of methylene chloride per L of brain tissue. The equivalent administered
human concentration in drinking water that will produce the same neurological effects was 565 mg of
methylene chloride/L. Using a daily drinking water consumption value of 2L and an average human body
weight of 70 kg, the LOAEL was calculated to be 16 mg/kg/day. An acute oral MRL was calculated by
dividing the LOAEL (16 mg/kg/day) by an uncertainty factor of 100 (10 for the use of a LOAEL and
10 for human variability), to yield 0.2 mg/kg/day.
121 METHYLENE CHLORIDE
2. HEALTH EFFECTS
C An MRL of 0.06 mg/kg/day has been derived for chronic oral exposure ($365 days) to methylene
chloride. This MRL supersedes the previous MRL of 0.2 mg/kg/day derived in the 1998 draft for
public comment profile. Refer to chapter 7 for additional information.
The chronic oral MRL was derived from a study by Serota et al. (1986a) in which F344 rats (85/sex/dose)
were exposed to methylene chloride in deionized drinking water at concentrations to provide target doses
of 0, 5, 50, 125, or 250 mg/kg/day for 104 weeks. The nominal mean doses were 0, 6, 55, 131, and
249 mg/kg/day. There were no treatment-related effects on survival or on the incidence of adverse
clinical signs. Organ weights were not significantly affected by treatment. Histopathology was only
observed in the liver, which therefore is the critical target organ. Statistically significant cellular changes
(hepatic foci/areas of cellular alterations and fatty changes) were observed at dose levels $50 mg/kg/day.
Reduced weight gain was observed at 131 and 249 mg/kg/day, but quantitative data were not provided.
Hematological effects (increased mean hematocrit, hemoglobin, and erythrocyte count) were observed at
all dose levels except 6 mg/kg/day. Therefore, the lowest dose in rats was identified as the NOAEL.
Based on measured, mean drinking water consumption rates, this dose was calculated to be 6 mg/kg/day.
The chronic oral MRL was calculated by dividing the NOAEL (6 mg/kg/day) by an uncertainty factor of
100 (10 for extrapolation from animals to humans and 10 for human variability).
No intermediate oral MRL was derived because of an inadequate database.
Death. Acute inhalation exposure to methylene chloride has caused death in humans (Bakinson and
Jones 1985; Bonventre et al. 1977; Hall and Rumack 1990; Stewart and Hake 1976). Although exposure
levels were not measured, estimates suggest that a combination of high exposure levels and/or inadequate
ventilation has contributed to these lethal accidents. Measurements of methylene chloride in tissues or of
COHb in blood have corroborated high exposures in some cases (Manno et al. 1992; Moskowitz and
Shapiro 1952; Winek et al. 1981; Tay et al. 1995). The biologic cause of death was not verified in all
cases, but is thought to have been respiratory depression secondary to narcosis. Asphyxia accompanied
by bilateral pulmonary congestion and focal hemorrhage was reported in one case (Winek et al. 1981) and
myocardial infarction was reported in another (Stewart and Hake 1976). Mortality risk was not increased
in humans exposed occupationally to 30–120 ppm of methylene chloride for over 30 years (Friedlander et
al. 1978) and no excess mortality was found in workers exposed to 140–475 ppm for at least 3 months
(Lanes et al. 1993; Ott et al. 1983b). Inhalation exposure to concentrations of 3,500 ppm for longer
122 METHYLENE CHLORIDE
2. HEALTH EFFECTS
durations (14 weeks to 2 years) was lethal in some animals (Burek et al. 1984; MacEwen et al. 1972). No
mortality increase was noted in chronic inhalation studies at 500 ppm in the rat (Nitschke et al. 1988a).
In one suicide case, ingestion of paint remover containing 75–80% methylene chloride resulted in death
from corrosion of the gastrointestinal tract (Hughes and Tracey 1993). In animals, acute exposure to high
doses (2,100 mg/kg or greater) by gavage caused death (Kimura et al. 1971; Ugazio et al. 1973);
intermediate-duration exposures to 64 or 320 mg/kg/day by gavage significantly increased mortality in
female mice and male rats, respectively (Maltoni et al. 1988). However, exposure to methylene chloride
in drinking water (up to 250 mg/kg/day) did not significantly affect survival (Serota et al. 1986a, 1986b).
Gavage administration may result in more severe effects than ad libitum water ingestion since the
administered dose may temporarily saturate normal metabolic processes, and result in a different
metabolic profile. No data were found on death in humans or animals from dermal exposure.
Systemic Effects
Respiratory Effects. In one workplace accident, acute inhalation exposure to methylene chloride
(probably at a high concentration) resulted in bilateral pulmonary congestion with focal hemorrhage
(Winek et al. 1981). Less severe respiratory symptoms (cough, breathlessness, chest tightness) were
reported in occupational exposure incidents (concentrations unknown; Bakinson and Jones 1985).
Exposure to 18–1,200 mg/m
3
(5–340 ppm; 8-hour TWA) resulted in irritation of the respiratory tract in
one occupational study (Anundi et al. 1993). However, a study in humans found no effect on pulmonary
function following repeated exposures to methylene chloride vapors (up to 500 ppm) (Stewart et al.
1972). Studies in animals corroborated the severe pulmonary effects (congestion, edema, inflammation)
of acute or intermediate exposures at high concentrations (Heppel et al. 1944; NTP 1986). Resolution of
acute Clara cell damage in mice exposed to 4,000 ppm of methylene chloride was correlated with
cytochrome P-450 activity in the lung (Foster et al. 1992). No information was found on the respiratory
effects of low levels of methylene chloride in humans near hazardous waste sites or industrial urban areas
or in animals.
Cardiovascular Effects. Myocardial infarction occurred in one case of acute inhalation occupational
exposure (Stewart and Hake 1976). However, occupational studies in humans did not find any
association between exposure to methylene chloride at 75–475 ppm and cardiac abnormalities (Cherry et
al. 1981) or excess mortality due to ischemic heart disease (Hearne et al. 1990; Ott et al. 1983b). Further,
123 METHYLENE CHLORIDE
2. HEALTH EFFECTS
a cross-sectional study of workers showed no excess of electrocardiographic abnormalities among those
exposed to methylene chloride (Ott et al. 1983c). Another study in humans did not reveal effects on
cardiovascular functions at concentrations up to 500 ppm (NIOSH 1974). These findings suggest the
cardiovascular system is not a sensitive target for exposure to methylene chloride in humans. One study
in mice reported atrioventricular block following acute inhalation exposure to very high levels of
methylene chloride (>200,000 ppm) (Aviado and Belej 1974). The relevance of this finding is limited
since the exposure level was so high.
In an experiment with rats, using an ischemia-reperfusion model, intravenous methylene chloride infusion
(leading to blood concentrations of <0.1 mg/mL) markedly increased the atrioventricular block during the
reperfusion phase (Scholz et al. 1991). From these results, the authors concluded that the initial coma
resulting from methylene chloride-induced poisoning is likely to result not only from anesthetic effects,
but also from sudden onset of cardiac arrhythmias.
Gastrointestinal Effects. Nausea and vomiting were reported in 13 out of 33 occupational cases of acute
inhalation exposure to methylene chloride in the United Kingdom (Bakinson and Jones 1985); exposure
levels were not reported in these cases. In mice exposed to 4,000 ppm of methylene chloride for 2 years,
dilatation of the stomach was reported (NTP 1986). In humans attempting suicide, ingestion of a single
oral dose of Nitromors, a paint remover solvent containing 75–80% methylene chloride, resulted in severe
corrosion and ulceration of the gastrointestinal tract (Hughes and Tracey 1993), peritonitis and septicemia
occurred in the fatal case, whereas intestinal diverticuli developed during recovery in the another case
(Roberts and Marshall 1976). No other studies were located regarding gastrointestinal effects in humans
or animals after exposure to methylene chloride.
Hematological Effects. Metabolism of methylene chloride results in excess carbon monoxide and
increases in COHb, which contributes to hypoxia (Tomaszewski 1998). Blood COHb concentrations
were about 30% higher than normal in 2 lethal cases in which workers were estimated to be exposed to
extremely high concentrations (up to 168,000 ppm) of methylene chloride in a confined work space
(Manno et al. 1992). Several hours after an adult woman ingested a fatal oral dose of Nitromors, a paint
remover containing 75–80% methylene chloride, her COHb level was 9%. In all three fatal exposures,
cause of death was not associated with elevated COHb. In autoworkers who were exposed to methylene
chloride dermally and by inhalation (3–154 ppm), blood COHb measurements taken within 24 hours of
exposure were 1.2–11% for nonsmokers and 7.3 and 17.3% for two smokers (Kelly 1988). One-day
124 METHYLENE CHLORIDE
2. HEALTH EFFECTS
occupational exposures to methylene chloride at levels below ACGIH standard (50 ppm, 8-hour TWA)
produced small increases in COHb levels in both nonsmoking and smoking adults (Soden et al. 1996);
additional daily cumulative exposure to methylene chloride did not further increase COHb levels.
Increases in blood COHb levels between 5 and 6.8% were measured in nonsmoking volunteers exposed to
methylene chloride at concentrations up to 200 ppm for 4 to 7.5 hours (DiVincenzo and Kaplan 1981;
Putz et al. 1979).
Other studies have reported increases in red cell count, hemoglobin, and hematocrit in women, but not in
men, occupationally exposed to concentrations of up to 475 ppm during an 8-hour workday (Ott et al.
1983d); these effects were judged by the authors to be suggestive of compensatory hematopoietic effects.
Similar findings were not observed in rodents chronically exposed to methylene chloride by inhalation at
concentrations up to 3,500 ppm (Burek et al. 1984), but were observed in rodents exposed orally for
3 months at 480 mg/kg/day (Kirschman et al. 1986), or for 2 years at 55–249 mg/kg/day (Serota et al.
1986a, 1986b). Intravascular hemolysis was reported in the case of a man who attempted suicide by
ingesting the paint remover, Nitromors (Roberts and Marshall 1976) and in a high-dose acute gavage
study in rats (Marzotko and Pankow 1987).
Hepatic Effects. Human data are limited on the effects of methylene chloride on the liver. A slight
exposure-related increase in serum bilirubin (but not at levels of clinical significance) was observed in
workers with exposure up to an average of 475 ppm of methylene chloride, but serum levels of hepatic
enzymes (e.g., aspartate aminotransferase, alanine aminotransferase, lactate dehydrogenase, and alkaline
phosphatase) were not elevated (Ott et al. 1983a); another occupational study found no exposure-related
changes in hepatic enzymes (Anundi et al. 1993). In another study, methylene chloride vapors (up to
500 ppm) did not affect comparable serum enzyme activity in volunteers (Stewart et al. 1972). However,
the liver appears to be a major target organ following methylene chloride exposure in animals,
particularly at high exposure levels (>5,000 ppm; Heppel et al. 1944). Histomorphological and
biochemical changes of the liver occur following acute inhalation (6 hours to 7 days) at high
concentration levels (5,200 ppm) (Morris et al. 1979), while fatty changes and biochemical alterations
(altered cytochrome P-450 levels) were also observed at lower concentrations (100 ppm) for continuous,
24-hour intermediate-duration exposure (100 days) (Haun et al. 1972; Kjellstrand et al. 1986; Weinstein
and Diamond 1972). Cytoplasmic vacuolization was observed in rats at 25 ppm (Haun et al. 1972).
Using these data, ATSDR derived an intermediate inhalation MRL of 0.3 ppm, as calculated in Table 2-1.
Exposure to 1,000–4,000 ppm for 2 years resulted in an increased incidence of hemosiderosis and focal
125 METHYLENE CHLORIDE
2. HEALTH EFFECTS
hepatic necrosis in rats (NTP 1986). An increased incidence of fatty changes occurred following chronic
exposure at 500 ppm, but not at 200 ppm (Nitschke et al. 1988a); fatty changes were reversible when
exposure ceased. Using data from the Nitschke et al. (1988a) study, ATSDR derived a chronic inhalation
MRL of 0.3 ppm, as shown in Table 2-1. Necrosis or fatty changes in the liver have been observed in rats
given high oral doses (>1,000 mg/kg/day) of methylene chloride (Kirschmann et al. 1986; Ugazio et al.
1973). Chronic ingestion of methylene chloride in drinking water has been associated with fatty changes
in rats at 55 mg/kg/day or greater and in mice at 175 mg/kg/day or greater, but not at 6 mg/kg/day (Serota
et al. 1986a, 1986b). Based on this value (6 mg/kg/day), a chronic oral MRL of 0.06 mg/kg/day was
calculated as described in Table 2-2.
Endocrine Effects. No relevant information was located regarding endocrine effects in human or animals
associated with exposure to methylene chloride.
Renal Effects. Kidney function was not altered in humans repeatedly exposed to methylene chloride
vapors (up to 500 ppm) for 6 weeks (Stewart et al. 1972) and no alterations in urinary microglobulins or
N-acetyl-beta-glucosaminidase were detected in workers chronically exposed to methylene chloride
(Anundi et al. 1993). In rats, nonspecific renal tubular and degenerative changes occurred after
continuous intermediate-duration exposure to methylene chloride vapors (100–5,000 ppm) (Haun et al.
1972; MacEwen et al. 1972) or chronic exposure to 4,000 ppm (NTP 1986). Similar renal changes
occurred in dogs exposed to 1,000 ppm for 4 weeks (MacEwen et al. 1972). There were no studies
reporting renal effects following oral exposure to methylene chloride in humans. In rat oral studies using
methylene chloride at doses >1,300 mg/kg/day, a single dose inhibited diuresis (Marzotko and Pankow
1987) and treatment for 3 months increased kidney weights in females (Kirschman et al. 1986). At
$166 mg/kg/day, methylene chloride lowered the pH of urine in rats of both sexes (Kirschman et al.
1986). There are no data that would enable an assessment of the potential for renal effects in humans
living near hazardous waste sites.
Dermal Effects. No studies were located regarding dermal effects in human or animals associated with
inhalation or oral exposure to methylene chloride. However, in some occupational accidents, direct
contact with methylene chloride has resulted in second or third degree chemical burns within 30 minutes
(Hall and Rumack 1990; Wells and Waldron 1984; Winek et al. 1981).
126 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Ocular Effects. No studies were located on ocular effects in humans by oral or dermal exposure.
However, repeated direct exposure to methylene chloride vapors (up to 500 ppm) caused mild irritation to
the eyes of volunteers (Stewart et al. 1972). In one occupational accident, direct contact with the liquid
(duration unspecified) resulted in severe corneal burns (Hall and Rumack 1990). In animals, small
increases in corneal thickness and intraocular tension were reported after exposure to vapors of 490 ppm
of methylene chloride or greater, but effects were reversible within 2 days after exposure ceased
(Ballantyne et al. 1976). Inflammation of the conjunctivas and eyelids as well as increases in corneal
thickness and intraocular tension were observed following direct contact of methylene chloride (0.1 mL)
with the eyes of rabbits. Effects were reversible within 3–9 days (Ballantyne et al. 1976).
Immunological and Lymphoreticular Effects. No studies were located regarding immunological
effects in humans after inhalation, oral, or dermal exposure. Splenic atrophy was evident in dogs that
died following continuous intermediate-duration exposure to vapors of methylene chloride (1,000 ppm)
(MacEwen et al. 1972), but the incidence did not increase over control levels in rats and mice chronically,
but discontinuously, exposed at a level of 4,000 ppm or less (NTP 1986). Splenic fibrosis was observed in
rats after chronic inhalation exposure at $1,000 ppm methylene chloride (Mennear et al. 1988), but was
not observed in a recent intermediate-duration study in rats exposed to 5,187 ppm methylene choride
(Halogenated Solvent Industry Alliance, Inc. (2000). This latter study also found that the IgM antibody
response to SRBC was not significantly altered in exposed rats relative to controls. Due to the very
limited and inconsistent nature of the database, further studies are necessary before conclusions can be
drawn about the relevance of these findings to human health.
Neurological Effects. Studies in humans and animals indicate the central nervous system is an
important target for methylene chloride; in the case of acute exposures, anesthetic responses, which
subsided once exposure ceased, have been reported in humans and animals (Bakinson and Jones 1985;
Hall and Rumack 1990; Heppel et al. 1944; Snyder et al. 1992a, 1992b). Neurological effects in humans
have included headache, dizziness, confusion, memory loss, intoxication, incoordination, paresthesia, and
in severe cases, unconsciousness and seizures (Bakinson and Jones 1985; Hall and Rumack 1990; Kelly
1988). Degraded performance in various psychomotor tasks was reported in humans acutely exposed to
methylene chloride (200 ppm or greater) in experimental studies (Fodor and Winneke 1971; Putz et al.
1979; Stewart et al. 1972; Winneke 1974). Impaired performance during exposure to methylene chloride
was associated with a rise in blood levels COHb to about 5%, which occurred after 3 hours of exposure at
200 ppm (Putz et al. 1979). In more specific measures of cortical function, Winneke (1974) attributed
127 METHYLENE CHLORIDE
2. HEALTH EFFECTS
impaired performance in volunteers following 3–4 hours of exposure to 300 ppm of methylene chloride to
the properties of the parent compound; exposure to 50–100 ppm of carbon monoxide did not generate the
same adverse effects. Based on this value (300 ppm), an acute inhalation MRL of 0.6 ppm was calculated
as described in Table 2-1 (Winneke 1974). Studies in factory workers chronically exposed to methylene
chloride revealed no evidence of neurological or behavioral impairment at exposure levels of 75–100 ppm
(Cherry et al. 1981). There was a reduction in test scores pertaining to mood changes in workers in 1 of
3 rapid rotation shifts exposed to vapors of methylene chloride (28–173 ppm), but no effects were
observed on performance as determined by digit symbol substitution scores (Cherry et al. 1983). Mood
changes are very subjective and these results may reflect other causes.
Neurological effects have been noted in animals following exposure to methylene chloride independent of
the route. Narcotic effects of methylene chloride (incoordination, gait disturbances, reduced activity,
somnolence) were observed in monkeys, rabbits, rats and guinea pigs following acute exposure at
$10,000 ppm (Heppel and Neal 1944; Heppel et al. 1944); dogs exposed at this level became
uncoordinated, then excited and hyperactive (Heppel et al. 1944). These effects wore off when exposure
ceased. Studies in rats and gerbils suggest that methylene chloride induces regional alterations in DNA
concentrations, amino acids levels, and enzyme activities in the brain. There was a decrease in succinic
dehydrogenase activity in the cerebellum in rats exposed to vapors of methylene chloride at concen-
trations of 500 ppm or greater and signs of increased protein breakdown in the cerebrum at 1,000 ppm
(Savolainen et al. 1981). The DNA concentration decreased in the hippocampus and cerebellum in
gerbils exposed to $210 ppm of methylene chloride, indicating decreased cell density in these brain
regions, probably due to cell loss (Karlsson et al. 1987; Rosengren et al. 1986). Levels of aminobutyric
acid increased in the posterior cerebellar vermis of gerbils exposed to 210 ppm of methylene chloride;
however, the significance of this finding is uncertain (Briving et al. 1986). On the other hand, studies in
rats exposed to methylene chloride (2,000 ppm) for 13 weeks revealed that the compound did not cause
clinical, postural, sensory, locomotor, evoked potential, or pathological effects (Mattsson et al. 1990).
However, when rats were given an intraperitoneal injection of methylene chloride, 115 mg/kg was
sufficient to alter the pattern of flash evoked potentials (Herr and Boyes 1997); in this study, the
responses generated in methylene chloride-treated rats were different from those generated by other
solvents, leading the authors to conclude that lipid solubility alone was insufficient to predict the
neurotoxic effects. The mechanism by which methylene chloride exerts its effects on the central nervous
system is not clear. Anaesthetic effects of the parent compound and hypoxic effects of its metabolite
carbon monoxide may both contribute to the observed symptoms.
128 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Reproductive Effects. Data on reproductive toxicity in humans are limited; there are no studies
involving oral exposure. One group of cases reported genital pain, testicular atrophy, recent infertility,
and low/abnormal sperm counts in workers who inhaled vapors of methylene chloride and who had direct
contact with the liquid on the job for more than a year (Kelly 1988). Exposure to methylene chloride in
this group was confirmed by blood COHb analysis and the presence of typical neurological deficits. A
retrospective study of pregnancy outcomes among Finnish pharmaceutical workers reported a slightly
increased risk of spontaneous abortion (not quite statistically significant) associated with inhalation
exposure to methylene chloride (Taskinen et al. 1986). No animal reproductive studies were conducted
for oral or dermal exposures to methylene chloride, and the inhalation studies were negative. Methylene
chloride did not adversely affect fertility and litter size in rats that inhaled methylene chloride vapors at
concentrations up to 1,500 ppm or less for two generations (Nitschke et al. 1988b). After intermediate-
duration exposure (6 weeks) to vapors of methylene chloride (200 ppm or less), there were no
microscopic lesions in testes of rats (Raje et al. 1988). Uterine, ovarian, and testicular atrophy has been
noted in rats and mice chronically exposed for 2 years to 4,000 ppm of methylene chloride, but this was
reported to be secondary to malignant neoplasms (NTP 1986). Based on these data, methylene chloride
does not appear to pose a hazard to human reproduction, except at very high exposure levels.
Developmental Effects. There are few studies addressing developmental effects in humans. A
retrospective study of pregnancy outcomes among Finnish pharmaceutical workers reported a slightly
increased risk of spontaneous abortion associated with inhalation exposure to methylene chloride
(Taskinen et al. 1986). A study examining the effect of low environmental concentrations of methylene
chloride (<0.01 ppm) on over 90,000 births found no significant effect on birth weight (Bell et al. 1991).
Studies in rats demonstrated that methylene chloride crosses the placenta and that metabolism of
methylene chloride by the maternal liver elevates the blood levels of carbon monoxide in the fetus
(Anders and Sunram 1982). Animal studies demonstrated that inhalation of methylene chloride vapors at
concentrations of 1,250 ppm produced minor skeletal variants: delayed ossification of sternebrae in rats
and extra center of ossification in the sternum of mice and rats (Schwetz et al. 1975). Fetal weight was
reduced and behavioral changes occurred in rat pups following exposure of dams to 4,500 ppm of
methylene chloride (Bornschein et al. 1980; Hardin and Manson 1980). The significance of these
observations is uncertain since each of the three studies used only one concentration level and the
observed effects occurred at maternally toxic concentrations. Growth (yolk sac blood vessel growth,
body length, protein concentration) and development (somite addition) were significantly retarded in rat
embryos cultured in the presence of methylene chloride at $0.5 mg/mL, but not at 0.2 mg/mL
129 METHYLENE CHLORIDE
2. HEALTH EFFECTS
(Brown-Woodman et al. 1998); the effect on the yolk sac blood vessels may have interfered with the
ability of the embryos to take up nutrients. Methylene chloride had no effect on heart function in these
embryos. Although fetal body weights were decreased in these animal studies, the absence of other
fetotoxic effects, embryolethality, or major malformations, suggests that methylene chloride is not likely
to cause developmental effects or behavioral changes at levels normally encountered in the environment;
with current standards and procedures, workplace exposure of pregnant women is unlikely to be
hazardous to the fetus. The reduction in the rate of spontaneous abortions observed during the later years
of the Finnish study was attributed partly to improved industrial hygiene (Taskinen et al. 1986).
Genotoxic Effects. No studies were located regarding the genotoxic effects of methylene chloride in
humans after inhalation, oral, or dermal exposure. In vitro results were mixed in bacterial assays and in
tests employing mammalian cells (Table 2-3). Methylene chloride has caused chromosomal aberrations
in some studies, but not in others. Given the evidence of in vitro clastogenicity and its negative results in
unscheduled DNA-synthesis and DNA-binding studies, methylene chloride may be a weak mutagen in
mammalian systems. The chemical has been evaluated in several in vivo assay systems in animals to
assess its potential to induce gene mutation and cause chromosomal aberrations or DNA damage and
repair. Many studies were negative and some were positive (Table 2-4). Tissue-specific genetic damage
in mice following exposure to methylene chloride, suggests that the ability of tissues to metabolize
methylene chloride may determine its genotoxicity (Sasaki et al. 1998). Specific polymorphisms in the
metabolizing enzymes are associated with genotoxicity. In vitro, low concentrations of methylene
chloride were more mutagenic when a functional GSTT1-1 gene was present (DeMarini et al. 1997).
Human erythrocytes expressing the GSTT1-null phenotype had a higher incidence of sister chromatid
exchange than normal following exposure to methylene chloride (Hallier et al. 1994). Similarly,
expression of the DraI DD mutant of CYP2E1 was associated with an increase in bleomycin-induced
single-strand breaks in human lymphocyte DNA (El-Zein et al. 1997a); possibly methylene chloride
exposure would induce similar genetic damage in individuals with that genotype. Additional details are
presented in Section 2.10, Populations that are Unusually Susceptible.

130
Table 2-3. Genotoxicity of Methylene Chloride In Vitro
Results
Species (test system) End point
With
activation
Without
activation Reference
Mammalian cells:
Peripheral lymphocytes Chromosomal aberrations + 0 Thilagar et al.
1984a
Mouse/lymphoma L5178Y Chromosomal aberrations + + Thilagar et al. 1984a
Chinese hamster Chromosomal aberrations + + Thilagar et al. 1984a
Human/primary fibroblasts Unscheduled DNA synthesis Not tested – Jongen et al. 1981
Chinese hamster (V79) Unscheduled DNA synthesis Not tested – Jongen et al. 1981
Chinese hamster (V79) Sister chromatid exchanges (+) (+) Jongen et al. 1981
Human/peripheral lymphocytes Unscheduled DNA synthesis – – Perocco and Prodi 1981
Prokaryotic organisms:
Salmonella typhimurium
(TA98, TA100)
Gene mutation + + Gocke et al. 1981
S. typhimurium
(TA1535, TA1538, TA1537)
Gene mutation – – Gocke et al. 1981
DNA = deoxyribonucleic acid; + = positive result; – = negative result; (+) = weakly positive result
METHYLENE CHLORIDE
2. HEALTH EFFECTS

131
Table 2-4. Genotoxicity of Methylene Chloride In Vivo
Species (test system) End point Results Reference
Mammalian cells:
Mouse (bone marrow and lung cells) Chromosomal aberrations + Allen et al. 1990
Mouse (peripheral erythrocytes) Micronuclei + Allen et al. 1990
Mouse (peripheral lymphocytes and lung cells) Sister chromatid exchanges + Allen et al. 1990
Mouse (bone marrow cells) Micronuclei – Sheldon et al. 1987
Mouse Dominant lethality – Raje et al. 1988
Mouse (liver and lung) DNA breakage + Sasaki et al. 1998
Mouse (stomach, urinary bladder, kidney, brain DNA breakage _ Sasaki et al. 1998
and bone marrow)
Rat (liver) DNA breakage + Kitchin and Brown 1989
Rat Unscheduled DNA synthesis – Trueman and Ashby 1987
Mouse Unscheduled DNA synthesis – Trueman and Ashby 1987
Rat (bone marrow cells) Chromosomal aberrations – Burek et al. 1984
Mouse (bone marrow cells) Chromosomal aberrations – Gocke et al. 1981
Rat (liver and lung cells) DNA alkylation – Green et al. 1988
Mouse (liver and lung cells) DNA alkylation – Green et al. 1988
Eukaryotic organisms:
Insect:
Drosphila Gene mutation (sex-linked (+) Gocke et al. 1981
recessive lethal)
+ = positive result; – = negative result; (+) = weakly positive result; DNA = deoxyribonucleic acid
METHYLENE CHLORIDE
2. HEALTH EFFECTS
132 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Cancer. A significant increase in bile-duct cancer was observed within a cohort of workers who had
been exposed to methylene chloride (at #1,700 ppm, 8-hour TWA) for up to 28 years (Lanes et al. 1990).
Epidemiology studies have not revealed a causal relationship between deaths due to cancer and
occupational exposure to methylene chloride at lower levels (475 ppm or less) (Friedlander et al. 1978;
Hearne et al. 1987, 1990; Ott et al. 1983b). It should be noted that these latter studies had limited power
to detect very small increases in cancer and are not sufficient to rule out a carcinogenic potential of
methylene chloride. Studies in animals exposed via inhalation have demonstrated that methylene chloride
can increase the incidence of naturally-occurring tumors. When administered by inhalation, methylene
chloride (2,000 ppm or greater) increased the incidence of alveolar/bronchiolar neoplasms in
mice of both sexes (NTP 1986). Concentrations of 500 ppm or greater of methylene chloride increased
the incidence of benign mammary gland tumors per animal in females and male rats (Burek et al. 1984;
Nitschke et al. 1988a; NTP 1986). The incidence of liver tumors increased over concurrent control levels
in male mice and female rats administered methylene chloride (50–250 mg/kg/day) in drinking water;
however, the incidence of lesions in treated groups were within the historical range of control values and
showed no dose response (Serota et al. 1986a, 1986b). The results of recent toxicokinetics studies
suggest that the parent compound and/or reactive metabolites produced by the GST pathway are the
source of methylene chloride-induced tumor increases. Based on these findings, the EPA has ranked
methylene chloride as a Group B2 carcinogen (probable human carcinogen). The EPA has calculated that
an upper limit 10
-6
risk level corresponds to 0.0006 ppm and 0.001 mg/kg/day for inhalation and oral
70-year continuous exposures, respectively. OSHA (1997) determined that methylene chloride is a
potential occupational carcinogen, based on rodent inhalation studies and PBPK modeling, and
established an inhalation unit risk of 3.62 x 10
-3
(µg/m
3
)
-1
for occupational exposures.
The Department of Health and Human Services (NTP 1999) has determined that methylene chloride may
reasonably be anticipated to be a human carcinogen. IARC (1987) has classified methylene chloride in
Group 2B (possibly carcinogenic to humans). EPA (IRIS 1999) has determined that methylene chloride
is a probable human carcinogen. Section 2.10 discusses polymorphisms in genes that metabolize
methylene chloride that may be associated with an increased risk of cancer.
133 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2.6 ENDOCRINE DISRUPTION
Recently, attention has focused on the potential hazardous effects of certain chemicals on the endocrine
system because of the ability of these chemicals to mimic or block endogenous hormones, or otherwise
interfere with the normal function of the endocrine system. Chemicals with this type of activity are most
commonly referred to as endocrine disruptors. Some scientists believe that chemicals with the ability to
disrupt the endocrine system are a potential threat to the health of humans, aquatic animals, and wildlife.
Others believe that endocrine disrupting chemicals do not pose a significant health risk, particularly in
light of the fact that hormone mimics exist in the natural environment. Examples of natural hormone
mimics are the isoflavinoid phytoestrogens (Adlercreutz 1995; Livingston 1978; Mayr et al. 1992). These
compounds are derived from plants and are similar in structure and action as endogenous estrogen. While
there is some controversy over the public health significance of endocrine disrupting chemicals, it is
agreed that the potential exists for these compounds to affect the synthesis, secretion, transport, binding,
action, or elimination of natural hormones in the body that are responsible for the maintenance of
homeostasis, reproduction, development, and/or behavior (EPA 1997). As a result, endocrine disruptors
may play a role in the disruption of sexual function, immune suppression, and neurobehavioral function.
Endocrine disruption is also thought to be involved in the induction of breast, testicular, and prostate
cancers, as well as endometriosis (Berger 1994; Giwercman et al. 1993; Hoel et al. 1992).
There is no evidence suggesting that methylene chloride is an endocrine disruptor.
2.7 CHILDREN’S SUSCEPTIBILITY
This section discusses potential health effects from exposures during the period from conception to
maturity at 18 years of age in humans, when all biological systems will have fully developed. Potential
effects on offspring resulting from exposures of parental germ cells are considered, as well as any indirect
effects on the fetus and neonate resulting from maternal exposure during gestation and lactation.
Relevant animal and in vitro models are also discussed.
Children are not small adults. They differ from adults in their exposures and may differ in their
susceptibility to hazardous chemicals. Children’s unique physiology and behavior can influence the
extent of their exposure. Exposures of children are discussed in Section 5.6 Exposures of Children.
134 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Children sometimes differ from adults in their susceptibility to hazardous chemicals, but whether there is
a difference depends on the chemical (Guzelian et al. 1992; NRC 1993). Children may be more or less
susceptible than adults to health effects, and the relationship may change with developmental age
(Guzelian et al. 1992; NRC 1993). Vulnerability often depends on developmental stage. There are
critical periods of structural and functional development during both prenatal and postnatal life and a
particular structure or function will be most sensitive to disruption during its critical period(s). Damage
may not be evident until a later stage of development. There are often differences in pharmacokinetics
and metabolism between children and adults. For example, absorption may be different in neonates
because of the immaturity of their gastrointestinal tract and their larger skin surface area in proportion to
body weight (Morselli et al. 1980; NRC 1993); the gastrointestinal absorption of lead is greatest in infants
and young children (Ziegler et al. 1978). Distribution of xenobiotics may be different; for example,
infants have a larger proportion of their bodies as extracellular water and their brains and livers are
proportionately larger (Altman and Dittmer 1974; Fomon 1966; Fomon et al. 1982; Owen and Brozek
1966; Widdowson and Dickerson 1964). The infant also has an immature blood-brain barrier (Adinolfi
1985; Johanson 1980) and probably an immature blood-testis barrier (Setchell and Waites 1975). Many
xenobiotic metabolizing enzymes have distinctive developmental patterns. At various stages of growth
and development, levels of particular enzymes may be higher or lower than those of adults, and
sometimes unique enzymes may exist at particular developmental stages (Komori et al. 1990; Leeder and
Kearns 1997; NRC 1993; Vieira et al. 1996). Whether differences in xenobiotic metabolism make the
child more or less susceptible also depends on whether the relevant enzymes are involved in activation of
the parent compound to its toxic form or in detoxification. There may also be differences in excretion,
particularly in newborns who all have a low glomerular filtration rate and have not developed efficient
tubular secretion and resorption capacities (Altman and Dittmer 1974; NRC 1993; West et al. 1948).
Children and adults may differ in their capacity to repair damage from chemical insults. Children also
have a longer remaining lifetime in which to express damage from chemicals; this potential is particularly
relevant to cancer.
Certain characteristics of the developing human may increase exposure or susceptibility while others may
decrease susceptibility to the same chemical. For example, although infants breathe more air per
kilogram of body weight than adults breathe, this difference might be somewhat counterbalanced by their
alveoli being less developed, which results in a disproportionately smaller surface area for alveolar
absorption (NRC 1993).
135 METHYLENE CHLORIDE
2. HEALTH EFFECTS
There is no evidence from human toxicity studies that children are, or are likely to be, more susceptible to
health effects from inhalation, oral, or dermal exposure to methylene chloride than adults. No cases of
accidental poisoning in children due to methylene chloride exposure have been reported. The nervous
system is a sensitive acute target of methylene chloride exposure in adults and the response in children is
likely to be similar. Methylene chloride is neurotoxic at high concentrations, in part because of its
lipophilic characteristics. There are no data in the literature that suggest that children are more
susceptible to the acute or chronic neurotoxic effects of methylene chloride than adults. However, there is
little information available in the literature on the chronic low-concentration neurotoxic effects of
methylene chloride exposure in either children or adults. Methylene chloride did not produce any
toxicologically significant developmental effects in animals in a two-generation reproductive study with
rats (Nitschke et al. 1988b) or in mouse and rat developmental studies, except at very high, maternally
toxic doses (Bornschein et al. 1980; Hardin and Manson 1980; Schwetz et al. 1975).
The only animal study that is suggestive of potentially age-related vulnerability to methylene chloride is
that of Maronpot et al. (1995b), in which B6C3F
1
mice were exposed to 2,000 ppm of methylene chloride
by inhalation for 26, 52, or 78 weeks. The results of stop-exposure experiments showed that early
exposure to methylene chloride was more effective than late exposure in inducing pulmonary neoplasms.
However, the authors mentioned the possibility that the late-exposed animals were sacrificed too early for
tumors to have developed.
There is no information regarding the pharmacokinetics of methylene chloride in children or regarding
the nutritional factors that may influence the absorption of methylene chloride. A PBPK model has been
developed for estimating the amounts and concentrations of methylene chloride in human breast milk that
would result from inhalation exposures to methylene chloride (Fisher et al. 1997). Animal studies
demonstrate that methylene chloride and/or its metabolites distribute in the liver, kidney, lungs, brain,
muscle, and adipose tissues after inhalation exposures (Carlsson and Hultengren 1975; McKenna et al.
1982) and has also been shown to cross the placenta in rats (Anders and Sunram 1982). Toxicokinetic
data indicate that methylene chloride is rapidly cleared after cessation of exposure. Small amounts of
methylene chloride have been found in breast milk (EPA 1980d; Pellizzari et al. 1982).
It is not known whether the metabolism of methylene chloride in children is different than in adults, but
there are theoretical reasons to suspect it might be. Available data suggest that there are two pathways by
which methylene chloride is metabolized. One pathway utilizes the mixed function oxidase (MFO)
136 METHYLENE CHLORIDE
2. HEALTH EFFECTS
enzymes and produces carbon monoxide (CO). The other pathway involves the glutathione transferase
(GST) and produces carbon dioxide (CO
2
). No information was located regarding the possibility that the
metabolism of methylene chloride via the GST metabolic pathway is developmentally regulated.
CYP2E1 (the specific MFO pathway for methylene chloride) expression is low in fetal liver but increases
several hours after birth in humans and continues to increase during the first week of life (Carpenter et al.
1996, 1997; Jones et al. 1992; Viera et al. 1996). In the human fetal liver, CYP2E1 transcripts were not
detected at 10 weeks of gestation, but were detected as early as 19 weeks (Carpenter et al. 1996). The
CYP2E1 gene in the liver is modified by cytosine methylation at the 3' region during fetal development,
which contributes to the low expression of the gene during fetal life (Jones et al. 1992; Viera et al. 1996).
CYP2E1 expression in the fetal brain, however, has been detected near baseline levels at 46 days of
gestation, but at significant levels beginning at 58 days (Brzezinski et al. 1999). The early expression of
CYP2E1 in the fetal brain suggests that the brain may be particularly vulnerable to oxidative stress as a
result of xenobiotic metabolism.
In cases of heavy ethanol consumption by the mother, CYP2E1 is induced in the placenta (Rasheed et al.
1997) and in the liver (Carpenter et al. 1997). In vitro tests demonstrated that ethanol treatment of human
fetal hepatocytes upregulated expression of CYP2E1 (Carpenter et al. 1996). This presumably would
increase the rate of metabolism of methylene chloride in the fetus.
Evidence from pregnant rats exposed to methylene chloride indicates that the maternal liver metabolizes
methylene chloride at a higher rate than the fetus, but that the metabolite carbon monoxide equilibrates
between the dam and fetus (Anders and Sunram 1982). The high affinity of fetal hemoglobin for both
carbon monoxide and oxygen suggests that fetuses may be at risk from hypoxia following maternal
exposures to high levels of methylene chloride (Anders and Sunram 1982; Longo 1977). The greater risk
of hypoxia and the potential for greater neurological damage would last until the expression of adult
hemoglobin during the first year of life.
As mentioned above, methylene chloride has been isolated from human breast milk (EPA 1980d;
Pellizzari et al. 1982), so it is possible that maternal exposures could transmit the compound to infants.
However, PBPK modeling suggests that lactating females who breast feed their infants will not deliver
methylene chloride in significant quantities (Fisher et al. 1997). Simulation of a workday maternal
inhalation exposure to methylene chloride at 50 ppm yielded a predicted daily intake in the infant of only
0.213 mg, which is significantly less than the 2.0 mg/day equivalent EPA Health Advisory Intake.
137 METHYLENE CHLORIDE
2. HEALTH EFFECTS
There are no biomarkers of exposure or effect for methylene chloride that have been validated in children
or in adults exposed as children. There are no biomarkers in adults that identify previous childhood
exposure. No information was located regarding pediatric-specific methods for reducing peak absorption
following exposure to methylene chloride, reducing body burden or interfering with the mechanism of
action for toxic effects. In addition, no data were located regarding whether methods for reducing toxic
effects might be contraindicated in children.
2.8 BIOMARKERS OF EXPOSURE AND EFFECT
Biomarkers are broadly defined as indicators signaling events in biologic systems or samples. They have
been classified as markers of exposure, markers of effect, and markers of susceptibility (NAS/NRC 1989).
Due to a nascent understanding of the use and interpretation of biomarkers, implementation of biomarkers
as tools of exposure in the general population is very limited. A biomarker of exposure is a xenobiotic
substance or its metabolite(s) or the product of an interaction between a xenobiotic agent and some target
molecule(s) or cell(s) that is measured within a compartment of an organism (NAS/NRC 1989). The
preferred biomarkers of exposure are generally the substance itself or substance-specific metabolites in
readily obtainable body fluid(s), or excreta. However, several factors can confound the use and
interpretation of biomarkers of exposure. The body burden of a substance may be the result of exposures
from more than one source. The substance being measured may be a metabolite of another xenobiotic
substance (e.g., high urinary levels of phenol can result from exposure to several different aromatic
compounds). Depending on the properties of the substance (e.g., biologic half-life) and environmental
conditions (e.g., duration and route of exposure), the substance and all of its metabolites may have left the
body by the time samples can be taken. It may be difficult to identify individuals exposed to hazardous
substances that are commonly found in body tissues and fluids (e.g., essential mineral nutrients such as
copper, zinc, and selenium). Biomarkers of exposure to methylene chloride are discussed in
Section 2.8.1.
Biomarkers of effect are defined as any measurable biochemical, physiologic, or other alteration within an
organism that, depending on magnitude, can be recognized as an established or potential health
impairment or disease (NAS/NRC 1989). This definition encompasses biochemical or cellular signals of
tissue dysfunction (e.g., increased liver enzyme activity or pathologic changes in female genital epithelial
138 METHYLENE CHLORIDE
2. HEALTH EFFECTS
cells), as well as physiologic signs of dysfunction such as increased blood pressure or decreased lung
capacity. Note that these markers are not often substance specific. They also may not be directly
adverse, but can indicate potential health impairment (e.g., DNA adducts). Biomarkers of effects caused
by methylene chloride are discussed in Section 2.8.2.
A biomarker of susceptibility is an indicator of an inherent or acquired limitation of an organism's ability
to respond to the challenge of exposure to a specific xenobiotic substance. It can be an intrinsic genetic
or other characteristic or a preexisting disease that results in an increase in absorbed dose, a decrease in
the biologically effective dose, or a target tissue response. If biomarkers of susceptibility exist, they are
discussed in Section 2.10 “Populations That Are Unusually Susceptible”.
2.8.1 Biomarkers Used to Identify or Quantify Exposure to Methylene Chloride
Measurements of parent methylene chloride and its metabolites in expired air, blood, and urine have been
used as indicators of exposure. Elimination of methylene chloride from the body occurs primarily
through pulmonary excretion; approximately 70–75% is excreted unchanged at concentrations from 50 to
200 ppm (DiVincenzo and Kaplan 1981). Pulmonary excretion was rapid during the first hour, then
began to decline as steady state was approached. By 7 hours postexposure, expired air contained less than
1 ppm of methylene chloride for exposures between 50 and 150 ppm. The 7-hour values for an exposure
of 200 ppm were twice that for the other concentrations. At 16 hours, negligible levels of methylene
chloride were detected in all concentration groups. Therefore, measurements of methylene chloride in
expired air as a biomarker are useful only if they occur within 6–8 hours of the most recent exposure
(DiVincenzo and Kaplan 1981).
Methylene chloride can be detected in blood. Because it is cleared from blood very rapidly, this method
is only useful for monitoring recent exposures. A plasma half-life of inhaled methylene chloride in
humans is estimated to be 40 minutes (DiVincenzo et al. 1972).
Methylene chloride has been detected in adipose tissue in humans, but animal studies suggest that
methylene chloride is cleared so rapidly (90% decrease in 2 hours), that it is useful for measuring only
recent exposures (Carlsson and Hultengren 1975). Methylene chloride was detected in the breast milk of
mothers living in urban industrial areas, but the route of exposure was not analyzed (EPA 1980e;
Pellizzari et al. 1982).
139 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Methylene chloride is also excreted in urine. Humans exposed to 100 ppm of methylene chloride vapor
for 2 hours averaged 26.6 µg methylene chloride in the urine within 24 hours after exposure and 85.5 µg
at exposure levels of 200 ppm (DiVincenzo et al. 1972).
Levels of COHb in the blood may also be used as an indicator of exposure to methylene chloride. Levels
of COHb are concentration- and time-dependent (Stewart et al. 1972). Human subjects exposed to
concentrations of 500 ppm or less for 1 hour experienced elevation in COHb levels (1–4%). These levels
rose to an average of 10% saturation within 1 hour after exposure to higher concentrations (1,000 ppm for
2 hours) (Stewart et al. 1972). Exposure to concentrations as low as 100 ppm for 7.5 hours or 200 ppm
for 4 hours resulted in COHb elevation above 5% in nonsmokers (Putz et al. 1979; Stewart et al. 1972).
Levels of COHb can remain elevated above preexposure levels for more than 21 hours post exposure
(Stewart et al. 1972). DiVincenzo and Kaplan (1981) showed recovery to baseline each day for
5 consecutive days. COHb saturation levels were lower than in Stewart et al. (1972). Different methods
of COHb detection were used by the authors. Although COHb levels can be used as an indicator of
exposure to methylene chloride, this biomarker is not specific. Exposure to other sources of carbon
monoxide, such as tobacco smoke, incomplete organic fuel combustion, and automobile exhaust, will also
increase COHb levels.
Ghittori et al. (1993) examined several methods for conducting biological monitoring of workers exposed
occupationally to methylene chloride. Twenty males (12 smokers and 8 nonsmokers), employed at
different jobs in a pharmaceutical factory where methylene chloride was used to wash gelatin capsules,
wore a personal passive dosimeter in the respiratory zone during half of work days (4 hours) in order to
measure the weighted mean-inspired environmental concentration of methylene chloride.
Tetrachloroethylene was also present in the work environment. Immediately after the end of the
exposure, a urine sample was collected using gas-tight samplers. Carbon monoxide (CO) was determined
at the end of the shift using a portable instrument.
Ambient exposures ranged from 49 to 168 ppm. No significant correlation was observed among the CO
of all subjects and the concentration of methylene chloride in ambient air. When those workers who
smoked were removed from analysis, a correlation between the methylene chloride concentration in air
and the CO concentration in alveolar air was found ® = 0.87). Significant linear correlation was found
between the environmental concentration of methylene chloride in the breathing zone and methylene
chloride concentration in urine ® = 0.9 uncorrected, 0.72 corrected for creatinine). The authors suggest
140 METHYLENE CHLORIDE
2. HEALTH EFFECTS
that measuring methylene chloride in the urine was a more useful measure of exposure, since unlike
COHb in the blood or CO in exhaled breath, it was unaffected by smoking.
Concentrations of CO in exhaled air studied in other groups of volunteers were shown to reach a
maximum at 1–2 hours after exposure and were also directly proportional to the magnitude of exposure
both during and after exposure. These findings are consistent with those studies.
The authors concluded that the results indicated that methylene chloride urinary concentration could be
used as a possible biological index to evaluate methylene chloride air exposure, according to what has
been observed for other solvents. However, at high ambient exposures (i.e., those exceeding 5–7 times
the threshold limit value-time weighted average [TLV-TWA] of 49 ppm), the use of methylene chloride
as a biological index might be problematic because exposure to very high concentrations gives
disproportionately less COHb in the blood and more unmetabolized methylene chloride is produced.
2.8.2 Biomarkers Used to Characterize Effects Caused by Methylene Chloride
As discussed in Section 2.2, the effects that are most often observed in humans exposed to methylene
chloride vapors are central nervous system depression and behavioral effects. Clinical signs and
symptoms which may be monitored include irritability, narcosis, and fatigue. Impairment of visual,
auditory, and psychomotor functions can also be evaluated to detect early effects on the central nervous
system. Since these effects also occur following exposure to numerous other chemicals, they are not
specific for methylene chloride exposure and evaluation is often subjective.
Honma and Suda (1997) have investigated the effects of a single intraperitoneal administration of several
short-chain chlorinated compounds, including methylene chloride, on lipoproteins in plasma and liver in
male Fischer F344 rats. Changes in lipoproteins caused by these solvents were compared with
hepatotoxicity markers such as GPT (ALT). Following administration of methylene chloride in olive oil,
concentrations of lipoproteins (VLDL, LDL, HDL), triglyceride, cholesterol, and GPT in plasma were
determined, as were changes in liver weight and amounts of triglyceride and glutathione in liver tissue.
For methylene chloride, peaks of changes in these endpoints were observed at 8 or 19 hours following
compound administration. The dose dependency of these changes were investigated after intraperitoneal
dosing of 300 and 1,000 mg/kg methylene chloride. HDL decreased significantly at a dose of 300 mg/kg,
whereas a marked increase in LDL occurred at 1,000 mg/kg. GPT also increased significantly at a dose
141 METHYLENE CHLORIDE
2. HEALTH EFFECTS
of 1,000 mg/kg. The authors conclude that changes in some lipoproteins in plasma occurred at lower
doses than those causing elevation of GPT activity; therefore, these lipoproteins may be able to serve as
sensitive and simple markers of adverse liver effects (i.e., biomarkers of effects). However, further
investigation is necessary to determine whether these endpoints can serve as biomarkers of effects in
humans when exposure is via inhalation and confounding variables are be numerous.
No other biomarkers have been identified to characterize effects associated with exposure to methylene
chloride.
For more information on biomarkers for renal and hepatic effects of chemicals see ATSDR/CDC
Subcommittee Report on Biological Indicators of Organ Damage (1990) and for information on
biomarkers for neurological effects see OTA (1990).
2.9 INTERACTIONS WITH OTHER CHEMICALS
Limited studies were found in the available literature on the interactions of methylene chloride with other
chemicals. Exposure of adult human subjects to 500 ppm of methylene chloride resulted in levels of
COHb in the blood comparable with those produced by the TLV for CO (50 ppm) (Fodor and Roscovana
1976). A 4-hour exposure of human subjects to 200 ppm of methylene chloride or 70 ppm of CO resulted
in similar blood COHb levels (Putz et al. 1979). This suggests that simultaneous exposure to methylene
chloride and CO has an additive effect on blood COHb levels.
A possible additive effect of methylene chloride and toluene (as well as mineral spirits and methyl ethyl
ketone) may have been operative in an occupational mixed-solvent exposure study in which manual
dexterity, and visual memory task performances were impaired, even though the concentrations of the
specific chemicals were below the recommended their threshold limit values (White et al. 1995); the
reported adverse neurobehavioral effects are similar to those reported for individual exposures of
methylene chloride at higher concentrations (Putz et al. 1979). In a rat study, methylene chloride and
toluene interacted in an additive manner to cause retardation of embryonic growth and development in
vitro (Brown-Woodman et al. 1998). In rats, combined CO and methylene chloride exposure yielded
additive increases in the COHb levels (ACGIH 1986).
142 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Methylene chloride decreases the nerve conduction velocity in animals when used alone but when the
compound is administered with ethanol there is a more pronounced decrease of nerve conduction velocity
(Glatzel et al. 1987). The additive or synergist effect of ethanol coadministration decreases at higher
doses. Rats maintained on drinking water containing 10% (v/v) ethanol show increased levels of COHb
after a single oral dose or during inhalation exposure to methylene chloride compared to rats that have not
been exposed to ethanol (Wirkner et al. 1997). This interaction has been attributed to the induction of
CYP2E1 by ethanol and a resulting increased production of carbon monoxide from methylene chloride.
Wirkner et al. (1997) indicate that ethanol has no synergistic effect when methylene chloride is
administered at high doses because the cytochrome P-450-dependent monooxygenase pathway becomes
saturated at exposures >500 ppm.
Interactions resulting from the inhibition of metabolism of methylene chloride have been described in
rats. Blood COHb levels in rats, after an oral dose of methylene chloride or during inhalation exposure to
methylene chloride, are decreased by a concurrently administered oral dose of toluene (Ciuchta et al.
1979; Pankow et al. 1991a, 1991b). This interaction has been attributed to inhibition of the metabolism
of methylene chloride to carbon monoxide, possibly as a result of both chemicals being a substrate for the
2E1 isoenzyme of cytochrome P-450 (Pelekis and Krishnan 1997).
In a rat study, methylene chloride (coinjected intraperitoneally) potentiated the hepatotoxic effect of
carbon tetrachloride (Kim 1997). Coadministration reduced the methylene-chloride-induced rise in blood
COHb, but augmented the carbon tetrachloride-induced increase in liver microsomal enzyme activities 3-
to 8-fold. Methylene chloride increased the covalent binding of carbon tetrachloride metabolites to lipids,
which the author suggests may be a cause of enhanced hepatotoxicity of the mixture.
2.10 POPULATIONS THAT ARE UNUSUALLY SUSCEPTIBLE
A susceptible population will exhibit a different or enhanced response to methylene chloride than will
most persons exposed to the same level of methylene chloride in the environment. Reasons may include
genetic makeup, age, health and nutritional status, and exposure to other toxic substances (e.g., cigarette
smoke). These parameters result in reduced detoxification or excretion of methylene chloride, or
compromised function of organs affected by methylene chloride. Populations who are at greater risk due
to their unusually high exposure to methylene chloride are discussed in Section 5.7, Populations With
Potentially High Exposures.
143 METHYLENE CHLORIDE
2. HEALTH EFFECTS
There are certain subgroups of the general population that may be more susceptible to methylene chloride
than others. One basis for this concern is the potential effect of COHb, produced from CO, a metabolite
of methylene chloride. The COHb generated from methylene chloride is expected to be additive to COHb
from other sources. Thus, methylene chloride exposure at high concentrations may pose an additional
human health burden. Of particular concern are smokers (who maintain significant constant levels of
COHb), and persons with existing cardiovascular disease. In addition, higher than normal levels of CO
may result when alcoholics are exposed to methylene chloride, since ethanol increases the expression and
activity of CYP2E1 (Carpenter et al. 1996). Similarly, enhanced expression of CY2E1 occurs in the
condition of diabetes, although insulin erases that effect (Thomas et al. 1987).
Varying susceptibility to methylene chloride may be correlated with polymorphism in its metabolizing
enzymes. Genetic polymorphisms have been identified for both GSTT1 and CYP2E1 (Garte and Crosti
1999), and the health consequences of these variants are being explored (d’Errico et al. 1999; Lang and
Pelkonen 1999; Pelkonen et al. 1999; Strange and Fryer 1999; Stubbins and Wolf 1999). In the case of
GSTT1, which is the major metabolic pathway for methylene chloride at concentrations >500 ppm, there
is, in addition to the wild type gene, a nonfunctioning allele with a deletion, so that three phenotypes
exist: “high conjugators” homozygous for the wild type allele; “nonconjugators” homozygous for the
deletion (“null) allele; and “low conjugators”, heterozygotes with one of each (Thier et al. 1998). Thier et
al. (1998) demonstrated that tissue samples representing the three phenotypes varied in the ability to
metabolize methylene chloride. The health implications of the homozygous GSTT1-null genotype for
people exposed to methylene chloride exposure are not clear. A potentially positive effect is that no toxic
reactive GST-intermediates of methylene chloride would be produced, as demonstrated by the absence of
RNA-formaldehyde adduct formation in GSTT1-null human hepatocytes treated with methylene chloride
in vitro (Casanova et al. 1997). However, the overall rate of methylene chloride metabolism and
elimination would be reduced in GSTT1-null individuals, so that narcotic effects of the parent compound
would last longer. Furthermore, increased genotoxic damage (sister chromatid exchange) occurs
following exposure to methylene chloride, in null-phenotype, compared to wild type lymphocytes (Hallier
et al. 1994). Variations in the incidence of GSTT1 polymorphisms have been identified in different
ethnic groups (Strange and Fryer 1999). In one study, the proportion of individuals carrying functional
GSTT1 alleles was 36% among Chinese, 80% among Caucasians, and 90% among Mexican-Americans
(Nelson et al. 1995). In a study of 416 subjects, the GSTT1-null genotype occurred among 24.1% of the
blacks and 15% of the whites (Chen et al. 1996). The GSTT1-null phenotype was detected in 21.1% of
144 METHYLENE CHLORIDE
2. HEALTH EFFECTS
lung cancer patients with either squamous cell carcinoma or adenocarcinoma (El-Zein et al. 1997a).
Polymorphisms have also been detected in CYP2E1 (Garte and Crosti 1999; Lang and Pelkonen 1999).
One polymorphism involves a mutation in the 5' flanking region of the CYP2E1 gene that leads to a
higher rate of transcription, a higher level of protein, and higher enzyme activity compared to cells
containing the wild type allele (Wan et al. 1998). The frequency of the mutant allele was 16% in a group
of 203 Mexican-Americans (Wan et al. 1998), which is considerably higher than the 1–5% frequency
measured in Caucasians (Stephens et al. 1994). Presumably, this allele would result in higher rates of
metabolism of methylene chloride, and possibly more severe toxic effects, but this has not been assayed.
A number of studies have tried to associate specific polymorphisms with cancer incidence (d’Errico et al.
1999). The CYP2E1 DraI DD genotype was found to be associated with a significantly higher risk of
lung cancer among Mexican-Americans and African-Americans (Wu et al. 1998); expression of this
genotype was also associated with an increase in bleomycin-induced single-strand breaks in lymphocytes
tested in vitro. In another study, a rare CYP2E1 PstI variant (mutated in the transcription regulation
region) was found in 7/57 lung cancer patients and 2/48 controls (El-Zein et al. 1997a); all seven patients
developed adenocarcinoma. In a case control study, combined homozygosity for the CYP2E1 PstI
variant and the GSTT1-null genotype was associated with an increased risk of developing lung cancer
(El-Zein et al. 1997b). Presumably, individuals expressing these mutant genotypes would be susceptible
to adverse effects following exposure to methylene chloride.
2.11 METHODS FOR REDUCING TOXIC EFFECTS
This section will describe clinical practice and research concerning methods for reducing toxic effects of
exposure to methylene chloride. However, because some of the treatments discussed may be
experimental and unproven, this section should not be used as a guide for treatment of exposures to
methylene chloride. When specific exposures have occurred, poison control centers and medical
toxicologists should be consulted for medical advice. The following texts provide specific information
about treatment following exposures to methylene chloride: Ellenhorn 1997; Stewart and Dodd 1964.
145 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2.11.1 Reducing Peak Absorption Following Exposure
Human exposure to methylene chloride may occur by inhalation, ingestion, or by dermal contact.
Mitigation approaches to reduce absorption of methylene chloride have included general
recommendations of separating contaminated food, water, air, or clothing from the exposed individual.
Externally, exposed eyes and skin are flushed with a clean neutral solution such as water or normal saline.
If the victim is alert and oriented and has an intact gag reflex, water or milk may be administered after
ingestion of small amount of methylene chloride to wash residual chemical through the esophagus and
dilute the contents in the stomach. In cases where persons have ingested more than several swallows,
emesis can be induced with ipecac syrup after it has been determined that the victim is alert and oriented
and precautions have been taken to protect the respiratory tract from aspiration of gastric contents. Once
the individual is placed in the care of a health professional, gastric lavage can be performed within 1 hour
of the exposure. Activated charcoal and cathartics are frequently recommended; however, no data exist to
support their efficacy (Ellenhorn 1997).
Once methylene chloride has been inhaled or ingested, it is readily absorbed through the lungs or
gastrointestinal tract. Dermal exposure also results in absorption, although at a slower rate than the other
exposure routes and some references question whether the amount absorbed would be sufficient to cause
systemic toxicity (Ellenhorn 1997; Stewart and Dodd 1964). Once absorbed, methylene chloride is
rapidly metabolized in the liver in part to carbon monoxide. Because of the affinity of hemoglobin for
carbon monoxide, COHb levels in the blood will increase, but free circulating carbon monoxide will not
increase.
2.11.2 Reducing Body Burden
Inhalation data demonstrate that a portion of the absorbed methylene chloride is stored in fat tissue as
evidenced by continued increase in COHb levels. It is likely to occur only with long-term exposure and
at high concentrations (DiVincenzo et al. 1972). Investigations in humans following oral exposure to
methylene chloride have failed to detect significant retention in fat or other tissue stores (Angelo et al.
1986a, 1986b; Ellenhorn 1997).
Following absorption, methylene chloride is distributed mainly to the liver, brain and subcutaneous
adipose tissue (Carlsson and Hultengren 1975). The liver is the primary site of metabolism, although
146 METHYLENE CHLORIDE
2. HEALTH EFFECTS
additional transformation occurs in the lungs and kidneys. In the liver, methylene chloride may undergo
metabolism by two pathways. The first pathway produces CO and CO
2
and is saturable at a few
hundred ppm. The second pathway yields formaldehyde and formic acid and shows no indication of
saturation at inhaled concentrations up to 10,000 ppm. Acute toxic effects (central nervous system
depression) may persist for hours after removal from the source of exposure because of continued
metabolism of methylene chloride released from tissue storage (ATSDR 1990). COHb levels can
continue to rise, peaking 5–6 hours after exposure. Peak levels of 12 (NIOSH 1974) and 50% (Ellenhorn
1997) have been reported. Most of the effects seen following acute high-concentration exposure are due
to the anesthetic properties of the parent compound. Fatalities have occurred with COHb levels less than
10%; thus, it is not likely that carboxyhemoglobinea was the cause of death. A study by Scholz et al.
(1991) suggests that, in addition to anesthetic effects, the sudden onset of cardiac arrhythmia induced by
acute exposure to methylene chloride may contribute to its lethality.
The metabolic contribution of each pathway appears to vary in humans, particularly with the exposure
level, and therefore toxicity extrapolation between high and low doses is complex (Gargas et al. 1986).
Furthermore, recent studies suggest that the second pathway is considerably more active in certain animal
species, particularly mice, a finding that complicates interspecies comparisons (ATSDR 1990; Green et
al. 1986b, 1986c).
The body eliminates methylene chloride primarily through the lungs. A small amount of unchanged
methylene chloride is also eliminated in the urine and feces. At low doses, a large percentage of
methylene chloride is metabolized to form COHb and eliminated as carbon monoxide, while at higher
doses more of the unchanged parent compound is exhaled (ATSDR 1990). The administration of 100%
oxygen is efficacious in reducing the half-life of carbon monoxide and should be continued until COHb is
less than 5%.
The prompt application of hemoperfusion and diuretic therapy eliminated metabolic acidosis and
hemoglobinuria in the case of an acute oral exposure to paint remover containing methylene chloride
(Roberts and Marshall 1976). This treatment was thought to have prevented renal damage, but did not
address the problem of gastrointestinal ulceration.
147 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2.11.3 Interfering with the Mechanism of Action for Toxic Effects
The primary effects of exposure to methylene chloride appear to be hepatotoxicity and neurotoxicity.
However, mechanisms of action for these toxic effects are not well characterized and it is difficult to
speculate concerning specific methods for preventing these effects.
Because one consequence of metabolism of methylene chloride is production of COHb, methylene
chloride exposure victims are sometimes given supplemental oxygen. The administration of oxygen
increases the dissociation of carbon monoxide from hemoglobin and hastens the reduction of COHb. The
half-life of COHb is normally approximately 5.3 hours but this can be reduced to 60–90 minutes with
inhalation of 100% oxygen. Hyperbaric oxygen as used in carbon monoxide poisoning from direct
carbon monoxide inhalation may be useful when high COHb levels are present (Haddad and Winchester
1990). At high COHb levels, hyperbaric oxygen will reduce the half-life to 20–40 minutes (ATSDR
1990). Because carbon monoxide is generated metabolically, this often necessitates a longer duration of
oxygen therapy after methylene chloride poisoning than with carbon monoxide poisoning.
Specific mechanisms of action relating to neurological effects are not well understood. Steroids and
mannitol have been used to decrease cerebral edema caused by methylene chloride toxicity, but their
value in preventing later neurologic sequelae remains unproven (ATSDR 1990).
Victims with cardiopulmonary disease or workers who are exposed to carbon monoxide sources could
have an elevated risk from methylene chloride metabolism and should be monitored closely. One
reference suggests that physical exercise and smoking be avoided because these activities may have an
additive effect on the COHb level (Ellenhorn 1997).
2.12 ADEQUACY OF THE DATABASE
Section 104(i)(5) of CERCLA, as amended, directs the Administrator of ATSDR (in consultation with the
Administrator of EPA and agencies and programs of the Public Health Service) to assess whether
adequate information on the health effects of methylene chloride is available. Where adequate
information is not available, ATSDR, in conjunction with the National Toxicology Program (NTP), is
required to assure the initiation of a program of research designed to determine the health effects (and
techniques for developing methods to determine such health effects) of methylene chloride.
148 METHYLENE CHLORIDE
2. HEALTH EFFECTS
The following categories of possible data needs have been identified by a joint team of scientists from
ATSDR, NTP, and EPA. They are defined as substance-specific informational needs that if met would
reduce the uncertainties of human health assessment. This definition should not be interpreted to mean
that all data needs discussed in this section must be filled. In the future, the identified data needs will be
evaluated and prioritized, and a substance-specific research agenda will be proposed.
2.12.1 Existing Information on Health Effects of Methylene Chloride
The existing data on health effects of inhalation, oral, and dermal exposure of humans and animals to
methylene chloride are summarized in Figure 2-5. The purpose of this figure is to illustrate the existing
information concerning the health effects of methylene chloride. Each dot in the figure indicates that one
or more studies provide information associated with that particular effect. The dot does not necessarily
imply anything about the quality of the study or studies, nor should missing information in this figure be
interpreted as a “data need”. A data need, as defined in ATSDR’s Decision Guide for Identifying
Substance-Specific Data Needs Related to Toxicological Profiles (ATSDR 1989), is substance-specific
information necessary to conduct comprehensive public health assessments. Generally, ATSDR defines a
data gap more broadly as any substance-specific information missing from the scientific literature.
As shown in Figure 2-5, studies of humans exposed to methylene chloride by the inhalation route are
available. These have focused mainly on neurological, cancer, and systemic effects (e.g., cardiovascular).
One report focused on possible reproductive effects following inhalation (and possible dermal) exposure.
Other end points (immunological, developmental, and genotoxic effects) have not been evaluated. There
are no studies on the effects of methylene chloride in humans after ingestion. Other than one study
evaluating potential reproductive effects following inhalation and possibly dermal exposure, no other end
points have been evaluated following dermal exposure.
Studies in animals have also focused mainly on inhalation exposure and several end points have been
evaluated. Effects of oral exposure have focused on death, systemic effects after intermediate and chronic
exposure, genotoxicity, and cancer. Other end points have not been evaluated. There are limited reports
on the effects of methylene chloride after direct ocular exposure of animals.
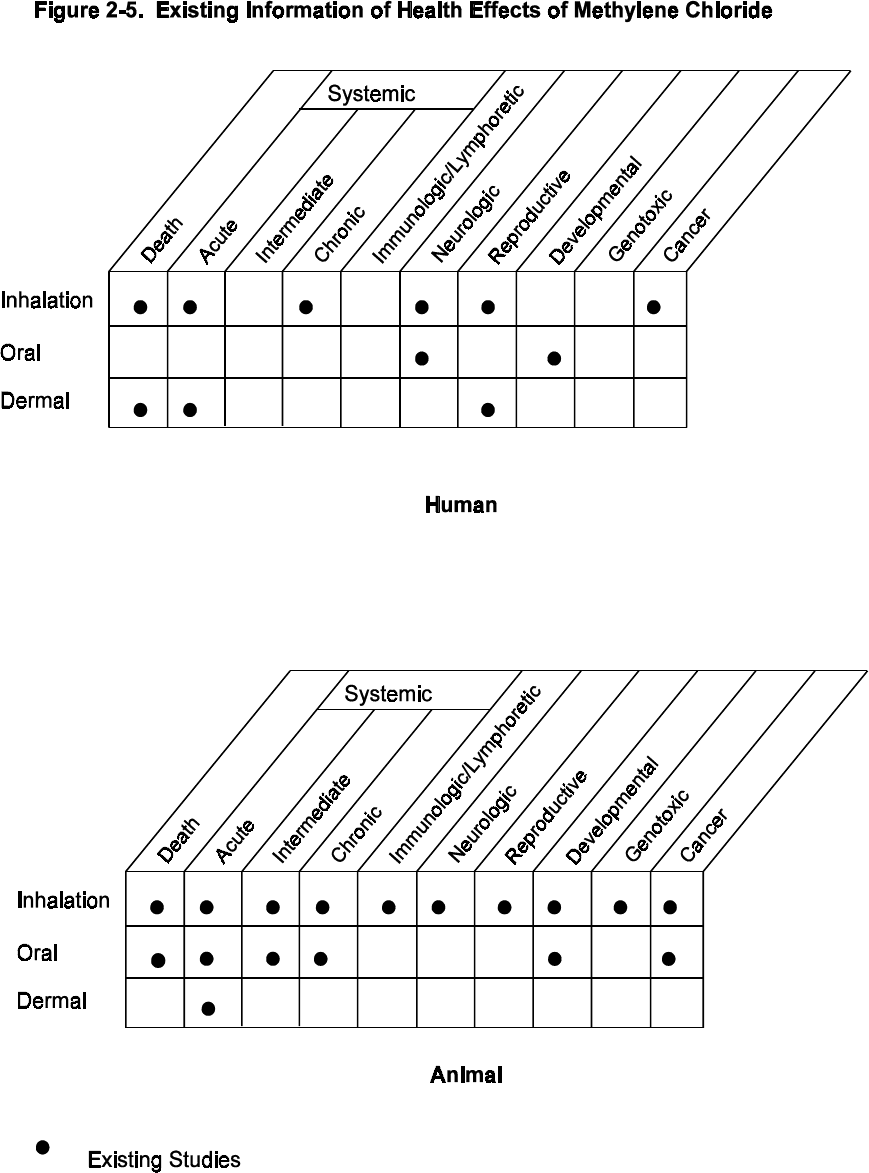
149 METHYLENE CHLORIDE
2. HEALTH EFFECTS
150 METHYLENE CHLORIDE
2. HEALTH EFFECTS
2.12.2 Identification of Data Needs
Acute-Duration Exposure. Available data indicate that the central nervous system is the primary
target of inhaled methylene chloride in humans (Bakinson and Jones 1985; Fodor and Winneke 1971;
Hall and Rumack 1990; Putz et al. 1979; Stewart et al. 1972; Winneke 1974), rats (Heppel and Neal 1944;
Rebert et al. 1989; Savolainen et al. 1981), guinea pigs, rabbits, dogs, and monkeys (Heppel et al. 1944).
An acute inhalation MRL was derived for methylene chloride based on a LOAEL of 300 ppm for
neurological effects in humans (Winneke 1974). Respiratory effects (cough, breathlessness, and in one
fatal case, pulmonary congestion with focal hemorrhage) were observed in several cases of acute
occupational exposure to methylene chloride in humans (Bakinson and Jones 1985; Snyder et al. 1992a,
1992b; Winek et al. 1981). No cardiovascular effects were seen in humans acutely exposed to
concentrations between 100 and 475 ppm (Cherry et al. 1981; Ott et al. 1983c). Gastrointestinal effects
(nausea and vomiting) were reported in several cases of acute occupational exposure to methylene
chloride (Bakinson and Jones 1985). Acute inhalation exposure to methylene chloride increased the
blood COHb level in humans (DiVincenzo and Kaplan 1981; Manno et al. 1992; Putz et al. 1979; Soden
et al. 1996). An acute inhalation study in mice (Aviado and Belej 1974) suggests that the cardiovascular
system may be a target for methylene chloride toxicity; however, the effects were observed at lethal
concentrations. An acute study in guinea pigs demonstrated an increase in hepatic triglycerides, but no
histopathological effects on the liver following exposure to methylene chloride at 5,200 ppm (Morris et
al. 1979). Since the most sensitive acute effects in humans are neurological, additional animal studies
that evaluate changes in analogous functions (visual, auditory discrimination tasks) at low levels of
exposure might be needed. These experiments should monitor CO/COHb levels in the animals to
correlate exposure and effect. Furthermore, the animals should be genotyped with respect to for GSTT1
and CYP2E1 to evaluate the mechanism of toxicity.
The only studies in humans on the effects of acute oral exposure to methylene chloride are reports of
suicide attempts by ingestion of paint removers. One of these studies reported suppression of the central
nervous system and metabolic acidosis (Roberts and Marshall 1976). Corrosion of the gastrointestinal
tract was also reported (Hughes and Tracey 1993; Roberts and Marshall 1976). Hematological effects
included elevated COHb (Hughes and Tracey 1993) and intravascular hemolysis leading to
hemoglobinuria (Roberts and Marshall 1976). No acute respiratory, cardiovascular, hepatic, endocrine, or
immunological effects have been reported in humans following ingestion of methylene chloride, and there
are no acute oral studies on the effect of low doses in humans. Limited studies in animals involved
151 METHYLENE CHLORIDE
2. HEALTH EFFECTS
exposure of rats to high doses of methylene chloride via gavage, resulting in increased mortality (Kimura
et al. 1971; Ugazio et al. 1973); as in suicide cases, respiratory failure as a result of suppression of the
central nervous system was the cause of death in these animal studies. Hemolysis occurred and diuresis
was inhibited in rats gavaged with 1,325 mg/kg of methylene chloride (Marzotko and Pankow 1987); in
rats given single oral doses $526 mg/kg, the only endocrine effects noted were dilatation of capillaries of
the adrenal medulla and an increase in the secretion of catecholamines (Marzotko and Pankow 1987). In
rats gavaged twice with 1,275 mg/kg, DNA damage and an increase in the activity of ornithine
decarboxylase were detected in the liver (Kitchin and Brown 1989). Genotoxicity (DNA breakage) was
detected in nuclei from selected rat organs following a single dose of methylene chloride (1,720 mg/kg)
(Sasaki et al. 1998). Since there is a potential for oral exposure for humans near hazardous waste sites
(through contaminated groundwater sources where volatization is restricted), additional animal data on
the effects of acute oral exposure to lower doses of methylene chloride would strengthen the database.
An acute oral MRL was derived by using a PBPK model to extrapolate human inhalation data from
Winneke (1974) to estimate the concentration of methylene chloride in drinking water needed to produce
a tissue specific concentration equivalent to that produced by exposure to 300 ppm of methylene chloride
(Reitz et al. 1997).
Case reports indicate that methylene chloride can cause eye and skin damage following direct contact
with the liquid (Hall and Rumack 1990; Wells and Waldron 1984; Winek et al. 1981). An acute study in
rabbits demonstrated adverse ocular effects (Ballantyne et al. 1976). Dermal absorption of methylene
chloride has been demonstrated in animals (McDougal et al. 1986), and therefore, is possible in humans.
Additional dermal studies in animals are needed to provide further insight into the potential risks of living
near hazardous waste sites.
Intermediate-Duration Exposure. Suggestive evidence for liver effects in humans exists, as
workers exposed to methylene chloride (up to 475 ppm) via inhalation have shown increased serum
bilirubin levels, but not at clinically significant levels (Ott et al. 1983a). Other liver injury parameters
were normal. Repeated exposures to methylene chloride vapors (up to 500 ppm) did not alter serum
enzyme activity, pulmonary function, or urine chemistry significantly in exposed and control groups
(NIOSH 1974). However, irritation of the respiratory tract, but no changes in urinary chemistry were
noted after exposure to a TWA of 5–340 ppm in an occupational study (Anundi et al. 1993).
152 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Available data for rats, dogs, monkeys, rabbits, guinea pigs, gerbils, and mice indicate that repeated
exposure to methylene chloride via inhalation produces central nervous system effects (Briving et al.
1986; Karlsson et al. 1987; Rosengren et al. 1986), respiratory effects (Heppel et al. 1944; NTP 1986)
hepatic effects (Haun et al. 1972; Heppel et al. 1994; Kjellstrand et al. 1986; MacEwen et al. 1972;
Norpoth et al. 1974; NTP 1986; Weinstein and Diamond 1972), and kidney effects (Haun et al. 1972;
MacEwen et al. 1972). An intermediate inhalation MRL was based on a LOAEL of 25 ppm for hepatic
and renal effects in rats (Haun et al. 1972).
No quantitative data are available for oral exposures of intermediate duration in humans. Hepatic damage
and lowered urinary pH in rats was reported following oral exposure $166 mg/kg/day for 3 months
(Kirschman et al. 1986), but the reporting of results was incomplete in this study. Furthermore, since the
lowest dose was a LOAEL, additional intermediate oral studies are needed in animals to evaluate specific
tissue and organ effects at lower doses and to determine threshold levels and dose-response relationships.
Reitz et al. (ATSDR 1997) developed a PBPK model to extrapolate inhalation data for an oral MRL,
based on the inhalation study of Haun et al. (1972), but the reporting of results in this study was
incomplete; a new intermediate oral animal study would be preferable. No intermediate oral MRL was
derived because of lack of adequate data.
There are no quantitative intermediate dermal exposure data available for humans or animals. Given the
potential for human exposure to methylene chloride through paint strippers, adhesives, glues, paint
thinners, wood stain, varnishes, spray paint, and automobile spray primers, information on the effects of
dermal exposures are needed to estimate more accurately the risks to human health from methylene
chloride.
Chronic-Duration Exposure and Cancer. No data are available on the non-neoplastic effects in
humans after chronic exposure to methylene chloride via the oral or dermal routes. Neurological and
reproductive effects have been reported in workers following combined inhalation/dermal exposures for
up to 3 years (Kelly 1988). Studies in animals suggest that the liver is a target organ following chronic
inhalation (Burek et al. 1984; Nitschke et al. 1988a; NTP 1986) and oral (Serota 1986a, 1986b) exposure.
Both a chronic inhalation MRL and a chronic oral MRL were derived using hepatotoxicity as the critical
endpoint (Nitschke et al. 1988a; Serota et al. 1986a). Renal effects were also observed following chronic
inhalation of methylene chloride in rodents (NTP 1986). Dermal data in animals after chronic exposure
153 METHYLENE CHLORIDE
2. HEALTH EFFECTS
to methylene chloride are not available. Additional dermal studies in animals are needed to enhance our
understanding of potential risk to people living near hazardous waste sites.
Epidemiological studies of human occupational cohorts show no increase in cancer of the lung, liver, or
any other organs from occupational inhalation exposures (Friedlander et al. 1978; Hearne et al. 1987,
1990; Ott et al. 1983a). There are no human oral or dermal exposure data for the cancer endpoint.
Inhalation studies in animals show a concentration-dependent, statistically significant increase in liver and
lung adenomas and carcinomas in mice exposed to high concentration of methylene chloride (Mennear et
al. 1988; NTP 1986) and benign mammary gland tumors in rats (Mennear et al. 1988; NTP 1986)
following 2 years of exposure to methylene chloride. The evidence for carcinogenicity in animals from
oral exposures (Serota et al. 1986a, 1986b) is inconclusive, and there are no dermal data available.
Therefore, additional chronic oral and dermal studies are needed to clarify the cancer risk of ingested
methylene chloride. The carcinogenic mechanism of inhaled methylene chloride in animals is not yet
understood despite an extensive database on toxicokinetics and potential mode(s) of action. Additional
information on the mechanisms of carcinogenesis is needed to provide further insight regarding current
findings that (1) the concentration-response may be nonlinear at low exposure concentrations and there is
a concentration range below which carcinogenicity is unlikely to occur and (2) the mouse is not an
appropriate model for investigation of potential human carcinogenicity because it is much more sensitive
than any other species.
Genotoxicity. Genotoxicity data show mixed results. Generally, mutagenesis assays are negative in
mammalian in vivo studies (Burek et al. 1984; Sheldon et al. 1987). Methylene chloride is mutagenic in
bacteria (Gocke et al. 1981), but results for Drosophila (Gocke et al. 1981) are contradictory. This
compound has shown a dose-response relationship for clastogenesis in mammalian cells in vitro (Thilagar
et al. 1984a) but rats exposed in vivo (Burek et al. 1984) have shown no effects. In mice exposed in vivo,
tissue-specific DNA breaks were observed in mice (Sasaki et al. 1998), which suggests that genotoxicity
of methylene chloride may be dependent on the expression of metabolizing enzymes. In view of the
unknown carcinogenic mechanisms, additional in vivo studies of clastogenesis are needed to provide
useful markers, both for dosimetry and specific mechanisms. A recent in vitro test using strains of
Salmonella with or without recombinant GSTT1 has demonstrated that methylene chloride may have
more than one mechanism of genotoxicity (De Marini et al. 1997). In bacteria expressing mammalian
GSTT1, methylene chloride, at moderate doses, caused a single type of genetic lesion. In bacteria without
GSTT1, higher doses were required to induce genetic lesions and the results were more varied. These
154 METHYLENE CHLORIDE
2. HEALTH EFFECTS
results may explain the variability observed in previous studies. Therefore, additional genotoxicity
studies should be carried out on mammalian cells for which the expression of GSTT1 and/or CYP2E1
isoenzymes is defined.
Reproductive Toxicity. Data on reproductive toxicity in humans are limited. One case study reported
genital pain, and low sperm counts (testicular atrophy in some cases) in workers who inhaled vapors of
methylene chloride and had direct contact with the liquid on the job for up to 3 years (Kelly 1988); they
were also exposed to styrene at low levels. Exposure to methylene chloride was confirmed by blood
COHb levels. In another study, workers exposed to methylene chloride for a shorter time showed no
reproductive effects (Wells et al. 1989b). A case-control occupational study reported a nearly significant
association of methylene chloride exposure and the incidence of spontaneous abortion (Taskinen et al.
1986). There are no oral or dermal exposure data that pertain to reproductive effects in humans. A two-
generation inhalation study in rats reported no effects on fertility and litter size (Nitschke et al. 1988b). In
addition, no effects on the testes were observed in dominant lethal tests involving male mice that inhaled
concentrations of methylene chloride up to 200 ppm for up to 6 weeks (Raje et al. 1988). Uterine,
ovarian, and testicular atrophy were observed in rodents following chronic exposure to methylene
chloride at 4,000 ppm (NTP 1986). There are no data on reproductive effects in animals following oral or
dermal exposure. Intermediate-duration oral and dermal studies that incorporate histopathological
analysis of the reproductive organs are needed to address this data need. Part of the analysis should
include a determination of whether the reproductive organs express GSTT1 and/or CYP2E1, as a first step
in evaluating their possible role in the reproductive toxicity of methylene chloride.
Developmental Toxicity. No data are available on developmental toxicity of methylene chloride
following inhalation, oral, or dermal exposure in humans, aside from the case-control study relating
exposure and the rate of spontaneous abortion mentioned above (Taskinen et al. 1986). Exposure to
0.01 ppm of methylene chloride had no effect on birth outcome in a study of over 90,000 pregnancies
(Bell et al. 1991). Several inhalation studies in animals indicate that methylene chloride can cross the
placenta (Anders and Sunram 1982). Some of these studies showed statistically nonsignificant
malformations in rats and mice or decreased fetal weight at maternally toxic concentrations (Bornschein
et al. 1980; Hardin and Manson 1980; Schwetz et al. 1975). However, there was a statistically significant
increase in the incidence of delayed ossification of sternebrae (Schwetz et al. 1975). The use of only one
concentration in these studies precludes any evaluation of concentration-response relationships.
155 METHYLENE CHLORIDE
2. HEALTH EFFECTS
However, Reitz et al. (1997) developed an inhalation route-to-oral route extrapolation and rodent-to-
human species extrapolation using PBPK modeling of the developmental toxicity data in Schwetz et al.
(1975). The resulting LOAEL was an intermediate oral dose of 142 mg/kg/day. Additional studies for
inhalation and dermal exposures in two species would be useful in clarifying the developmental toxicity
potential of this chemical. Conducting studies on animals with known genotypes with respect to
metabolizing enzymes GSTT1 and CYP2E1 are needed to evaluate the risk of exposure to methylene
chloride.
Immunotoxicity. No human data are available on immunotoxicity of methylene chloride. Animal data
are limited on the immunotoxicity of methylene chloride. One study was located that indicated that
methylene chloride caused splenic atrophy in dogs following continuous intermediate-duration inhalation
exposure (MacEwen et al. 1972). Splenic fibrosis was observed in a chronic rat study (Mennear et al.
1988), but occurred only at the highest concentration tested (1,000 ppm) and only in one sex. A recent
intermediate-duration study, also in rats, found no evidence of gross or microscopical alterations in the
spleen at a concentration of 5,187 ppm methylene chloride (Halogenated Solvent Industry Alliance, Inc.
(2000). This study also found no effect on IgM response to SRBC. Additional immunotoxicity studies
including evaluation of humoral- and cell-mediated immunity are needed to determine whether this
system is susceptible to methylene chloride, as some chlorinated hydrocarbons do affect the immune
system.
Neurotoxicity. The central nervous system is a target for both short- and long-term inhalation
exposures in humans. These data are derived from experimental (Fodor and Winneke 1971; Putz et al.
1979; Stewart et al. 1972; Winneke 1974;) and occupational studies (Lash et al.1991; White et al. 1995)
that reported alterations in behavioral performance and various psychomotor tasks following exposure to
methylene chloride. Other neurotoxic effects noted in occupational studies included dizziness, headaches,
nausea, memory loss, paresthesia, tingling in hands and feet, and loss of consciousness (Bakinson and
Jones 1985; Hall and Rumack 1990; Kelly 1988). It should be noted that some of these workers were
exposed to other unspecified solvents at the same time. Winneke (1974) attributed the neurological
effects in volunteers following a 3–4 hour inhalation exposure to 300–800 ppm of methylene chloride to
the anaesthetic properties of the parent compound since exposures to 50–100 ppm of carbon monoxide
alone did not produce these effects. The specific parameters in the Winneke (1974) study, critical flicker
fusion frequency, auditory vigilance, and other psychomotor tasks, were considered to be specific
measures of ‘cortical alertness’ or central nervous system depression. Putz et al. (1979) correlated
156 METHYLENE CHLORIDE
2. HEALTH EFFECTS
neurological deficits following inhalation exposure to methylene chloride (200 ppm) with the
accumulation of COHb in the blood and carbon monoxide in the exhaled breath; performance did not
deteriorate until 3 hours of exposure had elapsed and blood COHb levels exceeded 5%. Although the
Putz et al. (1979) study seems to implicate the metabolite CO as the neurotoxic agent, there is a need for
studies to determine the relative contributions of parent compound and metabolite to the neurotoxic
effects of methylene chloride. The Winneke (1974) study, which was deemed to have used more specific
measures of central nervous system depression than the Putz et al. (1979) study, was used to derive an
acute inhalation MRL and , by route-to-route extrapolation, also to derive an acute oral MRL (Reitz et al.
1997).
Subtle neurological effects were reported in rats after acute exposure to methylene chloride. Changes in
somatosensory-evoked potentials were observed after 1 hour exposure to 5,000 ppm of methylene
chloride (Rebert et al. 1989), and changes in cerebellar enzymes were detected following 2 weeks of
exposure at 500 ppm (Savolainen et al. 1981). Inhalation studies in rats did not reveal neurobehavioral
effects after intermediate-duration exposure at 4,500 ppm of methylene chloride vapors (Bornschein et al.
1980). In other studies, neurochemical changes were reported in gerbils at 210 ppm (Briving et al. 1986;
Karlsson et al. 1987; Rosengren et al. 1986). There were no treatment-related neurophysiological or
neuropathological effects in rats exposed to concentrations of 2,000 ppm for 13 weeks (Mattsson et al.
1990).
There are no oral or dermal studies in either humans or animals with regard to neurotoxicity of methylene
chloride. Data on electrophysiology, functional observational batteries, and neuropathology from 90-day
oral or dermal exposures in at least two different species would be useful to further evaluate neurotoxic
effects from these routes of exposure, since human exposure can occur from all three routes at hazardous
waste sites.
Epidemiological and Human Dosimetry Studies. Information on the effects of methylene
chloride in humans comes from occupational studies, case reports of acute high exposure, and studies
with volunteers. Asphyxia and eventually death occurred in a subject acutely exposure to a high but
undetermined concentration of methylene chloride in the air (Winek et al. 1981). Exposure to lower
concentrations affects primarily the central nervous system; signs and symptoms observed include
dizziness, incoordination, loss of balance, unconsciousness, and decreased performance in tests of sensory
and motor functions (Bakinson and Jones 1985; Fodor and Winneke 1971; Hall and Rumack 1990; Putz
157 METHYLENE CHLORIDE
2. HEALTH EFFECTS
et al. 1979; Stewart et al. 1972; Winneke 1974). These effects are likely to be caused by a combination of
the anaesthetic properties of the parent compound and accumulation of COHb which forms as a result of
methylene chloride metabolism. There is a potential for low-level general population exposure from
ambient methylene chloride emissions from paint removal, aerosol use, metal degreasing, electronics and
pharmaceutical manufacturing, and food processing (Callahan 1981; CPSC 1987, 1990; NAS 1978).
Such exposure levels are unlikely to produce adverse neurological effects. Hazardous waste sites release
methylene chloride into the air, groundwater, surface water, and soil. There are very few monitoring data
for methylene chloride outside the occupational setting (Singh et al. 1981). Because methylene chloride
evaporates readily from water and soil, inhalation is the main route of potential exposure. In the unlikely
event that long-term exposure of the general population (in the past or present) to low levels of primarily
methylene chloride is identified, individuals should be monitored for hematological and hepatic effects, as
these have been observed in studies in animals.
Biomarkers of Exposure and Effect.
Exposure. The presence of methylene chloride in the postexposure expired breath is the most commonly
used biomarker of exposure. Methylene chloride is easily detected in expired air for 24 hours following a
vapor exposure (NIOSH 1974). Methylene chloride can be detected in blood, but because clearance is so
rapid, this method is only useful for monitoring recent exposures (DiVincenzo and Kaplan 1980). The
half-life of COHb following methylene chloride exposure is twice that following exposure to carbon
monoxide (NIOSH 1974; Stewart and Hake 1976). COHb in blood may be monitored; however, COHb
is cleared so rapidly (plasma half-life of 40 minutes) that it is unlikely to be useful for monitoring
environmental exposures (DiVincenzo et al. 1972). Urinary excretion of methylene chloride is
measurable for several hours postexposure (DiVincenzo et al. 1972).
Effect. Changes in the nervous system are sensitive, but not specific, biomarkers of methylene chloride
effects. Clinical signs and symptoms which may be monitored include narcosis, fatigue, and analgesia
(Bakinson and Jones 1985; Hall and Rumack 1990; Putz et al. 1979; Winneke 1974). Impairment of
neurobehavioral functions and electromyographic measurements of nerve conduction velocity and
amplitude can be monitored to detect early signs of neurotoxicity in people exposed to methylene chloride
(Glatzel et al. 1987; White et al. 1995). Additional studies that couple measurement of methylene
chloride with tests for determining nervous system effects are needed to correlate exposure with adverse
health effects of this chemical.
158 METHYLENE CHLORIDE
2. HEALTH EFFECTS
While neurobehavioral tests are not specific for methylene chloride-induced toxicity, they do identify
potential health impairment. Studies to develop more specific biomarkers of methylene chloride-induced
effects are needed to assess the potential health risk of methylene chloride exposure near hazardous waste
sites.
Absorption, Distribution, Metabolism, and Excretion. Methylene chloride is a volatile liquid
with high lipid solubility and modest solubility in water. In humans, it is rapidly absorbed by the
inhalation (DiVincenzo and Kaplan 1981; McKenna et al. 1980) and ingestion routes of exposure
(Roberts and Marshall 1976). Dermal absorption is suspected to occur in humans, but quantitative data
are lacking. Methylene chloride is also readily absorbed by animals following inhalation (DeVincenzo et
al. 1972; MacEwen et al. 1972; McKenna et al. 1982) and oral exposure (Angelo et al. 1986a); dermal
absorption data in animals are also lacking. Once absorbed, methylene chloride is quickly distributed to a
wide range of tissues and body fluids. Absorption and distribution of methylene chloride can be affected
by a number of factors, including dose level, vehicle, physical activity, duration of exposure, and amount
of body fat (Astrand et al. 1975; DiVincenzo et al. 1972; Engstrom and Bjorstrom 1977).
Distribution data in humans are lacking, but it has been found in human breast milk (EPA 1980e;
Pellizzari et al. 1982). Methylene chloride is widely distributed in animal tissues after inhalation
exposure (Carlsson and Hultengren 1975; McKenna et al. 1982). The highest concentrations are found in
adipose tissue and liver. Methylene chloride has been found in blood from rats’ fetuses (Anders and
Sunram 1982). After acute exposure, methylene chloride disappears rapidly from fat (Carlsson and
Hultengren 1975). No studies were located regarding distribution of methylene chloride following
dermal exposure in animals. Distribution of methylene chloride does not seem to be route-dependent and
it does not bioaccumulate in tissues.
Methylene chloride is metabolized via two pathways, the MFO pathway, which produces CO and CO
2
,
and the GST pathway which yields only CO
2
(Gargas et al. 1986). Methylene chloride is metabolized
almost exclusively by the MFO pathway at low exposures. The only data available for humans on the
toxicokinetics parameters are for inhalation exposures. These data show that the GST activity is low in
humans relative to other animal species (Andersen et al. 1987). It has been postulated that the activity of
the GST pathway in rats is less than that of mice and might only become significant when the P-450
pathway (MFO) has been saturated (Green et al. 1986b, 1986c). Toxicokinetic data for oral and dermal
exposures would be useful because some exposures to humans are expected to be from contaminated
159 METHYLENE CHLORIDE
2. HEALTH EFFECTS
drinking water and soils. Toxicokinetic models (Andersen and Krishnan 1994; Andersen et al. 1987,
1991; Dankovic and Bailer 1994; Ramsey and Andersen 1984; Reitz 1990) have been developed that
account for the nonlinearities in the internal dose across exposure levels that arise from concentration-
dependent changes in absorption, distribution, excretion, and saturation of metabolism following
inhalation exposure to methylene chloride. Reitz et al. (1997) have developed an inhalation route-to-oral
route extrapolation for methylene chloride. The metabolism of methylene chloride in humans appears to
be qualitatively similar to that in animals (Fodor et al. 1973; Wirkner et al. 1997).
Methylene chloride is excreted via expired air primarily as CO and CO
2
at low concentrations (McKenna
et al. 1982). As concentration increases, more unchanged parent compound is exhaled. A small fraction
of absorbed methylene chloride or metabolites has been detected in urine of humans occupationally
exposed (DiVincenzo et al. 1972) and in the urine and feces of experimental animals (McKenna at al.
1982). Additional data on excretion parameters as functions of concentration are needed to understand
the toxicokinetics of methylene chloride.
Comparative Toxicokinetics. Generally, animal data on toxicokinetics parameters substantiate
those from humans. There is rapid absorption through the lung (DiVincenzo and Kaplan 1981;
DiVincenzo et al. 1972; MacEwen et al. 1972; McKenna et al. 1982). The metabolites of methylene
chloride in different species indicate that they are metabolized by the same pathways to CO or to CO
2
(Fodor et al. 1973; Gargas et al. 1986; Wirnker et al. 1997). The data from both human and animal
studies indicate that the target organs of methylene chloride toxicity are the same across species.
Interesting differences do exist, however, in some metabolic parameters. The rate of clearance from rat
tissues was markedly slower than in the mouse following inhalation exposures. In contrast to findings of
a direct proportional relationship between inspired air concentration of methylene chloride and blood
level in man and other animals, there is a report that the ratio at steady state between blood levels of
methylene chloride and exposure increases as the concentration increases in rats (Green et al. 1986b,
1986c). Distribution of methylene chloride across tissues in rats and humans has been shown to be
lipophilic except for one rat study that showed less of the chemical in adipose tissue (McKenna and
Zempel 1981). Species differences in placental anatomy and physiology may contribute to variations in
fetal exposure to methylene chloride or its metabolites following maternal exposure. Additional data on
the impact of differences in species sensitivity to specific internal doses by different routes of exposure
are needed to resolve questions about extrapolation of predicted effects from different routes in different
species.
160 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Methods for Reducing Toxic Effects. There is no specific treatment or antidote for methylene
chloride intoxication. Available methods and treatments (e.g., gastric lavage with the airway protected, or
the use of oxygen as in carbon monoxide poisoning) for reducing peak absorption have been shown to be
beneficial (Shih 1998; Tomaszewski 1998). There is no evidence that activated charcoal or cathartics are
effective (Shih 1998). Because methylene chloride is metabolized to carbon monoxide, mitigation
strategies (e.g., hyperbaric oxygen) used for carbon monoxide intoxication have been effective in
increasing the oxygen carrying capacity of the blood (Tomaszewski 1998).
Children’s Susceptibility. Data needs relating to both prenatal and childhood exposures, and
developmental effects expressed either prenatally or during childhood, are discussed in detail in the
Developmental Toxicity subsection above.
There are no studies that specifically addressed effects of exposure to methylene chloride in children.
There is no evidence from human toxicity studies that children are, or are likely to be, more susceptible to
health effects of exposure to methylene chloride than adults. No cases of accidental poisoning in children
due to methylene chloride exposure have been reported. The nervous system is a sensitive target for acute
exposure to methylene chloride in adults, and the response in children is likely to be similar.
Although developmental studies in animals indicate that methylene chloride is not a teratogen
(Bornschein et al. 1980; Hardin and Manson1980), there is a need to evaluate neurological/neuro-
behavioral effects in animals exposed in utero. Subtle neurological effects could result from hypoxia
(CO-mediated) or from reactive intermediates of metabolism that would only be revealed by appropriate
behavioral testing of the offspring. It is not clear that the "wheel running activity" and"avoidance
learning" tests that Bornschein et al. (1980) employed in rats exposed in utero were adequate to reveal
neurological deficits. Acute effects observed in an adult human study involved degraded performance on
visual and auditory discrimination tasks (Putz et al.1979). If neurological effects were detected in mice, it
would be useful to conduct additional developmental studies using mice in which functional genes for
GSTT1 and/or CYP2E1 have been knocked-out, to discover which metabolic pathway is implicated in
developmental neurological effects.
There is no direct evidence regarding pharmacokinetics or the mechanism of toxicity of methylene
chloride in children. However, considering what is known about the expression of metabolizing enzymes
(see Section 2.7 Children’s Susceptibility), there is no reason to expect that children over the age of
161 METHYLENE CHLORIDE
2. HEALTH EFFECTS
6 months will be qualitatively different from adults; by 6 months, the last of the fetal hemoglobin has
been replaced by the adult isoforms. At fetal and early postnatal stages, however, children will be more
vulnerable to methylene-chloride-generated CO because of its effect on fetal hemoglobin (Longo 1977).
CO binding to Hb causes oxygen to be held more tightly, so that tissues receive less oxygen; the net effect
is that the CO clears more slowly from the circulation. It is not known to what extent methylene chloride
(or metabolites other than CO or CO
2
) can cross the placenta in humans or accumulate in breast milk. A
study in animals showed that methylene chloride crosses the placental barrier (Anders and Sunram 1982),
but quantitative data are lacking. There are no animal studies testing whether methylene chloride can
pass into breast milk. There are no studies quantifying maternal exposure and output into breast milk.
The Fisher et al. (1997) PBPK model predicts a relatively low transfer into breast milk following maternal
inhalation exposure.
In adults, the biomarker of exposure to methylene chloride is the parent compound, and the biomarkers of
effect are CO in the exhaled breath or COHb in the blood. There is no reason to expect that biomarkers in
children would be any different.
It is expected that any chemical interactions involving methylene chloride would have the same effects in
children as in adults. The exception would be the fetal period, during which concentrations of
metabolizing enzymes are significantly lower than in the adult. A fetus exposed to methylene chloride
and another chemical that requires CYP2E1 for metabolism, might be at higher risk than an adult.
Treatment with 100% oxygen has been used to enhance the rate of clearance of CO from the bloodstream
after acute methylene chloride exposure. Although there is no specific information regarding pediatric
treatments for methylene chloride exposure, 100% oxygen has been used for treating pregnant women
exposed to CO (Longo 1977). In these cases, it is recommended that the oxygen treatment be extended
3.5–5 times longer than the time required to bring the maternal COHb level down to 3%, to ensure that
the fetus is adequately treated. If an exposed mother has neurological symptoms or a COHb level above
15%, a single hyperbaric oxygen treatment is indicated (Tomaszewski 1998). It does not appear that any
pediatric-specific methods need to be developed.
Since methylene chloride may be genotoxic, depending on the genotype with respect to GSTT1 and
CYP2E1 (see Section 2.10), it is possible that parental exposure could affect the fetus. At issue would be
whether the gonads express alleles that are associated with genotoxicity, since the effect is intracellular.
162 METHYLENE CHLORIDE
2. HEALTH EFFECTS
Child health data needs relating to exposure are discussed in 5.8.1 Identification of Data Needs:
Exposures of Children.
2.12.3 Ongoing Studies
A number of research projects are in progress investigating the health effects and mechanism of action of
methylene chloride. These projects have been identified from FEDRIP (1999).
Dr. J.B. Wheeler, at Vanderbilt University, Nashville, Tennessee, is comparing the ability of rat and
human glutathione S-transferases (GST) to form mutagenic DNA adducts with glutathione in the presence
of different haloalkanes. He will examine the mechanism of activation by GST, the stability of the
glutathione conjugates, and the DNA adducts produced. This research is sponsored by the National
Cancer Institute.
Dr. T.R. Devereux, at the NIEHS, is identifying critical target genes and genetic alterations that may be
important in chemical carcinogenesis. He is characterizing genetic alterations in oncogenes (e.g., ras) and
tumor suppressor genes (e.g., p53 and p16) from rodent tumors and human cancers generated by exposure
to chemical agents. He is also examining polymorphisms in the human p16 gene that may predispose to
lung cancer.
Dr. D.A. Bell, of the Genetic Risk Group at the NIEHS, is conducting case-control studies of
5,000 genotyped individuals to test the impact of cancer susceptibility genes on cancer of the bladder,
lung, liver, colon, stomach, prostate, and breast. He and his colleagues have identified ethnic differences
in the frequency of at-risk genotypes for glutathione transferase (M1, theta 1, and Pi), and
N-acetyltransferase (1 and 2).
163 METHYLENE CHLORIDE
3. CHEMICAL AND PHYSICAL INFORMATION
3.1 CHEMICAL IDENTITY
Information regarding the chemical identity of methylene chloride is located in Table 3-1. Methylene
chloride is a halogenated hydrocarbon. It is also commonly known as dichloromethane.
Table 3-1 lists common synonyms, trade names, and other pertinent identification information for
methylene chloride.
3.2 PHYSICAL AND CHEMICAL PROPERTIES
Information regarding the physical and chemical properties of methylene chloride is located in Table 3-2.
Methylene chloride is a colorless liquid with a sweet, pleasant odor.

164 METHYLENE CHLORIDE
3. CHEMICAL AND PHYSICAL INFORMATION
Table 3-1. Chemical Identity of Methylene Chloride
Characteristic Information Reference
Chemical Name
Synonyms
Registered trade name(s)
Chemical formula
Chemical structure
Identification numbers:
CAS
NIOSH RTECS
EPA hazardous waste
OHM/TADS
DOT/UN/NA/IMCO shipping
HSDB
NCI
Methylene chloride
Dichloromethane
Narkotil; Solaesthin; Solmethine;
and others
CH
2
Cl
2
Cl
H H
Cl
75-09-2
PA8050000
U080, F002
7217234
UN1593, IMCO 6.1
66
C50102
Lide 1994
Lide 1994
OHM/TADS 1998
Lide 1994
Lide 1994
Lide 1994
RTECS 1999
Lewis 1996
OHM/TADS 1998
HSDB 1999
HSDB 1999
HSDB 1999
CAS = Chemical Abstracts Services; DOT/UN/NA/IMCO = Department of Transportation/United Nations/North
America/International Maritime Dangerous Goods Code; EPA = Environmental Protection Agency;
HSDB = Hazardous Substances Data Bank; NCI = National Cancer Institute; NIOSH = National Institute for
Occupational Safety and Health; OHM/TADS = Oil and Hazardous Materials/Technical Assistance Data System;
RTECS = Registry of Toxic Effects of Chemical Substances

165 METHYLENE CHLORIDE
3. CHEMICAL AND PHYSICAL INFORMATION
Table 3-2. Physical and Chemical Properties of Methylene Chloride
Property Information Reference
Molecular weight 84.93 Lide 1994
Color Colorless Lewis 1996
Physical state Liquid Lide 1994
Melting point -95.1 EC Weast 1985
Boiling point 40 EC Lide 1994
Density:
at 25 EC 1.3182 g/mL Lide 1994
Vapor density 2.93 (Air =1) Verscheuren 1983
Odor Sweet, pleasant Verschueren 1983
Odor threshold:
Water 9.1 ppm Amoore and Hautala 1983
Air 540–2,160 mg/m
3
Ruth 1986
(160–620 ppm)
Solubility:
Water at 20 EC 20,000 mg/L Verschueren 1983
at 25 EC 16,700 mg/L Verschueren 1983
Organic solvent(s) Soluble in alcohol, ether, Lewis 1996
acetone, chloroform, and carbon
tetrachloride
Partition coefficients:
Log K
ow
1.3 Hansch and Leo 1979
Log K
oc
1.4 Roy and Griffin 1982
Vapor pressure:
at 20 EC 349 mmHg Verscheuren 1983
at 30 EC 500 mmHg Verscheuren 1983
Henry’s law constant 2.03x10
-3
atm-m
3
/mol at 25 EC EPA 1982e
Autoignition temperature 1,139 EF (615 EC) Lewis 1996
Flashpoint Nonflammable Sax and Lewis 1987
Flammability limits Nonflammable Sax and Lewis 1987
Coversion factors 1 mg/m
3
=0.28 ppm WHO 1996
1 ppm=3.53 mg/m
3
WHO 1996
Explosive limits Not explosive Sax and Lewis 1987
167 METHYLENE CHLORIDE
4. PRODUCTION, IMPORT/EXPORT, USE, AND DISPOSAL
4.1 PRODUCTION
Table 4-1 lists the facilities in each state that manufacture or process methylene chloride, the intended
use, and the range of maximum amounts of methylene chloride that are stored on site. There are currently
461 facilities that reported producing or processing methylene chloride in the United States. The data
listed in Table 4-1 are derived from the Toxics Release Inventory (TRI98 2000). Only certain types of
facilities were required to report (461 facilities reported this type of information out of a total of 714
facilities that reported methylene chloride releases to the environment). Therefore, this is not an
exhaustive list.
Methylene chloride is produced by the chlorination of methane with chlorine or by the chlorination of
methanol with hydrogen chloride followed by chlorination of methyl chloride (Mannsville Chemical
Products Corporation 1988). Production of methylene chloride grew steadily through the 1970s and early
1980s at about 3% each year, with a peak production of about 620 million pounds in 1984. By 1988,
methylene chloride production volume had dropped due to declining demand to about 500 million pounds
(Mannsville Chemical Products Corporation 1988; USITC 1989). This decline in the demand for
methylene chloride was expected to continue at a rate of about 1–2% per year through 1993, as more
manufacturers move toward water-based aerosol systems in anticipation of further regulation of
methylene chloride (HSDB 1999; NTP 1989). In 1994, the latest year for which data are available,
403 million pounds of methylene chloride were produced (C&EN 1996). As of October 1, 1996, the
International Trade Commission ceased to collect or publish annual synthetic organic chemicals data.
The National Petroleum Refiners Association, which currently collects such data, does not include
methylene chloride on its list of organic chemicals.
The Toxics Release Inventory (TRI98 2000) reports that methylene chloride is produced at 23 facilities in
14 states. Table 4-1 summarizes information on the U.S. companies that reported the manufacture and
use of methylene chloride in 1998 (TRI98 2000). The TRI data should be used with caution since only
certain types of facilities are required to report. This is not an exhaustive list.
The Stanford Research Institute (SRI 1999) reports methylene chloride production by two companies at
four U.S. facilities with a total manufacturing capacity of 545 million pounds. These facilities are Dow

168 METHYLENE CHLORIDE
4. PRODUCTION, IMPORT/EXPORT, USE, AND DISPOSAL
Table 4-1. Facilities that Manufacture or Process Methylene Chloride
Range of maximum amounts
on site in thousands of
State
a
Number of facilities pounds
b
Activities and uses
c
reporting
AL 12 100-999999 1,4,7,8,10,11,12,13
AR 7 1000-999999 2,3,7,10,11,12,13
AZ 5 100-999999 8,10,11,12,13
CA 23 0-9999999 2,3,4,8,9,10,11,12,13
CO 4 1000-999999 8,10,11,13
CT 8 1000-99999 2,3,10,11,12,13
DE 2 10000-99999 8,11
FL 12 100-999999 8,10,11,12,13
GA 18 0-999999 2,3,8,9,10,11,12,13
IA 10 100-999999 8,10,11,12,13
IL 23 0-9999999 1,2,3,5,7,8,9,10,11,12,13
IN 21 100-9999999 1,2,3,5,8,9,10,10,11,12,13
KS 8 100-999999 1,2,3,4,8,10,11,12,13
KY 9 100-99999 1,5,7,8,10,11,13
LA 8 1000-9999999 1,4,5,6,7,8,10,12,13
MA 10 1000-999999 2,3,8,9,10,11,12,13
MD 5 1000-99999 8,10,12,13
ME 1 1000-9999 13
MI 17 100-999999 1,2,3,4,5,7,8,10,11,12,13
MN 8 100-99999 2,3,8,11,12,13
MO 13 1000-999999 4,8,10,11,12,13
MS 11 100-999999 1,2,3,8,11,12,13
MT 1 1000-9999 12
NC 17 100-999999 1,2,4,5,7,8,10,11,12,13

169 METHYLENE CHLORIDE
4. PRODUCTION, IMPORT/EXPORT, USE, AND DISPOSAL
Table 4-1. Facilities that Manufacture or Process
Methylene Chloride (continued)
Range of maximum amounts
on site in thousands of
State
a
Number of facilities pounds
b
Activities and uses
c
NE 3 1000-999999 11,13
NH 3 1000-99999 8,12,13
NJ 19 0-999999 1,2,3,4,7,8,9,10,11,12,13
NM 2 1000-99999 8,12
NV 3 100-9999 1,2,3,8,11,13
NY 14 1000-9999999 2,3,4,7,8,9,10,11,12,13
OH 17 100-9999999 2,3,7,8,10,11,12,13
OK 9 100-999999 8,10,11,13
OR 6 1000-999999 2,4,8,10,11,12,13
PA 17 1000-999999 8,9,10,11,12,13
PR 12 1000-9999999 1,2,5,6,8,9,10,11,12,13
RI 2 10000-99999 10,13
SC 14 100-999999 7,8,10,11,12,13
TN 15 100-9999999 8,9,10,11,12,13
TX 27 0-49999999 1,2,3,4,5,6,7,8,9,10,11,12,
13
UT 6 1000-999999 8,10,11,12,13
VA 10 1000-999999 8,10,11,12,13
WA 10 0-99999 1,6,7,10,11,12,13
WI 14 1000-999999 1,4,5,8,10,11,12,13
WV 5 0-9999999 8,11,13
TRI98 (2000 )
a
Post office state abbreviations
b
Data in TRI are maximum amounts on site at each facility
c
Activities/Uses:
1. Produce
2. Import
3. For on-site use/processing
4. For sale/distribution
5. As a byproduct
6. As an impurity
7. As a reactant
8. As a formulation component
9. As an article component
10. For repackaging only
11. As a chemical processing aid
12. As a manufacturing aid
13. Ancillary or other use
170 METHYLENE CHLORIDE
4. PRODUCTION, IMPORT/EXPORT, USE, AND DISPOSAL
Chemical Company in Freeport, Texas and Plaquemine, Louisiana; and Vulcan Materials Company in
Geismar, Louisiana and Wichita, Kansas.
4.2 IMPORT/EXPORT
Imports of methylene chloride had been decreasing from 60 million pounds in 1985 to 25–27 million
pounds in 1988–1989 (Mannsville Chemical Products Corporation 1988; NTP 1989). During 1992, the
import of methylene chloride had dropped to about 15.5 million pounds, but has since increased to
30 million pounds in 1996 (NTDB 1998). Methylene chloride exports have ranged from about
60–130 million pounds over the period of 1975–1988 (Mannsville Chemical Products Corporation 1988)
and ranged from about 132 to 145 million pounds over the period of 1992–1996 (NTDB 1998).
4.3 USE
Methylene chloride is used as a solvent in paint strippers and removers (25%), as a propellant in aerosols
(25%), as a process solvent in the manufacture of drugs, pharmaceuticals, and film coatings (20%), as a
metal cleaning and finishing solvent (10%), in electronics manufacturing (10%), and as an agent in
urethane foam blowing (10%) (NTP 1989). Aerosol products in which methylene chloride may be found
include paints, automotive products, and insect sprays. However, because of labeling regulations and
concerns over health and environmental issues, the use of methylene chloride in consumer aerosol
products has declined (Mannsville Chemical Products Corporation 1988). Methylene chloride was once
used in hair sprays but this use was banned in 1989 (FDA 1989).
Methylene chloride is also used as an extraction solvent for spice oleoresins, hops, and for the removal of
caffeine from coffee. These uses of methylene chloride are approved by the Food and Drug
Administration (see Chapter 7). However, it has been reported that because of concern over residual
solvent, most decaffeinators no longer use methylene chloride (Mannsville Chemical Products
Corporation 1988). Methylene chloride is also approved for use as a post-harvest fumigant for grains and
strawberries and as a degreening agent for citrus fruit (Hearne et al. 1990; Mannsville Chemical Products
Corporation 1988; Meister 1989; NTP 1989).
171 METHYLENE CHLORIDE
4. PRODUCTION, IMPORT/EXPORT, USE, AND DISPOSAL
4.4 DISPOSAL
Methylene chloride is listed as a toxic substance under Section 313 of the Emergency Planning and
Community Right to Know Act (EPCRA) under Title III of the Superfund Amendments and
Reauthorization Act (SARA) (EPA 1995). Disposal of wastes containing methylene chloride is
controlled by a number of federal regulations (see Chapter 7).
Methylene chloride and wastes containing methylene chloride are considered hazardous wastes and as
such, are subject to handling, transport, treatment, storage, and disposal requirements as mandated by law
(see Chapter 7) (IRPTC 1990; NTP 1989).
Methylene chloride wastes may be disposed of by controlled incineration (liquid injection, rotary kiln, or
fluidized bed) at the appropriate temperatures. No information was found regarding the amount of
methylene chloride disposed of by each incineration method. The percent removal of methylene chloride
by incineration is mandated by law depending upon the quantity of methylene chloride in the waste
(HSDB 1999; IRPTC 1990).
According to the Toxics Release Inventory (TRI98 2000), about 0.37 million pounds of methylene
chloride were transferred to landfills and/or other treatment/disposal facilities including publicly-owned
treatment works in 1998 (see Section 5.2). No information was found on disposal method trends or past
disposal practices.
173 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
5.1 OVERVIEW
Methylene chloride has been identified in at least 882 of the 1,569 hazardous wastes sites that have been
proposed for inclusion on the EPA National Priorities List (NPL) (HazDat 1999). However, the number
of sites evaluated for methylene chloride is not known. The frequency of these sites can be seen in
Figure 5-1. Of these sites, 882 are located in the United States, and none are located in the
Commonwealth of Puerto Rico (not shown).
Methylene chloride is a widely used industrial chemical with reported emissions to air of more than
40 million pounds annually in the United States. Most of the methylene chloride released to the
environment will partition to the atmosphere, where it will degrade with a lifetime of 6 months by
reaction with photochemically produced hydroxyl radicals (WHO 1996). The compound is expected to
be highly mobile in soil and to volatilize rapidly from surface water to the atmosphere. Biodegradation
may be important, but bioconcentration does not appear to be significant.
The principal route of exposure for the general population to methylene chloride is inhalation of ambient
air. Average daily intake of methylene chloride from urban air has been estimated to range from about
33 to 309 µg. Occupational and consumer exposure to methylene chloride in indoor air may be much
higher, especially from spray painting or other aerosol uses.
5.2 RELEASES TO THE ENVIRONMENT
According to the Toxics Release Inventory (TRI), in 1998, a total of about 41 million pounds
(18.6 million kg) of methylene chloride was released to the environment from 714 facilities (714 facilities
reported environmental releases of methylene chloride to TRI, another 113 facilities reported no releases
at all for a total of 827 facilities) (TRI98 2000). This total release volume is approximately 10% of the
total reported for 1997 (TRI97 1999). Table 5-1 lists amounts released from these facilities. In addition,
an estimated 0.37 million pounds (0.17 million kg) were released by manufacturing and processing
facilities to publicly owned treatment works (POTWs) or other off-site facilities (TRI98 2000). The TRI
data should be used with caution because only certain types of facilities are required to report. This is not
an exhaustive list.

174
Table 5-1. Releases to the Environment from Facilities that Manufacture or Process Methylene Chloride
Total reported amounts released in pounds per year
a
Number Total
State
b
of
facilities Air
c
Water Land
Underground
injection
Total
on-site release
d
off-site
release
e
Total on and
Off-site release
AL 14 948907 0 0 0 948907 470 949377
AZ 6 65837 0 0 0 65837 0 65837
AR 12 125869 12 0 360837 486718 269 486987
CA 44 986871 3 0 0 986874 9768 996642
CO 6 37077 0 0 0 37077 0 37077
CT 31 784114 755 3 0 784872 7450 792322
DE 2 4100 0 0 0 4100 0 4100
FL 16 932255 0 0 0 932255 759 933014
GA 31 973595 1160 169543 0 1144298 14935 1159233
IL 45 1745258 57 0 0 1745315 3295 1748610
IN 47 4630842 639 5 0 4631486 1152 4632638
IA 13 1156800 0 5 0 1156805 500 1157305
KS 12 288373 0 0 41323 329696 1799 331495
KY 15 2157130
57 0 0 2157187 11435 2168622
LA 11 41136 1020 0 9954 52110 2236 54346
ME 1 3394 0 0 0 3394 0 3394
MD 5 118361 0 0 0 118361 0 118361
METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE

175
Table 5-1. Releases to the Environment from Facilities that Manufacture or Process Methylene Chloride
(continued)
Total reported amounts released in pounds per year
a
Number
State
b
of
facilities Air
c
Water Land
Underground
injection
Total on-site
release
d
Total off-site
release
e
Total on and
Off-site release
MA 29 139895 0 0 0 139895 4772 144667
MI 32 422374 177 0 54800 477351 1954 479305
MN 17 183564 290 0 0 183854 0 183854
MS 15 3355695 0 0 0 3355695 900 3356595
MO 26 302907 110 5 0 303022 0 303022
MT 1 42300 0 0 0 42300 0 42300
NE 3 41152 0 0 0 41152 2085 43237
NV 2 6834 0 0 0 6834 0 6834
NH 3 38696 0 0 0 38696 40 38736
NJ 35 243166 548 3 0 243717 5977 249694
NM 1 105 0 0 0 105 0 105
NY 24 2038566 4305 0 0 2042871 29091 2071962
NC 29 2599415 4271 450 0 2604136 750 2604886
OH 54 1864209 257
0 0 1864466 21070 1885536
OK 12 250064 0 0 0 250064 32 250096
OR 7 79509 0 0 0 79509 2987 82496
METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE

176
Table 5-1. Releases to the Environment from Facilities that Manufacture or Process Methylene Chloride
(continued)
Total reported amounts released in pounds per year
a
Number
State
b
of
facilities Air
c
Water Land
Underground
injection
Total on-site
release
d
Total off-site
release
e
Total on and
Off-site release
PA 36 3545433 37 0 1 3545471 47945 3593416
PR 18 2441482 0 0 0 2441482 7555 2449037
RI 2 18677 0 0 0 18677 0 18677
SC 22 1934986 1050 12127 0 1948163 6020 1954183
TN 26 2738055 0 0 0 2738055 17200 2755255
TX 54 822273 567 1990 23749 848579 110891 959470
UT 7 56753 0 0 0 56753 8376 65129
VA 20 1412207 0 5 0 1412212 422 1412634
WA 14 176129 294 176 0 176599 45145 221744
WV 5 67789 147 38 0 67974 0 67974
WI 22 565148 0 0 0 565148 0 565148
Total 827 40387302 15756 184350 490664 41078072 367280 41445352
Source: TRI98 2000
a
Data in TRI are maximum amounts released by each facility.
b
Post office state abbreviations are used.
c
The sum of fugitive and stack releases are included in releases to air by a given facility.
d
The sum of all releases of the chemical to air, land, water, and underground injection wells.
e
Total amount of chemical transferred off-site, including to publicly owned treatment works (POTW)
METHYLENE CHLORIDE
5.
POTENTIAL FOR HUMAN EXPOSURE
177 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
1-4
5-8
9-16
21-34
52-72
94
Frequency of
NPL Sites
Derived from HazDat 2000
Figure 5-1. Frequency of NPL Sites with Methylene Chloride Contamination
178 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
Methylene chloride has been identified in a variety of environmental media (air, surface water,
groundwater, soil, and sediment) collected at 882 of the 1,569 NPL hazardous waste sites (HazDat 1999).
5.2.1 Air
According to the Toxics Release Inventory, in 1998, the estimated releases of methylene chloride of
40 million pounds (18.3 million kg) to air from 714 large processing facilities accounted for about 97.4%
of total environmental releases (TRI98 2000). Table 5-1 lists amounts released from these facilities. The
TRI data should be used with caution because only certain types of facilities are required to report. This
is not an exhaustive list.
Methylene chloride has been identified in air samples collected at 108 of the 1,569 NPL hazardous waste
sites where it was detected in some environmental media (HazDat 1999).
Because methylene chloride is a highly volatile substance, most environmental releases are into the
atmosphere. Methylene chloride is released to the atmosphere during its production, storage, and
transport, but most (more than 99%) of the atmospheric releases result from industrial and consumer uses
(EPA 1983c, 1985e). In 1992, 32.5% of the total global emission of methylene chloride was attributed to
North America (McCulloch and Midgley 1996). It has been estimated that 85% of the total amount of
methylene chloride produced in the United States is lost to the environment (EPA 1985e), about 86% of
which is released to the atmosphere (EPA 1982a). Thus, about 73% (370 million pounds) of the U.S.
production volume for 1988 (500 million pounds), of methylene chloride was lost to the atmosphere in
1988. Manufacturers, processors, and users of methylene chloride are required to report the quantities of
methylene chloride released to environmental media or transferred to off-site disposal facilities annually
(EPA 1998j). The data currently available, compiled in the Toxics Release Inventory (TRI98 2000), are
for releases in 1998 and are summarized in Table 5-1. Methylene chloride was the 14
th
-largest release
and transfer chemical reported in the United States for 1988 (EPA 1990c). The TRI data should be used
with caution because only certain types of facilities are required to report. This is not an exhaustive list.
Industrial methylene chloride emissions to the atmosphere reported to EPA for the 1988 TRI totaled about
127 million pounds (TRI88 1990). Emissions from the use of consumer products and other sources such
as hazardous waste sites may be estimated to be 243 million pounds by subtracting industrial emissions
(127 million pounds) from total atmospheric loss of methylene chloride (370 million pounds). Consumer
179 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
products containing methylene chloride in wide use include paint strippers, aerosols, adhesives and glues,
and cleaning fluids and degreasers (CPSC 1990). Virtually all the methylene chloride in these products is
released to the atmosphere during use. Its use in hair sprays was banned in 1989 by the FDA (1989).
Methylene chloride is formed during water chlorination (NAS 1977) and is emitted to the air from waste
waters in treatment plants (Corsi et al. 1987; Dunovant et al. 1986; Namkung and Rittmann 1987).
Methylene chloride emissions from treatment plants in California are estimated to exceed 400,000 pounds
annually (Corsi et al. 1987). Declining production amounts of methylene chloride will result in a
decrease in the volume emitted to the atmosphere.
5.2.2 Water
According to the Toxics Release Inventory, in 1998, the estimated releases of methylene chloride of about
15,756 pounds (7,090 kg) to water from 714 facilities accounted for < 0.04% of total environmental
releases (TRI98 2000). Table 5-1 lists amounts released from these facilities. The TRI data should be
used with caution because only certain types of facilities are required to report. This is not an exhaustive
list.
Methylene chloride has been identified in surface water and groundwater samples collected at 633 and
218, respectively, of the 1,569 NPL hazardous waste sites where it was detected in some environmental
media (HazDat 1999).
About 2% of environmental releases of methylene chloride are to water (EPA 1982a). Industrial releases
of methylene chloride to surface water and underground injection (potential groundwater release)
reported to the TRI in 1998 totaled 506,420 pounds (TRI98 2000). Methylene chloride has been
identified in industrial and municipal waste waters from several sources at concentrations ranging from
0.08 ppb to 3,400 ppm (Dunovant et al. 1986; EPA 1979a, 1982a; Namkung and Rittmann 1987). It has
also been reported in the leachate from industrial and municipal landfills at concentrations from 0.01 to
184 ppm (Brown and Donnelly 1988; Sawhney 1989). The levels of methylene chloride in surface water
have been reported to vary from nondetectable to 10 ppb (California Environmental Protection Agency
1992).
180 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
Methylene chloride has been detected in both surface water and groundwater samples taken at hazardous
waste sites. Data from the Contract Laboratory Program (CLP) Statistical Database indicate methylene
chloride was found at geometric mean concentrations of 68 and 98 ppb in surface water and groundwater
samples, respectively, at about 30% of the sites sampled (CLPSD 1990). Note that the information used
from the CLP Statistical Database includes data from both NPL and non-NPL sites.
5.2.3 Soil
According to the Toxics Release Inventory, in 1998, the estimated releases of methylene chloride of about
184,350 pounds (82,958 kg) to soil from 714 facilities accounted for about <0.44% of total environmental
releases (TRI98 2000). Table 5-1 lists amounts released from these facilities. The TRI data should be
used with caution because only certain types of facilities are required to report. This is not an exhaustive
list.
Methylene chloride has been identified in soil (412 sites) and sediment (179 sites) samples collected at
the 1,569 NPL hazardous waste sites where it was detected in some environmental media (HazDat 1999).
The principal sources of methylene chloride releases to land are disposal of methylene chloride products
and containers to landfills. Substantial reduction to industrial disposal of methylene chloride to the land
is likely since the inception of the Land Disposal Restrictions (EPA 1998c). However, disposal of
containers for consumer products containing residues of methylene chloride may continue to occur. It is
estimated that about 12% of methylene chloride losses to the environment are to land (EPA 1982a).
Methylene chloride has been detected in soil/sediment samples taken at 36% of the hazardous waste sites
included in the CLP Statistical Database at a geometric mean concentration of 104 ppb (CLPSD 1990).
5.3 ENVIRONMENTAL FATE
5.3.1 Transport and Partitioning
Methylene chloride is a volatile liquid, with a boiling point of 40 EC (Lide 1994) and a vapor pressure of
349 mm Hg at 20 EC (Verschueren 1983). Therefore, methylene chloride tends to volatilize to the
atmosphere from water and soil. The half-life of methylene chloride volatilization from water has been
181 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
found to be 21 minutes under experimental conditions (Dilling et al. 1975), but actual volatilization from
natural waters will depend on the rate of mixing, wind speed, temperature, and other factors (Dilling et al.
1975; EPA 1979b). The Henry's law constant value (H) of 0.002 atm/m
3
/mol (EPA 1982e; Gossett 1987)
indicates that methylene chloride will volatilize rapidly from moist soil and water surfaces (Thomas
1990).
Methylene chloride is not strongly sorbed to soils or sediments (Dilling et al. 1975; Dobbs et al. 1989).
Based on its low soil organic carbon partitioning coefficient (K
oc
) of 25, methylene chloride is likely to be
very highly mobile in soils (Bahnick and Doucette 1988; Roy and Griffin 1985) and may be expected to
leach from soils into groundwater.
Based on a reported log octanol/water partition coefficient (K
ow
) of 1.3 (Hansch and Leo 1979), an
estimated bioconcentration factor (BCF) of 2.3 was derived (EPA 1980a, 1984). There is no evidence of
biomagnification, but because the estimated BCF is low, significant biomagnification of methylene
chloride in aquatic food chains is not expected.
5.3.2 Transformation and Degradation
5.3.2.1 Air
The main degradation pathway for methylene chloride in air is its reaction with photochemically
generated hydroxyl radicals (Cox et al. 1976; Crutzen and Fishman 1977; Davis et al. 1976; EPA 1980f,
1987i). Thus, the atmospheric lifetime of methylene chloride may be predicted from the hydroxyl radical
concentration in air and the rate of reaction. Most reported rates for hydroxyl radical reaction with
methylene chloride range from 1.0x10
-13
to 1.5x10
-13
cm
3
/mol/sec, and estimates of average atmospheric
hydroxyl radical concentration range from 2.5x10
5
to 1x10
6
mol/cm
3
(Cox et al. 1976; Crutzen and
Fishman 1977; Davis et al. 1976; EPA 1980f, 1985e, 1985g). Using this information, an average
atmospheric lifetime for methylene chloride may be calculated to be 130 days. Other estimates range
from 100 to 500 days (Altshuller 1980; Cox et al. 1976; Davis et al. 1976; EPA 1987i; Sidebottom and
Franklin 1996). Because this degradation pathway is relatively slow, methylene chloride may become
widely dispersed but is not likely to accumulate in the atmosphere. The small amount of methylene
chloride which reaches the stratosphere (about 1%) may undergo direct photolytic degradation; however,
photolysis in the troposphere is not expected (Howard et al. 1990). Reactions of methylene chloride with
182 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
ozone or other common atmospheric species (e.g., oxygen atoms, chlorine atoms, and nitrate radicals) are
not believed to contribute to its breakdown (EPA 1985g, 1987i; WHO 1996).
5.3.2.2 Water
Methylene chloride undergoes slow hydrolysis in water. The experimental half-life reported for the
hydrolysis reaction, at neutral conditions, is approximately 18 months at 25 EC (Dilling et al. 1975).
However, the rate of reaction varies greatly with changes in temperature and pH. A hydrolytic half-life of
14 days was reported for methylene chloride in acidic solutions at 80–150 EC (EPA 1979b, 1985e). This
experimental value, when extrapolated to 25 EC, is about 700 years. Different mechanisms of hydrolyses
may be responsible for these two widely different values.
Both aerobic and anaerobic biodegradation may be an important fate process for methylene chloride in
water (Brunner et al. 1980; Davis et al. 1981; EPA 1985e; Stover and Kincannon 1983; Tabak et al.
1981). In the laboratory, methylene chloride can be biodegraded by aerobic bacteria such as
Methylobacterium sp. and Methylophilus sp. and anaerobic bacteria such as Hyphomicrobium sp. and
Dehalobacterium sp. (Leisinger et al. 1994; Magli et al. 1998). These bacteria are able to efficiently
utilize methylene chloride as a carbon and energy source. The Acinebacter sp. has been shown to use
oxygen or nitrate ion as the electron acceptor (Sheehan and Freedman 1996). Methylene chloride has
been observed to undergo degradation at a rapid rate under aerobic conditions. Reported total methylene
chloride loss was 100% after 7 days in a static culture flask biodegradability screening test (Tabak et al.
1981) and 92% after 6 hours in a mixed microbial system (Davis et al. 1981). Volatilization loss was not
more than 25% (Tabak et al. 1981).
5.3.2.3 Sediment and Soil
Degradation of methylene chloride was found to occur in soils with concentrations ranging from
approximately 0.1 to 5.0 ppm (Davis and Madsen 1991). The rate of biodegradation was found to be
dependent on soil type, substrate concentration, and redox state of the soil. Methylene chloride
biodegradation has been reported to occur under both aerobic conditions and anaerobic conditions (Davis
and Madsen 1991). The biodegradation of methylene chloride appears to be accelerated by the presence
of elevated levels of organic carbon (Davis and Madsen 1991).
183 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
Methylene chloride has a low tendency to absorb to soil (Dilling et al. 1975; Dobbs et al. 1989);
therefore, there is a potential for leaching to groundwater. Also, because of the high vapor pressure,
volatilization to air is also a likely fate process from dry soil. Its high Henry’s law constant
(0.002 atm/m
3
/mol) indicates that volatilization from moist soil is also likely.
5.4 LEVELS MONITORED OR ESTIMATED IN THE ENVIRONMENT
Reliable evaluation of the potential for human exposure to methylene chloride depends in part on the
reliability of supporting analytical data from environmental samples and biological specimens. In
reviewing the data on methylene chloride levels monitored or estimated in the environment, it should also
be noted that the amount of chemical identified analytically is not necessarily equivalent to the amount
that is bioavailable. The analytical methods available for monitoring methylene chloride in a variety of
environmental media are detailed in Chapter 6 (Analytical Methods).
5.4.1 Air
Methylene chloride has been detected in ambient air samples taken from around the world, as shown in
Table 5-2. Background levels are usually at about 50 parts per trillion (0.17 µg/m
3
) (Singh et al. 1982).
Methylene chloride was among the chemicals monitored in a statewide survey of hazardous air pollutants
by the Arizona Hazardous Air Pollutants Monitoring Program. The average amount of methylene
chloride detected in air ranged from 0.61 ppm on a hillside in Yavapai County to 1.62 ppm in Phoenix
(Zielinska et al. 1998). Concentrations of methylene chloride in urban areas and in the vicinity of
hazardous waste sites are generally one to two orders of magnitude higher. These values are all much
lower than the concentrations which may be encountered inside buildings where products containing
methylene chloride are being used (NAS 1978; Otson et al. 1983).
5.4.2 Water
Methylene chloride has been detected in surface water, groundwater, and finished drinking water
throughout the United States. It was detected in 30% of 8,917 surface water samples recorded in the
STORET database, at a median concentration of 0.1 ppb (Staples et al. 1985). In a New Jersey survey
(Page 1981), methylene chloride was found in 45% of 605 surface water samples, with a maximum
concentration of 743 ppb. Methylene chloride has also been identified in surface waters in Maryland

184 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
Table 5-2. Summary of Methylene Chloride Levels in Air
Concentration (ppb)
a
Location Maximum Mean Reference
Background No data 0.05 Singh et al. 1982
Oceanic:
Northern Hemisphere No data 10.37 Koppmann et al. 1993
Southern Hemisphere No data 5.18 Koppmann et al. 1993
Rural/suburban United States No data 0.05
c
–0.60
c
EPA 1988d
Urban United States 22–200 0.23–1.93
b
EPA 1980e, 1988d; Harkov
et al. 1984, 1985; Shikiya
et al. 1984; Singh et al.
1982
Source dominated No data 0.58
c
EPA 1988d
Hazardous waste sites 10–190 0.09–11.23
b
Harkov et al. 1985;
La Regina et al. 1986
Indoor (nonresidential) 19,000 0.06–5,472
b
Harsch 1977; Otson et al.
1983
a
1 ppb = 3.47 µg/m
3
b
Range of values
c
Median value
d
Range of individual values
185 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
(Helz and Hsu 1978), in Lakes Erie and Michigan (Konasewich et al. 1978), and at hazardous waste sites
(Hauser and Bromberg 1982). Methylene chloride (0.2 ppm) was also detected in the shallow ground-
water beneath Denver, Colorado (Bruce and McMahon 1996). Seawater also contains small amounts of
methylene chloride; the mean reported concentration is 2.2x10
-3
ppb (Singh et al. 1983).
Since volatilization is restricted in groundwater, concentrations of methylene chloride are often higher
there than in surface water. Occurrence of methylene chloride in groundwater has been reported in
several surveys across the United States, with concentrations ranging from 0 to 3,600 ppb (Dyksen and
Hess 1982; EPA 1985h; Page 1981). Based on CERCLA records compiled during 1987, the compound
was the sixth most frequently detected organic contaminant found in groundwater during hazardous waste
disposal site investigations with a detection frequency of 19% (Plumb 1987).
Methylene chloride has been detected in drinking water supplies in numerous U.S. cities (Coleman et al.
1976; Dowty et al. 1975; EPA 1975b; Kool et al. 1982; Kopfler et al. 1977). Reported mean concentra-
tions are generally less than 1 ppm (EPA 1975b). It was detected in 2.2% of 182 samples of bottled water
surveyed with a mean concentration of positive samples at 0.059 ppb (Page et al. 1993). Water
chlorination in treatment plants appears to increase both the concentration and the frequency of
occurrence of methylene chloride in drinking water supplies (Bellar et al. 1974; EPA 1975b; NAS 1977).
5.4.3 Sediment and Soil
No studies were located on methylene chloride levels in soil. However, methylene chloride was detected
in 20% of 338 sediment samples recorded in the STORET database, at a median concentration of
13 µg/kg (Staples et al. 1985).
5.4.4 Other Environmental Media
Although methylene chloride is used in food processing (solvent extraction of coffee, spices, hops) and as
a post-harvest fumigant for some foods (strawberries, grain), little information was located on levels in
foods. Residual levels of methylene chloride in decaffeinated coffee beans ranging from 0.32 to
0.42 ppm were reported in the United States (IARC 1986), and levels ranging from 0.01 to 0.1 ppm were
reported by a major coffee processor (FDA 1985). These levels are well within the FDA limits of 10 ppm
for methylene chloride in decaffeinated coffee. Although the FDA considers residues at or below the
186 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
FDA limit to be safe (FDA 1985), it has been reported that methylene chloride is no longer used as a
decaffeinating agent by most coffee decaffeinators (Mannsville Chemical Products Corporation 1988).
The FDA has also set a limit of 30 ppm for methylene chloride in spice oleoresins, but EPA has exempted
methylene chloride from the requirement of a tolerance for fumigated food materials (see Chapter 7).
Oysters and clams from Lake Pontchartrain in Louisiana had mean methylene chloride levels of 7.8 and
27 ppb, respectively (Ferrario et al. 1985).
5.5 GENERAL POPULATION AND OCCUPATIONAL EXPOSURE
Inhalation of methylene chloride from ambient air is the predominant route of exposure for the general
population in the United States. As calculated by Singh et al. (1981), the average daily doses of
methylene chloride from ambient air in three U.S. cities range from 33 to 309 µg/day, based on 1979
monitoring data and a daily air intake of 23 m
3
by an adult. Since the amount of methylene chloride
manufactured has recently dropped below the production levels of the late 1970s, average daily doses
may currently be somewhat less than these values. Methylene chloride was detected in the blood of fewer
than 10% of the 600 samples obtained from people who participated in the Third National Health and
Nutrition Examination Survey (NHANES III) (Ashley et al. 1994). Inhalation exposure may also occur
through the use of consumer products containing methylene chloride (CPSC 1987) (see Section 5.7).
Since concentrations of methylene chloride in water and food are generally quite low, it appears that
exposure from sources other than air is unlikely to be important. For example, drinking water containing
1 ppb methylene chloride would provide an additional intake of 2 µg per day for an adult drinking 2 L of
water per day.
Methylene chloride has been identified, but not quantified, in eight out of eight samples of human breast
milk collected in four urban areas in the United States (EPA 1980d; Pellizzari et al. 1982).
Occupational exposure to methylene chloride may be important in numerous industries. Workers may be
exposed during the production and processing of methylene chloride. They may also be exposed to
methylene chloride during a variety of industrial activities including spray painting, spray gluing, metal
painting, paint stripping, and aerosol packing (IARC 1986; OSHA 1986; Whitehead et al. 1984). The
NIOSH estimated that the number of workers exposed to methylene chloride increased from about
187 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
1 million in the early 1970s to 1.4 million in the early 1980s (NIOSH 1986; NOES 1990). Since
production and use of methylene chloride has decreased (as have threshold limit values) since the
mid-1980s (see Sections 4.1 and 4.3), the number of workers exposed to methylene chloride has
decreased accordingly.
Monitoring data for methylene chloride in workplace air from 1968 to 1982 indicate that concentrations
in the general work area ranged from 0.086 to 964.8 ppm while samples in the breathing zone of workers
ranged up to 1,411 ppm (IARC 1986). Installation of appropriate ventilation systems has been found to
lower the workers’ exposure to methylene chloride in the breathing zone from 600–1,150 ppm to
28–34 ppm (Estill and Spencer 1996). The current OSHA Permissible Exposure Limit (PEL) for
methylene chloride for an 8-hour workday is 25 ppm (88.25 mg/m
3
) (OSHA 1997). An estimated
237,496 workers are potentially exposed while involved with methylene chloride manufacturing, paint
manufacturing, metal cleaning, polyurethane foam manufacturing, plastics and adhesives manufacturing,
ink use, pharmaceuticals, construction, and shipyards. The largest numbers of occupationally exposed
individuals occur in areas of metal cleaning, industrial paint stripping, and ink solvent uses (OSHA 1997).
The American Conference of Governmental Industrial Hygienists (ACGIH) reduced their Threshold
Limit Value (TLV) (the recommended 8-hour exposure limit) from 100 to 50 ppm (174 mg/m
3
) in 1988
(ACGIH 1990), which continues to be the recommended TLV level (ACGIH 1998).
5.6 EXPOSURES OF CHILDREN
This section focuses on exposures from conception to maturity at 18 years in humans. Differences from
adults in susceptibility to hazardous substances are discussed in 2.7 Children's Susceptibility.
Children are not small adults. A child's exposure may differ from an adult's exposure in many ways.
Children drink more fluids, eat more food, breathe more air per kilogram of body weight, and have a
larger skin surface in proportion to their body volume. A child's diet often differs from that of adults.
The developing human's source of nutrition changes with age: from placental nourishment to breast milk
or formula to the diet of older children who eat more of certain types of foods than adults. A child's
behavior and lifestyle also influence exposure. Children crawl on the floor, put things in their mouths,
sometimes eat inappropriate things (such as dirt or paint chips), and spend more time outdoors. Children
also are closer to the ground, and they do not use the judgment of adults in avoiding hazards (NRC
1993).
188 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
Young children often play close to the ground and frequently play in dirt, which increases their dermal
exposure to toxicants in dust and soil. They also tend to ingest soil, either intentionally through pica or
unintentionally through hand-to-mouth activity. Children, thus, may be orally and dermally exposed to
methylene chloride present as a contaminant in soil and dust. It has been demonstrated that methylene
chloride is rapidly absorbed by the skin (McDougal et al. 1986). Methylene chloride has a K
oc
of 25,
indicating a very low adsorption to soil (Bahnick and Doucette 1988; Roy and Griffin 1985; Swann et al.
1983). Most of the methylene chloride present in the upper layers of the soil will be rapidly volatilized to
air (vapor pressure=349 mmHg at 20 EC). Loss of methylene chloride from the soil decreases the
potential of dermal and oral exposure to children, but its rapid volatilization results in inhalation being the
most likely route of exposure during play on the ground.
The higher ventilation rate in children compared to adults means that for a brief period, children will be
more vulnerable than adults to acute neurological effects from short-term inhalation exposures. However,
differences between children and adults are eliminated as steady-state concentrations are reached, i.e.,
within 2–4 hours (see Section 2.3.1.1). Thus, children would not be expected to be more vulnerable than
adults in the case of intermediate- or chronic-duration exposures. Young children are closer to the ground
or floor because of their shorter stature. The methylene chloride vapors being heavier than air (vapor
density = 2.93) tend to concentrate near the ground. Children, therefore, are at a greater risk of exposure
than adults during accidental spills or through indoor use of methylene chloride in an unventilated area.
Exposures of the embryo or fetus to volatile organic compounds such as methylene chloride may occur if
the expectant mother is exposed. A newborn infant may be exposed by breathing contaminated air and by
ingestion of mother’s milk, which can contain small amounts of methylene chloride. Children may be
exposed through accidental ingestion of products containing methylene chloride. Older children and
adolescents may be exposed to methylene chloride in their jobs or hobbies, or through deliberate solvent
abuse by “sniffing.” Human epidemiological studies and case reports discussing reproductive and/or
developmental toxicity of methylene chloride in humans have been reviewed. Exposure routes included
occupational duties and sniffing of paint removers. Inhalant abuse during pregnancy poses significant
risks to the pregnancy and endangers both the mother and the fetus. Solvent abuse of methylene chloride
for euphoric effects results in exposure levels that equal or exceed those producing adverse effects in
animals.
189 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
There are no existing studies that have monitored the level of exposure from methylene chloride to
children. Most uses of methylene chloride are associated with occupational purposes, so it is unlikely that
children will receive significant doses. Under extreme conditions where paint thinners or other mixtures
containing high concentrations of methylene chloride are used in the presence of children in an enclosed
area with little or no ventilation, children could receive significant exposure. There are studies that
examine the exposure to children from parents’ work clothes, skin, hair, tools, or other objects removed
from the workplace (NIOSH 1995); however, this type of “take home” or secondary exposure is unlikely
due to the high volatility of methylene chloride. Additional exposure from consumer products can occur,
but is unlikely to be significant, although little data are available at this time.
Methylene chloride has been identified, but not quantified, in eight out of eight samples of human breast
milk collected in four urban areas in the United States (EPA 1980d; Pellizzari et al. 1982).
It is not known whether children differ in their weight-adjusted intake of methylene chloride. However,
children drink more fluids per kilogram of body weight than adults (NRC1993) and methylene chloride
has been detected in drinking water (Section 5.4.2).
5.7 POPULATIONS WITH POTENTIALLY HIGH EXPOSURES
In addition to individuals who are occupationally exposed to methylene chloride (see Section 5.5), there
are several groups within the general population that could have potentially high exposures (higher than
background levels) to methylene chloride. These populations include individuals living in proximity to
sites where methylene chloride was produced or sites where methylene chloride was disposed, and
individuals living near 1 of the 1,569 NPL hazardous waste sites where methylene chloride has been
detected in some environmental media (HazDat 1996).
Individuals using consumer products containing substantial amounts of methylene chloride have the
potential for high exposure to this compound (CPSC 1987). Paint strippers, adhesive removers, spray
shoe polishes, adhesives, glues, paint thinners, and many other household products contain enough
methylene chloride to expose consumers to significant amounts of methylene chloride vapor when the
products are used, especially indoors (CPSC 1987, 1990). Indoor air concentrations resulting from the
use of methylene chloride-containing consumer products have been estimated to range from 0.06 to
5,472 ppb (Callahan 1981; NAS 1978; Otson et al. 1983). Previous estimates indicated that a concentra
190 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
tion of 50 ppm (174 mg/m
3
) of methylene chloride would have been expected in the breathing zone of
consumers following hair spray use (FDA 1985), resulting in a time-weighted average exposure of
0.174 ppm. Hair care specialists would have been exposed to 10 times this level (FDA 1985). However,
this source of exposure has been virtually eliminated since the FDA (1989) banned the use of methylene
chloride in hairsprays.
Coffee drinkers who consume large amounts of decaffeinated coffee which has been extracted with
methylene chloride may be exposed from this source. Assuming all the methylene chloride is extracted
from the beans during brewing and none is volatilized (FDA 1985), the maximum daily intake for a
person drinking 5 cups of decaffeinated coffee is about 12 µg, a fraction of intake from ambient air.
Since volatilization during brewing is very likely, the actual intake is probably much lower. In addition,
this source of exposure is becoming less important, since most decaffeination processes no longer use
methylene chloride.
People living near industrial or hazardous waste sites with higher than average levels of methylene
chloride in the air or water would have potential above-average exposure. However, the magnitude of
this exposure can only be evaluated on a site-by-site basis.
Currently, workers in the industries identified above (Section 5.5) have the highest potential exposure to
methylene chloride. OSHA has determined that a PEL TWA of 25 ppm substantially reduces significant
risk of cancer from methylene chloride in occupational settings. OSHA also believes that a lower limit
would further reduce risk and has set an action level measured as an 8-hour TWA to 12.5 ppm (OSHA
1997).
5.8 ADEQUACY OF THE DATABASE
Section 104(i)(5) of CERCLA, as amended, directs the Administrator of ATSDR (in consultation with the
Administrator of EPA and agencies and programs of the Public Health Service) to assess whether
adequate information on the health effects of methylene chloride is available. Where adequate
information is not available, ATSDR, in conjunction with the National Toxicology Program (NTP), is
required to assure the initiation of a program of research designed to determine the health effects (and
techniques for developing methods to determine such health effects) of methylene chloride.
191 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
The following categories of possible data needs have been identified by a joint team of scientists from
ATSDR, NTP, and EPA. They are defined as substance-specific informational needs that if met would
reduce the uncertainties of human health assessment. This definition should not be interpreted to mean
that all data needs discussed in this section must be filled. In the future, the identified data needs will be
evaluated and prioritized, and a substance-specific research agenda will be proposed.
5.8.1 Identification of Data Needs
Physical and Chemical Properties. Knowledge of the physical and chemical properties is essential
for estimating the partitioning of the chemical and the fate in the environment. Information about the
physical and chemical properties of methylene chloride is adequate (EPA 1982e; Hansch and Leo 1979;
Lide 1994; Roy and Griffin 1982; Verschueren 1983) and can be used to determine the behaviors of the
chemical. No further investigation is required.
Production, Import/Export, Use, Release, and Disposal. As of October 1, 1996, the Interna-
tional Trade Commission ceased to collect or publish annual synthetic organic chemicals data. The
National Petroleum Refiners Association, which currently collects such data, does not include methylene
chloride on its list of organic chemicals. The available production data for methylene chloride are out of
date. It is essential that these data be updated regularly to allow a more accurate determination of the
potential for human exposure.
Remedial investigations and feasibility studies at NPL sites that contain methylene chloride are needed to
provide further information on environmental concentrations and human exposure levels near these sites.
According to the Emergency Planning and Community Right-to-Know Act of 1986, 42 U.S.C. Section
11023, industries are required to submit chemical release and off-site transfer information to the EPA.
The Toxics Release Inventory (TRI), which contains this information for 1996, became available in May
of 1998. This database will be updated yearly and should provide a list of industrial production facilities
and emissions.
Data on the production and uses of methylene chloride in the United States are available (Mannsville
Chemical Products Corporation 1988; NTP 1989; SRI 1999; TRI98 2000), however the parameters in
which companies are monitored for the production of methylene chloride could be better characterized.
192 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
Production of methylene chloride has decreased due to declining demands (HSDB 1990; Mannsville
Chemical Products Corporation 1988; USITC 1989). Import/export data are available (Mannsville
Chemical Products Corporation 1988; NTDB 1998) as well as data on land disposal (TRI98 2000), but
more information is needed on disposal by incineration.
Environmental Fate. Methylene chloride is a highly volatile chemical and tends to volatilize from
water and soil to the atmosphere (Dilling et al. 1975; EPA 1979b; Gossett 1987). The half-life of
methylene chloride volatilization from water has been measured (Dilling et al. 1975). Methylene chloride
does not strongly sorb to soils or sediments and can be expected to leach from soils to ground water
(Dilling et al. 1975; Dobbs et al. 1989). Atmospheric lifetimes have been estimated from the reactions of
methylene chloride with hydroxyl radicals and the concentration of such radicals (Altshuller 1980; Cox et
al. 1976; Davis et al. 1976; EPA 1987i). Half-life values for the hydrolysis of methylene chloride in
water has been reported (Dilling et al. 1975). Degradation of methylene chloride has been found to occur
(Davis and Madsen 1991). Biodegradation of methylene chloride has been found to occur under both
aerobic and anaerobic conditions (Leisinger et al. 1994; Magli et al. 1998; Sheehan and Freedman 1996).
More information and data are needed for this process.
Bioavailability from Environmental Media. Data indicates the predominant route of exposure to
methylene chloride is through inhalation (Angelo et al. 1986a; DiVincenzo et al. 1972). Studies also
indicate the compound is readily absorbed following dermal exposure and ingestion (DiVincenzo and
Kaplan 1981; McDougal et al. 1986; Stewart and Dodd 1964). Little information was found on levels of
methylene chloride in food and no studies were found on the levels of methylene chloride in soil.
Additional data on absorption in humans following ingestion and dermal exposures are needed to
establish the importance of consumption of contaminated drinking water and foodstuffs and dermal
contact with consumer products containing methylene chloride as exposure pathways for the general
population.
Food Chain Bioaccumulation. The bioconcentration factor (2.3) estimated for methylene chloride is
low (EPA 1980a, 1984). More data is needed on the bioaccumulation of methylene chloride by plants,
aquatic organisms, and animals, and the biomagnification of methylene chloride in terrestrial and aquatic
food chains. Data on bioaccumulation and biomagnification would aid in determining if levels of
methylene chloride in the environment affect food chains and potentially impact human exposure.
193 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
Exposure Levels in Environmental Media. Studies are available documenting levels of methylene
chloride in air (EPA 1988d; Harkov et al. 1984; Otson et al. 1983; Singh et al. 1982), water (Dyksen and
Hess 1982; EPA 1975b, 1985h; Page 1981; Singh et al. 1983; Staples et al. 1985), and sediments (Staples
et al. 1985). More information is needed regarding the levels of methylene chloride in soils and
sediments since there is little information at this time. Since production and use of methylene chloride
have decreased in recent years and are projected to continue the downward trend, it would be valuable to
have recent data to better estimate current human exposure levels from these media. More information is
needed on methylene chloride levels measured in food.
Reliable monitoring data for the levels of methylene chloride in contaminated media at hazardous waste
sites are needed so that the information obtained on levels of methylene chloride in the environment can
be used in combination with the known body burdens of methylene chloride to assess the potential risk of
adverse health effects in populations living in the vicinity of hazardous waste sites.
Exposure Levels in Humans. Methylene chloride has been identified and quantified in human breast
milk (EPA 1980d; Pellizzari et al. 1982). More data is needed to determine the levels of methylene
chloride in blood, urine, and other tissues in the general population, particularly for populations living in
the vicinity of hazardous waste sites that contain methylene chloride. This information is needed to
establish levels of methylene chloride to which the general population has been exposed through contact
with contaminated air, drinking water, and consumer products. This information is necessary for
assessing the need to conduct health studies on these populations.
Exposures of Children. There are no studies monitoring the level of exposure of children to
methylene chloride. This data gap requires future studies to determine the exposure of methylene
chloride to children and if the significant exposures can be decreased by any means. Additional studies
are needed to examine various exposure pathways that are unique to children. Studies are also needed to
examine children’s weight-adjusted intake of methylene chloride.
Despite their higher ventilation rate per kilogram of body weight compared to adults, children are not at a
greater risk of inhalation exposure to methylene chloride, except, initially for acute exposures. Steady
state considerations tend to eliminate the difference between children and adults after a short time (see
Section 2.3.1.1). They also spend more time closer to ground because of their height. Methylene
chloride vapors, being heavier than air, tend to concentrate closer to the ground, thereby increasing the
194 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
risk for children. No data are available on the exposure of children to methylene chloride present in the
air.
A study on usefulness of intervention methods in cases of inhalant abuse by pregnant women is required.
More research is needed to rule out concomitant risk factors and to identify specific chemicals and
patterns of use associated with adverse effects.
Child health data needs relating to susceptibility are discussed in 2.12.2, Identification of Data Needs:
Children’s Susceptibility.
Exposure Registries. No exposure registries for methylene chloride were located. This substance is
not currently one of the compounds for which a subregistry has been established in the National Exposure
Registry. The substance will be considered in the future when chemical selection is made for
subregistries to be established. The information that is amassed in the National Exposure Registry
facilitates the epidemiological research needed to assess adverse health outcomes that may be related to
exposure to this substance.
5.8.2 Ongoing Studies
As part of the ongoing Third National Health and Nutrition Evaluation Survey (NHANES III), the
Environmental Health Laboratory Sciences Division of the National Center for Environmental Health,
Centers for Disease Control and Prevention, is analyzing human blood samples for methylene chloride
and other volatile organic compounds. These data will give an indication of the frequency of occurrence
and background levels of these compounds in the general population.
A number of ongoing research efforts may provide data regarding the potential for human exposure to
methylene chloride. These projects are summarized in Table 5-3.

195 METHYLENE CHLORIDE
5. POTENTIAL FOR HUMAN EXPOSURE
Table 5-3. Ongoing Studies on the Potential for Human Exposure to
Methylene Chloride
Investigator Affiliation Research Description Sponsor
Argonne National
Laboratory,
Lemont, IL
Destruction/conversion of
hazardous waste chlorocarbons
EM
Smith, G.B. New Mexico State
University,
Las Cruces, NM
Pollutant toxicity reduction through
biodegradation
NIGMS
Anders, M.W. University of
Rochester,
Rochester, NY
Metabolism and toxicity of
halogenated hydrocarbons
NIEHS
Hemingway,
R.W.
Forest Service,
Pineville, LA
Knot VOCs USDA, CRGO
Morra, M.J. University of Idaho,
Moscow, ID
Biotic and abiotic degradation of
glucosinolates and halogenated
hydrocarbons in soil
USDA, CRGO
Scow, K.M. University of California,
Davis, CA
Microbial biodegradation of
organic chemicals in soil
USDA, CRGO
Cook, R. TDA Research, Inc.
Wheat Ridge, CO
Catalysts for the oxidation of
chlorinated hydrocarbons
EPA
Dragan, A. Dragan Engineering,
Encino, CA
Biofilteration of volatile organic
compounds emitted a industrial
waste treatment
AF
Wijmans, J.G. Membrane Technology
and Research,
Menlo Park, NJ
Recovery of liquid hazardous
wastes from carbon adsorption
steam regeneration streams
EPA
Pfefferle, W. Precision Combustion,
New Haven, CT
Catalytically stabilized thermal
incineration
NSF
Bohn, M.S. National Renewable
Energy Laboratory,
Golden, CO
Incineration of hazardous
materials with carbonate salts
USDOE
Strand, S.E. University of
Washington,
Seattle, WA
Using trees to remediate
groundwaters contaminated with
chlorinated hydrocarbons
Bernhard, R.A. University of California,
Davis, CA
Isolation, identification, and
significance of micro-organic
constituents in foods
AF = Air Force; EPA = Environmental Protection Agency; NIEHS = National Institute of Environmental Health
Sciences; NIGMS = National Institute of General Medical Sciences; NSF = National Science Foundation;
USDA, CRGO = U.S. Department of Agriculture, Competitive Research Grant Office; USDOE = U.S. Department of
Energy
197 METHYLENE CHLORIDE
6. ANALYTICAL METHODS
The purpose of this chapter is to describe the analytical methods that are available for detecting,
measuring, and/or monitoring methylene chloride, its metabolites, and other biomarkers of exposure and
effect to methylene chloride. The intent is not to provide an exhaustive list of analytical methods.
Rather, the intention is to identify well-established methods that are used as the standard methods of
analysis. Many of the analytical methods used for environmental samples are the methods approved by
federal agencies and organizations such as EPA and the National Institute for Occupational Safety and
Health (NIOSH). Other methods presented in this chapter are those that are approved by groups such as
the Association of Official Analytical Chemists (AOAC) and the American Public Health Association
(APHA). Additionally, analytical methods are included that modify previously used methods to obtain
lower detection limits and/or to improve accuracy and precision.
6.1 BIOLOGICAL SAMPLES
Analysis of biological materials for methylene chloride is performed most frequently by gas
chromatography with a flame ionization detector (GC/FID) (DiVincenzo et al. 1971; Engstrom and
Bjurstrom 1977). Table 6-1 summarizes the data for specific methods for biological fluids and tissues.
Separation of methylene chloride from biological samples in preparation for GC/FID analysis is most
often achieved by heating the sample in a sealed flask until the analyte concentration is in equilibrium in
the sample matrix and the headspace vapor. The headspace vapor is then drawn off for analysis by gas
chromatography (GC). Headspace sampling eliminates the need for a solvent extraction procedure, thus
simplifying sample preparation and improving sensitivity (DiVincenzo et al. 1971). However,
partitioning of the analyte between the headspace and the sample matrix depends on the nature of the
matrix and must be determined separately for each different kind of matrix (Walters 1986). Acid
hydrolysis of adipose tissue is required prior to headspace sampling (Engstrom and Bjurstrom 1977).
6.2 ENVIRONMENTAL SAMPLES
Several methods are available for the determination of methylene chloride in air, water, soil/sediments,
and food. Table 6-2 summarizes several representative methods appropriate for quantifying methylene In
most analytical methods, methylene chloride is trapped on a solid sorbent such as activated charcoal. Air

198
Table 6-1. Analytical Methods for Determining Methylene Chloride in Biological Materials
Analytical
Sample
detection Percent
Sample matrix Preparation method method limit recovery Reference
Blood Heat sample, collect headspace vapor GC/FID 0.022 ppm 49.8±1.33 DiVincenzo et al. 1971
Urine Heat sample, collect headspace vapor GC/FID No data 59±2.75 DiVincenzo et al. 1971
Urine Heat sample, inject headspace air, loop GC/MS 0.5 ppb 95% Ghittori et al. 1993
Breath Heat sample, inject into gas sample, loop GC/FID 0.2±0.1 ppm No data DiVincenzo et al. 1971
Adipose tissue Hydrolyze with acid, heat sample, collect
headspace vapor
GC/FID 1.6 ppm
a
No data Engstrom and Bjurstrom 1977
Human milk Purge with helium, trap on sorbent trap,
desorb thermally
GC/MS No data No data Pellizzari et al. 1982
a
Lowest reported concentration
FID = flame ionization detector; GC = gas chromatography; MS = mass spectrometry
METHYLENE CHLORIDE
6. ANALYTICAL METHODS

199
Table 6-2. Analytical Methods for Determining Methylene Chloride in Environmental Samples
Sample matrix Preparation method
Analytical
method
Sample
detection
limit
Percent
recovery Reference
Air Adsorb on charcoal, desorb with carbon
disulfide
GC/FID 25 ppb
a
90–100
b
APHA 1977
Air Adsorb on charcoal, desorb with carbon
disulfide
GC/MS No data No data Ghittori et al. 1993
Air Adsorb on charcoal, desorb with carbon
disulfide
GC/MS No data No data Xiao et al. 1993
Air Adsorb on charcoal, desorb with carbon
disulfide
GC/FID 2,900 ppb
0.01 mg/sample
95.3 NIOSH 1994
[Method 1005]
Air Extract with methylene chloride, purge with
helium, desorb thermally
GC/MS 0.05 ppm -100 Savard et al. 1992
Air Adsorb on charcoal, desorb with benzyl
alcohol
GC/ECD .0.5 ppb No data Woodrow et al. 1988
Air Purge with nitrogen, direct beam with
mirrors to detect signal
RS-FTIR 0.57 ppm-m 99.7–100 Xiao et al. 1993
Air Adsorb on activated charcoal, desorb with
carbon disulfide
Toxic Gas
Vapor Detector
Tube
No data No data EMMI 1999a
Air Adsorb on activated charcoal, desorb with
carbon disulfide
GC/FID No data No data EMMI 1999b
Air Aspirate with a pump through detector tube Toxic Gas
Vapor Detector
Tube
50 ppm No data EMMI 1999c
Stack gas
effluents
Sorption onto Tenax
®
, thermal desorption GC/MS No data 50–150 EPA 1986e
OSW 5041A
METHYLENE CHLORIDE
6. ANALYTICAL METHODS

200
Table 6-2. Analytical Methods for Determining Methylene Chloride in Environmental Samples(continued)
Analytical
Sample
detection Percent
Sample matrix Preparation method method limit recovery Reference
Water Purge with inert gas, trap on sorbent trap,
desorb thermally
Water Purge with inert gas, trap on sorbent trap,
desorb thermally
Water Purge with inert gas, trap on sorbent trap,
desorb thermally
Water Purge with inert gas, trap on sorbent trap,
desorb thermally
Water Purge with inert gas, trap on sorbent trap,
desorb thermally
Water Purge with inert gas, trap on sorbent trap,
desorb thermally
Water Purge with helium, trap on sorbent trap,
desorb thermally
Water Purge with helium, trap on sorbent trap,
desorb thermally
Waste water Purge with helium, trap on sorbent trap,
desorb thermally
Waste water Purge with inert gas, trap on sorbent trap,
desorb thermally
Waste water Purge with inert gas, trap on sorbent trap,
desorb thermally
GC/HSD
GC/ELCD
GC/MS
HRGC/MS
HRGC/ELCD
HRGC/MS
GC/MS
GC/MS
GC/MS or
GC/PID or
GC/ECD
GC/MS
GC/MS
No data
0.01 ppb
1.0 ppb
0.03–0.09 ppb
0.01–0.05 ppb
0.02–0.2 ppb
No data
0.099 ppb
0.5 ppb
0.25 ppb
2.8 ppb
85
97–100
99
95–97
97±28
95±5
99–105
85
80–120
25–162
D–221
EPA 1989f
EPA 1989g
EPA 1989c
EPA1989b
APHA 1989a
APHA 1989b
Michael et al. 1988
APHA 1998a
APHA 1998b
EPA 1998h [Method 601]
EPA 1998i [Method 624]
METHYLENE CHLORIDE
6. ANALYTICAL METHODS

201
Table 6-2. Analytical Methods for Determining Methylene Chloride in Environmental Samples(continued)
Sample matrix Preparation method
Analytical
method
Sample
detection
limit
Percent
recovery Reference
Solid/
Solid Waste
Purge with inert gas, trap on sorbent trap,
desorb thermally
GC/MS 0.03 ppb 95 EPA 1996b [Method 8260]
Solid/
Solid Waste
Purge with inert gas, trap on sorbent trap,
desorb thermally; or inject directly into GC
GC/PID/HECD 0.02 ppb 95±2.8 EPA 1996c [Method 8021]
Soil/sediment/
solid waste
Headspace extraction GC/FID or
GC/PID/ELCD
No data No data EPA 1986e
OSW 5021
Soil/sediment Extract with methanol, purge and trap GC/ECD No data 25–162 EPA 1986e
OSW 8010B
Solid waste
matrices
Purge and trap or direct injection GC/ECD/PID 0.02 ppb 97 EPA 1986e
OSW 8021B-PID
Solid waste
matrices
Headspace extraction, purge and trap GC/MS 5 ppb 0–0221 EPA 1986e
OSW 8240B-S
Solid waste
matrices
Purge and trap or direct injection GC/MS 0.03 ppb 95 EPA 1986e
OSW 8260B
Food Equilibrate in heated sodium sulfate
solution, collect headspace vapor
GC/ELCD 0.05 ppm No data Page and Charbonneau
1984
Food Isolate solvent by closed system vacuum
distillation with toluene as carrier solvent
GC/ELCD 7 ng 94 Page and Charbonneau
1984
Food Isolate solvent by closed system vacuum
distillation with toluene as carrier solvent
GC/ECD 7 ng 100 Page and Charbonneau
1984
METHYLENE CHLORIDE
6. ANALYTICAL METHODS

202
Table 6-2. Analytical Methods for Determining Methylene Chloride in Environmental Samples(continued)
Sample matrix Preparation method
Analytical
method
Sample
detection
limit
Percent
recovery Reference
Food Purge with nitrogen, trap on sorbent trap,
elute with hexane
GC/ELCD 1.2 ppb
c
84–96 Heikes 1987
Food Extract with acetone-water, back extract
with isooctane
GC/ELCD 4 ppb 66 Daft 1987
a
Lowest value for various compounds reported during collaborative testing of this method.
b
Estimated accuracy of the method when the personal sampling pump is calibrated with a charcoal tube in the line.
c
Lowest reported concentration.
D = detected; ECD = electron capture detector; ELCD = electrolytic conductivity detector; FID = flame ionization detector; GC = gas chromatography; HECD =
electrolytic conduction detector; HRGC = high resolution gas chromatography; HSD = halogen specific detector; MS = mass spectrometry; PID = photo ionization
detector; RS-FTIR = remote sensing Fourier transform infrared
METHYLENE CHLORIDE
6. ANALYTICAL METHODS
203 METHYLENE CHLORIDE
6. ANALYTICAL METHODS
samples are drawn directly through the sorbent (APHA 1977; NIOSH 1984). For water, soil, or solid
chloride in each of these environmental media. The EPA and NIOSH methods are standardized to
comply with regulatory requirements.
waste samples, methylene chloride is purged from the sample with an inert gas such as helium, and then
passed through the sorbent (APHA 1989a, 1989b; EPA 1989c, 1989f, 1989g). Desorption is thermal or
by carbon disulfide. Vacuum distillation or solvent extraction are sometimes used for separation of
methylene chloride from food samples (Daft 1987; Page and Charbonneau 1977).
Following separation of the organic compounds by GC, methylene chloride is detected by one of several
types of instruments: a flame ionization detector (FID), electron capture detector (ECD), electrolytic
conductivity detector (ELCD) or halogen specific detector (HSD). A mass spectrometer (MS) may be
used for unequivocal identification. While the ELCD appears to be most sensitive, detection limits for all
these methods are well below levels of health concern, but are not below EPA calculated cancer risk
levels or all MRLs.
Several physical parameters may interfere with analytical accuracy. High sampling flow rates and high
temperature and humidity may cause decreased adsorption of methylene chloride vapor on the solid
sorbent (APHA 1977). In addition, methylene chloride is a common laboratory contaminant and is
frequently found in laboratory blanks and in the environment (e.g., soil and water samples).
6.3 ADEQUACY OF THE DATABASE
Section 104(i)(5) of CERCLA, as amended, directs the Administrator of ATSDR (in consultation with the
Administrator of EPA and agencies and programs of the Public Health Service) to assess whether
adequate information on the health effects of methylene chloride is available. Where adequate
information is not available, ATSDR, in conjunction with the National Toxicology Program (NTP), is
required to assure the initiation of a program of research designed to determine the health effects (and
techniques for developing methods to determine such health effects) of methylene chloride.
The following categories of possible data needs have been identified by a joint team of scientists from
ATSDR, NTP, and EPA. They are defined as substance-specific informational needs that if met would
reduce the uncertainties of human health assessment. This definition should not be interpreted to mean
204 METHYLENE CHLORIDE
6. ANALYTICAL METHODS
that all data needs discussed in this section must be filled. In the future, the identified data needs will be
evaluated and prioritized, and a substance-specific research agenda will be proposed.
6.3.1 Identification of Data Needs
Methods for Determining Biomarkers of Exposure and Effect. Exposure to methylene chloride
may be evaluated by measuring the levels of this compound in blood (Ashley et al. 1994), urine, breath
(DiVincenzo et al. 1971), adipose tissue (Engstrom and Bjurstrom 1977), and human milk (Pellizari et al.
1982). Sensitive methods such as gas chromatography with a flame ionization detector (GC/FID) and gas
chromatography with a mass spectrometry detector (GC/MS) are available for the determination of
methylene chloride in biological materials. However, additional data on detection limits and accuracy are
needed to determine whether these methods are sufficiently sensitive and specific to identify individuals
with low-level exposures.
Neurological or neurobehavioral effects are characteristic markers of effects of methylene chloride.
These effects can occur in people exposed to low levels. No specific tests are known that measure
specific biomarkers of methylene chloride effects. The tests are standard for a wide range of chemicals
and are not very sensitive. Therefore, other methods are needed to identify specific biomarkers of
methylene chloride exposure.
Methods for Determining Parent Compounds and Degradation Products in Environmental
Media. Air is the environmental medium of most concern for human exposure to methylene chloride.
Exposure from drinking water may also be of concern in some areas, such as near hazardous waste sites.
Existing analytical methods can measure methylene chloride in these and other environmental media at
background levels. Analytical methods such as gas chromatography with flame ionization detector
(GC/FID), electron capture detector (GC/ECD), and mass spectrometry detector (GC/MS) are available
for determining methylene chloride in environmental media. High resolution gas chromatography with
mass spectrometry (HLGC/MS) and with electrolytic conductivity detector (HLGC/ELCD) as well as
Remote Sensing Fourier Transform Infrared (RS-FTIR) spectroscopy are also reliable. Exposure to
methylene chloride may be evaluated by measuring the levels of this compound in air (APHA, NIOSH),
water (APHA 1989a, 1989b; EPA 1989a, 1989b, 1989c, 1989d, 1989e, 1989f, 1989g), waste water (EPA
1998a, 1998b, 1998c, 1998d, 1998e, 1998f, 1998g, 1998h, 1998i, 1998j, 1998k, 1998l), soil/solid waste
205 METHYLENE CHLORIDE
6. ANALYTICAL METHODS
(EPA 1996a, 1996b, 1996c, 1996d), and food (Daft 1987; Heikes 1987; Page and Charbonneau 1977,
1984). The accuracy and precision of the methods are well documented and mass spectrometry provides
adequate specificity. Development of methods to improve the accuracy, sample preparation, and transfer
techniques are needed to monitor environmental media, especially how to preclude false positives.
6.3.2 Ongoing Studies
The following information was found as a result of a search of Federal Research in Progress (FEDRIP
1996).
The Environmental Health Laboratory Sciences Division of the National Center for Environmental
Health, Centers for Disease Control and Prevention, is developing methods for the analysis of methylene
chloride and other volatile organic compounds in blood. These methods use purge and trap methodology,
high resolution gas chromatography, and magnetic sector mass spectrometry which gives detection limits
in the low parts per trillion (ppt) range.
The Environmental Health Laboratory Sciences Division of the National Center for Environmental
Health, Centers for Disease Control and Prevention, is developing methods for the analysis of methylene
chloride and other phenolic compounds in urine. These methods use high resolution gas chromatography
and magnetic sector mass spectrometry which gives detection limits in the low parts per trillion (ppt)
range.
Other on-going studies to improve analytical methods for methylene chloride and related compounds
include the EPA "Master Analytical Scheme" being developed for organic compounds in water (Michael
et al. 1988), the research in supercritical fluid extraction (King 1989) which is applicable to organohalide
analytes, and the development of whole column cryotrapping techniques (Pankow and Rosen 1988).
These improvements are designed to overcome problems with purge and trap sample preparation and
increase sensitivity, reliability, and speed of the analyses.
207 METHYLENE CHLORIDE
7. REGULATIONS AND ADVISORIES
The international, national, and state regulations and guidelines regarding methylene chloride in air,
water, and other media are summarized in Table 7-1.
The International Agency for Research on Cancer (IARC) classifies methylene chloride as a Group 2B
carcinogen (possibly carcinogenic to humans) (IARC 1987). The Department of Health and Human
Services (DHHS) has determined that methylene chloride may reasonably be anticipated to be a
carcinogen. The National Institute for Occupational Safety and Health has listed methylene chloride as a
possible human carcinogen (NIOSH 1997).
OSHA requires employers of workers who are occupationally exposed to methylene chloride to institute
engineering controls and work practices to reduce and maintain employee exposure at or below
permissible exposure limits (PEL). The employer must use engineering and work practice controls, if
feasible, to reduce exposure to or below an 8-hour TWA of 25 ppm (OSHA 1998a, 1998b). Respirators
must be provided and used during the time period necessary to install or implement feasible engineering
and work practice controls, or where controls are not yet sufficient. Respirators are also required when
the employer determines that compliance with the TWA or PEL is not feasible with engineering or work
practice controls, such as maintenance and repair activities, vessel cleaning, or other operations where
exposures are intermittent and limited in duration, and in emergencies (OSHA 1987).
The EPA has calculated a chronic oral reference dose (RfD) of 0.06 mg/kg/day for methylene chloride
based on a NOAEL of approximately 6 mg/kg/day for rats in a 2-year drinking water bioassay (IRIS
1999; Serota et al. 1986a). The critical effect was liver toxicity. ATSDR has calculated an acute
inhalation MRL of 0.6 ppm based on a LOAEL of 300 ppm in an acute study in humans that evaluated
the effects of methylene chloride on the central nervous system (Winneke 1974). This MRL supersedes
the previous acute inhalation MRL of 3 ppm derived in the 1998 draft for public comment version of this
profile. In the new derivation of this MRL, a PBPK model (Reitz et al. 1997) was applied to adjust the
dosage yielding an adjusted LOAEL of 60 ppm. An intermediate-duration inhalation MRL of 0.3 ppm
was calculated based on a 100-day inhalation study in rats that identified a LOAEL of 25 ppm for liver
effects (Haun et al. 1972). A chronic inhalation MRL of 0.3 ppm was calculated based on a NOAEL of
50 ppm for liver effects (Nitschke et al. 1988a). Using a PBPK model for inhalation-to-oral extrapolation
of the Winneke (1974) data, an acute oral MRL of 0.2 mg/kg/day was calculated, based on a LOAEL of
208 METHYLENE CHLORIDE
7. REGULATIONS AND ADVISORIES
16 mg/kg/day for neurological effects (Reitz et al. 1997). This MRL supersedes the previous acute oral
MRL of 0.5 mg/kg/day published in the 1998 draft for public comment version of this profile. In the new
derivation, the uncertainty factor for human variability was increased from 3 to 10. ATSDR has also
calculated a chronic oral MRL of 0.06 mg/kg/day based on a NOAEL of 6 mg/kg/day for liver effects
(Serota et al. 1986a). This MRL supersedes the previous chronic oral MRL of 0.2 mg/kg/day published
in the 1998 draft for public comment version of this profile. In the new derivation, the uncertainty factor
for extrapolation from animals to humans was increased from 3 to 10.

209 METHYLENE CHLORIDE
7. REGULATIONS AND ADVISORIES
Table 7-1. Regulations and Guidelines Applicable to Methylene Chloride
Agency Description Information References
INTERNATIONAL
IARC Carcinogenic classification Group 2B
a
IARC 1987
NATIONAL
Regulations:
a. Air:
ACGIH TLV-TWA 50 ppm ACGIH 1999
NIOSH REL Lowest feasible NIOSH 1999
concentration
OSHA PEL 8hr-TWA 25 ppm 29 CFR 1910.1052
STEL determined over a 15 125 ppm OSHA 1999
minute sampling period
b. Water:
EPA MCL applying to community 0.005 mg/L 40 CFR 141.61
and non-transient, non- EPA 1999g
community water systems
MCLG Zero 40 CFR 141.50
EPA 1999f
Health Advisories EPA 1996a
1-day (10-kg child) 10 mg/L
10-day (10-kg child) 2 mg/L
Longer-term child ND
b
Longer-term adult ND
DWEL 2 mg/L
Water Quality Criteria EPA 1999j
water + organisms 4.7 µg/L
organisms only 1600 µg/L
c. Food:
EPA Indirect food additive: No tolerance limit when 40 CFR 180.1001
ingredient in pesticide used as an inert EPA 1999i
formulations as a solvent ingredient
used on growing crops
Exemption from tolerance for Yes 40 CFR 180.1010
fumigant uses EPA 1998b
FDA Ban on use of methylene Yes 21 CFR 700.19
chloride in cosmetic products FDA 1999c

210 METHYLENE CHLORIDE
7. REGULATIONS AND ADVISORIES
Table 7-1. Regulations and Guidelines Applicable to
Methylene Chloride (continued)
Agency Description Information References
FDA (contd)
Methylene chloride present in
foods with the following limits:
Spice oleoresins, residue
Limit in hops extract
Hops extract, residue
Decaffeinated roasted
coffee and soluble coffee
extract (instant coffee)
Indirect food additive
component of adhesives
used in food packaging or
transporting
Indirect food additive: used in
the production of
polycarbonate resins which
are used for food packaging
and transport
Indirect food additive: used in
ink for marking fruits and
vegetables
d. Other:
ACGIH Carcinogenic classification
Biological Exposure Indices
Methemoglobin in blood
EPA RfD (oral)
Carcinogen Classification
Cancer slope factor (q1*)
Inhalation unit risk
Reportable quantity for
hazardous substances
Methylene chloride -
designated CERCLA
hazardous substance
under sections 307(a) of
the Clean Water Act and
RCRA section 3001
Toxic Chemical Release
Reporting— effective date
Identification and listing as a
hazardous waste
30 ppm
2.2%
5 ppm
10 ppm
Yes
Yes
Yes
A3
c
1.5% of hemoglobin
6x10
-2
mg/kg/day
Group B2
d
7.5x10
-3
(mg/kg)/day
4.7x10
-7
(µg/m
3
)
-1
1,000 lbs
1/1/87
Yes
21 CFR 173.255
FDA 1999b
21 CFR 175.105
FDA 1999a
21 CFR 177.1580
FDA 1999d
21 CFR 73.1
FDA 1999e
ACGIH 1999
IRIS 1999
40 CFR 302.4
EPA 1999a
40 CFR 372.65
EPA 1999b
40 CFR 261.33
EPA 1999c

211 METHYLENE CHLORIDE
7. REGULATIONS AND ADVISORIES
Table 7-1. Regulations and Guidelines Applicable to
Methylene Chloride (continued)
Agency Description Information References
d. Other:
EPA (contd)
List of toxic pollutants
designated pursuant to
section 307(a)(1) of the Act
OSHA Inhalation Unit Risk
USC List of hazardous air
pollutants
Universal treatment
standards
wastewater
non-wastewater
STATE
Regulations and
guidelines:
a. Air:
Idaho Acceptable ambient
concentration for a
carcinogen
Kansas Concentration limits for
hazardous air emissions
Massachusetts Acceptable ambient air
concentrations
New Jersey Acceptable ambient air
concentrations
New York Acceptable ambient air
concentrations
North Carolina Acceptable ambient air
concentrations
Rhode Island Acceptable ambient air
concentrations
Vermont Acceptable ambient air
concentrations
Washington Acceptable ambient air
concentrations
Wisconsin Acceptable emission levels
<25 feet
25 feet
Yes
3.62x10
-3
(µg/m
3
)
-1
Yes
0.089 mg/L
30 mg/L
2.4x10
-1
µg/m
3
10 tons/year
0.24 µg/m
3
(annual)
4.7x10
-7
µg/m
3
27.0 µg/m
3
(annual)
2.4x10
-2
mg/m
3
(annual)
2.0 µg/m
3
(annual)
0.02 µg/m
3
(annual)
2.0 µg/m
3
(annual)
29 lbs/hr
122 lbs/hr
40 CFR 401.15
EPA 1998h
OSHA 1997
42 USC 7412
USC 1999
40 CFR 268.48
EPA 1999d
ID Dept Health
Welfare 1999
KS Dept. Health
Env 1998
MA Dept. Env.
Protect. 1998
NJ Dept Env.
Protect. 1998
NY Dept. Env.
Conserv. 1998
NC Div. Env.
Manage. 1998
RI Dept. Env
Management 1992
VT Nat. Res.
Agency 1998
WA Dept. Ecology
1998
WI Dept Natural
Resources 1997

212 METHYLENE CHLORIDE
7. REGULATIONS AND ADVISORIES
Table 7-1. Regulations and Guidelines Applicable to
Methylene Chloride (continued)
Agency Description Information References
b. Water.
Alabama Human health criteria for the AL Dept Env
consumption of: Management 1998
water and organism
e
3.4x10-4 µg/L
organism only
e
1.48x10-2 µg/L
Alaska Maximum contaminant level 0.005 mg/L AK Dept Env
Conserv 1999
New Jersey Ground water quality 2 µg/L NJ Dept Env Protec
1993
Oklahoma Ground water quality criteria 10.0 µg/L OK Dept Env
Quality 1997
South Dakota Maximum contaminant 0.005 mg/L SD Dept Env
levels— apply to community Natural Resources
and non-transient and non- 1998
community water systems
a
: Group 2B - The agent is possibly carcinogenic to humans
b
: ND - Not data
c
: A3 - Confirmed animal carcinogen with unknown relevance to humans
d
: B2 - Probable human carcinogen
e
: The following equations were used to calculate the values as given in the Alabama State laws:
Consumption of water and organism:
Concentration (mg/l) = (HBW x RL)/(CPF x [(FCR x BCF) + WCR])
Consumption of organism only:
Concentration (mg/l) = (HBW x RL)/(CPF x FCR x BCF)
HBW = human body weight, set at 70 kg
RL = risk level, set at 1 x 10
-5
CPF = cancer potency factor, 0.0075 (kg-day)/mg
FCR = fish consumption rate, set at 0.030 kg/day
BCF = bioconcentration factor, 0.9 L/kg
WCR = water consumption rate, set at 2 L/day
ACGIH = American Conference of Governmental Industrial Hygienists; DWEL = drinking water equivalent level;
EPA = Environmental Protection Agency; FDA = Food and Drug Administration; IARC = International Agency for
Research on Cancer; IRIS = Integrated Risk Information System; MCL = maximum contaminant level;
MCLG = maximum contaminant level goal; NIOSH = National Institute of Occupational Safety and Health;
OSHA = Occupational Safety and Health Administration; PEL = permissible exposure limit; REL = recommended
exposure release; RfD = reference dose; TLV= threshold limit value; TWA = time-weighted average
___________________
213 METHYLENE CHLORIDE
8. REFERENCES
Abelson PH. 1993. Health risk assessment. Regul Toxicol Pharmacol 17:219-223.
*ACGIH. 1986. Methylene chloride. Documentation of the threshold limit values and biological
exposure indices. American Conference of Governmental Industrial Hygienists. Cincinnati, OH.
*ACGIH. 1990. Methylene chloride. Threshold limit values for chemical substances and physical
agents and biological exposure indices. American Conference of Governmental Industrial Hygienists.
Cincinnati, OH.
*ACGIH. 1998. Threshold limit values for chemical substances and physical agents and biological
exposure indices. American Conference of Governmental Industrial Hygienists. Cincinnati, OH.
*ACGIH 1999. Methylene chloride. Documentation of the threshold limit values and biological
exposure indices. American Conference of Governmental Industrial Hygienists. Cincinnati, OH.
*Adinolfi M. 1985. The development of the human blood-CSF-brain barrier. Dev Med Child Neurol
27:532-537.
*Adlercreutz H. 1995. Phytoestrogens: Epidemiology and a possible role in cancer protection. Environ
Health Perspect Suppl 103(7):103-112.
*AK Dept Env Conserv. 1999. Drinking water. Alaska Department of Environmental Conservation.
Rule 18 AAC 80. http://www.state.ak.us/local/akpages/ENV.CONSERV/home.htm
*AL Dept Env Management. 1998. Water quality criteria. Alabama Department of Environmental
Management, Water Division. http://www.adem.state.al.us/
*Allen J, Kligerman A, Campbell J, et al. 1990. Cytogenetic analyses of mice exposed to
dichloromethane. Environ Mol Mutagen 15:221-228.
Allen MR, Braithwaite A, Hills CC. 1997. Trace organic compounds in landfill gas at seven U.K. waste
disposal sites. Environ Sci Technol 31:1054-1061.
*Altman PK, Dittmer DS. 1974. Biological handbooks: Biology data book, Vol 3. 2nd ed. Bethesda,
MD: Federation of American Societies for Experimental Biology 1987-2008, 2041.
*Altshuller AP. 1980. Lifetimes of organic molecules in the troposphere and lower stratosphere. Adv
Environ Sci Technol 10:181-219.
*Amoore JE, Hautala E. 1983. Odor as an aid to chemical safety: Odor thresholds limit values and
volatilities for 214 industrial chemicals in air and water dilution. J Appl Toxicol 3:272-290.
* Cited in text
*Anders MW, Sunram JM. 1982. Transplacental passage of dichloromethane and carbon monoxide.
Toxicol Lett 12:231-234.
214 METHYLENE CHLORIDE
8. REFERENCES
Anders MW, Kubic VL, Ahmed AE. 1977. Metabolism of halogenated methanes and macromolecular
binding. J Environ Pathol Toxicol 1:117-124.
Andersen ME. 1995. Development of physiologically based pharmacokinetic and physiologically based
pharmacodynamic models for applications in toxicology and risk assessment. Toxicol Lett 79:35-44.
*Andersen ME, Krishnan K. 1994. Physiologically based pharmacokinetics and cancer risk assessment.
Environ Health Perspect 1:103-108.
*Andersen ME, Clewell HJ, Gargas ML, et al. 1987. Physiologically based pharmacokinetics and the
risk assessment process for methylene chloride. Toxicol Appl Pharm 87:185-205.
*Andersen ME, Clewell HJ, Gargas ML, et al. 1991. Physiologically based pharmacokinetic modeling
with dichloromethane, its metabolite, carbon monoxide, and blood carboxyhemoglobin in rats and
humans. Toxicol Appl Pharmacol 108:14-27.
*Andersen ME, Clewell HJ, Mahle DA, et al. 1994. Gas uptake studies of deuterium isotope effects on
dichloromethane metabolism in female B6C3F1 mice in vivo. Toxicol Appl Pharmacol 128:158-165.
Andersen MW, Maronpot RR. 1993. Methylene chloride-induced turmorigenesis. Carcinogenesis
14:787-788.
Andrae U, Wolff T. 1983. Dichloromethane is not genotoxic in isolated rat hepatocytes. Arch Toxicol
52:287-290.
Angelo MJ, Pritchard AB. 1984. Simulations of methylene chloride pharmacokinetics using a
physiologically based model. Regul Toxicol Pharm 4:329-339.
*Angelo MJ, Pritchard AB, Hawkins DR, et al. 1986a. The pharmacokinetics of dichloromethane. I.
Disposition in B6C3F
1
mice following intravenous and oral administration. Food Chem Toxicol
24:965-974.
*Angelo MJ, Pritchard AB, Hawkins DR, et al. 1986b. The pharmacokinetics of dichloromethane. II.
Disposition in Fischer 344 rats following intravenous and oral administration. Food Chem Toxicol
24(9):975-980.
Antoine SR, DeLeon IR, O'Dell-Smith RM. 1986. Environmentally significant volatile organic
pollutants in human blood. Bull Environ Contam Toxicol 36:364-371.
*Anundi H, Lind ML, Friis L, et al. 1993. High exposures to organic solvents among graffiti removers.
Int Arch Occup Environ Health 65:247-251.
*APHA. 1977. Methods of air sampling and analysis. 2nd ed. Washington, DC: American Public
Health Association, 894-902.
*APHA. 1989a. Purge and trap capillary-column gas chromatographic method. In: Standard methods
for the examination of water and wastewater. 17th ed. Washington, DC: American Public Health
Association.
215 METHYLENE CHLORIDE
8. REFERENCES
*APHA. 1989b. Purge and trap capillary-column gas chromatographic/mass spectrometric method. In:
Standard methods for the examination of water and wastewater. 17th ed. Washington, DC: American
Public Health Association.
*APHA. 1998a. Method - 6210B. Standard methods for the evaluations of water and wastewater. 20
th
ed. Washington, DC: American Public Health Association.
*APHA. 1998b. Method - 6230B. Standard methods for the evaluations of water and wastewater. 20
th
ed. Washington, DC: American Public Health Association.
Arulgnanendran VRJ, Nirmalakhandan N. 1997. Microbial toxicity in soil medium. Ecotoxicol Environ
Saf 39:48-56.
*Ashley DI, Bonin MA, Cardinali FL, et al. 1994. Blood concentrations of volatile organic compounds
in a non occupationally exposed US population and in groups with suspected exposure. Clin Chem
40:1401-1404.
*Åstrand I, Övrum P, Carlsson A. 1975. Exposure to methylene chloride: I. Its concentration in
alveolar air and blood during rest and exercise and its metabolism. Scand J Work Environ Health
1:78-94.
*ATSDR. 1989. Agency for Toxic Substances and Disease Registry. Decision guide for identifying
substance-specific data needs related to toxicological profiles; Notice. Federal Register 54(174):37618-
37634.
*ATSDR. 1990. Methylene chloride toxicity. Case studies in environmental medicine. Atlanta, GA:
Agency for Toxic Substances and Disease Registry, U.S. Department of Health and Human Services.
PB 85-241529.
*ATSDR. 1993. Toxicological profile for methylene chloride. Atlanta, GA: Agency for Toxic
Substances and Disease Registry, U.S. Department of Health and Human Services. PB 93-182483.
*ATSDR/CDC. 1990. Biomarkers of organ damage or dysfunction for the renal, hepatobiliary, and
immune systems. Subcommittee on Biomarkers of Organ Damage and Dysfunction, Agency for Toxic
Substances and Disease Registry, Atlanta, GA.
*Aviado DM, Belej MA. 1974. Toxicity of aerosol propellants on the respiratory and circulatory
systems: I. Cardiac arrhythmia in the mouse. Toxicology 2:31-42.
Aviado DM, Zakhari S, Watanabe T. 1977. Methylene chloride. In: Goldberg L, ed. Non-fluorinated
propellants and solvents for aerosols. Cleveland, OH: CRC Press, 19-45.
Bader R, Leisinger T. 1994. Isolation and characterization of the Methylophilus sp. strain DM11 gene
encoding dichloromethane dehalogenase/glutathione s-transferase. J Bacteriol 176:3466-3473.
*Bahnick DA, Doucette WJ. 1988. Use of molecular connectivity indices to estimate soil sorption
coefficients for organic chemicals. Chemosphere 17(9):1703-1715.
*Bakinson MA, Jones RD. 1985. Gassings due to methylene chloride, xylene, toluene, and styrene
reported to Her Majesty’s factory inspectorate 1961-80. Br J Ind Med 42:184-190.
216 METHYLENE CHLORIDE
8. REFERENCES
*Ballantyne B, Gazzard MF, Swanson DW. 1976. Ophthalmic toxicology of dichloromethane.
Toxicology 6:173-187.
Balmer FM, Smith FA, Leach LJ, et al. 1972. Effects in the liver of methylene chloride inhaled alone
and with ethyl alcohol. Am Ind Hyg Assoc J 37:345-352.
Balogh S, Onayiga R, Edblad D. 1998. Evaluation of an automatic composite sampler for volatile
organic compounds in raw wastewater. J Air Waste Manage Assoc 48:271-275.
Banerjee S, Howard PH. 1988. Improved estimation of solubility and partitioning through correction of
UNIFAC-derived activity coefficients. Environ Sci Technol 22:839-841.
*Barnes DG, Dourson M. 1988. Reference dose (RfD): Description and use in health risk assessments.
Regul Toxicol Pharmacol 8:471-486.
*Barrowcliff DF, Knell AJ. 1979. Cerebral damage due to endogenous chronic carbon monoxide
poisoning caused by exposure to methylene chloride. J Soc Occup Med 29:12-14.
*Bell BP, Franks P, Hildreth N, et al. 1991. Methylene chloride exposure and birthweight in Monroe
County, New York. Environ Res 55:31-39.
Bell DA, Taylor JA, Paulson DF, et al. 1993. Genetic risk and carcinogen exposure: A common
inherited defect of the carcinogen-metabolism gene glutathione S-transferase M1 (GSTM1) that increases
susceptibility to bladder cancer. J Natl Cancer Inst 85:1159-1164.
*Bellar TA, Lichtenberg JJ, Kroner RC. 1974. The occurrence of organohalides in chlorinated drinking
waters. J Am Water Works Assoc (December):703-706.
*Berger GS. 1994. Epidemiology of endometriosis. In: Berger GS, ed. Endometriosis: Advanced
management and surgical techniques. New York, NY: Springer-Verlag.
Berger M, Fodor GG. 1968. CNA disorders under the influence of air mixtures containing
dichloromethane. Zentrabl Bakteriol 215:417.
Blancato JN. 1991. Physiologically-based pharmacokinetic models in risk and exposure assessment.
Ann Ist Super Sanita 27(4):601-608.
*Blocki FA, Logan MSP, Baoli C, et al. 1994. Reaction of rat liver glutathione s-transferase and
bacterial dichloromethane dehalogenase with dihalomethanes. J Biol Chem 269:8826-8830.
*Bogaards JJP, van Ommen B, van Bladeren PJ. 1993. Interindividual differences in the in vitro
conjugation of methylene chloride with glutathione by cytosolic glutathione S-transferase in 22 human
liver samples. Biochem Pharmacol 45:2166-2169.
*Bonventre J, Brennan O, Juson D, et al. 1977. Two deaths following accidental inhalation of
dichloromethane and 1,1,1- trichloroethane. J Anal Toxicol 1:158-160.
Borisover MD, Graber ER. 1997. Specific interactions of organic compounds with soil; Organic carbon.
Chemosphere 34(8):1761-1776.
217 METHYLENE CHLORIDE
8. REFERENCES
*Bornschein RL, Hastings L, Mason JM. 1980. Behavioral toxicity in the offspring of rats following
maternal exposure to dichloromethane. Toxicol Appl Pharmacol 52:29-37.
Brack W, Rottler H, Frank H. 1998. Volatile fractions of landfill leachates and their effect on
chlamydomonas reinhardtii: in vivo chlorophyll a fluorescence. Environ Toxicol Chem
17(10):1982-1991.
Brewer WE, Galipo RC, Morgan SL, et al. 1997. The confirmation of volatiles by solid-phase
microextraction and GC/MS in the investigation of two traffic fatalities. J Anal Toxicol 21: 286-290.
*Briving C, Hamberger A, Kjellstrand P, et al. 1986. Chronic effects of dichloromethane on amino
acids, glutathione and phosphoethanolamine in gerbil brain. Scand J Work Environ Health 12:216-220.
Brodbelt JS, Cooks RG, Tou JC, et al. 1987. In vivo mass spectrometric determination of organic
compounds in blood with a membrane probe. Anal Chem 59:454-458.
Brodzinski R, Singh HB. 1983. Volatile organic chemicals in the atmosphere: An assessment of
available data. Research Triangle Park, NC: U.S. Environmental Protection Agency, Environmental
Sciences Research Laboratory. EPA-600/S/3-83-027.
Brooke D, Howe P. 1994. Environmental hazard assessment: Dichloromethane. London, England:
Toxic Substances Division, Department of the Environment.
*Brown KW, Donnelly KC. 1988. An estimation of the risk associated with the organic constituents of
hazardous and municipal waste landfill leachates. Hazardous Waste and Hazardous Materials 5(1):1-30.
*Brown-Woodman PDC, Hayes LC, Huq F, et al. 1998. In vitro assessment of the effect of halogenated
hydrocarbons: chloroform, dichloromethane, and dibromoethane on embryonic development of the rat.
Teratology 57:321-333.
*Bruce BW, McMahon PB. 1996. Shallow ground-water quality beneath a major urban center: Denver,
Colorado, USA. J Hydrol 186:129-151.
*Brunner WD, Staub D, Leisinger T. 1980. Bacterial degradation of dichloromethane. Appl Environ
Microbiol 40(5):950-958.
*Brzezinski MR, Boutelet-Bochan H, Person RE, et al. 1999. Catalytic and quantitation of cytochrome
P-450 2E1 in prenatal human brain. J Pharmacol Exp Ther 289(3):1648-1653.
Budavari S, O'Neil M, Smith A, eds. 1989. The Merck index: An encyclopedia of chemicals, drugs and
biologicals. 11th ed. Rahway, NJ: Merck and Company, Inc.
*Bukowski JA, Sargent EV, Pena BM. 1992. Evaluation of the utility of a standard history questionnaire
in assessing the neurological effects of solvents. Am J Ind Med 22:337-345.
Burek JD, Nitschke KD, Bell TJ, et al. 1980. Methylene chloride: A two-year inhalation toxicity and
oncogenicity study in rats and hamsters. Dow Chemical Company, Health and Environmental Sciences,
Toxicology Research Laboratory, Midland, MI.
*Burek JD, Nitschke KD, Bell TJ, et al. 1984. Methylene chloride: A two-year inhalation toxicity and
oncogenicity study in rats and hamsters. Fund Appl Toxicol 4:30-47.
218 METHYLENE CHLORIDE
8. REFERENCES
Bus JS, Reitz RH. 1992. Dose-dependent metabolism and dose setting in chronic studies. Toxicol Lett
64/65:669-676.
Butler FE, Coppedge EA, Suggs JC, et al. 1988. Development of a method for determination of
methylene chloride emissions at stationary sources. JAPCA 38:272-277.
Butler R, Solomon IJ, Snelson A. 1978. Rate constants for the reaction of OH with halocarbons in the
presence of O
2
+ N
2
. J Air Pollut Control Assoc 28:1131-1133.
Cabbar HC, Varol N, McCoy BJ. 1998. Sorption and diffusion of chlorinated methanes in moist clay.
AIChE J 44(6):1351-1355.
*California Environmental Protection Agency. 1992. Health risk assessment of dichloromethane
(methylene chloride) in California ground water. California: Lawrence Livermore National Laboratory,
U.S. Department of Energy. NTIS no. DE93-018480.
*Callahan MA. 1981. Written communication to Marilyn C. Bracken, Toxic Substances Priorities
Committee, regarding the final report of TSPC solvents work group #2. Office of Pesticides and Toxic
Substances, U.S. Environmental Protection Agency, Washington, DC.
*Cantor KP, Stewart PA, Brinton LA, et al. 1995. Occupational exposures and female breast cancer
mortality in the United States. J Occ Env Med 37(3):336-348.
*Carlsson A, Hultengren M. 1975. Exposure to methylene chloride: III. Metabolism of
14
C-labeled
methylene chloride in rat. Scand J Work Environ Health 1:104-108.
*Carpenter SP, Lasker JM, Raucy JL. 1996. Expression, induction, and catalytic activity of the ethanol-
inducible cytochrome P450 (CYP2E1) in human fetal liver and hepatocytes. Mol Pharmacol 49:260-268.
*Carpenter SP, Savage DD, Schultz ED, et al. 1997. Etanol-mediated transplantational induction of
CYP2E1 in fetal rat liver. J Pharmacol Exp Ther 282:1028-1036.
Casanova M. 1996. Are you a man or a mouse? Regul Toxicol Pharmacol 24:106.
*Casanova M, Deyo DF, Heck HA. 1992. Dichloromethane (Methylene chloride): Metabolism to
formaldehyde and formation of DNA-protein cross-links in B6C3F1 mice and Syrian golden hamsters.
Toxicol Appl Pharmacol 114:162-165.
*Casanova M, Bell DA, Heck HA. 1997. Dichloromethane metabolism to formaldehyde and reaction of
formaldehyde with nucleic acids in hepatocytes of rodents and humans with and without glutathione
S-transferase T1 and M1 genes. Fund Appl Toxicol 37:168-180.
*Casanova M, Conolly RB, Heck HA. 1996. DNA-protein crosslinks (DPX) and cell proliferation in
B6C3F
1
mice but not Syrian golden hamsters exposed to dichloromethane: Pharmacokinetics and risk
assessment with DPX as dosimeter. Fundam Appl Toxicol 31:103-116.
CCRIS. 1998. Chemical Carcinogenesis Research Information System. National Library of Medicine,
National Toxicology Information Program, Bethesda, MD. May 11, 1998.
*C&EN 1996. Production by the U.S. chemical industry. Chemical and Engineering News June24, 1996.
219 METHYLENE CHLORIDE
8. REFERENCES
Chang H-L, Alvarez-Cohen L. 1996. Biodegradation of individual and multiple chlorinated aliphatic
hydrocarbons by methane-oxidizing cultures. Appl Environ Microbiol 62(9):3371-3377.
Charnley G. 1992. Cancer dose-response modeling and methylene chloride. In: Zervos C., ed.
Oncogene and transgenics correlates of cancer risk assessments. New York, NY: Plenum Press, 231-240.
Chellman GJ, Hurtt ME, Bus JS, et al. 1987. Role of testicular versus epididymal toxicity in the
induction of cytotoxic damage in Fischer-344 rat sperm by methyl chloride. Reprod Toxicol 1:25-35.
Chemical Exposure. 1990. Dialog Information Systems, Inc., Palo Alto, CA. July 19, 1990.
Chemical Regulations and Guidelines. 1990. Dialog Information Systems, Inc., Palo Alto, CA.
July 19, 1990.
Chemline. 1990. Chemical dictionary online. National Library of Medicine, National Toxicology
Information Program, Bethesda, MD. July 5, 1990.
*Chen C-L, Liu Q, Relling MV. 1996. Simultaneous characterization of glutathione S-transferase M1
and T1 polymorphisms by polymerase chain reaction in American whites and blacks. Parmacogenetics
6:187-191.
*Cherry N, Venables H, Waldron HA, et al. 1981. Some observations on workers exposed to methylene
chloride. Br J Ind Med 38:351-355.
*Cherry N, Venables H, Waldron HA. 1983. The acute behavioral effects of solvent exposure. J Soc
Occup Med 33:13-18.
*Ciuchta HP, Savell GM, Spiker RC. 1979. The effect of alcohols and toluene upon methylene chloride-
induced carboxyhemoglobin in the rat and monkey. Toxicol Appl Pharmacol 49:347-354.
Clewell HJ. 1993. Coupling of computer modeling with in vitro methodologies to reduce animal usage
in toxicity testing. Toxicol Lett 68:101-117.
Clewell HJ. 1995. Incorporating biological information in quantitative risk assessment: An example with
methylene chloride. Toxicol 102:83-94.
*Clewell HJ, Andersen ME. 1985. Risk assessment extrapolations and physiological modeling. Toxicol
Ind Health 1(4):111-131.
Clewell HJ, Lee T, Carpenter RL. 1994. Sensitivity of physiologically based pharmacokinetic models to
variation in model parameters: Methylene chloride. Risk Anal 14:521-531.
Clewell HJ, Gentry PR, Gearhart JM. 1997. Investigation of the potential impact of benchmark dose and
pharmacokinetic modeling in noncancer risk assessment. J Toxicol Environ Health 52:475-515.
*CLPSD. 1990. Contract Laboratory Program Statistical Database. Viar and Company, Management
Service Division, Alexandria, VA. July 1990.
Cocco P, Dosemeci M, Gomez MR, et al. 1994. A retrospective evaluation of exposure to
dichloromethane by using a job-exposure matrix. Med Lav 85:84-87.
220 METHYLENE CHLORIDE
8. REFERENCES
*Cocco P, Heineman EF, Dosemeci M. 1999. Occupational risk factors for cancer of the central nervous
system (CNS) among US women. Am J Ind Med 36:70-74.
Cohen N, Benson SW. 1987. Empirical correlations for rate coefficients for reactions of OH with
haloalkanes. J Phys Chem 91:171-175.
*Coleman WE, Lingg RD, Melton RG, et al. 1976. The occurrence of volatile organics in five drinking
water supplies using gas chromatography/mass spectrometry. In: Keith LH, ed. Identification and
analysis of organic pollutants in water. Ann Arbor, MI: Ann Arbor Science Publishers Inc., 305-327.
Coleman JJ, Blake DR, Rowland FS. 1998. Atmospheric residence time of CH
3
Br estimated from the
Junge spatial variability relation. Science 281:392-396.
*Corsi RL, Chang, DPY, Schroeder ED, et al. 1987. Emissions of volatile and potentially toxic organic
compounds from municipal wastewater treatment plants. Proceedings of the 80th annual meeting of the
Air Pollution Control Association, New York, NY, June 21-26, 1987.
Cox RA, Denwent RC, Eggleton AEJ, et al. 1976. Photochemical oxidation of halocarbons in the
troposphere. Atmos Environ 10:305-308.
*CPSC. 1987. Labeling of certain household products containing methylene chloride; statement of
interpretation and enforcement policy. Consumer Product Safety Commission. Federal Register
52(177):34698-34703.
*CPSC. 1990. Information on methylene chloride containing products; general order for submission.
Consumer Product Safety Commission. Federal Register 55(153):32282-32283.
Crebelli R, Benigni R, Franekic J, et al. 1988. Induction of chromosome malsegregation by halogenated
organic solvents in Aspergillus nidulans: Unspecific or specific mechanism? Mutat Res 201:401-411.
CRISP. 1990. Computer Retrieval of Information on Scientific Projects. National Institutes of Health,
Division of Research Grants, Bethesda, MD. July 16, 1990.
CRIS/USDA. 1998. Current Research Information System, U.S. Department of Agriculture. Dialog
Information Systems, Inc., Palo Alto, CA. May 11, 1998.
Cronn DR, Rasmussen RA, Robinson E. 1977. Report for phase II. Measurement of tropospheric
halocarbons by gas chromatography/mass spectrometry. Report to U.S. Environmental Protection
Agency.
Cronn DR, Robinson E. 1979. Determination of trace gases in Learjet and U-2 whole-air samples
collected during the intertropical convergence zone study. In: Poppoff IG, Page WA, Margozzi AP, eds.
Intertropical convergence zone experiment. NASA TMX78577.
*Crutzen PJ, Fishman J. 1977. Average concentrations of OH in the troposphere and the budgets of CH
4
,
CO, H
2
and CH
3
CCl
3
. Geophys Res Lett 4(8):321-324.
*Daft J. 1987. Determining multifumigants in whole grains and legumes, milled and low-fat grain
products, spices, citrus fruit, and beverages. J Assoc Off Anal Chem 70(4):734-739.
221 METHYLENE CHLORIDE
8. REFERENCES
Daft JL. 1989. Determination of fumigants and related chemicals in fatty and nonfatty foods. J Agric
Food Chem 37:560-564.
Dai Y, Rashba-Step J, Cederbaum AI. 1993. Stable expression of human cytochrome P4502E1 in
HepG2 cells: Characterization of catalytic activities and production of reactive oxygen intermediates.
Biochemistry 32:6928-6937.
*Dankovic DA, Bailer AJ. 1994. The impact of exercise and intersubject variability on dose estimates
for dichloromethane derived from a physiologically based pharmacokinetic model. Fundam Appl Toxicol
22:20-25.
DART. 1990. Developmental and Reproductive Toxicology. National Library of Medicine, National
Toxicology Information Program, Bethesda, MD. July 6, 1990.
*Davis DD, Machado G, Conaway B, et al. 1976. A temperature-dependent kinetics study of the
reaction of OH with CH
3
Cl, CH
2
Cl
2
, CHCl
3
and CH
3
. Br J Chem Phys 65:1268-1274.
*Davis EM, Murray HE, Liehr JG, et al. 1981. Basic microbial degradation rates and chemical
byproducts of selected organic compounds. Water Res 15:1125-1127.
*Davis JW, Madsen SS. 1991. The biodegredation of methylene chloride in soils. Environ Toxicol
Chem 10:463-474.
*DeJohgn J, Blaauboer BJ. 1996. Simulation of toluene kinetics in the rat by a physiologically based
pharmacokinetic model with application of biotransformation parameters derived independently in vitro
and in vivo. Fundam Appl Toxicol 32:260-268.
*DeJohgn J, Verhaar HJM, Hermens JLM. 1998. Role of kinetics in acute lethality of nonreactive
volatile organic compounds (VOCs). Toxicol Sci 45:26-32.
*DeMarini DM, Shelton ML, Warren SH, et al. 1997. Glutatione S-transferase-mediated induction of
GCÿAT transitions by halomethanes in Salmonella. Environ Mol Mutagen 30:440-447.
DeMedinilla J, Espigares M. 1988. Contamination by organic solvents in auto paint shops. Ann Occup
Hyg 32:509-513.
*d'Errico A, Malats N, Vineis P, et al. 1999. review of studies of selected metabolic polymorphisms and
cancer. In: Vineis P, Malats N, Lang M, et al., ed. Metabolic polymorphisms and susceptibility to
cancer. Lyon, France: International Agency for Research on Cancer, 323-393.
Derwent RG, Jenkin ME, Saunders SM. 1996. Photochemical ozone creation potentials for a large
number of reactive hydrocarbons under European conditions. Atmos Environ 30(2):181-199.
*Devereux TR, Foley JF, Maronpot RR, et al. 1993. Ras proto-oncogene activation in liver and lung
tumors from B6C3F1 mice exposed chronically to methylene chloride. Carcinogenesis 14(5): 795-801.
Dhanya S, Saini RD. 1997. Rate constants of OH radical reactions in gas phase with some fluorinated
compounds: a correlation with molecular parameters. Int J Chem Kinet 29:187-194.
Dhillon S, Burg RV. 1995. Methylene chloride. J Appl Toxicol 15:329-335.
222 METHYLENE CHLORIDE
8. REFERENCES
Dilling HPA, Shillaker RO. 1985. Toxicity review 12-dichloromethane (methylene chloride). Health
and Safety Executive. Her Majesty's Stationary Office, London, England.
*Dilling WL, Tefertiller NB, Kallos GJ. 1975. Evaporation rates of methylene chloride, chloroform,
1,1,1-trichloroethane, trichloroethylene, tetrachloroethylene, and other chlorinated compounds in dilute
aqueous solutions. Environ Sci Technol 9(9):833-838.
Dillon D, Edwards I, Combes R, et al. 1992. The role of glutathione in the bacterial mutagenicity of
vapour phase dichloromethane. Environ Mol Mutagen 20:211-217.
*DiVincenzo GD, Kaplan CJ. 1981. Uptake, metabolism, and elimination of methylene chloride vapor
by humans. Toxicol Appl Pharmacol 59:130-140.
*DiVincenzo GD, Yanno FJ, Astill BD. 1971. The gas chromatographic analysis of methylene chloride
in breath, blood, and urine. Am Ind Hyg Assoc J 32:387-391.
*DiVincenzo GD, Yanno FJ, Astill BD. 1972. Human and canine exposure to methylene chloride vapor.
Am Ind Hyg Assoc J 33:125-135.
*Dobbs RA, Wang L, Govind R. 1989. Sorption of toxic organic compounds on wastewater solids:
Correlation with fundamental properties. Environ Sci Technol 23:1092-1097.
Domenico PA, Palciauskas VV. 1982. Alternative boundaries in solid waste management. Ground
Water 20:303-311.
dos Santos LMF, Livingston AG. 1997. Mineralisation of 1,2-dibromoethane and other brominated
aliphatics under aerobic conditions. Water Sci Technol 36(10):17-25.
Dosemeci M, Cocco P, Gomez M, et al. 1994. Effects of three features of a job-exposure matrix on risk
estimates. Epidemiology 5:124-127.
Dow Chemical Company. 1961. The results of chronic skin absorption studies on chlorothene and
methylene chloride. Midland, Mississippi. OTS84003A.
Dow Chemical Company. 1982. Ninety day percutaneous absorption study in rabbits with Dow
Chemical Company - chlorothene; Dow Chemical Company - Methylene chloride; Dow Chemical
Company - Isopropyl Alcohol. Midland, Mississippi. OTS84003A.
Dow Chemical Company. 1994. Initial submission: Toxicological properties and industrial handling
hazards of (1) bromochloromethane; (2) methylene chloride; (3) flux oil; (4) bottoms from DBDPO
process. EPA/OTS Doc No. FYI-OTS-0794-1085.
*Dowty BJ, Carlisle DR, Laseter JL. 1975. New Orleans drinking water sources tested by gas
chromatography-mass spectrometry. Environ Sci Technol 9(8):762-765.
*Dunovant VS, Clark CS, QueHee SS, et al. 1986. Volatile organics in the wastewater and air spaces of
three wastewater treatment plants. J Water Pollut Control Fed 58(9):886-895.
Dupont Chemicals. 1970. Initial submission: toxicity tests on Project Victor samples with cover letter
dated 10/15/92. OTS0571515.
223 METHYLENE CHLORIDE
8. REFERENCES
*Dyksen JE, Hess AF. 1982. Alternatives for controlling organics in groundwater supplies. J Am Water
Works Assoc (74):394-403.
ECSA. 1995. Methylene chloride properties, uses and impact on the environment and health. European
Chlorinated Solvent Association. Brussels, Belgium. September 1995.
E. I. Dupont de Nemours & Co. 1992. Inhalation toxicity studies of methylene chloride with cover letter.
TSCATS-440754. NTIS/OTS 0555748.
Ellenhorn MJ, Barceloux DG. 1988. Medical toxicology. Diagnosis and treatment of human poisoning.
New York, NY: Elsevier Science Inc., 983-985.
*Ellenhorn MJ. 1997. Ellenhorn’s medical toxicology: Diagnosis and treatment of human poisoning.
New York, NY: Elsevier Science Inc., 1422t, 1423t, 1437t, 1438t, 1465-1466, 1408.
Elovaara E, Hemminki K, Vainio H. 1979. Effects of methylene chloride, trichloroethane,
trichloroethylene, tetrachloroethylene, and toluene on the development of chick embryos. Toxicology
12:111-119.
*El-Zien RA, Zwischenberger JB, Abdel-Rahman SZ, et al. 1997a. Polymorphism of metabolizing
genes and lung cancer histology: prevalence of CYP2E1 in adenocarcinoma. Cancer Lett 112:71-78.
*El-Zein RA, Zwischenberger JB, Wood TG, et al. 1997b. Combined genetic polymorphism and risk for
development of lung cancer. Mutat Res 381:189-200.
*EMMI. 1999a. Environmental monitoring methods index. Version 1.1. PC#4082. Rockville, MD:
U.S. Environmental Protection Agency, Government institutes. ASTM D3686.
*EMMI. 1999b. Environmental monitoring methods index. Version 1.1. PC#4082. Rockville, MD:
U.S. Environmental Protection Agency, Government institutes. ASTM D3687.
*EMMI. 1999c. Environmental monitoring methods index. Version 1.1. PC#4082. Rockville, MD:
U.S. Environmental Protection Agency, Government institutes. ASTM D4490.
Enfield CG, Carsel RF, Cohen SZ, et al. 1982. Approximating pollutant transport to ground water.
Ground Water 20:711-722.
*Engström J, Bjurström R. 1977. Exposure to methylene chloride: Content in subcutaneous adipose
tissue. Scand J Work Environ Health 3:215-224.
Environment Canada. 1993. Priority substances list assessment report: Dichloromethane. Ottawa,
Canada: Canada Communication Group Publishing.
EPA. 1975a. Preliminary assessment of suspected carcinogens in drinking water. Washington, DC:
U.S. Environmental Protection Agency, Office of Toxic Substances. EPA-560/14-75-005. PB 260961.
*EPA. 1975b. Region V joint federal/state survey of organics and inorganics in selected drinking water
supplies. Chicago, IL: U.S. Environmental Protection Agency and Illinois Environmental Protection
Agency.
224 METHYLENE CHLORIDE
8. REFERENCES
EPA. 1975c. Identification of organic compounds in effluents from industrial sources. Washington, DC:
U.S. Environmental Protection Agency, Office of Toxic Substances. EPA-560/3-75-002. NTIS no.
PB-241641.
*EPA. 1979a. Identification of organic compounds in industrial effluent discharges. Athens, GA:
US Environmental Protection Agency, Environmental Research Laboratory. EPA-600/4-79-016.
PB 294794.
*EPA. 1979b. Water-related environmental fate of 129 priority pollutants. Vol. II. Halogenated
aliphatic hydrocarbons, halogenated ethers, monocyclic aromatics, phthalate esters, polycyclic aromatic
hydrocarbons, nitrosamines, and miscellaneous compounds. Washington, DC: U.S. Environmental
Protection Agency, Office of Water Planning and Standards. EPA-440/4-79-029b. NTIS no.
PB 80-204381.
*EPA. 1980a. Ambient water quality criteria for halomethanes. Washington, DC: U.S. Environmental
Protection Agency, Office of Water Regulations and Standards. EPA 440/5-80-051. PB 81 117 624.
EPA. 1980b. U.S. Environmental Protection Agency. Federal Register 45:33132-33133.
EPA. 1980c. U.S. Environmental Protection Agency. Part V. Federal Register 45(231):79334-79335.
EPA. 1980d. Acquisition and chemical analysis of mother’s milk for selected toxic substances.
Research Triangle Park, NC: U.S. Environmental Protection Agency. EPA 560/13-80-029.
PB 81-231029.
*EPA. 1980e. Atmospheric measurements of selected hazardous organic chemicals. Research Triangle
Park, NC: US Environmental Protection Agency, Environmental Sciences Research Laboratory.
*EPA. 1980f. Fate of toxic and hazardous materials in the air environment. Research Triangle Park,
NC: U.S. Environmental Protection Agency, Environmental Sciences Research Lab. NTIS
no. PB80-221948. EPA 600/3-80-084.
EPA. 1981. Environmental risk assessment of dichloromethane. Draft report. Washington, DC: U.S.
Environmental Protection Agency, Office of Toxic Substances.
*EPA. 1982a. Chlorinated organic solvents: Trichloroethene, tetrachloroethene, 1,1,1-trichloroethane,
dichloromethane, and tetrachloromethane. In: Intermedia priority pollutant guidance documents.
Washington, DC: U.S. Environmental Protection Agency, Office of Pesticides and Toxic Substances.
EPA. 1982b. Purgeable halocarbons-method 601. In: Methods for organic chemical analysis of
municipal and industrial wastewater. Cincinnati, OH: U.S. Environmental Protection Agency,
Environmental Monitoring and Support Laboratory. EPA 600/4-82-057.
EPA. 1982c. Purgeables-method 624. In: Methods for organic chemical analysis of municipal and
industrial wastewater. Cincinnati, OH: U.S. Environmental Protection Agency, Environmental
Monitoring and Support Laboratory. EPA 600/4-82-057.
EPA. 1982d. U.S. Environmental Protection Agency. Federal Register 47:26992,27007-27008.
*EPA. 1982e. Aquatic fate process data for organic priority pollutants. Washington, DC:
US Environmental Protection Agency, Office of Water Regulations and Standards. EPA 440/4-81-014.
225 METHYLENE CHLORIDE
8. REFERENCES
EPA. 1983a. Methods for chemical analysis of water and wastes. Cincinnati, OH: U.S. Environmental
Protection Agency, Office of Research and Development, Environmental Monitoring and Support
Laboratory. EPA 600/4-79-020.
EPA. 1983b. Treatability manual. Vol. 1. Treatability data. Washington, DC: U.S. Environmental
Protection Agency, Office of Research and Development. EPA 600/2-82-001A.
*EPA. 1983c. Human exposure to atmospheric concentrations of selected chemicals. Vol. 2. Research
Triangle Park, NC: U.S. Environmental Protection Agency. NTIS no. PB83-265249.
*EPA. 1984. Health effects assessment for methylene chloride. Cincinnati, OH: U.S. Environmental
Protection Agency, Environmental Criteria and Assessment Office. ECAO-CIN-H028. NTIS no.
PB86-134392.
EPA. 1985a. Addendum to the health assessment document for dichloromethane (methylene chloride):
Updated carcinogen assessment of dichloromethane (methylene chloride): Final report. Washington,
DC: U.S. Environmental Protection Agency, Office of Research and Development. EPA
600/8-82-0044FF.
*EPA. 1985b. Analysis of the applicability of TSCA Section 4(f) to methylene chloride.
U.S. Environmental Protection Agency.
EPA. 1985c. Criteria document on dichloromethane. Final draft. Washington, DC: U.S. Environmental
Protection Agency, Office of Drinking Water.
EPA. 1985d. Environmental profiles and hazard indices for constituents of municipal sludge: Methylene
chloride. Washington, DC: U.S. Environmental Protection Agency, Office of Water Regulations and
Standards.
*EPA. 1985e. Health assessment document for dichloromethane (methylene chloride): Updated
carcinogen assessment of dichloromethane (methylene chloride). Washington, DC: U.S. Environmental
Protection Agency, Office of Health and Environmental Assessment. EPA/600/8-82/004FA.
EPA. 1985f. Summary of environmental profiles and hazard indices for constituents of municipal
sludge: Methods and results. Washington, DC: U.S. Environmental Protection Agency, Office of Water
Regulations and Standards.
*EPA. 1985g. Atmospheric fates of organic chemicals: Prediction of ozone and hydroxyl radical
reaction rates and mechanisms. Research Triangle Park, NC: U.S. Environmental Protection Agency,
Atmospheric Sciences Research Laboratory. EPA/600/3-85/063. NTIS no. PB85-241529.
*EPA. 1985h. Synthetic organic compound sampling survey of public water supplies. Washington, DC:
U.S. Environmental Protection Agency. NTIS no. PB85-214427.
EPA. 1986a. Gas chromatography/mass spectrometry for volatile organics-method 8240. In: Test
methods for evaluating solid waste. 3rd ed. SW-846. Washington, DC: U.S. Environmental Protection
Agency, Office of Solid Waste and Emergency Response.
EPA. 1986b. Halogenated volatile organics-method 8010. In: Test methods for evaluating solid waste.
3rd ed. SW-846. Washington, DC: U.S. Environmental Protection Agency, Office of Solid Waste and
Emergency Response.
226 METHYLENE CHLORIDE
8. REFERENCES
EPA. 1986c. Superfund public health evaluation manual. Washington, DC: U.S. Environmental
Protection Agency, Office of Emergency and Remedial Response. EPA 540/1-86-060.
EPA. 1986d. U.S. Environmental Protection Agency. Federal Register 51:33992-34003.
*EPA. 1986e. Test methods for evaluating solid waste. Washington, DC: Office of Solid Waste, U.S.
Environmental Protection Agency. OSW Methods 5041A, 5021, 8010B, 8021B, 8240B, 8260B.
*EPA. 1987a. Dichloromethane health advisory. Washington, DC: U.S. Environmental Protection
Agency, Office of Drinking Water. PB 87-235578. 150-164.
*EPA. 1987b. Health advisory for dichloromethane: Draft. Washington, DC: U.S. Environmental
Protection Agency, Office of Drinking Water.
EPA. 1987c. Household solvent products: A national usage survey. Washington, DC:
U.S. Environmental Protection Agency.
EPA. 1987d. Technical analysis of new methods and data regarding dichloromethane hazard
assessments. Draft document. U.S. Environmental Protection Agency.
EPA. 1987e. Update to health assessment document and addendum for dichloromethane (methylene
chloride): Pharmacokinetics, mechanism of action, and epidemiology. Draft Report. U.S.
Environmental Protection Agency. EPA 600/8-87/1030A.
EPA. 1987f. U.S. Environmental Protection Agency. Part II. Federal Register 52(130):25709-25710.
EPA. 1987g. U.S. Environmental Protection Agency. Part II. Federal Register 52:25942-25953.
EPA. 1987h. U.S. Environmental Protection Agency. Part V. Federal Register 52:25760-25763, 25791.
*EPA. 1987i. Atmospheric persistence of eight air toxics: Project summary. Research Triangle Park,
NC: U.S. Environmental Protection Agency, Atmospheric Sciences Research Laboratory.
EPA 600/53-87/004. HC/MF PB 87-145306.
*EPA. 1987j. Environmental Protection Agency policy and guidance statement on petitions under the
emergency planning and community right-to-know act. U.S. Environmental Protection Agency. Federal
Register. 52 FR 107970.
EPA. 1988a. U.S. Environmental Protection Agency. Part II. Federal Register 53:4500-4501.
EPA. 1988b. U.S. Environmental Protection Agency. Part II. Federal Register 53:31138-31142,
31211-31222.
EPA. 1988c. U.S. Environmental Protection Agency. Part IV. Federal Register 53:38642-38654.
*EPA. 1988d. National ambient volatile organic compounds (VOCs): Data base update. Research
Triangle Park, NC: US Environmental Protection Agency, Atmospheric Sciences Research Laboratory.
EPA/600/3-88/010a.
EPA. 1989a. Interim methods for development of inhalation reference doses. Washington, DC: U.S.
Environmental Protection Agency, Office of Health and Environmental Assessment.
227 METHYLENE CHLORIDE
8. REFERENCES
EPA 600/8-86-032a.
*EPA. 1989b. Measurement of purgeable organic compounds in water by capillary column gas
chromatography/mass spectrometry-method 524.2. In: Methods for the determination of organic
compounds in drinking water. Cincinnati, OH: U.S. Environmental Protection Agency, Environmental
Monitoring Systems Laboratory. EPA/600/4-88/039.
*EPA. 1989c. Measurement of purgeable organic compounds in water by packed column gas
chromatography/mass spectrometry-method 524.1. In: Methods for the determination of organic
compounds in drinking water. Cincinnati, OH: U.S. Environmental Protection Agency, Environmental
Monitoring Systems Laboratory. EPA/600/4-88/039.
*EPA. 1989d. Updated health effects assessment for methylene chloride. Cincinnati, OH:
U.S. Environmental Protection Agency, Environmental Criteria and Assessment Office.
EPA/600-8-89/092. NTIS no. PB90-142449.
*EPA. 1989e. U.S. Environmental Protection Agency. Part V. Federal Register 54:33426, 33461.
*EPA. 1989f. Volatile halogenated organic compounds in water by purge and trap gas chromatography-
method 502.1. In: Methods for the determination of organic compounds in drinking water. Cincinnati,
OH: U.S. Environmental Protection Agency, Environmental Monitoring Systems Laboratory. EPA
600/4-88-039.
*EPA. 1989g. Volatile organic compounds in water by purge and trap capillary column gas
chromatography with protoionization and electrolytic conductivity detectors in series-method 502.2. In:
Methods for the determination of organic compounds in drinking water. Cincinnati, OH:
U.S. Environmental Protection Agency, Environmental Monitoring Systems Laboratory. EPA
600/4-88/039.
EPA. 1990a. U.S. Environmental Protection Agency. Part II. Federal Register 55:22520-22536,
22683-22714.
EPA. 1990b. U.S. Environmental Protection Agency. Part II. Federal Register 55:30370-30373,
30386-30387.
*EPA. 1990c. Toxics in the community: National, and local perspectives. Washington, DC: U.S.
Environmental Protection Agency, Office of Pesticides and Toxic Substances. EPA 560/4-90-017.
*EPA. 1990d. Interim methods for development of inhalation reference concentrations. Washington,
DC: U.S. Environmental Protection Agency, Office of Health and Environmental Assessment, Office of
Research and Development, Environmental Criteria and Assessment Office. EPA 600/8-90/066A.
EPA. 1991. Evaluations of VOC emissions from heated roofing asphalt. Research Triangle Park, NC:
U.S. Environmental Protection Agency, Control Technology Center. EPA-600/2-91-061.
EPA. 1992a. Final quantification of toxicological effects for dichloromethane. Washington, DC: U.S.
Environmental Protection Agency, Office of Water. NTIS no. PB92-173335.
EPA. 1992b. U.S. Environmental Protection Agency. Part III. Federal Register 57:31789-31790.
228 METHYLENE CHLORIDE
8. REFERENCES
EPA. 1993. Locating and estimating air emissions from sources of methylene chloride. U.S.
Environmental Protection Agency, Office of Air Quality. EPA 454/R-93-006.
*EPA. 1994. Methods for derivation of inhalation reference concentrations and application of inhalation
dosimetry. Washington, DC: Office of Research and Development, U.S. Environmental Protection
Agency. EPA/600/8-90/066F.
*EPA. 1996a Drinking water regulations and health advisories. U.S. Environmental Protection Agency,
Office of Water. http://www.epa.gov/ostwater/tools/dwstds3.html. Accessed on May 20, 1998.
*EPA. 1996b. Volatile organic compounds by gas chromatography/mass spectrometry (GC/MS)-
method 8260B. In: Test methods for evaluating solid waste. 3rd ed. SW-846. Washington, DC: U.S.
Environmental Protection Agency, Office of Solid Waste and Emergency Response.
*EPA. 1996c. Aromatic and halogenated volatile by gas chromatography using photoionization and/or
electrolytic conductivity detectors-method 8021B. In: Test methods for evaluating solid waste. 3rd ed.
SW-846. Washington, DC: U.S. Environmental Protection Agency, Office of Solid Waste and
Emergency Response.
*EPA. 1996d. Involvement of dichloromethane in the intrinsic Biodegredation of chlorinated ethenes
and ethanes. In: Symposium of natural attenuation of chlorinated organics in ground water. U.S.
Environmental Protection Agency. EPA\540\R-96\509.
*EPA. 1997. Special report on environmental endocrine disruption: An effects assessment and analysis.
Washington, DC: U.S. Environmental Protection Agency, Risk Assessment Forum. EPA/630/R-96/012.
EPA. 1998a. Effluent guidelines and standards: General provisions. U.S. Environmental Protection
Agency. Code of Federal Regulations. 40 CFR 401.
*EPA. 1998b. Exemption from tolerances: Methylene chloride; exemption from the requirement of a
tolerance. Environmental Protection Agency. Code of Federal Regulations. 40 CFR 180.1010.
*EPA. 1998c. Ground-water monitoring list: Appendix IX. U.S. Environmental Protection Agency.
Code of Federal Regulations. 40 CFR 268.
*EPA. 1998d. Hazardous waste constituent: Appendix VIII. U.S. Environmental Protection Agency.
Code of Federal Regulations. 40 CFR 261.
*EPA. 1998e. Health and safety data reporting: Substances and listed mixtures to which this subpart
applies. U.S. Environmental Protection Agency. Code of Federal Regulations. 40 CFR 716.120
*EPA. 1998f. List of hazardous substances and reportable quantities. U.S. Environmental Protection
Agency. Code of Federal Regulations. 40 CFR 302.4.
*EPA. 1998g. Maximum containment level goals for organic containments. U.S. Environmental
Protection Agency. Code of Federal Regulations. 40 CFR 141.50.
*EPA. 1998h. Methods for organic chemical analysis of municipal and industrial wastewater-method
601. Appendix A to 40 Part 136. http://www.epa.gov/ostwater/Tools/guide/methods.html. Accessed on
June 2, 1998.

229 METHYLENE CHLORIDE
8. REFERENCES
*EPA. 1998i. Methods for organic chemical analysis of municipal and industrial wastewater-method
624. Appendix A to 40 Part 136. Http://www.epa.gov/ostwater/Tools/guide/methods.html. Accessed on
June 2, 1998.
*EPA. 1998j. NPDES permit application testing requirements. U.S. Environmental Protection Agency.
Code of Federal Regulations. 40 CFR 122.21.
*EPA. 1998k. Pollutants eligible for a removal credit. U.S. Environmental Protection Agency. Code of
Federal Regulations. 40 CFR 403.
*EPA. 1998l. Toxic chemical release reporting: Community right-to-know. U.S. Environmental
Protection Agency. Code of Federal Regulations. 40 CFR 372.
*EPA 1999a. U. S. Environmental Protection Agency. Code of Federal Regulations. 40 CFR 302.4.
*EPA 1999b. U. S. Environmental Protection Agency. Code of Federal Regulations. 40 CFR 372.65.
*EPA 1999c. U. S. Environmental Protection Agency. Code of Federal Regulations. 40 CFR 261.33.
*EPA 1999d. U. S. Environmental Protection Agency. Code of Federal Regulations. 40 CFR 268.48.
EPA 1999e. U. S. Environmental Protection Agency. Code of Federal Regulations. 40 CFR 141.6.
*EPA 1999f. U. S. Environmental Protection Agency. Code of Federal Regulations. 40 CFR 141.50.
*EPA 1999g. U. S. Environmental Protection Agency. Code of Federal Regulations. 40 CFR 141.61.
EPA 1999h. U. S. Environmental Protection Agency. Code of Federal Regulations. 40 CFR 142.62.
*EPA 1999i. U. S. Environmental Protection Agency. Code of Federal Regulations. 40 CFR 180.1001.
*EPA 1999j. National Recommended Water Quality Criteria- Correction. U. S. Environmental
Protection Agency, Office of Water. EPA 822-Z-99-001.
*Estill CF, Spencer AB. 1996. Case study: Control of methylene chloride exposures during furniture
stripping. Am Ind Hyg Assoc J 57:43-49.
Ewing BB, Chian ESK, Cook JC, et al. 1977. Monitoring to detect previously unrecognized pollutants in
surface waters. Washington, DC: U.S. Environmental Protection Agency, Office of Pesticides and Toxic
Substances. EPA 560/6-77-015.
FAO and WHO Working Groups. 1993. Dichloromethane. WHO food additive series 30:135-82.
Farmer PB, Sepai O, Lawrence R, et al. 1996. Biomonitoring human exposure to environmental
carcinogenic chemicals. Mutagenesis 11:363-381.
*FDA. 1985. Cosmetics; proposed ban on the use of methylene chloride as an ingredient of aerosol
cosmetic products. Food and Drug Administration. Federal Register 50(243):51551-51559.
*FDA. 1989. Cosmetics; ban on the use of methylene chloride as an ingredient of cosmetic products.
Food and Drug Administration. Federal Register 54(124):27328-27342.
230 METHYLENE CHLORIDE
8. REFERENCES
FDA. 1997a. Diluents in color additive mixtures for food use exempt from certification. U.S. Food and
Drug Administration. Code of Federal Regulations. 21 CFR 73.1.
FDA. 1997b. Food additives permitted for direct addition to food for human consumption: Modified hop
extract. U.S. Food and Drug Administration. Code of Federal Regulations. 21 CFR 560.
FDA. 1997c. Indirect food additives: Adhesives and components of coating: Adhesives. U.S. Food and
Drug Administration. Code of Federal Regulations. 21 CFR 175.105.
FDA. 1997d. Indirect food additives: Polymers: Polycarbonate resins. U.S. Food and Drug
Administration. Code of Federal Regulations. 21 CFR 177.1580
FDA. 1997e. Requirements for specific cosmetic products. U.S. Food and Drug Administration. Code
of Federal Regulations. 21 CFR 700.19.
FDA. 1997f. Secondary direct food additives permitted in food for human consumption: Solvents,
lubricants, release agents and related substances. U.S. Food and Drug Administration. Code of Federal
Regulations. 21 CFR 173.255.
FDA. 1997g. Food labeling; Statement of identity, nutrition labeling and ingredient labeling of dietary
supplements; compliance policy guide, revocation; Correction. Department of Health and Human
Services, Food and Drug Administration. Federal Register 62(243): 66275-66277.
*FDA. 1999a. Food and Drug Administration. Code of Federal Regulations. 21 CFR 175.105.
*FDA. 1999b. Food and Drug Administration. Code of Federal Regulations. 21 CFR 173.255.
*FDA. 1999c. Food and Drug Administration. Code of Federal Regulations. 21 CFR 700.19.
*FDA. 1999d. Food and Drug Administration. Code of Federal Regulations. 21 CFR 177.1580.
*FDA. 1999e. Food and Drug Administration. Code of Federal Regulations. 21 CFR 73.1.
*FEDRIP. 1996. Federal Research in Progress. Dialog Information Systems, Inc., Palo Alto, CA.
FEDRIP. 1998. Federal Research in Progress. Dialog Information Systems, Inc., Palo Alto, CA.
*FEDRIP. 1999. Federal Research in Progress. Dialog Information Systems, Inc., Palo Alto, CA.
December 1999.
*Ferrario JB, Lawler GC, DeLeon IR, et al. 1985. Volatile organic pollutants in biota and sediments of
Lake Pontchartrain. Bull Environ Contam 34:246-255.
*Fisher J, Mahle D, Bankston L, et al. 1997. Lactational transfer of volatile chemicals in breast milk.
Am Ind Hyg Assoc J 58:425-431.
*Fisher JW, Whittaker TA, Taylor DH, et al. 1989. Physiologically based pharmacokinetic modeling of
the pregnant rat: A multiroute exposure model for trichloroethylene and its metabolite, trichloroacetic
acid. Toxicol Appl Pharmacol 99:395-414.
Flury F, Zernik F. 1931. Harmful gases, vapors, fog, smoke, and dust. Berlin: Julius Springer, 311-312.
231 METHYLENE CHLORIDE
8. REFERENCES
*Fodor GG, Roscovana G. 1976. [Increased blood-CO-content in humans and animals by incorporated
halogenated hydrocarbons.] Zentrabl Bakteriol (Orig B). 162:34-40 (German).
*Fodor GG, Winneke G. 1971. Nervous system disturbances in men and animals experimentally
exposed to industrial solvent vapors in England. Proceedings of the 2nd international clean air congress.
New York, NY: Academic Press.
*Fodor GG, Prajsnar D, Schlipkoter HW. 1973. Endogenous conformation by incorporated halogenated
hydrocarbons of the methane series. Staubreinhalt Luft 33:260-261.
Foley JF, Tuck PD, Ton TT, et al. 1993. Inhalation exposure to a hepatocarcinogenic concentration of
methylene chloride does not induce sustained replicative DNA synthesis in hepatocytes of female
B6C3F1 mice. Carcinogenesis 14:811-817.
*Foman SJ. 1966. Body composition of the infant. Part I: The male reference infant. In: Falkner F, ed.
Human development. Philadelphia, PA: WB Saunders, 239-246.
*Foman SJ, Haschke F, Ziegler EE, et al. 1982. Body composition of reference children from birth to
age 10 years. Am J Clin Nutr 35:1169-1175.
Forster HV, Graff S, Hake CL, et al. 1974. Pulmonary-hematologic studies on humans during exposure
to methylene chloride. NIOSH-MCOW-ENVM-MC-74-4.
*Foster JR, Green T, Smith LL, et al. 1992. Methylene chloride -- An inhalation study to investigate
pathological and biochemical events occurring in the lungs of mice over an exposure period of 90 days.
Fundam Appl Toxicol 18:376-388.
Foster JR, Green T, Smith LL, et al. 1994. Methylene chloride: An inhalation study to investigate
toxicity in the mouse lung using morphological, biochemical and Clara cell culture techniques. Toxicol
91:221-234.
Freedman DL, Gossett JM. 1991. Biodegredation of dichloromethane and its utilization as a growth
substrate under methanogenic conditions. Appl Environ Microbiol 57:2847-2857.
Freedman DL, Smith CR, Noguera DR. 1997. Dichloromethane Biodegredation under nitrate-reducing
conditions. Water Environ Res 69:115-122.
*Friedlander BR, Hearne FT, Hall S. 1978. Epidemiologic investigation of employees chronically
exposed to methylene chloride: Mortality analysis. J Occup Med 20(10):657-666.
FSTRAC. 1990. Summary of state and federal drinking water standards and guidelines. Washington,
DC: U.S. Environmental Protection Agency.
*Gargas ML, Burgess RJ, Voisard DE, et al. 1989. Partition coefficients of low-molecular-weight
volatile chemicals in various liquids and tissues. Toxicol Appl Pharmacol 98:87-99.
*Gargas ML, Clewell HJ, Andersen ME. 1986. Metabolism of inhaled dihalomethanes in vivo:
Differentiation of kinetic constants for two independent pathways. Toxicol Appl Pharm 82:211-223.
232 METHYLENE CHLORIDE
8. REFERENCES
*Garte S, Crosti F. 1999. A nomenclature system for metabolic gene polymorphisms. In: Vineis P,
Malats N, Lang M, et al., ed. Metabolic polymorphisms and susceptibility to cancer. Lyon, France:
International Agency for Research on Cancer, 5-12.
General Electric. 1982. Fourteen day range finding study in rats. Pittsfield, Massachusetts.
OTS84003A.
*Ghittori S, Marraccini P, Franco G, et al. 1993. Methylene chloride exposure in industrial workers.
Am Ind Hyg Assoc J 54(1):27-31.
*Gibbs GW, Amsel J, Soden K. 1996. A cohort mortality study of cellulose triacetate-fiber workers
exposed to methylene chloride. J Occup Environ Med 38(7):693-697.
Giroux D, Lapointe G, Baril M. 1992. Toxicological index and the presence in the workplace of
chemical hazards for workers who breast-feed infants. Amer Ind Hyg Assoc J 53:471-474.
*Giwercman A, Carlsen E, Keiding N, et al. 1993. Evidence for increasing incidence of abnormalities of
the human testis: A review. Environ Health Perspect Suppl 101(2):65-71.
*Glatzel W, Tietze K, Gutewort R, et al. 1987. Interaction of dichloromethane and ethanol in rats:
Toxicokinetics and nerve conduction velocity. Alcohol Clin Exp Res 11:450-452.
*Gocke E, King M-T, Eckhardt K, et al. 1981. Mutagenicity of cosmetic ingredients licensed by
European Communities. Mutat Res 90:91-109.
Gold LS, Slone TH, Ames BN. 1998. What do animal cancer tests tell us about human cancer risk?:
overview of analysis of the carcinogenic potency database. Drug Metab Rev 30(2):359-404.
Goss K-U. 1997. Conceptual model for the adsorption of organic compounds from the gas phase to
liquid and solid surfaces. Environ Sci Technol 31(12):3600-3605.
Gosselin RE, Smith RP, Hodge HC, et al. 1984. Clinical toxicology of commercial products. 5th ed.
Baltimore, MD: Williams and Wilkins, II-161.
*Gossett JM. 1987. Measurement of Henry's Law constants for C
1
and C
2
chlorinated hydrocarbons.
Environ Sci Technol 21:202-208.
Gradiski D, Bonnet G, Raoult G, et al. 1978. [Compared acute pulmonary toxicity of the main
chlorinated aliphatic solvents.] Arch Mal Prof Med Trav Secus Soc 39:249. (French).
Graham DR. 1990. Solvent abuse. In: Haddad LM, Winchester JF, ed. Clinical management of
poisoning and drug overdose. Philadelphia, PA: W.B. Saunders Company, 1216-1222.
*Graves RJ, Green T. 1996. Mouse liver glutathione S-transferase mediated metabolism of methylene
chloride to a mutagen in the CHO/HPRT assay. Mutat Res 367:143-150.
*Graves RJ, Callander RD, Green T. 1994a. The role of formaldehyde and S-chloromethylglutathione in
the bacterial mutagenicity of methylene chloride. Mutat Res 320:235-243.
*Graves RJ, Coutts C, Eyton-Jones H, et al. 1994b. Relationship between hepatic DNA damage and
methylene chloride-induced hepaticarcinogenicity in B6C3F1 mice. Carcinogenesis 15(5):991-996.
233 METHYLENE CHLORIDE
8. REFERENCES
*Graves RJ, Coutts C, Green T. 1995. Methylene chloride-induced DNA damage: An interspecies
comparison. Carcinogenesis 16(8):1919-1926.
*Graves RJ, Trueman P, Jones S, et al. 1996. DNA sequence analysis of methylene chloride-induced
HPRT mutations in Chinese hamster ovary cells: Comparison with the mutation spectrum obtained for
1,2-dibromoethane and formaldehyde. Mutagenesis 11(3):229-233.
*Green T. 1991. Species differences in carcinogenicity: The role of metabolism and pharmacokinetics in
risk assessment. Ann Ist Super Sanita 27:595-600.
*Green T. 1997. Methylene chloride induced mouse liver and lung tumours: An overview of the role of
mechanistic studies in human safety assessment. Hum Exp Toxicol 16:3-13.
Green T, Nash JA, Mainwaring G, eds. 1986a. Methylene chloride: In vitro metabolism in rat, mouse,
and hamster liver and lung fractions and in human liver fractions. ICI Central Toxicology Laboratory,
Report no. CTL/R/879, September 22. TSCATS 305692. OTS 0514367. EPA 86-880000289.
*Green T, Provan WM, Collinge DC, et al., eds. 1986b. Methylene chloride: Interaction with the rat and
mouse liver and lung DNA in vivo. ICI Central Toxicology Laboratory, Report no. CTL/R/851, January
22. TSCATS 305690. OTS 0514364. EPA 86-880000286.
*Green T, Proven WM, Nash JA, et al., eds. 1986c. Methylene chloride (dichloromethane): In vivo
inhalation pharmacokinetics and metabolism in F344 rats and B6C3F1 mice. ICI Central Toxicology
Laboratory, Report No. CTL/R/880, September 22. TSCATS 305694. NTIS/OTS 0514368. EPA
86-880000290.
*Green T, Provan WM, Collinge DC, et al. 1988. Macromolecular interactions of inhaled methylene
chloride in rats and mice. Toxicol Appl Pharmacol 93:1-10.
Grimsrud EP, Rasmussen RA. 1975. Survey and analysis of halocarbons in the atmosphere by gas
chromatography-mass spectrometry. Atmos Environ 9:1014-1017.
Guengerich FP. 1997. Mechanisms of mutagenicity of DNA adducts derived from alkyl and vinyl
halides. Jpn J Toxicol Environ Health (Eisei Kagaku) 43(2):69-82.
*Guzelian PS, Henry CJ, Olin SS, eds. 1992. Similarities and differences between children and adults:
Implications for risk assessment. Washington, DC: International Life Sciences Institute Press.
Haag WR, Johnson MD, Scofield R. 1996. Direct photolysis of trichloroethene in air: effect of
cocontaminants, toxicity of products, and hydrothermal treatment of products. Environ Sci Technol
30:414-421.
*Haddad LM, Winchester JF. 1990. Clinical management of poisoning and drug overdose. Second
edition. Philadelphia, PA: W.B. Saunders Company.
Hakkola J, Pelkonen O, Pasanen M, et al. 1998. Xenobiotic-metabolizing cytochrome P450 enzymes in
the human feto-placental unit: Role in intrauterine toxicity. Crit Rev Toxicol 28(1):35-72.
*Hall AH, Rumack BH. 1990. Methylene chloride exposure in furniture-stripping shops: Ventilation and
respirator use practices. J Occup Med 32(1):33-41.
234 METHYLENE CHLORIDE
8. REFERENCES
Hall RM, Martinez KF, Jensen PA. 1995. Control of methylene chloride furniture stripping dip tank.
Appl Occup Environ Hyg 10:188-195.
*Hallier E, Schroder KR, Asmuth K, et al. 1994. Metabolism of dichloromethane (methylene chloride to
formaldehyde in human erythrocytes: Influence of polymorphism of glutathione transferase theta (GST
T1-1). Arch Toxicol 68:423-427.
*Hansch C, Leo A. 1979. Substituent constants for correlation analysis in chemistry and biology. New
York, NY: John Wiley and Sons.
*Hardin BD, Manson JM. 1980. Absence of dichloromethane teratogenicity with inhalation exposure to
rats. Toxicol Appl Pharmacol 52:22-28.
*Harkov R, Kebbekus B, Bozzelli JW, et al. 1984. Comparison of selected volatile organic compounds
during the summer and winter at urban sites in New Jersey. Sci Total Environ 38:259-274.
*Harkov R, Giante SJ, Bozelli JW, et al. 1985. Monitoring volatile organic compounds at hazardous and
sanitary landfills in New Jersey. J Environ Sci Health A20(5):491-501.
*Harsch D. 1977. Study of halocarbon concentrations in indoor environments. Final report. Report to
U.S. Environmental Protection Agency, Office of Research and Development, Washington, DC, by
Washington State University, College of Engineering, Pullman, WA. Project 1505. Contract
WA-6-99-2922-J.
Hashmi M, Dechert S, Dekant W, et al. 1994. Bioactivation of [
13
C]dichloromethane in mouse, rat, and
human liver cytosol:
13
C nuclear magnetic resonance spectroscopic studies. Chem Res Toxicol 7:291-
296.
Hastings SE. 1984. Written communication (April 24) to T. Knutson, Office of Toxic Substances (TS-
793-I), regarding TSCA section 8(d) health and safety study submissions. Stauffer Chemical Company,
Westport, Connecticut. OTS84003A.
*Haun CC, Vernot EH, Darmer KI, et al. 1972. Continuous animal exposure to low levels of
dichloromethane. In: Proceedings of the 3
rd
annual conference on environmental toxicology. Wright
Patterson Air Force Base, OH: Aerospace Medical Research Laboratory, 199-208. AMRL-TR-72-130.
AD 773766.
*Hauser TR, Bromberg SM. 1982. EPA's monitoring program at Love Canal 1980. Environ Monit
Assess 2:249-271.
*HazDat. 1996. Agency for Toxic Substances and Disease Registry (ATSDR), Atlanta, GA.
www.atsdr.cdc.gov/hazdat.html.
HazDat. 1998. Agency for Toxic Substances and Disease Registry (ATSDR), Atlanta, GA.
June 8, 1998. www.atsdr.cdc.gov/hazdat.html.
*HazDat. 1999. Agency for Toxic Substances and Disease Registry (ATSDR), Atlanta, GA.
December 14, 1999. www.atsdr.cdc.gov/hazdat.html.
*HazDat. 2000. Agency for Toxic Substances and Disease Registry (ATSDR), Atlanta, GA.
www.atsdr.cdc.gov/hazdat.html.
235 METHYLENE CHLORIDE
8. REFERENCES
*Hearne FT, Grose F, Pifer JW, et al. 1987. Methylene chloride mortality study: Dose-response
characterization and animal model comparison. J Occup Med 29(3):217-228.
*Hearne FT, Pifer JW, Grose F. 1990. Absence of adverse mortality effects in workers exposed to
methylene chloride: An update. J Occup Med 32(3):234-240.
HEAST. 1990. Health Effects Assessment Summary Tables. Third quarter FY-1990. Washington, DC:
U.S. Environmental Protection Agency.
*Hegi ME, Soderkvist P, Foley JF, et al. 1993. Characterization of p53 mutations in methylene chloride-
induced lung tumors from B6C3F1 mice. Carcinogenesis 14(5):803-810.
*Hegi ME, Devereux TR, Dietrich WF, et al. 1994. Allelotype analysis of mouse lung carcinomas
reveals frequent allelic losses on chromosome 4 and an association between allelic imbalances on
chromosome 6 and K-ras activation. Cancer Res 54:6257-6264.
*Heikes DL. 1987. Purge and trap method for determination of volatile halocarbons and carbon disulfide
in table-ready foods. J Assoc Off Anal Chem 70:215-226.
Heikes DL, Hopper ML. 1986. Purge and trap method for determination of fumigants in whole grains,
milled grain products, and intermediate grain-based foods. J Assoc Off Anal Chem 69:990-998.
*Heineman EF, Cocco P, Gómez MR, et al. 1994. Occupational exposure to chlorinated aliphatic
hydrocarbons and risk of astrocytic brain cancer. Am J Ind Med 26:155-169.
*Helz GR, Hsu RY. 1978. Volatile chloro- and bromocarbons in coastal waters. Limnol Oceanogr
23(5):858-869.
*Heppel LA, Neal PA. 1944. Toxicology of dichloromethane (methylene chloride): II. Its effects upon
running activity in the male rat. J Ind Hyg Toxicol 26(1):17-21.
*Heppel LA, Neal PA, Perrin ML, et al. 1944. Toxicology of dichloromethane (methylene chloride): I.
Studies on effects of daily inhalation. J Ind Hyg Toxicol 26(1):8-16.
*Herr DW, Boyes WK. 1997. A comparison of the acute neuroactive effects of dichloromethane, 1,3-
dichloropropane, and 1,2-dichlorobenzene on rat flash evoked potentials (FEPs)1,2. Fundam Appl
Toxicol 35:31-48.
Hertz-Picciotto I, Neutra RR. 1994. Resolving discrepancies among studies: The influence of dose on
effect size. Epidemiology 5:156-163.
Hetrick DM, Jarabek AM, Travis CC. 1991. Sensitivity analysis for physiologically based
pharmacokinetic models. J Pharmacokinet Biopharm 19(1):1-20.
Hiatt MH. 1981. Analysis of fish and sediment for volatile priority pollutants. Anal Chem
53:1541-1543.
*Hoel DG, Davis DL, Miller AB, et al. 1992. Trends in cancer mortality in 15 industrialized countries,
1969-1986. J Natl Cancer Inst 84(5):313-320.
236 METHYLENE CHLORIDE
8. REFERENCES
Hoff JT, Wania F, Mackay D, et al. 1995. Sorption of nonpolar organic vapors by ice and snow.
Environ Sci Technol 29:1982-1989.
Hoffmann P, Heinroth K, Richards D, et al. 1994. Depression of calcium dynamics in cardiac
myocytes–a common mechanism of halogenated hydrocarbon anesthetics and solvents. J Mol Cell
Cardiol 26:579-589.
Hoffmann P, Muller SP, Heinroth K, et al. 1996. Calcium dynamics in cardiac myocytes as a target of
dichloromethane cardiotoxicity. Arch Toxicol 70:158-163.
*Honma T, Suda M. 1997. Changes in plasma lipoproteins as toxicity markers for carbon tetrachloride,
chloroform, and dichloromethane. Ind Health 35:519-531.
Hovorka S, Dohnal V. 1997. Determination of air-water partitioning of volatile halogenated
hydrocarbons by the inert gas stripping method. J Chem Eng Data 42:924-933.
*Howard PH, Sage GW, Jarvis WF, et al., eds. 1990. Handbook of environmental fate and exposure data
for organic chemicals. Vol. II. Solvents. Chelsea, MI: Lewis Publishers, Inc., 176-183.
Hoyer ME, Keeler GJ, Ball JC. 1992. Detection of oxidative mutagens in an urban air-particulate
extract: A preliminary study. Mutat Res 283:295-299.
*HSDB. 1990. Hazardous Substances Data Bank. National Library of Medicine, National Toxicology
Information Program, Bethesda, MD. July 18, 1990.
HSDB. 1998. Hazardous Substances Data Bank. National Library of Medicine, National Toxicology
Information Program, Bethesda, MD. May 11, 1998.
*HSDB. 1999. Hazardous Substances Data Bank. National Library of Medicine, National Toxicology
Information Program, Bethesda, MD.
HSIA. 1988. Halogenated Solvents Industry Alliance. Public comments on scientific and technical
issues on the draft toxicological profile for methylene chloride. Submitted to Agency for Toxic
Substances and Disease Registry (ATSDR), March 8.
*HSIA. 2000. Methylene chloride: 28-day inhalation toxicity study in the rat to assess potential
immunotoxicity. 1-82.
Hughes CS. 1985. CEH product review: Chlorinated methanes. In: Chemical economics handbook.
Menlo Park, CA: SRI International.
*Hughes NJ, Tracey JA. 1993. A case of methylene chloride (nitromors) poisoning, effects on
carboxyhaemoglobin levels. Hum Exp Toxicol 12:159-160.
*IARC. 1986. Dichloromethane. IARC monograph on the evaluation of the carcinogenic risk of
chemicals to humans. Some halogenated hydrocarbons and pesticide exposures. Vol. 41. World Health
Organization, International Agency for Research on Cancer, Lyon, France, 43-85.
*IARC. 1987. IARC monographs on the evaluation of carcinogenic risk of chemicals to humans. Suppl.
7. Overall evaluations of carcinogenicity: An updating of IARC monographs volumes 1 to 42. World
Health Organization, International Agency for Research on Cancer, Lyon, France, 29-33, 62.
237 METHYLENE CHLORIDE
8. REFERENCES
*ID Dept Health Welfare. 1999. Air pollution control. Idaho Department of Health and Welfare. Rule
16.01.01. http://www.state.id.us/
IJC. 1983. An inventory of chemical substances identified in the Great Lakes ecosystem. Vol. 1.
Summary. Report to the Great Lakes Water Quality Board by the International Joint Commission,
Windsor, Ontario.
IRIS. 1998. Integrated Risk Information System. U.S. Environmental Protection Agency, Washington,
DC. May 11, 1998.
*IRIS. 1999. Integrated Risk Information System. U.S. Environmental Protection Agency, Washington,
DC. December 13, 1999.
*IRPTC. 1990. International Register of Potentially Toxic Chemicals. United Nations Environment
Programme, Geneva, Switzerland. July, 1990.
James KJ, Stack MA. 1997. The impact of leachate collection on air quality in landfills. Chemosphere
34(8):1713-1721.
Jansson T, Romert L, Magnusson J, et al. 1991. Genotoxicity testing of extracts of a Swedish moist oral
snuff. Mutat Res 261: 101-115.
Jensen AA. 1983. Chemical contaminants in human milk. Residue Rev 89:1-108.
*Johanson CE. 1980. Permeability and vascularity of the developing brain: Cerebellum vs cerebral
cortex. Brain Res 190:3-16.
Jones DL, Burklin CE, Seaman JC, et al. 1996. Models to estimate volatile organic hazardous air
pollutant emissions from municipal sewer systems. J Air Waste Manage Assoc 46:657-666.
*Jones DP, Thor H, Andersson B, et al. 1978. Detoxification reactions in isolated hepatocytes: Role of
gluthathione peroxidase, catalase, and formaldehyde dehydrogenase in reactions relating to N-
demethylation by the cytochrome P-450 system. J Biol Chem 253(17):6031-6037.
*Jones SM, Boobis AR, Moore GE, et al. 1992. Expression of CYP2E1 during fetal development:
methylation of the CYP2E1 gene in human fetal and adult liver samples. Biochem Pharmacol
43(8):1876-1879.
*Jongen WMF, Lohman PHM, Kottenhagen MJ, et al. 1981. Mutagenicity testing of dichloromethane in
short-term mammalian test systems. Mutat Res 81:203-213.
Jury WA, Winer AM, Spencer WF, et al. 1987. Transport and transformations of organic chemicals in
the soil-air-water ecosystem. Rev Environ Contam Toxicol 99:120-164.
Kadry AM, Skowronski GA, Abdel-Rahman MS. 1995. Evaluation of the use of uncertainty factors in
deriving RfDs for some chlorinated compounds. J Toxicol Environ Health 45:83-95.
Kaiser J. 1996. New data help toxicologists home in on assessing risks. Science 272:200.
238 METHYLENE CHLORIDE
8. REFERENCES
Kan AT, Tomson MB. 1996. UNIFAC prediction of aqueous and nonaqueous solubilities of chemicals
with environmental interest. Environ Sci Technol 30:1369-1376.
*Kanno J, Foley JF, Kari F, et al. 1993. Effect of methylene chloride inhalation on replicative DNA
synthesis in the lungs of female B6C3F
1
mice. Environ Health Perspec 101(suppl5):271-276.
*Kari FW, Foley JF, Seilkop SK, et al. 1993. Effect of varying exposure regimens on methylene
chloride-induced lung and liver tumors in female B6C3F1 mice. Carcinogenesis 14(5):819-826.
*Karlsson J-E, Rosengren LE, Kjellstrand P, et al. 1987. Effects of low-dose inhalation of three
chlorinated aliphatic organic solvents on deoxyribonucleic acid in gerbil brain. Scand J Work Environ
Health 13:453-458.
Kawata K, Tanabe A, Saito S, et al. 1997. Screening of volatile organic compounds in river sediment.
Bull Environ Contam Toxicol 58:893-900.
Keaton BF. 1990. Chlorinated hydrocarbons. In: Haddad LM, Winchester JF, ed. Clinical management
of poisoning and drug overdose. Philadelphia, PA: W.B. Saunders Company, 1216-1222.
Kedderis GL. 1997. Extrapolation of in vitro enzyme induction data to humans in vivo. Chem Biol
Interact 107:109-121.
*Kelly M. 1988. Case reports of individuals with oligospermia and methylene chloride exposures.
Reprod Toxicol 2:13-17.
*Kim C, Manning RO, Brown RP, et al. 1996b. Use of the vial equilibrium technique for determination
of metabolic rate constants for dichloromethane. Toxicol Appl Pharmacol 139:243-251.
Kim NY, Park, SW, Suh JK. 1996a. Two fatal cases of dichloromethane of chloroform poisoning. J
Forensic Sci 41: 527-529.
Kim SK, Kim YC. 1996. Effect of a single administration of benzene, toluene or m-xylene on
carboxyhaemoglobin elevation and metabolism of dichloromethane in rats. J Appl Toxicol 16:437-444.
*Kim YC. 1997. Dichloromethane potentiation of carbon tetrachloride hepatotoxicity in rats. Fundam
Appl Toxicol 35:138-141.
Kim YC, Carlson GP. 1986. The effect of an unusual workshift on chemical toxicity. I. Studies on the
exposure of rats and mice to dichloromethane. Fundam Appl Toxicol 6:162-171.
*Kimura ET, Ebert DM, Dodge PW. 1971. Acute toxicity and limits of solvent residue for sixteen
organic solvents. Toxicol Appl Pharmacol 19:699-704.
*King JW. 1989. Fundamentals and applications of supercritical fluid extraction in chromatographic
science. J Chromatogr 27:355-364.
*Kirschman JC, Brown NM, Coots RH, et al. 1986. Review of investigations of dichloromethane
metabolism and subchronic oral toxicity as the basis for the design of chronic oral studies in rats and
mice. Food Chem Toxicol 24(9):943-949.
239 METHYLENE CHLORIDE
8. REFERENCES
*Kitchin KT, Brown JL. 1989. Biochemical effects of three carcinogenic chlorinated methanes in rat
liver. Teratogenesis Carcinog Mutagen 9:61-69.
*Kjellstrand P, Bjerkemo M, Adler-Maihofer M, et al. 1986. Effects of methylene chloride on body and
organ weight and plasma butyrylcholinesterase activity in mice. Acta Pharmacol Toxicol 59:73-79.
Klecka GM. 1982. Fate and effects of methylene chloride in activated sludge. Appl Environ Microbiol
44:701-707.
*Komori M, Nishio K, Kitada M, et al. 1990. Fetus-specific expression of a form of cytochrome P-450
in human liver. Biochemistry 29:4430-4433.
*Konasewich D, Traversy, WJ, Zar H, et al. 1978. Status report on organic and heavy metal
contaminants in the Lakes Erie, Michigan, Huron, and Superior basins. Great Lake Water Quality Board,
1-14.
Kong ZM, Yu LW, Liu ZT, et al. 1996. Mutagenicity of organic pollutants and their active components
in the Xi River water at Shenyang. Bull Environ Contam Toxicol 56:803-808.
*Kool HJ, vanKreijl CF, Zoetman BCJ. 1982. Toxicology assessment of organic compounds in drinking
water. CRC Crit Rev Environ Control 12(4):307-350.
*Kopfler FC, Melton RG, Mullaney JL. et al. 1977. Human exposure to water pollutants. Adv Environ
Sci Technol 8:419-433.
Koppel C, Arndt I, Arendt U, et al. 1985. Acute tetrachloroethylene poisoning blood elimination kinetics
during hyperventilation therapy. Clin Toxicol 23:103-115.
*Koppmann R, Johnen FJ, Plass-Dulmer C, et al. 1993. Distribution of methylchloride, dichloromethane,
trichloroethene and tetrachloroethene over the north and south Atlantic. J Geophys Res 98(D11):20,517-
520,526.
Kramers PGN, Mout HCA, Bissumbhar B, et al. 1991. Inhalation exposure in Drosophila mutagenesis
assays: experiments with aliphatic halogenated hydrocarbons, with emphasis on the genetic activity
profile of 1,2-dichloroethane. Mutat Res 252:17-33.
*Krishnan K, Andersen ME. 1994. Physiologically based pharmacokinetic modeling in toxicology. In:
Hayes AW, ed. Principles and methods of toxicology. 3rd ed. New York, NY: Raven Press, Ltd., 149-
188.
Krishnan K, Pelekis M. 1995. Hematotoxic interactions: Occurrence, mechanisms and predictability.
Toxicology 105:355-364.
*Krishnan K, Andersen ME, Clewell HJ III, et al. 1994. Physiologically based pharmacokinetic
modeling of chemical mixtures. In: Yang RSH, ed. Toxicology of chemical mixtures: Case studies,
mechanisms, and novel approaches. San Diego, CA: Academic Press, 399-437.
Krishnan K, Haddad S, Pelekis M. 1995. A simple index for representing the discrepancy between
simulations of physiological pharmacokinetic models and experimental data. Toxicol Ind Health
11:413-422.
240 METHYLENE CHLORIDE
8. REFERENCES
*KS Dept Health Env. 1998. Ambient air quality standards and air pollution control. Kansas
Department of Health and Environment, Kansas Administrative Rules. Rule 28-19.
http://www.kdhe.state.ks.us/
KY Dept. for Env. Protect. 1998. Existing Sources: Threshold ambient limits and significant emission
levels of toxic pollutants. Kentucky Department for Environmental Protection, Division of Air Quality.
401 KY Admin. Regs. 53:010.
Laham S, Potvin M. 1976. Microdetermination of dichloromethane in blood with a syringeless gas
chromatographic injection system. Chemosphere 6:403-411.
*Lanes SF, Cohen A, Rothman KJ, et al. 1990. Mortality of cellulose fiber production workers. Scand J
Work Environ Health 16:247-251.
*Lanes SF, Rothman KJ, Dreyer NA, et al. 1993. Mortality update of cellulose fiber production workers.
Scand J Work Environ Health 19:426-428.
*Lang M, Pelkonen O. 1999. Metabolism of xenobiotics and chemical carcinogenesis. In: Vineis P,
Malats N, Lang M, et al., ed. Metabolic polymorphisms and susceptibility to cancer. Lyon, France:
International Agency for Research on Cancer, 13-22.
LaPat-Polasko LT, McCarty PL, Zehnder AJB. 1984. Secondary substrate utilization of methylene
chloride by an isolated strain of Pseudomonas. Appl Environ Microbiol 47:825-830.
*LaRegina J, Bozzelli JW, Harkov R, et al. 1986. Volatile organic compounds at hazardous waste sites
and a sanitary landfill in New Jersey: An up-to-date review of the present situation. Environ Progr
5(1):18-27.
*Lash AA, Becker CE, So Y, et al. 1991. Neurotoxic effects of methylene chloride: Are they long
lasting in humans? Br J Ind Med 48:418-426.
*Leeder JS, Kearns, GL. 1997. Pharmacogenetics in pediatrics: Implications for practice. Pediatr Clin
North Am 44:55-77.
Lefevre PA, Ashby J, eds. 1986. Methylene chloride: Induction of S-phase hepatocytes in the mouse
after in vivo exposure. ICI Central Toxicology Laboratory, Report no. CTL/R/885, September 22.
TSCATS 305696. OTS 0514369. EPA 86-880000286.
Lefevre PA, Ashby J. 1989. Evaluation of dichloromethane as an inducer of DNA synthesis in the
B6C3F1 mouse liver. Carcinogenesis 10:1067-1072.
Leiber CS. 1997. Cytochrome P-4502E1: Its physiological and pathological role. Physiol Rev
77(2):517-544.
*Leisinger T, Bader R, Hermann R, et al. 1994. Microbes, enzymes and genes involved in
dichloromethane utilization. Biodegredation 5:237-248.
*Leung H-W. 1993. Physiologically-based pharmacokinetic modelling. In: Ballentine B, Marro T,
Turner P, eds. General and applied toxicology. Vol. 1. New York, NY: Stockton Press, 153-164.
Lewis DFV, Ioannides C, Parke DV. 1998. Cytochromes P450 and species differences in xenobiotic
metabolism and activation of carcinogen. Environ Health Perspect 106(10):633-641.
241 METHYLENE CHLORIDE
8. REFERENCES
*Lewis RJ. 1996. Sax’s dangerous properties of industrial materials. 9th ed. New York, NY: Van
Nostrand Reinhold, 2230-2231.
Liang GB, Ritto CJ, Gellman SH. 1992. Thermodynamic analysis of β-turn formation in pro-ala, pro-
gly, and pro-val model peptides in methylene chloride. J Am Chem Soc 114:4440-4442.
*Lide DR. 1994. CRC handbook of chemistry and physics. Boca Raton, FL: CRC Press, 6-57, 15-45.
Liniger B, Sigrist T. 1994. Carboxyhaemoglobinaemia resulting from exposure to dichloromethane with
dermatological effects. Hautarzt 45:8-11.
Liteplo RG, Long GW, Meek ME. 1998. Relevance of carcinogenicity bioassays in mice assessing
potential health risks associated with exposure to methylene chloride. Hum Exp Toxicol 17:84-87.
*Livingston, AL. 1978. Forage plant estrogens. J Toxicol Environ Health 4:301-324.
Logemann E, van der Gunther S. 1991. Intoxication with a paint-stripper containing dichloromethane.
Arch Kriminol 188:159-166.
Long G, Meek ME, Caldwell I, et al. 1994. Dichloromethane: Evaluation of risks to health from
environmental exposure in Canada. J Environ Sci Health C12:305-318.
*Longo LD. 1977. The biological effects of carbon monoxide on the pregnant woman, fetus, and
newborn infant. Am J Obstet Gynecol 129:69-103.
Lopez S, Topalian JH, Mitra SK, et al. 1989. Absorption and desorption of dichloromethane vapor by
water drops in air. An experimental test of scavenging theory. J Atmos Chem 8:175-188.
Lundberg I, Ekdahl M, Kronevi T, et al. 1986. Relative hepatotoxicity of some industrial solvents after
intraperitoneal injection or inhalation exposure in rats. Environ Res 40:411-420.
Lynge E, Anttila A, Hemminiki K. 1997. Organic solvents and cancer. Cancer Causes and Control
8:406-419.
*MA Dept. Env. Protect. 1998. Allowable ambient limit (AAL), 24-hour averages. Massachusetts
Department of Environmental Protection, Division of Air Quality Control. Mass. Reg. Code Tit. 310,
Chapter 6.00.
*MacEwen JD, Vernot EH, Haun CC. 1972. Continuous animal exposure to dichloromethane. Wright-
Patterson Air Force Base, OH: Aerospace Medical Research Laboratory. AMRL-TR-72-28.
AD 746295.
*Mägli A, Messmer M, Leisinger T. 1998. Metabolism of dichloromethane by the strict anaerobe
dehalobacterium formicoaceticum. Appl Environ Microbiol 64(2):646-650.
Mahmud M, Kales SN. 1999. Methylene chloride poisoning in a cabinet worker. Environ Health
Perspect 107(9):769-772.
Mainwaring GW, Nash J, Davidson M, et al. 1996a. Isolation of a mouse Theta glutathione S-transferase
active with methylene chloride. Biochem J 314:445-448.
242 METHYLENE CHLORIDE
8. REFERENCES
*Mainwaring GW, Wiliams SM, Foster JR, et al. 1996b. The distribution of theta-class gluthione S-
transferases in the liver and lung of mouse, rat and human. Biochem J 318:297-303.
*Maltoni C, Cotti G, Perino G, et al. 1988. Long-term carcinogenicity bioassays on methylene chloride
administered by ingestion to Sprague-Dawley rats and Swiss mice and by inhalation to Sprague-Dawley
rats
a
. Ann NY Acad Sci 534:352-366.
*Manno M, Rugge M, Cocheo V. 1992. Double fatal inhalation of dichloromethane. Hum Exp Toxicol
11:540-545.
*Mannsville Chemical Products Corporation. 1988. Chemical products synopsis: Methylene chloride.
Mannsville Chemical Products Corp., Asbury Park, NJ.
Maronpot RR, Anna CH, Devereux TR, et al. 1995a. Considerations concerning the murine
hepatocarcinogenicity of selected chlorinated hydrocarbons. Prog Clin Biol Res 391:305-323.
*Maronpot RR, Devereux TR, Hegi M, et al. 1995b. Hepatic and pulmonary carcinogenicity of
methylene chloride in mice: A search for mechanisms. Toxicol 102:73-81.
*Marzotko D, Pankow D. 1987. Effect of single dichloromethane administration on the adrenal medulla
of male albino rats. Acta Histochem (Jena) 82:177-183.
*Mattsson JL, Albee RR, Eisenbrandt DL. 1990. Neurotoxicologic evaluation of rats after 13 weeks of
inhalation exposure to dichloromethane or carbon monoxide
1,2
. Pharmacol Biochem Beh 36:671-681.
*Mayr U, Butsch A, Schneider S. 1992. Validation of two in vitro test systems for estrogenic activities
with zearalenone, phytoestrogens and cereal extracts. Toxicology 74:135-149.
McCarroll NE, Cortina TA, Zieo MJ, et al. 1983. Evaluation of methylene chloride and vinylidene
chloride in mutational assays. Environ Mutagen 5:426-427.
McConnell FE, Solleveld HA, Swenberg JA, et al. 1986. Guidelines for combining neoplasms for
evaluation of rodent carcinogenesis studies. J Natl Can Inst 76:283-289.
*McCulloch A, Midgley PM. 1996. The production and global distribution of emissions of
trichloroethane, tetrachloroethene, and dichloromethane over the period 1988-1992. Atmos Environ
30(4):601-606.
*McDougal JN, Jepson GW, Clewell HJ, et al. 1986. A physiological pharmacokinetic model for dermal
absorption of vapors in the rat. Toxicol Appl Pharmacol 85:286-294.
*McKenna MJ, Zempel JA. 1981. The dose-dependent metabolism of [
14
C] methylene chloride
following oral administration to rat. Food Cosmet Toxicol 19:73-78.
*McKenna MJ, Saunders JH, Boeckler WH, et al. 1980. The pharmacokinetics of inhaled methylene
chloride in human volunteers [Abstract]. Toxicol Appl Pharm A59.
*McKenna MJ, Zempel JA, Braun WH. 1982. The pharmacokinetics of inhaled methylene chloride in
rats. Toxicol Appl Pharmacol 65:1-10.
243 METHYLENE CHLORIDE
8. REFERENCES
McRobie DJ, Glover DD, Tracy TS. 1998. Effects of gestational and overt diabetes on human placental
cytochromes P450 and glutathione S-transferase. Drug Metab Dispos 26(4):367-371.
*Meister RT, ed. 1989. Farm chemicals handbook. Willoughby, OH: Meister Publishing Company,
C193, C344.
*Mennear JH, McConnell EE, Huff JE, et al. 1988. Inhalation toxicity and carcinogenesis studies of
methylene chloride (dichloromethane) in F344/N rats and B6C3F
1
mice. Ann NY Acad Sci 534:343-351.
*Meyer DJ, Coles B, Pemble SE et al. 1991. Theta, a new class of glutathione transferases purified from
rat and man. Biochem J 274:409-414.
*Meylan WM, Howard PH. 1995. Atom/fragment contribution method for estimating octanol-water
partition coefficients. J Pharm Sci 84(1):83-175.
*Michael LC, Pellizzari ED, Wiseman RW. 1988. Development and evaluation of a procedure for
determining volatile organics in water. Environ Sci Technol 22(5):565-570.
Miller JL, Sardo MA, Thompson TL, et al. 1997. Effect of application solvents on heterotrophic and
nitrifying populations in soil microcosms. Environ Toxicol Chem 16(3):447-451.
Miller MS, Juchau MR, Guengerich P, et al. 1996. Drug metabolic enzymes in developmental
toxicology. Fundam Appl Toxicol 34:165-175.
Miller JL, Sardo MA, Thompson TL, et al. 1997. Effect of application solvents on heterotrophic and
nitrifying populations in soil microcosms. Environ Toxicol Chem 16(3):447-451.
Mills WB, Cheng JJ, Droppo JG, et al. 1997. Multimedia benchmarking analysis for three risk
assessment models: RESRAD, MMSOILS, and MEPAS. Risk Anal 17:187-201.
MIS. 1990. Agency for Toxic Substances and Disease Registry, Office of External Affairs, Exposure
and Disease Registry Branch, Atlanta, GA. September 24, 1990.
Miyamoto K, Urano K. 1996. Reaction rates and intermediates of chlorinated organic compounds in
water and soil. Chemosphere 32(12):2399-2408.
Mokrauer JE, Kosson DS. 1989. Electrophysical sorption of single carbon halogenated solvents onto
soil. Environ Prog 8:1-5.
Moody DE. 1981. Correlations among changes in hepatic microsomal components after intoxication
with alkyl halides and other hepatotoxins. Mol Pharmacol 20:685-693.
Morgenroth E, Schroeder ED, Chang DPY, et al. 1996. Nutrient limitation in a compost biofilter
degrading hexane. J Air Waste Manage Assoc 46:300-308.
*Morris JB, Smith FA, Garman RH. 1979. Studies on methylene chloride-induced fatty liver. Exp Mol
Path 30:386-393.
*Morselli PL, Franco-Morselli R, Bossi L. 1980. Clinical pharmacokinetics in newborns and infants.
Clin Pharmacokinet 5:485-527.
244 METHYLENE CHLORIDE
8. REFERENCES
*Moskowitz S, Sharpio H. 1952. Fatal exposure to methylene chloride vapor. Ind Hyg Occ Med
6:116-123.
Muller S, Weise M, Krug T, et al. 1991. Adrenergic cardiovascular actions in rats as affected by
dichloromethane exposure. Biomed Biochim Acta 50:307-311.
Myers RAM. 1990. Carbon monoxide poisoning. In: Haddad LM, Winchester JF, ed. Clinical
management of poisoning and drug overdose. Philadelphia, PA: W.B. Saunders Company, 1216-1222.
*Namkung E, Rittmann BE. 1987. Estimating volatile organic compound emissions from publicly
owned treatment works. J Water Pollut Control Fed 59(7):670-678.
*NAS. 1977. Drinking water and health. Washington, DC: National Academy of Sciences. National
Academy Press, 743-745.
*NAS. 1978. Scientific and technical assessments of environmental pollutants: Nonfluorinated
halomethanes in the environment. Washington, DC: National Academy of Sciences.
NAS. 1980. National Academy of Sciences. Drinking water and health. Vol. 3. Washington, DC:
National Academy Press.
*NAS/NRC. 1989. Biologic markers in reproductive toxicology, National Academy of
Sciences/National Research Council, Washington, DC: National Academy Press, 15-35.
NATICH. 1989. National Air Toxics Information Clearinghouse: NATICH database report on state,
local and EPA air toxics activities. Report to U.S. Environmental Protection Agency, Office of Air
Quality Planning and Standards. Research Triangle Park, NC, by Radian Corporation, Austin, TX. EPA-
450/3-89-29.
NCA. 1982. 24-month chronic toxicity and oncogenicity study of methylene chloride in rats. Final
report. Vols. I-IV. Report to National Coffee Association by Hazleton Laboratories America, Inc.
[Unpublished] 2112-2101.
NCA. 1983. 24-month oncogenicity study of methylene chloride in mice. Final report. Vol. I. Report
to National Coffee Association by Hazleton Laboratories America, Inc. [Unpublished] 2112-2106.
*NC Div. Env. Manage. 1998. Toxic air pollutant guidelines. North Carolina Division of Environmental
Management, Air Quality Section. NC Admin. Code Tit. 15A, R. 2D.1100.
*Nelson HH, Wiencke JK, Christiani DC, et al. 1995. Ethnic differences in the prevalence of the
homozygous deleted genotype of glutathione S-transferase theta. Carcinogenesis 16:1243-1245.
Newsom JM. 1985. Transport of organic compounds dissolved in ground water. Environmental
Monitoring Review 5:28-36.
NFPA. 1978. Fire protection guide for hazardous materials. National Fire Protection Association.
*NIOSH. 1974. Methylene chloride: Development of a biologic standard for the industrial worker by
breath analysis. Cincinnati, OH: National Institute of Occupational Safety and Health. NTIS No.
PB83-245860. NIOSH-MCOW-ENVM-MC-74-9.
245 METHYLENE CHLORIDE
8. REFERENCES
NIOSH. 1976. National Institute for Occupational Safety and Health. Criteria for a recommended
standard...occupational exposure to methylene chloride. Cincinnati, OH: U.S. Department of Health,
Education and Welfare, Public Health Service, Centers for Disease Control, National Institute for
Occupational Safety and Health. DHEW publ. no 76-138.
*NIOSH. 1984. Methylene chloride-method 1005. In: NIOSH manual of analytical methods. 3rd ed.
Cincinnati, OH: Department of Health and Human Services, National Institute of Occupational Safety
and Health. DHHS publ. no. 84-100.
*NIOSH. 1986. Current intelligence bulletin 46 - methylene chloride. Cinncinati, OH: National
Institute for Occupational Safety and Health. Department of Health and Human Services. PB 86-114.
NIOSH. 1992. Recommendations for occupational safety health, compendium of policy documents and
statements. U.S. Department of Health and Human Services, Centers for Disease Control, National
Institute for Occupational Safety and Health, Cincinnati, OH.
NIOSH. 1993. Posthearing comments from the National Institute for Occupational Safety and Health on
the Occupational Safety and Health Administration proposed rule on occupational exposure to methylene
chloride. NTIS-PB95-200432.
*NIOSH. 1994. Methylene chloride - method no. 1005. In: NIOSH manual of analytical methods.
4
th
ed. Cincinnati, OH: Department of Health and Human Services, National Institute of Occupational
Safety and Health. DHHS publication no. 94-113.
*NIOSH. 1995. Report to Congress on workers’ home contamination study conducted under the
workers’ family protection act. Cincinnati, OH: National Institute of Occupational Safety and Health.
DHHS publication no. 95-123.
*NIOSH. 1997. National Institute for Occupational Safety and Health pocket guide to chemical hazards.
Washington, DC: Department of Health and Human Services, 208-209.PB 97-177604. Pub No. 97-140.
*NIOSH 1999. Methylene chloride. Online pocket guide to chemical hazards.
Wysiwyg://58/http://www.cdc.gov/niosh/npg/npg.html
*Nitschke KD, Burek JD, Bell TJ, et al. 1988a. Methylene chloride: A 2-year inhalation toxicity and
oncogenicity study in rats. Fundam Appl Toxicol 11:48-59.
*Nitschke KD, Eisenbrandt DL, Lomax LG, et al. 1988b. Methylene chloride: Two-generation
inhalation reproductive study in rats. Fundam Appl Toxicol 11:60-67.
*NJ Dept Env Protec. 1993. Ground water quality standards. New Jersey Department of Environmental
Protection, Division of Water Quality. N.A.J.C. 7:9-6. http://www.state.nj.us/dep/
*NJ Dept. Env. Protect. 1998. Unit risk factors for inhalation. New Jersey Department of Environmental
Protection, Air Management. NJ Admin Code Tit. 7, Chapter 7.27, Subchapter 7.
*NOES. 1990. National Occupational Exposure Survey, National Institute of Occupational Safety and
Health, Cincinnati, OH. July 16, 1990.
NOHS. 1990. National Occupational Hazard Survey, National Institute of Occupational Safety and
Health, Cincinnati, OH. July 16, 1990.

246 METHYLENE CHLORIDE
8. REFERENCES
Nordic Chemicals Control Group. 1992. Nordic criteria for reproductive toxicity: Dichloromethane
(methylene chloride). 31-33.
Norman WC. 1996a. Exposure to methylene chloride. Science 274:3271.
Norman WC. 1996b. Flawed estimates of methylene chloride exposures. Am J Ind Med 30:504-505.
*Norpoth K, Witting U, Springorum M, et al. 1974. Induction of microsomal enzymes in the rat liver by
inhalation of hydrocarbon solvents. Int Arch Arbeitsmed 33:315-321.
*NRC. 1993. National Research Council. Pesticides in the diets of infants and children. Washington
DC, National Academy Press.
*NTDB. 1998. Dichloromethane. National Trade Data Bank: The Export Connection.
NTP. 1985. National Toxicology Program. 1985. NTP technical report on the toxicology and
carcinogenesis studies of dichloromethane in F-344 in rats B6C3F1 mice/inhalation studies). February.
NTP-TR-306. Board draft.
*NTP. 1986. National Toxicology Program. NTP technical report on the toxicology and carcinogenesis
studies of dichloromethane (methylene chloride) (CAS No. 75-09-2) in F344/N rats and B6C3F
1
mice
(inhalation studies). Research Triangle Park, NC: U.S. Department of Health and Human Services.
NTP-TR-306. NIH Pub No. 86-2562.
*NTP. 1989. National Toxicology Program. Dichloromethane (methylene chloride) CAS no. 75-90-2.
Fifth annual report on carcinogens: 1989 Summary. Report of the National Institute of Environmental
Health Sciences, Research Triangle Park, NC, by Technical Resources, Inc., Rockville, MD, 110-113.
NTP 89-239.
NTP. 1998. National Toxicology Program. Dichloromethane (methylene chloride) CAS no. 75-90-2.
Eighth annual report on carcinogens: 1998 Summary. Research Triangle Park, NC: National Institute of
Environmental Health Sciences.
NTP. 1999. Report on carcinogens. National Toxicology Program. http://ntp-
server.niehs.nih.gov/NewHomeRoc/AboutRoC.html.
*NY Dept. Env. Conserv. 1998. High toxicity air contaminants. New York Department of
Environmental Conservation, Division of Air Resources. NY Comp. Codes R. & Regs. Tit. 6, Part 257.
Nylen P. 1996. Differing non-additive alterations in different parts of the nervous system of the rat.
Food Chem Toxicol 34:1121-1123.
*Oda Y, Yamazaki H, Thier R, et al. 1996. A new Salmonella typhimurium NM5004 strain expressing
rat glutathione S-transferase 5-5: Use in detection of genotoxicity of dihaloalkanes using an SOS/umu test
system. Carcinogenesis 17:297-302.
*OHM/TADS. 1998. Oil and Hazardous Materials/Technical Assistance Data System. Baltimore, MD:
Chemical Information System, Inc. May 12, 1998.
*OK Dept Env Quality. 1997. Water quality standards. Oklahoma Department of Environmental
Quality, Water Resources Board. Chapter 45. Http://www.deq.state.ok.us/
247 METHYLENE CHLORIDE
8. REFERENCES
ORVWSC. 1982. Ohio River Valley Water Sanitary Commission. Assessment of water quality
conditions. Ohio River Mainstream. 1981-1982. Table 13.
OSHA. 1979. General industry standards. Washington, DC: Occupational Safety and Health
Administration, Department of Labor. OSHA 2206. Revised January 1978.
*OSHA. 1986. Occupational Safety and Health Administration. Occupational exposure to methylene
chloride. Federal Register 51:42257.
OSHA. 1989. Occupational Safety and Health Administration: Part III. Federal Register 54:2332-2335,
2923, 2959.
OSHA. 1991. Occupational Safety and Health Administration: Part II. Federal Register
56:57036-57139.
*OSHA. 1997. Occupational exposure to methylene chloride. Occupational Safety and Health
Administration, Department of Labor. Federal Register 62(7):1494-1600.
*OSHA. 1998a. Methylene chloride; Final rule. Department of Labor, Occupational Safety and Health
Administration. Federal Register 63(183): 50712-50732.
*OSHA. 1998b. Substance safety data sheet and technical guidelines for methylene chloride.
Occupational Safety and Health Administration, Department of Labor. Code of Federal Regulations. 29
CFR 1910.1052.
*OSHA. 1999. U. S. Department of Labor. Occupational Safety and Health Administration. Code of
Federal Regulations. 29 CFR 1910.1052.
*OTA. 1990. Neurotoxicity: Identifying and controlling poisons of the nervous system. Washington,
DC: Office of Technology Assessment. OTA-BA-436.
*Otson R, Doyle EE, Williams DT, et al. 1983. Survey of selected organics in office air. Bull Environ
Contam Toxicol 31:222-229.
*Ott, MG, Skory LK, Holder BB, et al. 1983a. Health evaluation of employees occupationally exposed
to methylene chloride: Clinical laboratory evaluation. Scand J Work Environ Health 9 (Suppl 1):17-25.
*Ott MG, Skory LK, Holder BB, et al. 1983b. Health evaluation of employees occupationally exposed
to methylene chloride: Mortality. Scand J Work Environ Health 9(suppl1):8-16.
*Ott MG, Skory LK, Holder BB, et al. 1983c. Health evaluation of employees occupationally exposed to
methylene chloride: Twenty-four hour electrocardiographic monitoring. Scand J Work Environ Health
9(suppl1):26-30.
*Ott MG, Skory LK, Holder BB, et al. 1983d. Health evaluation of employees occupationally exposed
to methylene chloride: Metabolism data and oxygen half-saturation pressure. Scand J Work Environ
Health 9(suppl1):31-38.
248 METHYLENE CHLORIDE
8. REFERENCES
Ottenwalder H, Jager R, Thier R, et al. 1989. Influence of cytochrome P-450 inhibitors on the inhalative
uptake of methyl chloride and methylene chloride in male B6C3F1 mice. Arch Toxicol Suppl
13:258-261.
*Owen GM, Brozek J. 1966. Influence of age, sex, and nutrition on body composition during childhood
and adolescence. In: Falkner F, ed. Human development, Philadelphia, PA, WB Saunders, 22-238.
*Page BD, Charbonneau CF. 1977. Coffee and tea: Gas chromatographic determination of residual
methylene chloride and trichloroethylene in decaffeinated instant and ground coffee with electrolytic
conductivity and electron capture detection. J Assoc Off Anal Chem 60(3):710-715.
*Page BD, Charbonneau CF. 1984. Coffee and tea: Headspace gas chromatographic determination of
methylene chloride in decaffeinated tea and coffee, with electrolytic conductivity detection. J Assoc Off
Anal Chem 67(4):757-761.
Page BD, Kennedy BPC. 1975. Determination of methylene chloride, ethylene dichloride, and
trichloroethylene as solvent residues in spice oleoresins, using vacuum distillation and electron capture
gas chromatography. J Assoc Off Anal Chem 58:1062-1068.
*Page BD, Conacher HBS, Salminen J, et al. 1993. Survey of bottled drinking water sold in Canada.
Part 2: Selected volatile organic compounds. J AOAC Int 76(1):26-31.
*Page GW. 1981. Comparison of groundwater and surface water for patterns and levels of
contamination by toxic substances. Environ Sci Technol 15(12):1475-1481.
Pankow D, Hoffman P. 1989. Dichloromethane metabolism to carbon monoxide can be induced by
isoniazid, acetone and fasting. Arch Toxicol Suppl 13:302-303.
*Pankow D, Jagielki S. 1993. Effect of methanol or modifications of the hepatic glutathione
concentration on the metabolism of dichloromethane to carbon monoxide in rats. Hum Exp Toxicol
12:227-231.
Pankow D, Marzotko D. 1987. [To the acute hepatic toxicity of dichloromethane.] Z Gesamte Hyg Ihre
Grenzgeb 33:518-519. (German).
*Pankow JF, Rosen ME. 1988. Determination of volatile compounds in water by purging directly to a
capillary column with whole column cryotrapping. Environ Sci Technol 22:398-405.
*Pankow D, Kretschmer, Weise M. 1991a. Effect of pyrazole on dichloromethane to carbon monoxide.
Arch Toxicol Suppl 14:246-248.
*Pankow D, Matschiner F, Weigmann HJ. 1991b. Influence of aromatic hydrocarbons on the
metabolism of dichloromethane to carbon monoxide in rats. Toxicology 68:89-100.
Pankow D, Weise M, Hoffmann P. 1992. Effect of isoniazid or phenobarbital pretreatment on the
metabolism of dihalomethanes to carbon monoxide. Pol J Occup Med Environ Health 5:245-250.
249 METHYLENE CHLORIDE
8. REFERENCES
Paone DA, Weinreb HG, Bauer MJ, et al. 1996. A pilot-scale fluid bed reactor for treatment of
methlyene chloride process stream. In: Hickey RF, Smith G, ed. Biotechnological industrial waste
treatment bioremediation: International symposium on the implementation of biotechnological industrial
waste treatment bioremediation. Boca Raton, Florida: Lewis, 143-153.
Park JK, Kim JY, Edil TB. 1996. Mitigation of organic compound movement in landfills by shredded
tires. Water Environ Res 68(1):4-10.
Pearson CR, McConnell G. 1975. Chlorinated C
1
and C
2
hydrocarbons in the marine environment. Proc
R Soc London B 189:305-332.
*Pelekis M, Krishnan K. 1997. Assessing the relevance of rodent data on chemical interactions for
health risk assessment purposes: A case study with dichloromethane-toluene mixture. Regul Toxicol
Pharmacol 25:79-86.
Pelekis M, Poulin P, Krishnan K. 1995. An approach for incorporating tissue composition into
physiologically based pharmacokinetic models. Toxicol Ind Health 11:511-522.
*Pelkonen O, Raunio H, Rautio A, et al. 1999. Xenobiotic-metabolizing enzymes and cancer risk:
correspondence between genotype and phenotype. In: Vineis P, Malats N, Lang M, et al., ed. Metabolic
polymorphisms and susceptibility to cancer. Lyon, France: International Agency for Research on Cancer,
77-78.
Pellizzari ED. 1974. Electron capture detection in gas chromatography. J Chromatogr 98:323-361.
Pellizzari ED. 1977. Analysis of organic air pollutants by gas chromatography and mass spectroscopy.
U.S. Environmental Protection Agency, Office of Research and Development. EPA-600/2-77-100.
Pellizzari ED. 1978a. Measurement of carcinogenic vapors in ambient atmospheres. U.S. Environmental
Protection Agency, Office of Research and Development. EPA-600/7-78-062.
Pellizzari ED. 1978b. Quantification of chlorinated hydrocarbons in previously collected air samples.
Research Triangle Park, NC: U.S. Environmental Protection Agency, Office of Air Quality Planning and
Standards. EPA-450/3-78-112.
Pellizzari ED, Bunch JE. 1979. Ambient air carcinogenic vapors: Improved sampling and analytical
techniques and field studies. U.S. Environmental Protection Agency, Office of Research and
Development. EPA-600/2-79-081.
Pellizzari ED, Erickson MD, Zweidinger RA. 1979. Formulation of a preliminary assessment of
halogenated organic compounds in man and environmental media. Washington, DC: U.S.
Environmental Protection Agency, Office of Pesticides and Toxic Substances. EPA-560/13-179-006.
*Pellizzari ED, Hartwell TD, Harris BSH, et al. 1982. Purgeable organic compounds in mother's milk.
Bull Environ Contam Toxicol 28:322-328.
Pemble S, Schroeder KR, Spencer SR, et al. 1994. Human glutathione S-transferase Theta (GSTT1):
cDNA cloning and the characterization of a genetic polymorphism. Biochem J 300:271-276.
*Perocco P, Prodi G. 1981. DNA damage by halokanes in human lymphocytes cultured in vitro. Cancer
Lett 13:213-218.
250 METHYLENE CHLORIDE
8. REFERENCES
*Peterson JE. 1978. Modeling the uptake, metabolism, and excretion of dichloromethane by man. Am
Ind Hyg Assoc J 39:41-47.
*Plumb RH. 1987. A comparison of ground water monitoring data from CERCLA and RCRA sites.
Ground Water Monit Rev (Fall):94-100.
Post W, Kromhout H, Heederik D, et al. 1991. Semiquantitative estimates of exposure to methylene
chloride and styrene: The influence of quantitative exposure data. Appl Occup Environ Hyg 6:197-204.
*Poulin P, Krishnan K. 1996. Molecular structure-based prediction of the partition coefficients of
organic chemicals for physiological pharmacokinetic models. Toxicol Meth 6(3):117-137.
*Poulin P, Krishnan K. 1998. A quantitative structure-toxicokinetic relationship model for highly
metabolized chemicals. ATLA 26:45-59.
*Poulin P, Krishnan K. 1999. Molecular structure-based prediction of the toxicokinetics of inhaled
vapors in humans. Int J Toxicol 18:7-18.
*Putz VR, Johnson BL, Setzer JV. 1979. A comparative study of the effects of carbon monoxide and
methylene chloride on human performance. J Environ Pathol Toxicol 2:97-112.
*Raje R, Basso M, Tolen T, et al. 1988. Evaluation of in vivo mutagenicity of low-dose methylene
chloride in mice. J Am Coll Toxicol 7(5):699-703.
*Ramsey JC, Andersen ME. 1984. A physiologically based description of the inhalation
pharmacokinetics of styrene in rats and humans. Toxicol Appl Pharmacol 73:159-175.
*Rasheed A, Hines RN, McCarver-May DG. 1997. Variation in induction of human placental CYP2E1:
Possible role in susceptibility to fetal alcohol syndrome? Toxicol Appl Pharmacol 144:396-400.
Rasmussen RA, Harsch DE, Sweany PH, et al. 1977. Determination of atmospheric halocarbons by a
temperature programmed gas chromatographic freezeout concentration method. JAPCA 27:579-581.
Rastogi SC. 1994. Regulation of dichloromethane and 1,1,1-trichloroethane in cosmetic products. Sci
Total Environ 156:23-25.
Ratney RS, Wegman DH, Elkins HB. 1974. In vivo conversion of methylene chloride to carbon
monoxide. Arch Environ Health 28:223-226.
*Rebert CS, Matteucci MJ, Pryor GT. 1989. Acute effects of inhaled dichloromethane on the EEG and
sensory-evoked potentials of Fischer-344 rats. Pharmacol Biochem Beh 34:619-629.
*Reitz RH. 1990. Quantitating the production of biological reactive intermediates in target tissues:
Example, dichloromethane. In: Witmer CM, et al, ed. Biological reactives intermediates IV. New York:
Plenum Press, 649-655.
*Reitz RH. 1991. Estimating the risk of human cancer associated with exposure to methylene chloride.
Ann Ist Super Sanita 27(4):609-614.

251 METHYLENE CHLORIDE
8. REFERENCES
*Reitz RH, Smith FA, Andersen ME, et al. 1988. Use of physiological pharmacokinetics in cancer risk
assessments: A study of methylene chloride. In: Paustenbach DJ, ed. The risk assessment of
environmental and human health hazards. New York, NY: John Wiley & Sons, Inc., 238-265.
*Reitz RH, Mendrala AL, Guengerich FP. 1989. In vitro metabolism of methylene chloride in human
and animal tissues: Use in physiologically based pharmacokinetic models. Toxicol Appl Pharmacol
97:230-246.
*Reitz RH, Hays SM, Gargas ML. 1997. Addressing priority data needs for methylene chloride with
physiologically based pharmacokinetic modeling. Atlanta, GA: Agency for Toxic Substances and
Disease Registry.
Rhomberg L. 1995. Use of quantitative modelling in methylene chloride risk assessment. Toxicol
102:95-114.
Rhone-Poulenc, Inc. 1994. Notice of monitored concentrations of methylene chloride, 1,1,1-
trichloroethane, benzene, in local wells. TSCATS-441027. NTIS/OTS 0556009.
Rice D. 2000. Parallels between attention deficit hyperactivity disorder and behavioral deficits produced
by neurotoxic exposure in monkeys. Environ Health Perspect 108(suppl 3)405-408.
Rice D, Barone S. 2000. Critical periods of vulnerability for the developing nervous system: Evidence
from humans and animal models. Environ Health Perspect 108(suppl 3):511-533.
*RI Dept Env Management. 1992. Air toxics. Rhode Island Department of Environmental Management,
Divison of Air and Hazardous Materials. Air pollution control regulation No. 22.
Http://www.sec.state.ri.us/dem/.
RI Dept. Env. Management. 1998. Acceptable ambient levels. Rhode Island Department of
Environmental Management, Division of Air Resources. Http://www.sec.state.ri.us/dem/regs.htm.
November 2, 1999.
*Riley EC, Fasset DW, Sutton WL. 1966. Methylene chloride vapor in expired air of human subjects.
Am Ind Hyg Assoc J 27:341-348.
Rittman BE, McCarty PL. 1980. Utilization of dichloromethane by suspended and fixed-film bacteria.
Appl Environ Microbiol 39:1225-1226.
*Roberts CJC, Marshall FPF. 1976. Recovery after “lethal” quantity of paint remover. Brit Med J
(January):20-21.
Robinson E. 1978. Analysis of halocarbons in Antarctica. Report to the National Science Foundation,
Washington, DC. Report 78/13-42
Rodkey FL, Collison HA. 1977. Effect of dihalogenated methanes on the in vivo production of carbon
monoxide and methane by rats. Toxicol Appl Pharmacol 40:39-47.
*Rosengren LE, Kjellstrand P, Aurell A, et al. 1986. Irreversible effects of dichloromethane on the brain
after long term exposure: A quantitative study of DNA and the glial cell marker proteins S-100 and GFA.
Br J Ind Med 43:291-299.
252 METHYLENE CHLORIDE
8. REFERENCES
*Roth RP, Drew RT, Lo RJ, et al. 1975. Dichloromethane inhalation, carboxyhemoglobin
concentrations, and drug metabolizing enzymes in rabbits. Toxicol Appl Pharmacol 33:427-437.
*Roy WR, Griffin RA. 1985. Mobility of organic solvents in water-saturated soil materials. Environ
Geol Water Sci 7(4):241-247.
RTECS. 1998. Registry of Toxic Effects of Chemical Substances. National Library of Medicine,
Bethesda, MD. May 11, 1998.
*RTECS. 1999. Registry of Toxic Effects of Chemical Substances. National Library of Medicine,
Bethesda, MD.
*Ruch RJ, Crist KA, Klaunig JE. 1989. Effects of culture duration on hydrogen peroxide-induced
hepatocyte toxicity. Toxicol Appl Pharm 100:451-464.
Ruelle P, Kesselring UW. 1997. Aqueous solubility prediction of environmentally important chemicals
from the mobile order thermodynamics. Chemosphere 34(2):275-298.
*Ruth JH. 1986. Odor thresholds and initation levels of several chemical substances. A review. Am Ind
Hyg Assoc J 47:A142-A151.
Sakai T, Miyake K, Kurata M, et al. 1991. A case of severe hepatic microcirculatory failure with
jaundice and fibrosis around the sinusoid caused by long-term exposure of methylene chloride. Nippon
Shokakibyo Gakkai Zasshi 88:185-189.
*Sasaki YF, Saga A, Akasaka M, et al. 1998. Detection of in vivo genotoxicity of haloalkanes and
haloalkenes carcinogenic to rodents by the alkaline single cell gel electrophoresis (comet) assay in
multiple mouse organs. Mutat Res 419:13-20.
*Savard S, Otson R, Douglas GR. 1992. Mutagenicity and chemical analysis of sequential organic
extracts of airborne particulates. Mutat Res 276:101-115.
*Savolainen H, Pfäffli P, Tengén M, et al. 1977. Biochemical and behavioural effects of inhalation
exposure to tetrachloroethylene and dichloromethane. J Neuropath Exp Neurol 36:941-949.
*Savolainen H, Kurppa K, Pfäffli P, et al. 1981. Dose-related effects of dichloromethane on rat brain in
short-term inhalation exposure. Chem Biol Interact 34:315-322.
*Sawhney BL. 1989. Movement of organic chemicals through landfill and hazardous waste disposal
sites. In: Reactions and movement of organic chemicals in soils. Madison, WI: Soil Science Society of
America and American and American Society of Agronomy, 447-474.
*Sax NI, Lewis RJ. 1987. Hawley's condensed chemical dictionary. 11th ed. New York, NY: Van
Nostrand Reinhold Company, 768.
SC Dept. Health & Env. Control. 1998. Toxic air pollutants with maximum allowable concentrations.
South Carolina Department of Health & Environmental Control, Bureau of Air Quality. 24A SC Code
Ann. Regs. 61-62.5, Standard 8.
Schnaak W, Küchler T, Kujawa M, et al. 1997. Organic contaminants in sewage sludge and their
ecotoxicological significance in the agricultural utilization of sewage sludge. Chemosphere 35(½):5-11.
253 METHYLENE CHLORIDE
8. REFERENCES
*Scholz J, Klapperstuck M, Weise M, et al. 1991. Acute effects of dichloromethane on arrhythmia
development during the early phase of myocardial ischemia and reperfusion in the rat. Arch Toxicol
Suppl 14:128-131.
*Schröder KR, Hallier E, Meyer DJ. 1996. Purification and characterization of a new glutathione S-
transferase, class θ, from human erythrocytes. Arch Toxicol 70:559-566.
Schwartz LJ. 1991. Toxic effects of selected industrial solvents in batch and continuous anaerobic
reactors. Appl Biochem Biotech 28/29:297-305.
*Schwetz BA, Leong BKJ, Gehring PJ. 1975. The effect of maternally inhaled trichloroethylene,
perchloroethane, methyl chloroform, and methylene chloride on embryonal and fetal development in mice
and rats. Toxicol Appl Pharmacol 32:84-96.
*SD Dept Env Natural Resources. 1998. Water hygiene. South Dakota Department of Environment and
Natural Resources, Drinking Water Program. Article 74:04.
http://www.state.sd.us/state/executive/denr/denr.html
Seilkop SK. 1995. The effect of body weight on tumor incidence and carcinogenicity testing in B6C3F1
mice and F344 rats. Fundam Appl Toxicol 24:247-259.
Selan FM, Evans MA. 1987. The role of microtubules in chlorinated alkane-induced fatty liver. Toxicol
Lett 36:117-127.
Selevan SG, Kimmel CA, Mendola P. 2000. Identifying critical windows of exposure for children’s
health. Environ Health Perspect 108(suppl 3):451-455.
Semprini L. 1997. Strategies for the aerobic co-metabolism of chlorinated solvents. Curr Opin
Biotechnol 8:296-308.
Serota D, Ulland B, Carlborg F. 1984. Hazleton chronic oral study in mice. Food solvents workshop
no. 1. Methylene chloride, March 8-9, Bethesda, Maryland. Toxicol Appl Pharmacol 87:185-205.
*Serota DG, Thakur AK, Ulland BM, et al. 1986a. A two-year drinking-water study of dichloromethane
in rodents. I. Rats. Food Chem Toxicol 24(9):951-958.
*Serota DG, Thakur AK, Ulland BM, et al. 1986b. A two-year drinking-water study of dichloromethane
in rodents. II. Mice. Food Chem Toxicol 24(9):959-963.
Setchell BP, Waites GMH. 1975. The blood testis barrier. In: Geiger SR, ed. Handbook of physiology:
Endocrinology V. Washington, DC: American Physiological Society.
Shah JJ, Singh HB. 1988. Distribution of volatile organic chemicals in outdoor and indoor air. Environ
Sci Technol 22:1381-1388.
*Sheehan GC, Freedman DL. 1996. High rate treatment of dichloromethane in anoxic and aerobic
fluidized bed bioreactors. WEFTEC '96, The 69th Annual Conference and Exposition of the Water
Environment Federation, October 5-9, 1996, Dallas, Texas.
254 METHYLENE CHLORIDE
8. REFERENCES
Sheldon T, Richardson CR, Hamilton K, et al., eds. 1986. Methylene chloride: An evaluation in the
mouse micronucleus test. ICI Central Toxicology Laboratory, Report No. CTL/P/1603, September 19.
TSCATS 305690. OTS 0514366. EPA 86-880000288.
*Sheldon T, Richardson CR, Elliott BM. 1987. Inactivity of methylene chloride in the mouse bone
marrow micronucleus assay. Mutagenesis 2(1):57-59.
Shelley ML, Andersen ME, Fisher JW. 1989. A risk assessment approach for nursing infants exposed to
volatile organics through the mother's occupational inhalation exposure. Appl Ind Hyg 4:21-26.
Sherman J, Chin B, Huibers PDT, et al. 1998. Solvent replacement for green processing. Environ
Health Perspect Suppl 106(1):253-271.
*Sherratt PJ, Pulford DJ, Harrison DJ, et al. 1997. Evidence that human class Theta glutathione S-
transferase T1-1 can catalyse the activation of dichloromethane, a liver and lung carcinogen in the mouse:
Comparison of the tissue distribution of GST T1-1 with that of classes alpha, mu and pi GST in human.
Biochem J 326:837-846.
Sherratt PJ, Manson MM, Thomson AM, et al. 1998. Increased bioactivation of dihaloalkanes in rat liver
due to induction of class theta glutathione s-transferase T1-1. Biochem J 335:619-630.
*Shih RD. 1998. Hydrocarbons. In: Goldfrank LR, Flomenbaum NE, Lewin NA, et al., ed. Goldfrank's
toxicologic emergencies. Stamford, Connecticut: Appleton & Lange, 1383-1398.
*Shikiya J, Tsou G, Kowalski J, et al. 1984. Ambient monitoring of selected halogenate hydrocarbons
and benzene in the California south coast air basin. Proceedings of the 77th annual meeting of the Air
Pollution Control Association, San Francisco, CA, June 24-29, 1984.
Sidebottom H, Franklin J. 1996. The atmospheric fate and impact of hydro-chlorofluorocarbons and
chlorinated solvents. Pure Appl Chem 68(9):1757-1769.
*Sikkema J, de Bont JAM, Poolman B. 1995. Mechanisms of membrane toxicity of hydrocarbons.
Microbiol Rev 59(2):201-222.
*Simula TP, Glancey MJ, Wolf CR. 1993. Human glutathione S-transferase-expressing Salmonella
typhimurium tester strains to study the activation/detoxification of mutagenic compounds: Studies with
halogenated compounds, aromatic amines and aflatoxin B
1
. Carcinogenesis 14:1371-1376.
Singh HB. 1977. Atmospheric halocarbons. Evidence in favor of reduced average hydroxyl radical
concentrations in the troposphere. Geophys Res Lett 4:101-104.
Singh HB, Salas LJ, Shigeishi H, et al. 1979. Atmospheric distributions, sources, and sinks of selected
halocarbons, hydrocarbons, SF
6
, and N
2
O. Report to U.S. Environmental Protection Agency,
Environmental Sciences Research Laboratory, Research Triangle Park, NC, by SRI International, Menlo
Park, CA. EPA-600/3-79-107.
*Singh HB, Salas LJ, Smith AJ, et al. 1981. Measurements of some potentially hazardous organic
chemicals in urban environments. Atmos Environ 15:601-612.
*Singh HB, Salas LJ, Stiles RE. 1983. Selected man-made halogenated chemicals in the air and oceanic
environment. J Geophy Res 88:3675-3683.
255 METHYLENE CHLORIDE
8. REFERENCES
*Singh HB, Salas LJ, Stiles RE, et al. 1982. Distribution of selected gaseous organic mutagens and
suspect carcinogens in ambient air. Environ Sci Technol 16:872-880.
Sinkkonen S, Welling L, Vattulainen A, et al. 1996. Short chain aliphatic halocarbons and
polychlorinated biphenyls in pine needles: effects of metal scrap plant emissions. Chemosphere
32(10):1971-1982.
Sitting M. 1985. Handbook of toxic and hazardous chemicals and carcinogens. 2nd ed. Park Ridge,
NY: Noyes Publications, 598-601.
Snodgrass WR. 1992. Physiological and biochemical differences between children and adults as
determinants of toxic response to environmental pollutants. In: Guzelian PS, Henry CJ, Olin SS, ed.
Similarities and differences between children and adults: Implications for risk assessment. Washington,
DC: International Life Sciences Institute Press, 35-42.
*Snyder RW, Mishel HS, Christensen GC. 1992a. Pulmonary toxicity following exposure to methylene
chloride and its combustion product, phosgene. Chest 101:860-861.
*Snyder RW, Mishel HS, Christensen GC. 1992b. Pulmonary toxicity following exposure to methylene
chloride and its combustion product, phosgene. Chest 102:1921.
*Soden KJ. 1993. An evaluation of chronic methylene chloride exposure. J Occup Med 35(3):282-286.
*Soden KJ, Marras G, Amsel J. 1996. Carboxyhemoglobin levels in methylene chloride-exposed
employees. J Occup Environ Med 38(4):367-371.
Spivack JL, Shank GK, Nick RJ, et al. 1996. Biodegredation of methlyene chloride in industrial process
wastewater: evaluation of reactor configurations and comparison of a pure hypomicrobial culture with
wastewater treatment sludge. In: Hickey RF, Smith G, ed. Biotechnological industrial waste treatment
bioremediation: International symposium on the implementation of biotechnological industrial waste
treatment bioremediation. Boca Raton, Florida: Lewis, 111-142.
SRI. 1997. Directory of chemical producers: United States of America. Menlo Park, CA: SRI
International, 749.
*SRI. 1999. Directory of chemical producers: United States of America. Menlo Park, CA: SRI
International, 744, 122, 123, 415.
*Staats DA, Fisher JW, Connolly RB. 1991. Gastrointestinal absorption of xenobiotics in
physiologically based pharmacokinetic models: A two-compartment description. Drug Metab Dispos
19(1):144-148.
*Staples CA, Werner AF, Hoogheem TJ. 1985. Assessment of priority pollutant concentrations in the
United States using STORET database. Environ Toxicol Chem 4:131-142.
Stauffer Chemical Company. 1973. Toxicology laboratory report T-4171; Methlyene chloride.
Westport, Connecticut. OTS84003A.
Stayner LT, Bailer AJ. 1993. Comparing toxicologic and epidemiologic studies: Methylene chloride–A
case study. Risk Anal 13:667-673.
256 METHYLENE CHLORIDE
8. REFERENCES
*Steinmetz KL, Green CE, Bakke JP, et al. 1988. Induction of unscheduled DNA synthesis in primary
cultures of rat, mouse, hamster, monkey, and human hepatocytes. Mutat Res 206:91-102.
*Stephens EA, Taylor JA, Kaplan N, et al. 1994. Ethnic variation in the CYP2E1 gene: Polymorphism
analysis of 695 African-Americans and Taiwanese. Pharmacogenetics 4:185-192.
*Stewart RD, Dodd HC. 1964. Absorption of carbon tetrachloride, trichloroethylene,
tetrachloroethylene, methylene chloride and 1,1,1-trichloroethane through human skin. Am Ind Hyg
Assoc J 25:439-446.
*Stewart RD, Hake CL. 1976. Paint remover hazard. JAMA 235(4):398-401.
*Stewart RD, Fischer TN, Hosko MJ, et al. 1972. Experimental human exposure to methylene chloride.
Arch Environ Health 25:342-348.
Storm JE, Rozman KK. 1998. Derivation of an occupational exposure limit (OEL) for methylene
chloride based on acute CNS effects and relative potency analysis. Regul Toxicol Pharmacol 27:240-250.
*Stott WT, Dryzga MD, Ramsey JC. 1983. Blood flow distribution in the mouse. J Appl Toxicol
3(6):310-312.
*Stover EL, Kincannon DF. 1983. Biological treatability of specific organic compounds found in
chemical industry wastewaters. J Water Pollut Control Fed 55(1):97-109.
*Strange RC, Fryer AA. 1999. The glutathione S-transferases: influence of polymorphism on cancer
susceptibility. In: Vineis P, Malats N, Lang M, et al., ed. Metabolic polymorphisms and susceptibility to
cancer. Lyon, France: International Agency for Research on Cancer, 231-249.
*Stubbins MJ, Wolf CR. 1999. Additional polymorphisms and cancer. In: Vineis P, Malats N, Lang M,
et al., ed. Metabolic polymorphisms and susceptibility to cancer. Lyon, France: International Agency for
Research on Cancer, 271-301.
Stubin AI, Brosnan TM, Porter KD, et al. 1996. Organic priority pollutants in New York City municipal
wastewaters: 1989-1993. Water Environ Res 68:1037-1044.
*Svirbely JL, Highman B, Alford WF, et al. 1947. The toxicity and narcotic action of mono-chloro-
mono-bromo-methane with special reference to inorganic and volatile bromide in blood, urine and brain.
J Ind Hyg Toxicol 29(6):382-389.
*Swann RL, Laskowski DA, McCall PJ, et al. 1983. A rapid method for the estimation of the
environmental parameters octanol water partition coefficient, soil sorption constant, water to air ratio, and
water solubility. Res Rev 85:17-28.
Sylvestre M, Bertrand J-L, Viel G. 1997. Feasibility study for the potential use of biocatalytic systems to
destroy chlorofluorocarbons (CFCs). Crit Rev Environ Sci 27(2):87-111.
*Tabak HH, Quave SA, Mashni CI, et al. 1981. Biodegredability studies with organic priority pollutant
compounds. J Water Pollut Control Assoc 53(10):1503-1518.
*Tardif R, Laparé S, Krishnan K, et al. 1993. Physiologically based modeling of the toxicokinetic
interaction between toluene and m-xylene in the rat. Toxicol Appl Pharmacol 120:266-273.
257 METHYLENE CHLORIDE
8. REFERENCES
*Taskinen H, Lindbohm M-L, Hemminki K. 1986. Spontaneous abortions among women working in the
pharmaceutical industry. Br J Ind Med 43:199-205.
Tateishi T, Nakura H, Asoh M, et al. 1997. A comparison of hepatic cytochrome P450 protein
expression between infancy and postinfancy. Life Sci 61(26):2567-2574.
*Tay P, Tan KT, Sam CT. 1995. Fatal gassing due to methylene chloride–A case report. Singapore Med
J 36:444-445.
Teschke K, Ahrens W, Andersen A, et al. 1999. Occupational exposure to chemical and biological
agents in the nonproduction departments of pulp, paper, and paper product mills: an international study.
Am Ind Hyg Assoc J 60:73-83.
Testai E, Di Marzio S, di Domenico A, et al. 1995. An in vitro investigation of the reductive metabolism
of chloroform. Arch Toxicol 70:83-88.
Thiebaud H, Merlin G, Alary J, et al. 1991. Gas chromatographic determination of dichloromethane in
the water of model aquatic ecosystems. Analusis 19:208-213.
*Thier R, Foest U, Deutshmann S, et al. 1991. Distribution of methylene chloride in human blood. Arch
Toxicol Suppl 14:254-258.
Thier R, Taylor JB, Pemble SE, et al. 1993. Expression of mammalian glutathione S-transferase 5-5 in
Salmonella typhimurium TA1535 leads to base-pair mutations upon exposure to dihalomethanes. Proc
Natl Acad Sci 90:8576-8580.
*Thier R, Wiebel FA, Hinkel A, et al. 1998. Species difference in the glutathione transferase GSTT1-1
activity towards the model substrates methyl chloride and dichloromethane in liver and kidney. Arch
Toxicol 72:622-629.
Thilagar AK, Kumaroo V. 1983. Induction of chromosome damage by methylene chloride in CHO cells.
Mutat Res 116:361-367.
*Thilagar AK, Back AM, Kirby PE, et al. 1984a. Evaluation of dichloromethane in short term in vitro
genetic toxicity assays. Environ Mutagen 6:418-419.
Thilagar AK, Kumaroo PV, Clark JJ, et al. 1984b. Induction of chromosome damage by
dichloromethane in cultured human peripheral lymphocytes, CHO cells and mouse lymphoma L5178Y
cells. Environ Mutagen 6:422.
Thomas AA, Pinkerton MK, Warden JA. 1972. Effects of low-level dichloromethane exposure on the
spontaneous activity of mice. In: Proceedings of the 3rd annual conference on environmental toxicology.
Wright-Patterson Air Force Base, OH: Aerospace Medical Research Laboratory. AMRL-TR72-130,
185-189.
*Thomas PE, Bandiera S, Maines SL, et al. 1987. Regulation of cytochrome P-450j, a high-affinity N-
nitrosodimethylamine demethylase, in rat hepatic microsomes. Biochemistry 26(8):2280-2289.
258 METHYLENE CHLORIDE
8. REFERENCES
Thomas, RG. 1990. Volatilization from water. In: Lyman WJ, Reehl WF, Rosenblatt DH, eds.
Handbook of Chemical Property Estimation Methods. American Chemical Society, Washington DC, 2
nd
Printing. Chapter 15.
Thomas RS, Yang RSH, Morgan DG, et al. 1996. PBPK modeling/Monte Carlo simulation of methylene
chloride kinetic changes in mice in relation to age and acute, subchronic, and chronic inhalation exposure.
Environ Health Perspec 104: 858-865.
*Thomas V. 1975. Biological-mathematical modeling of chronic toxicity. Wright Patterson Air Force
Base Research Report. AMRL-TR-75-5.
Tiffany-Castiglioni E, Ehrich M, Dees L, et al. 1999. Bridging the gap between in vitro and in vivo
models for neurotoxicology. Toxicol Sci 51:178-183.
*Tomaszewski C. 1998. Carbon monoxide. In: Goldfrank LR, Flomenbaum NE, Lewin NA, et al., ed.
Goldfrank's toxicologic emergencies. Stamford, Connecticut: Appleton & Lange, 1551-1563.
*Tomenson JA, Bonner SM, Heijne CG, et al. 1997. Mortality of workers exposed to methylene
chloride employed at a plant producing cellulose triacetate film base. Occup Environ Med 54:470-476.
Travis CC, Hattemer-Frey HA, Arms AD. 1988. Relationship between dietary intake of organic
chemicals and their concentrations in human adipose tissue and breast milk. Arch Environ Contam
Toxicol 17:473-478.
*TRI88. 1990. Toxic chemical release inventory. National Library of Medicine, National Toxicology
Information Program, Bethesda, MD.
TRI96. 1998. Toxic chemical release inventory. National Library of Medicine, National Toxicology
Information Program, Bethesda, MD.
*TRI97. 1999. Toxic chemical release inventory. National Library of Medicine, National Toxicology
Information Program, Bethesda, MD.
*TRI98. 2000. Toxic chemical release inventory. National Library of Medicine, National Toxicology
Information Program, Bethesda, MD.
*Trueman RW, Ashby J. 1987. Lack of UDS activity in the livers of mice and rats exposed to
dichloromethane. Environ Mol Mutagen 10:189-195.
Truman RW, Ashby J, eds. 1986. Methylene chloride: In vivo and in vitro unscheduled DNA synthesis
studies in the mouse and the rat. ICI Central Toxicology Laboratory, Report no. CTL/P 1444, January 21.
Tsang JSH, Ho YB. 1998. Biodedegredation of halogenated compounds. In: Rama BC, ed. Damaged
ecosystem restoration. Singapore: World Scientific, 203-224.
*Ugazio G, Burdino E, Danni O, et al. 1973. Hepatoxicity and lethality of halogenoalkanes. Biochem
Soc Trans 1:968-972.
*USC. 1999. United States Code. 74 USC 7412.

259 METHYLENE CHLORIDE
8. REFERENCES
*USITC. 1989. Synthetic organic chemicals: United States production and sales, 1988. Washington,
DC: U.S. International Trade Commission. USITC Publication 2219, 15-7, 15-30.
VanHylckama Vlieg JET, De Koning W, Janssen DB. 1996. Transformation kinetics of chlorinated
ethenes by methylosinus trichosporium OB3b and detection of unstable epoxides by on-line gas
chromatography. Appl Environ Microbiol 62(9):3304-3312.
*Verschueren K. 1983. Handbook of environmental data on organic chemicals. 2nd ed. New York:
Van Nostrand Reinhold Company, 848-849.
*Vieira I, Sonnier M, Cresteil T. 1996. Developmental expression of CYP2E1 in the human liver:
Hypermethylation control of gene expression during the neonatal period. Eur J Biochem 238:476-483.
VIEW Database. 1989. Agency for Toxic Substances and Disease Registry, Office of External Affairs,
Exposure and Disease Registry Branch, Atlanta, GA. June 20, 1989.
Vincent R, Poirot P, Subra I. 1994. Occupational exposure to organic solvents during paint stripping and
painting operations in the aeronautical industry. Int Arch Occup Environ Health 65:377-380.
Vineis P, Faggiano F. 1991. Epidemiological models and prevention of cancer. Ann Ocol 2:559-563.
Vineis P, Malats N, Lang M, et al. 1999. Metabolic polymorphisms and susceptibility to cancer. IARC
Scientific Publications ed. Lyon, France: International Agency for Research on Cancer, .
Von Oettingen WF, Powell CC, Sharpless NE, et al. 1949. Relation between the toxic action of
chlorinated methanes and their chemical and physicochemical properties. NIH Bulletin 191. Federal
Security Agency, U.S. Public Health Service, National Institutes of Health.
*VT Nat. Res. Agency 1998. Hazard ambient air standards: Hazardous air contaminants. Vermont
Natural Resources Agency, Department of Environmental Conservation, Air Pollution Control Division.
http://www.anr.state.vt.us/dec/air/airtoxic.htm. November 2, 1999.
Vuilleumier S, Leisinger T. 1996. Protein engineering studies of dichloromethane
dehalogenase/glutathione S-transferase from Methylophilus sp. strain DM11: Ser12 but not Tyr6 is
required for enzyme activity. Eur J Biochem 239:410-417.
Vuilleumier S, Sorribas H, Leisinger T. 1997. Identification of a novel determinant of glutathione
affinity in dichloromethane dehalogenases/glutathione S-transferases. Biochem Biophys Res Commun
238:452-456.
*WA Dept. Ecology 1998. Class A toxic air pollutants: With established acceptable source impact levels.
Washington Department of Ecology, Air Quality Program. http://www.wa.gov/ecology/leg/laws-
etc.html. November 12, 1999.
Waalkes MP, Harvey MJ, Klaassen CD. 1984. Relative in vitro affinity of hepatic metallothionein for
metals. Toxicol Lett 20:33-39.
*Walters SM. 1986. Cleanup of samples. In: Zweig G, Sherma J, eds. Analytical methods for
pesticides and plant growth regulators. Vol. 15: Principles, statistics, and applications. New York, NY:
Academic Press Inc., 67-110.
260 METHYLENE CHLORIDE
8. REFERENCES
*Wan Y-JY, Poland RE, Lin K-M. 1998. Genetic polymorhism of CYP2E1, ADH2, and ALDH2 in
Mexican-Americans. Genetic Testing 2(1):79-83.
Watanabe K, Seno H, Ishii A, et al. 1997. Capillary gas chromatography with cryogenic oven
temperature for headspace samples: analysis of chloroform or methylene chloride in whole blood. Anal
Chem 69:5178-5181.
*Weast RC, ed. 1985. CRC handbook of chemistry and physics. 66th ed. Boca Raton, FL: CRC Press
Inc., C-349.
*Weinstein RS, Diamond SS. 1972. Hepatotoxicity of dichloromethane (methylene chloride) with
continuous inhalation exposure at a low dose level. In: Proceedings of the 3rd annual conference on
environmental toxicology. Wright-Patterson Air Force Base, OH: Aerospace Medical Research
Laboratory, 209-220. AMRL-TR-72-130.
Weinstein RS, Boyd DD, Back KC. 1972. Effects of continuous inhalation of dichloromethane in the
mouse-morphologic and functional observations. Toxicol Appl Pharmacol 23:660.
Weiss G. 1967. Toxic encephalosis as an occupational hazard with methylene chloride. Zentralbl
Arbeitsmed 17:282-285.
Welch L. 1987. Reports of clinical disease secondary to methylene chloride exposure--a collection of
141 cases. Unpublished study. Report to U.S. Environmental Protection Agency, Office of Pesticide and
Toxic Substances, Washington, DC.
Welke B, Ettlinger K, Riederer M. 1998. Sorption of volatile organic chemicals in plant surfaces.
Environ Sci Technol 32:1099-1104.
*Wells GG, Waldron HA. 1984. Methylene chloride burns. Br J Ind Med 41:420.
*Wells VE, Schrader SM, McCammon CS, et al. 1989. Letter to the editor. Reprod Toxicol 3:281-282.
*West JR, Smith HW, Chasis H. 1948. Glomerular filtration rate, effective renal blood flow, and
maximal tubular excretory capacity in infancy. J Pediatr 32a:10-18.
*White RF, Proctor SP, Echeverria D, et al. 1995. Neurobehavioral effects of acute and chronic
mixed-solvent exposure in the screen printing industry. Am J Ind Med 28:221-231.
White PA, Rasmusen JB, Blaise C. 1996. Comparing the presence, potency, and potential hazard of
genotoxins extracted from a broad range of industrial effluents. Environ Mol Mutagen 27:116-139.
*Whitehead LW, Ball GL, Fine LJ, et al. 1984. Solvent vapor exposures in booth spray painting and
spray glueing, and associated operations. Am Ind Hyg Assoc J 45(11):767-772.
*WHO. 1996. Environmental health criteria 164: Methylene chloride, 2nd ed. World Health
Organization, Geneva, Switzerland.
*WI Dept Natural Resources. 1997. Control of hazardous pollutants. Wisconsin Department of Natural
Resources. Chs. 400-499 NR 445. http://www.dnr.state.wi.us/org/aw/air/index.htm
261 METHYLENE CHLORIDE
8. REFERENCES
*Widdowson EM, Dickerson JWT. 1964. Chemical composition of the body. In: Comar CL, Bronner F,
eds. Mineral metabolism: An advanced treatise volume II The elements part A. New York, NY:
Academic Press.
Wiger R. 1991. Dichloromethane: Summary and evaluation of effects on reproduction. In: Freij L, ed.
Effects on reproduction of dichloromethane, n-hexane, and 1,1,1-trichloroethane. Solna, Sweden: Nordic
Chemicals Control Group, 9-27.
*Winek CL, Collom WD, Esposito F. 1981. Accidental methylene chloride fatality. Forensic Sci Int
18:165-168.
*Winneke, G. 1974. Behavioral effects of methylene chloride and carbon monoxide as assessed by
sensory and psychomotor performance. In: Xintaras C, Johnson BL, de Groot I, eds. Behavioral
toxicology: Early detection of occupational hazards. Washington, DC: U.S. Department of Health,
Education and Welfare, 130-144.
Winneke G. 1981. The neurotoxicity of dichloromethane. Neurobehav Toxicol Teratol 3:391-395.
*Wirkner K, Damme B, Poelchen W, et al. 1997. Effect of long-term ethanol pretreatment on the
metabolism of dichloromethane to carbon monoxide in rats. Toxicol Appl Pharmacol 143:83-88.
Wood PR, Parsons FZ, DeMarco J, et al. 1981. Introductory study of the biodegredation of the
chlorinated methane, ethane, and ethene compounds. Presented at the American Water Works
Association Meeting, June.
*Woodrow JE, McChesney MM, Seiber JN. 1988. Determination of methyl bromide in air samples by
headspace gas chromatography. Anal Chem 60:509-512.
Woodruff TJ, Axelrad DA, Caldwell J, et al. 1998. Public health implications of 1990 air toxics
concentrations across the United States. Environ Health Perspect 106:245-251.
Workman DJ, Woods SL, Gorby YA, et al. 1997. Microbial reduction of vitamin B
12
by shewanella
alga strain BrY with subsequent transformation of carbon tetrachloride. Environ Sci Technol
31:2292-2297.
*Wu X, Amos CI, Kemp BL, et al. 1998. Cytochrome P450 2E1 DraI polymorphisms in lung cancer in
minority populations. Cancer Epidemiol Biomarkers Prev 7:13-18.
*Xiao H, Levine SP, Nowak J, et al. 1993. Analysis of organic vapors in the workplace by remote
sensing fourier transform infrared spectroscopy. Am Ind Hyg Assoc J 54(9):545-556.
Yamamoto K, Fukushima M, Kakutani N, et al. 1997. Volatile organic compounds in urban rivers and
their estuaries in Osaka, Japan. Environ Pollut 95(1):135-143.
Yesair DW, Jacques D, Schepis P, et al. 1977. Dose-related pharmacokinetics of
14
C methylene chloride
in mice. Fed Proc 36:998.
Zeneca Central Toxicology Lab. 1995a. DNA sequence analysis of methylene chloride-induced HPRT
mutations in CHO cells: Comparison with the mutation spectrum obtained for 1,2-dibromoethane and
formaldehyde. TSCATS-452017. NTIS/OTS 0572586.
262 METHYLENE CHLORIDE
8. REFERENCES
Zeneca Central Toxicology Lab. 1995b. Mouse liver glutathione S-transferase mediated metabolism of
methylene chloride to a mutagen in the CHO\HPRT assay. TSCATS-452021. NTIS/OTS 0572590.
*Ziegler EE, Edwards BB, Jensen RL, et al. 1978. Absorption and retention of lead by infants. Pediatr
Res 12:29-34.
Zielenska M, Ahmed A, Pienkowska M, et al. 1993. Mutational specificities of environmental
carcinogens in the lacI gene of Escherichia coli. VI: Analysis of methylene chloride-induced mutational
distribution in Uvr
+
and UvrB
-
strains. Carcinogenesis 14:789-794.
*Zielinska B, Fujita E, Sagebiel J, et al. 1998. Arizona hazardous air pollutants monitoring program. J
Air Waste Manage Assoc 48:1038-1050.
Zoeteman BCJ, Harmsen K, Linders JBHJ, et al. 1980. Persistant organic pollutants in the river water
and ground water of the Netherlands. Chemosphere 9:231-249.
Zwart A, Lommen JGJ, Feron VJ. 1992. Multi-compartment model to study the effect of air-blood and
blood-tissue partition coefficients on concentration-time-effect relationships. Arch Toxicol Suppl 15:249-
252.
263 METHYLENE CHLORIDE
9. GLOSSARY
Absorption—The taking up of liquids by solids, or of gases by solids or liquids.
Acute Exposure—Exposure to a chemical for a duration of 14 days or less, as specified in the
Toxicological Profiles.
Adsorption—The adhesion in an extremely thin layer of molecules (as of gases, solutes, or liquids) to the
surfaces of solid bodies or liquids with which they are in contact.
Adsorption Coefficient (K
oc
)—The ratio of the amount of a chemical adsorbed per unit weight of
organic carbon in the soil or sediment to the concentration of the chemical in solution at equilibrium.
Adsorption Ratio (Kd)—The amount of a chemical adsorbed by a sediment or soil (i.e., the solid phase)
divided by the amount of chemical in the solution phase, which is in equilibrium with the solid phase, at a
fixed solid/solution ratio. It is generally expressed in micrograms of chemical sorbed per gram of soil or
sediment.
Benchmark Dose (BMD)—Usually defined as the lower confidence limit on the dose that produces a
specified magnitude of changes in a specified adverse response. For example, a BMD
10
would be the
dose at the 95% lower confidence limit on a 10% response, and the benchmark response (BMR) would be
10%. The BMD is determined by modeling the dose response curve in the region of the dose response
relationship where biologically observable data are feasible.
Benchmark Dose Model—A statistical dose-response model applied to either experimental toxicological
or epidemiological data to calculate a BMD.
Bioconcentration Factor (BCF)—The quotient of the concentration of a chemical in aquatic organisms
at a specific time or during a discrete time period of exposure divided by the concentration in the
surrounding water at the same time or during the same period.
Biomarkers—Broadly defined as indicators signaling events in biologic systems or samples. They have
been classified as markers of exposure, markers of effect, and markers of susceptibility.
Cancer Effect Level (CEL)—The lowest dose of chemical in a study, or group of studies, that produces
significant increases in the incidence of cancer (or tumors) between the exposed population and its
appropriate control.
Carcinogen—A chemical capable of inducing cancer.
Case-Control Study—A type of epidemiological study which examines the relationship between a
particular outcome (disease or condition) and a variety of potential causative agents (such as toxic
chemicals). In a case-controlled study, a group of people with a specified and well-defined outcome is
identified and compared to a similar group of people without outcome.
Case Report—Describes a single individual with a particular disease or exposure. These may suggest
some potential topics for scientific research but are not actual research studies.
264 METHYLENE CHLORIDE
9. GLOSSARY
Case Series—Describes the experience of a small number of individuals with the same disease or
exposure. These may suggest potential topics for scientific research but are not actual research studies.
Ceiling Value—A concentration of a substance that should not be exceeded, even instantaneously.
Chronic Exposure—Exposure to a chemical for 365 days or more, as specified in the Toxicological
Profiles.
Cohort Study—A type of epidemiological study of a specific group or groups of people who have had a
common insult (e.g., exposure to an agent suspected of causing disease or a common disease) and are
followed forward from exposure to outcome. At least one exposed group is compared to one unexposed
group.
Cross-sectional Study—A type of epidemiological study of a group or groups which examines the
relationship between exposure and outcome to a chemical or to chemicals at one point in time.
Data Needs—Substance-specific informational needs that if met would reduce the uncertainties of human
health assessment.
Developmental Toxicity—The occurrence of adverse effects on the developing organism that may result
from exposure to a chemical prior to conception (either parent), during prenatal development, or
postnatally to the time of sexual maturation. Adverse developmental effects may be detected at any point
in the life span of the organism.
Dose-Response Relationship—The quantitative relationship between the amount of exposure to a
toxicant and the incidence of the adverse effects.
Embryotoxicity and Fetotoxicity—Any toxic effect on the conceptus as a result of prenatal exposure to
a chemical; the distinguishing feature between the two terms is the stage of development during which the
insult occurs. The terms, as used here, include malformations and variations, altered growth, and in utero
death.
Environmental Protection Agency (EPA) Health Advisory—An estimate of acceptable drinking water
levels for a chemical substance based on health effects information. A health advisory is not a legally
enforceable federal standard, but serves as technical guidance to assist federal, state, and local officials.
Epidemiology—Refers to the investigation of factors that determine the frequency and distribution of
disease or other health-related conditions within a defined human population during a specified period.
Genotoxicity—A specific adverse effect on the genome of living cells that, upon the duplication of
affected cells, can be expressed as a mutagenic, clastogenic or carcinogenic event because of specific
alteration of the molecular structure of the genome.
Half-life—A measure of rate for the time required to eliminate one half of a quantity of a chemical from
the body or environmental media.
Immediately Dangerous to Life or Health (IDLH)—The maximum environmental concentration of a
contaminant from which one could escape within 30 minutes without any escape-impairing symptoms or
irreversible health effects.
265 METHYLENE CHLORIDE
9. GLOSSARY
Incidence—The ratio of individuals in a population who develop a specified condition to the total
number of individuals in that population who could have developed that condition in a specified time
period.
Intermediate Exposure—Exposure to a chemical for a duration of 15–364 days, as specified in the
Toxicological Profiles.
Immunologic Toxicity—The occurrence of adverse effects on the immune system that may result from
exposure to environmental agents such as chemicals.
Immunological Effects—Functional changes in the immune response.
In Vitro—Isolated from the living organism and artificially maintained, as in a test tube.
In Vivo—Occurring within the living organism.
Lethal Concentration
(LO)
(LC
LO
)—The lowest concentration of a chemical in air which has been
reported to have caused death in humans or animals.
Lethal Concentration
(50)
(LC
50
)—A calculated concentration of a chemical in air to which exposure for a
specific length of time is expected to cause death in 50% of a defined experimental animal population.
Lethal Dose
(LO)
(LD
LO
)—The lowest dose of a chemical introduced by a route other than inhalation that
has been reported to have caused death in humans or animals.
Lethal Dose
(50)
(LD
50
)—The dose of a chemical which has been calculated to cause death in 50% of a
defined experimental animal population.
Lethal Time
(50)
(LT
50
)—A calculated period of time within which a specific concentration of a chemical
is expected to cause death in 50% of a defined experimental animal population.
Lowest-Observed-Adverse-Effect Level (LOAEL)—The lowest exposure level of chemical in a study,
or group of studies, that produces statistically or biologically significant increases in frequency or severity
of adverse effects between the exposed population and its appropriate control.
Lymphoreticular Effects—Represent morphological effects involving lymphatic tissues such as the
lymph nodes, spleen, and thymus.
Malformations—Permanent structural changes that may adversely affect survival, development, or
function.
Minimal Risk Level (MRL)—An estimate of daily human exposure to a hazardous substance that is
likely to be without an appreciable risk of adverse noncancer health effects over a specified route and
duration of exposure.
Modifying Factor (MF)—A value (greater than zero) that is applied to the derivation of a minimal risk
level (MRL) to reflect additional concerns about the database that are not covered by the uncertainty
factors. The default value for a MF is 1.
266 METHYLENE CHLORIDE
9. GLOSSARY
Morbidity—State of being diseased; morbidity rate is the incidence or prevalence of disease in a specific
population.
Mortality—Death; mortality rate is a measure of the number of deaths in a population during a specified
interval of time.
Mutagen—A substance that causes mutations. A mutation is a change in the DNA sequence of a cell’s
DNA. Mutations can lead to birth defects, miscarriages, or cancer.
Necropsy—The gross examination of the organs and tissues of a dead body to determine the cause of
death or pathological conditions.
Neurotoxicity—The occurrence of adverse effects on the nervous system following exposure to a
chemical.
No-Observed-Adverse-Effect Level (NOAEL)—The dose of a chemical at which there were no
statistically or biologically significant increases in frequency or severity of adverse effects seen between
the exposed population and its appropriate control. Effects may be produced at this dose, but they are not
considered to be adverse.
Octanol-Water Partition Coefficient (K
ow
)—The equilibrium ratio of the concentrations of a chemical
in n-octanol and water, in dilute solution.
Odds Ratio (OR)—A means of measuring the association between an exposure (such as toxic substances
and a disease or condition) which represents the best estimate of relative risk (risk as a ratio of the
incidence among subjects exposed to a particular risk factor divided by the incidence among subjects who
were not exposed to the risk factor). An odds ratio of greater than 1 is considered to indicate greater risk
of disease in the exposed group compared to the unexposed.
Organophosphate or Organophosphorus Compound—A phosphorus containing organic compound
and especially a pesticide that acts by inhibiting cholinesterase.
Permissible Exposure Limit (PEL)—An Occupational Safety and Health Administration (OSHA)
allowable exposure level in workplace air averaged over an 8-hour shift of a 40-hour workweek.
Pesticide—General classification of chemicals specifically developed and produced for use in the control
of agricultural and public health pests.
Pharmacokinetics—The science of quantitatively predicting the fate (disposition) of an exogenous
substance in an organism. Utilizing computational techniques, it provides the means of studying the
absorption, distribution, metabolism and excretion of chemicals by the body.
Pharmacokinetic Model—A set of equations that can be used to describe the time course of a parent
chemical or metabolite in an animal system. There are two types of pharmacokinetic models: data-based
and physiologically-based. A data-based model divides the animal system into a series of compartments
which, in general, do not represent real, identifiable anatomic regions of the body whereas the
physiologically-based model compartments represent real anatomic regions of the body.
Physiologically Based Pharmacodynamic (PBPD) Model—A type of physiologically-based dose-
response model which quantitatively describes the relationship between target tissue dose and toxic end
267 METHYLENE CHLORIDE
9. GLOSSARY
points. These models advance the importance of physiologically based models in that they clearly
describe the biological effect (response) produced by the system following exposure to an exogenous
substance.
Physiologically Based Pharmacokinetic (PBPK) Model—A model comprising a series of
compartments representing organs or tissue groups with realistic weights and blood flows. These models
require a variety of physiological information: tissue volumes, blood flow rates to tissues, cardiac output,
alveolar ventilation rates and, possibly membrane permeabilities. The models also utilize biochemical
information such as air/blood partition coefficients, and metabolic parameters. PBPK models are also
called biologically based tissue dosimetry models.
Prevalence—The number of cases of a disease or condition in a population at one point in time.
Prospective Study—A type of cohort study in which the pertinent observations are made on events
occurring after the start of the study. A group is followed over time.
q
1
*—The upper-bound estimate of the low-dose slope of the dose-response curve as determined by the
multistage procedure. The q
1
* can be used to calculate an estimate of carcinogenic potency, the
incremental excess cancer risk per unit of exposure (usually µg/L for water, mg/kg/day for food, and
µg/m
3
for air).
Recommended Exposure Limit (REL)—A National Institute for Occupational Safety and Health
(NIOSH) time-weighted average (TWA) concentrations for up to a 10-hour workday during a 40-hour
workweek.
Reference Concentration (RfC)—An estimate (with uncertainty spanning perhaps an order of
magnitude) of a continuous inhalation exposure to the human population (including sensitive subgroups)
that is likely to be without an appreciable risk of deleterious noncancer health effects during a lifetime.
The inhalation reference concentration is for continuous inhalation exposures and is appropriately
expressed in units of mg/m
3
or ppm.
Reference Dose (RfD)—An estimate (with uncertainty spanning perhaps an order of magnitude) of the
daily exposure of the human population to a potential hazard that is likely to be without risk of deleterious
effects during a lifetime. The RfD is operationally derived from the no-observed-adverse-effect level
(NOAEL-from animal and human studies) by a consistent application of uncertainty factors that reflect
various types of data used to estimate RfDs and an additional modifying factor, which is based on a
professional judgment of the entire database on the chemical. The RfDs are not applicable to
nonthreshold effects such as cancer.
Reportable Quantity (RQ)—The quantity of a hazardous substance that is considered reportable under
the Comprehensive Environmental Response, Compensation, and Liability Act (CERCLA). Reportable
quantities are (1) 1 pound or greater or (2) for selected substances, an amount established by regulation
either under CERCLA or under Section 311 of the Clean Water Act. Quantities are measured over a
24-hour period.
Reproductive Toxicity—The occurrence of adverse effects on the reproductive system that may result
from exposure to a chemical. The toxicity may be directed to the reproductive organs and/or the related
endocrine system. The manifestation of such toxicity may be noted as alterations in sexual behavior,
fertility, pregnancy outcomes, or modifications in other functions that are dependent on the integrity of
this system.
268 METHYLENE CHLORIDE
9. GLOSSARY
Retrospective Study—A type of cohort study based on a group of persons known to have been exposed
at some time in the past. Data are collected from routinely recorded events, up to the time the study is
undertaken. Retrospective studies are limited to causal factors that can be ascertained from existing
records and/or examining survivors of the cohort.
Risk—The possibility or chance that some adverse effect will result from a given exposure to a chemical.
Risk Factor—An aspect of personal behavior or lifestyle, an environmental exposure, or an inborn or
inherited characteristic, that is associated with an increased occurrence of disease or other health-related
event or condition.
Risk Ratio—The ratio of the risk among persons with specific risk factors compared to the risk among
persons without risk factors. A risk ratio greater than 1 indicates greater risk of disease in the exposed
group compared to the unexposed.
Short-Term Exposure Limit (STEL)—The American Conference of Governmental Industrial
Hygienists (ACGIH) maximum concentration to which workers can be exposed for up to 15 min
continually. No more than four excursions are allowed per day, and there must be at least 60 min
between exposure periods. The daily Threshold Limit Value - Time Weighted Average (TLV-TWA) may
not be exceeded.
Target Organ Toxicity—This term covers a broad range of adverse effects on target organs or
physiological systems (e.g., renal, cardiovascular) extending from those arising through a single limited
exposure to those assumed over a lifetime of exposure to a chemical.
Teratogen—A chemical that causes structural defects that affect the development of an organism.
Threshold Limit Value (TLV)—An American Conference of Governmental Industrial Hygienists
(ACGIH) concentration of a substance to which most workers can be exposed without adverse effect.
The TLV may be expressed as a Time Weighted Average (TWA), as a Short-Term Exposure Limit
(STEL), or as a ceiling limit (CL).
Time-Weighted Average (TWA)—An allowable exposure concentration averaged over a normal 8-hour
workday or 40-hour workweek.
Toxic Dose
(50)
(TD
50
)—A calculated dose of a chemical, introduced by a route other than inhalation,
which is expected to cause a specific toxic effect in 50% of a defined experimental animal population.
Toxicokinetic—The study of the absorption, distribution and elimination of toxic compounds in the
living organism.
Uncertainty Factor (UF)—A factor used in operationally deriving the Minimal Risk Level (MRL) or
Reference Dose (RfD) or Reference Concentration (RfC) from experimental data. UFs are intended to
account for (1) the variation in sensitivity among the members of the human population, (2) the
uncertainty in extrapolating animal data to the case of human, (3) the uncertainty in extrapolating from
data obtained in a study that is of less than lifetime exposure, and (4) the uncertainty in using lowest-
observed-adverse-effect level (LOAEL) data rather than no-observed-adverse-effect level (NOAEL) data.
A default for each individual UF is 10; if complete certainty in data exists, a value of one can be used;
however a reduced UF of three may be used on a case-by-case basis, three being the approximate
logarithmic average of 10 and 1.
269 METHYLENE CHLORIDE
9. GLOSSARY
Xenobiotic—Any chemical that is foreign to the biological system.
A-1 METHYLENE CHLORIDE
APPENDIX A
ATSDR MINIMAL RISK LEVELS AND WORKSHEETS
The Comprehensive Environmental Response, Compensation, and Liability Act (CERCLA) [42 U.S.C.
9601 et seq.], as amended by the Superfund Amendments and Reauthorization Act (SARA) [Pub. L.
99–499], requires that the Agency for Toxic Substances and Disease Registry (ATSDR) develop jointly
with the U.S. Environmental Protection Agency (EPA), in order of priority, a list of hazardous substances
most commonly found at facilities on the CERCLA National Priorities List (NPL); prepare toxicological
profiles for each substance included on the priority list of hazardous substances; and assure the initiation
of a research program to fill identified data needs associated with the substances.
The toxicological profiles include an examination, summary, and interpretation of available toxicological
information and epidemiologic evaluations of a hazardous substance. During the development of
toxicological profiles, Minimal Risk Levels (MRLs) are derived when reliable and sufficient data exist to
identify the target organ(s) of effect or the most sensitive health effect(s) for a specific duration for a
given route of exposure. An MRL is an estimate of the daily human exposure to a hazardous substance
that is likely to be without appreciable risk of adverse noncancer health effects over a specified duration
of exposure. MRLs are based on noncancer health effects only and are not based on a consideration of
cancer effects. These substance-specific estimates, which are intended to serve as screening levels, are
used by ATSDR health assessors to identify contaminants and potential health effects that may be of
concern at hazardous waste sites. It is important to note that MRLs are not intended to define clean-up or
action levels.
MRLs are derived for hazardous substances using the no-observed-adverse-effect level/uncertainty factor
approach. They are below levels that might cause adverse health effects in the people most sensitive to
such chemical-induced effects. MRLs are derived for acute (1–14 days), intermediate (15–364 days), and
chronic (365 days and longer) durations and for the oral and inhalation routes of exposure. Currently,
MRLs for the dermal route of exposure are not derived because ATSDR has not yet identified a method
suitable for this route of exposure. MRLs are generally based on the most sensitive chemical-induced end
point considered to be of relevance to humans. Serious health effects (such as irreparable damage to the
liver or kidneys, or birth defects) are not used as a basis for establishing MRLs. Exposure to a level
above the MRL does not mean that adverse health effects will occur.
A-2 METHYLENE CHLORIDE
APPENDIX A
MRLs are intended only to serve as a screening tool to help public health professionals decide where to
look more closely. They may also be viewed as a mechanism to identify those hazardous waste sites that
are not expected to cause adverse health effects. Most MRLs contain a degree of uncertainty because of
the lack of precise toxicological information on the people who might be most sensitive (e.g., infants,
elderly, nutritionally or immunologically compromised) to the effects of hazardous substances. ATSDR
uses a conservative (i.e., protective) approach to address this uncertainty consistent with the public health
principle of prevention. Although human data are preferred, MRLs often must be based on animal studies
because relevant human studies are lacking. In the absence of evidence to the contrary, ATSDR assumes
that humans are more sensitive to the effects of hazardous substance than animals and that certain persons
may be particularly sensitive. Thus, the resulting MRL may be as much as a hundredfold below levels
that have been shown to be nontoxic in laboratory animals.
Proposed MRLs undergo a rigorous review process: Health Effects/MRL Workgroup reviews within the
Division of Toxicology, expert panel peer reviews, and agency wide MRL Workgroup reviews, with
participation from other federal agencies and comments from the public. They are subject to change as
new information becomes available concomitant with updating the toxicological profiles. Thus, MRLs in
the most recent toxicological profiles supersede previously published levels. For additional information
regarding MRLs, please contact the Division of Toxicology, Agency for Toxic Substances and Disease
Registry, 1600 Clifton Road, Mailstop E-29, Atlanta, Georgia 30333.

A-3 METHYLENE CHLORIDE
APPENDIX A
MINIMAL RISK LEVEL (MRL) WORKSHEET
Chemical name(s): Methylene Chloride
CAS number(s): 75-09-2
Date: July 28, 2000
Profile status: Draft 3 Post Public Comment
Route: [X] Inhalation [ ] Oral
Duration: [X] Acute [ ] Intermediate [ ] Chronic
Key to figure: 15
Species: Human
MRL: 0.6 [ ] mg/kg/day [X] ppm [ ] mg/m
3
Reference: Winneke G. 1974. Behavioral effects of methylene chloride and carbon monoxide as
assessed by sensory and psychomotor performance. In: Behavioral Toxicology. Early Detection of
Occupational Hazards, Eds. C Xintaras, B Johnson, I deGroot. U.S. Department of Health, Education,
and Welfare.
Reitz RH, Hays SM, Gargas ML. 1997. Assessing priority data needs for methylene chloride with
physiologically-based pharmacokinetic modeling. Halogenated Solvents Industry Alliance (HSIA),
prepared for ATSDR.
Experimental design: Winneke (1974) exposed from 6 to 20 volunteers in a randomized blind clinical
chamber experiment to either filtered air or to concentrations of 300, 500, or 800 ppm of methylene
chloride vapors. Subjects were exposed for 3–4 hours and tested at 45-minute intervals with standard
neurobehavioral tests measuring: (1) critical flicker fusion frequency (visual); (2) auditory vigilance
performance; and (3) performance on psychomotor tasks.
Effects noted in study and corresponding doses: A statistically significant depression in critical flicker
fusion frequency (CFF frequency) was observed at all concentrations. The magnitude of CFF depression
was similar at exposure concentrations of 300 and 500 ppm and was larger at 800 ppm. Thus, there was
no dose-response at the two lowest concentrations, and a dose-response was evident at the highest
concentration. A decrease in auditory vigilance performance was observed at 500 ppm and psychomotor
task performance was impaired at 800 ppm. Thus, of the neurological indicators tested, CFF frequency is
most sensitive to acute inhalation exposure to methylene chloride. Based on this end point, the LOAEL is
300 ppm.
The Reitz et al. (1997) PBPK model was used to convert the LOAEL to account for a 24-hour exposure
scenario, yielding a duration-adjusted LOAEL of 60 ppm.
Dose and end point used for MRL derivation: 60 ppm; the MRL was derived based on a LOAEL of 300
ppm for adverse neurological effects (decreased critical flicker frequency and auditory vigilance
performance), duration-adjusted to 60 ppm by Rietz et al. (1997) .
[ ] NOAEL [X] LOAEL:

A-4 METHYLENE CHLORIDE
APPENDIX A
Uncertainty factors used in MRL derivation:
[ ] 1 [ ] 3 [X] 10 (for use of a LOAEL)
[ ] 1 [ ] 3 [ ] 10 (for extrapolation from animals to humans)
[ ] 1 [ ] 3 [X] 10 (for human variability)
Was a conversion factor used from ppm in food or water to a mg/body weight dose?
No
If an inhalation study in animals, list conversion factors used in determining human equivalent dose: N/A
Was a conversion used from intermittent to continuous exposure? If so, explain:
The Reitz et al. (1997) PBPK model was used to convert the LOAEL of 300 ppm to 60 ppm to account
for a 24-hour scenario.
Other additional studies or pertinent information that lend support to this MRL: Fodor and Winneke
(1971) observed similar effects on critical flicker fusion frequency at the same concentration (300 ppm) in
a different group of volunteers. Stewart et al. (1972) observed altered visual evoked responses in humans
exposed to 515 ppm methylene chloride for 1–2 hours. Putz et al. (1979) reported impairment in some
visual and psychomotor tasks following 4 hours of exposure to 200 ppm of methylene chloride.
(Although Putz et al. (1979) identified a lower LOAEL, it was not chosen as the basis for the acute
inhalation MRL because ATSDR considered the neurological tests employed by Winneke (1974) to be
more specific.) OSHA’s (OSHA 1997) new occupational standard of 125 ppm as a 15-minute STEL
(short term exposure limit) is consistent with this inhalation MRL.

A-5 METHYLENE CHLORIDE
APPENDIX A
MINIMAL RISK LEVEL (MRL) WORKSHEET
Chemical name(s): Methylene Chloride
CAS number(s): 75-09-2
Date: July 28, 2000
Profile status: Draft 3 Post Public Comment
Route: [X ] Inhalation [ ] Oral
Duration: [ ] Acute [X ] Intermediate [ ] Chronic
Key to figure: 36r
Species: Rat
MRL: 0.3 [ ] mg/kg/day [X] ppm [ ] mg/m
3
Reference: Haun CC, Vernot EH, Darmer KI, et al. 1972. Continuous animal exposure to low levels of
dichloromethane. AMRL-TR-72-130, paper no. 12.
Experimental design: Groups of 20 rats (sex and strain not specified) were exposed to methylene chloride
vapors continuously for 14 weeks at chamber concentrations of either 0 (controls), 25, or 100 ppm. Body
weights and clinical signs were monitored throughout the study. Necropsy was performed and tissues
were examined histopathologically and relative organ weights were determined at the end of exposure.
The study also evaluated mice, monkeys, and dogs. Those results are described below as supporting
evidence.
Effects noted in study and corresponding dose: Cytoplasmic vacuolization and positive-oil-red stain
(indicative of fatty infiltration) were reported at 25 and 100 ppm. Nonspecific tubular degeneration and
regenerative changes of the kidney were also observed at both exposure levels. The study did not report
whether there were exposure-related differences in the incidence or severity of effects. There were no
exposure-related effects on organ weights.
Dose and end point used for MRL derivation: The MRL was derived based on a LOAEL of 25 ppm for
hepatic effects (cytoplasmic vacuolization and fatty infiltration).
[ ] NOAEL [X] LOAEL:
Uncertainty factors used in MRL derivation:
[ ] 1 [X ] 3 [ ] 10 (for use of a minimal LOAEL)
[ ] 1 [X] 3 [ ] 10 (for extrapolation from animals to humans)
[ ] 1 [ ] 3 [X] 10 (for human variability)
Was a conversion factor used from ppm in food or water to a mg/body weight dose?
No
If an inhalation study in animals, list conversion factors used in determining human equivalent dose:
The blood:gas partition coefficient (H
b/g
) for the Sprague-Dawley rat and human are 19.4 and 8.94,
respectively. The ratio of the rat H
b/g
to the human H
b/g
is:
(H
b/g
)
A
/(H
b/g
)
H
= 19.4/8.94 = 2.2
Since the ratio is greater than 1, a value of 1.0 was used (EPA, 1994).
NOAEL
[HEC]
= NOAEL
[ADJ]
x (H
b/g
)
A
/(H
b/g
)
H

A-6 METHYLENE CHLORIDE
APPENDIX A
= 25 x 1.0
= 25 ppm
Was a conversion used from intermittent to continuous exposure? If so, explain:
No. Dosing was continuous for 90 days.
Other additional studies or pertinent information that lend support to this MRL: Haun et al. (1972) also
evaluated the effects of methylene chloride in mice, dogs, and monkeys. For all three species, exposure
was continuous for 14 weeks to 0, 25, or 100 ppm of methylene chloride.
In mice (20/group, sex and strain not reported), exposure to 100 ppm methylene chloride significantly
decreased the activity of liver cytochrome P-450 and decreased b
5
at 30, 60, and 90 days. Cytochromes b
5
and P-420 were also reduced at 30 days, but were elevated at 90 days. The 25 ppm exposure level had no
significant effect on liver cytochrome activities. Hexobarbital sleeping time measured at 30, 60, and
90 days was not significantly altered by exposure to methylene chloride. Although the authors indicate
that at the end of the exposure period, the animals were killed and subjected to gross and histopathologic
examination, the only result reported is that mice exposed to 25 ppm of methylene chloride showed no
pathologic changes, although the livers of the animals in the 100 ppm exposure group did show positive
fat stains.
Four monkeys per exposure level (sex and strain not reported) were exposed to methylene chloride.
Hematologic and clinical chemistry tests were conducted at various times during the experiment. Gross
and histopathologic examinations were done at the end of the study, but the scope was not indicated (it is
assumed that at least the liver and kidneys were examined). Significant but non-toxic elevations in
carboxyhemoglobin were seen throughout the study. Carboxyhemoglobin rose from about 0.5% in
controls to 1.7 and 4.5% in the low- and high-exposure groups, respectively. Hematology and clinical
chemistry values showed no significant differences from controls. The authors also reported that
exposure to methylene chloride did not cause any significant gross or histopathologic alterations.
The protocol used with the monkeys was also used with dogs. Sixteen dogs per exposure level were used
(sex and strain not reported). Hematology and clinical chemistry results showed no significant
differences from control dogs and there were no exposure-related gross or histopathologic alterations.
One additional study that reported effects at similar exposure levels as those as those used by Haun et al.
(1972) is that of Kjellstrand et al. (1986) who observed fatty infiltration and increased liver weight in
mice exposed continuously to 75 ppm of methylene chloride (the lowest level tested) for 90 days.

A-7 METHYLENE CHLORIDE
APPENDIX A
MINIMAL RISK LEVEL (MRL) WORKSHEET
Chemical name(s): Methylene Chloride
CAS number(s): 75-09-2
Date: July 28, 2000
Profile status: Draft 3 Post Public Comment
Route: [X] Inhalation [ ] Oral
Duration: [ ] Acute [ ] Intermediate [X] Chronic
Key to figure: 63r
Species: Rat
MRL: 0.3 [ ] mg/kg/day [X] ppm [ ] mg/m
3
Reference: Nitschke KD, Burek JD, Bell TJ, et al. 1988a. Methylene Chloride: A 2-year inhalation
toxicity and oncogenicity study in rats. Fundam Appl Toxicol 11:60-67.
Experimental design: The goal of the Nitchke et al. (1988a) study was to investigate the toxicity of
inhaled methylene chloride at lower concentrations than those in the other bioassays, in order to
determine a NOAEL for both toxicity and carcinogenicity. Exposure concentrations of 0, 50, 200, and
500 ppm of methylene chloride were selected for this bioassay. Groups of 90 male and 108 female
Sprague-Dawley rats were exposed to these concentrations for 6 hours/day, 5 days/week for 2 years. A
number of satellite groups were also exposed to assess the temporal relationship between methylene
chloride exposure and the expression of toxicity; subgroups of females in the main study were sacrificed
after 6, 12, 15, and 18 months of methylene chloride exposure. End points evaluated included: body
weight, food consumption rates, organ weights; hematology, clinical chemistry, urinalysis; pathology;
histopathology; and blood carboxyhemoglobin levels.
Effects noted in study and corresponding doses: No exposure-related gross or histopathologic changes
were observed in animals from interim sacrifice groups. At terminal sacrifice, the incidences of both
hepatocellular cytoplasmic vacuolization consistent with fatty changes and multinuceated hepatocytes
were statistically elevated in female rats exposed to 200 and 500 ppm of methylene chloride; a slight
increase in the incidence of hepatocellular vacuolization was also observed in male rats exposed to 500
ppm. Histopathological changes in the liver were not found in the male rats exposed to 50 or 200 ppm of
methylene chloride. No other pathologic or histopathologic nontumor findings were reported.
Dose and end point used for MRL derivation: Based on liver histopathology in female rats, a NOAEL of
50 ppm is used for MRL derivation.
[X] NOAEL [ ] LOAEL:
Uncertainty factors used in MRL derivation:
[ ] 1 [ ] 3 [ ] 10 (for use of a LOAEL)
[ ] 1 [X] 3 [ ] 10 (for extrapolation from animals to humans)
[ ] 1 [ ] 3 [X] 10 (for human variability)
Was a conversion factor used from ppm in food or water to a mg/body weight dose?
No
If an inhalation study in animals, list conversion factors used in determining human equivalent dose:

A-8 METHYLENE CHLORIDE
APPENDIX A
The blood:gas partition coefficient (H
b/g
) for the Sprague-Dawley rat and human are 19.4 and 8.94,
respectively. The ratio of the rat H
b/g
to the human H
b/g
is:
(H
b/g
)
A
/(H
b/g
)
H
= 19.4/8.94 = 2.2
Since the ratio is greater than 1, a value of 1.0 was used (EPA, 1994).
NOAEL
[HEC]
= NOAEL
[ADJ]
x (H
b/g
)
A
/(H
b/g
)
H
= 8.92 x 1.0
= 8.92 ppm
Was a conversion used from intermittent to continuous exposure? If so, explain:
Yes. The NOAEL was multiplied by 6/24 hours and 5/7 days.
NOAEL
[ADJ]
= 50 ppm x 6/24 x 5/7 = 8.92 ppm
Other additional studies or pertinent information that lend support to this MRL: The results of this study
are consistent with the body of data on methylene chloride toxicity and toxicokinetics. Studies by Burek
et al. (1984) and NTP (1986) demonstrate concordance and complementarity with the Nitschke et al.
(1988a) study. At doses of 500 ppm or higher, these other studies show a clear dose-response with liver
histopathology as the end point in rats. The findings of the Nitschke et al. (1988a) bioassay are also
supported by what is known about the toxicokinetics and mechanisms of action of methylene chloride
toxicity (and carcinogenicity).

A-9 METHYLENE CHLORIDE
APPENDIX A
MINIMAL RISK LEVEL (MRL) WORKSHEET
Chemical name(s): Methylene Chloride
CAS number(s): 75-09-2
Date: July 28, 2000
Profile status: Draft 3 Post Public Comment
Route: [ ] Inhalation [X] Oral
Duration: [X] Acute [ ] Intermediate [ ] Chronic
Key to figure: 6
Species: Human
MRL: 0.2 [X] mg/kg/day [ ] ppm [ ] mg/m
3
Reference: Reitz RH, Hays SM, and Gargas ML. 1997. Assessing Priority Data Needs for Methylene
Chloride with Physiologically-Based Pharmacokinetic Modeling. Halogenated Solvents Industry Alliance
(HSIA), prepared for ATSDR, using human volunteer data from Winneke (1974).
Experimental design: Reitz et al. (1997) modeled inhalation data from Winneke (1974) to predict the unit
concentration of methylene chloride in drinking water needed to produce a tissue-specific dose equivalent
to that produced by inhaling 300 ppm of methylene chloride. Volunteers were exposed via inhalation to
300-800 ppm of methylene chloride for approximately 4 hours and tested for neurobehavioral effects that
would reflect cortical function.
Reitz et al. (1997) modified the basic PBPK methylene chloride model developed by Andersen et al.
(1987), Reitz et al. (1988), and Andersen et al. (1992) in the following manner: (1) liver weights for
rodents were based on the actual organ weights of conrol laboratory animals which were sacrificed during
chronic toxicity studies at 6–18 months of age; (2) partition coefficients for methylene chloride derived
from in vitro experiments performed by Gargas et al. (1989), were used for liver, fat, muscle, and blood;
and (3) a brain compartment was added to the methylene chloride model so that central nervous system
effects could be assessed in female and male rodents and humans. Size of rodent brains, blood flow rates
to the brain, and partition coefficients for brain tissue were obtained from either published literature (e.g.,
Stott et al. 1983; Thomas 1975) or personal communication from the authors. The modified methylene
chloride model thus contained six tissue compartments: fat, muscle (slowly perfused tissue), rapidly
perfused tissue, liver, mammary tissue, and brain.
Effects noted in study and corresponding doses: Minimally adverse, dose-dependent effects in
neurobehavioral measurements were observed (visual critical flicker fusion frequency, auditory vigilance
performance, psychomotor tasks). Because similar exposures to 50–100 pm of carbon monoxide alone
did not produce these effects, the author (Winneke 1974) concluded that they were mediated by
methylene chloride directly and not by its oxidative metabolite carbon monoxide. A LOAEL of 300 ppm
from this study, based on decreased visual critical flicker fusion frequency, was used for the derivation of
the acute oral MRL. Auditory vigilance and psychomotor performance were only statistically decreased
at 500 and 800 ppm, respectively. Thus, critical flicker fusion frequency is the most sensitive end point
tested and can be considered to be a minimal effect.
Dose and end point used for MRL derivation: 16 mg/kg/day; route-to-route extrapolation. Reitz et al.
(1997) modeled the Winneke (1974) data to obtain the target organ (brain) concentrations of methylene
chloride associated with administered inhalation concentrations, and then calculate the human drinking
water concentrations (mg/L) that would result in the equivalent target organ-specific doses. Human
exposure patterns in the PBPK model simulated realistic human drinking water consumption patterns (i.e.,

A-10 METHYLENE CHLORIDE
APPENDIX A
consisting of bouts of drinking during the day, with and between meals, and little-to-no drinking during
the night). PBPK modeling predicted that peak concentrations of methylene chloride in the brain would
increase rapidly after each episode of drinking water consumption, and then drop sharply, to near-zero,
between bouts of dringing. Additionally, there would be no cumulative effects of repeated exposure.
For acute neurological effects, the associated dose measure was defined as the peak concentration of
methylene chloride in brain tissue (mg/L of brain tissue) of humans exposed to 300 ppm of methylene
chloride for 4 hours by inhalation. The modified PBPK model calculated that the administered inhalation
dose was equivalent to 3.95 mg of methylene chloride per liter of brain tissue. The equivalent
administered human concentration in drinking water that will produce the same neurological effects was
565 mg of methylene chloride/liter. Using a daily drinking water consumption value of 2 liters and an
average human body weight of 70 kg, the LOAEL was calculated to be 16 mg/kg/day.
[ ] NOAEL [X] LOAEL:
Uncertainty factors used in MRL derivation:
[ ] 1 [ ] 3 [X] 10 (for use of a LOAEL)
[ ] 1 [ ] 3 [ ] 10 (for extrapolation from animals to humans)
[ ] 1 [ ] 3 [X] 10 (for human variability)
Was a conversion factor used from ppm in food or water to a mg/body weight dose? If so, explain:
Yes. The PBPK model predicted a unit concentration in drinking water in mg/L. This value was
multiplied by 2 liters (default drinking water consumption rate) and divided by 70 kg (default human
body weight) to yield a mg/kg body weight dose.
Calculated LOAEL = unit concentration (mg/L) x water consumption (L) ÷ BW
= 565 mg/L x 2 L ÷ 70 kg
= 16 mg/kg/day
If an inhalation study in animals, list conversion factors used in determining human equivalent dose:
Was a conversion used from intermittent to continuous exposure? If so, explain:
No.
Other additional studies or pertinent information that lend support to this MRL: The PBPK model has
been validated.
A-11 METHYLENE CHLORIDE
APPENDIX A
MINIMAL RISK LEVEL (MRL) WORKSHEET
Chemical name(s): Methylene Chloride
CAS number(s): 75-09-2
Date: July 28, 2000
Profile status: Draft 3 Post Public Comment
Route: [ ] Inhalation [X] Oral
Duration: [ ] Acute [ ] Intermediate [X] Chronic
Key to figure: 11r
Species: Rat
MRL: 0.06 [X] mg/kg/day [ ] ppm [ ] mg/m
3
Reference: Serota D, Thakur, AK, Ulland BM, et al. 1986a. A two year drinking water study of
dichloromethane in rodents. I. Rats. Food Chem Toxicol 24:951-958.
Experimental design: Only one long-term bioassay has been conducted with methylene chloride (Serota et
al. 1986a, 1986b). Fischer-344 rats (85/sex/dose) and B6C3F
1
mice (50–200/sex/dose) were exposed to
methylene chloride in deionized drinking water at target concentrations aimed at exposing rats to 0, 5, 50,
125, or 250 mg/kg/day and mice to 0, 60, 125, 185, and 250 mg/kg/day for 104 weeks. Two untreated
control groups were run concurrently. The nominal mean doses were 0, 6, 55, 131, and 249 mg/kg/day. A
satellite group was exposed to nominal daily doses of 250 mg/kg/day for 78 weeks followed by a
24-week recovery period. Subgroups of animals were sacrificed at 26, 52, and 78 weeks in treated groups
and one control group. Body weights and food and water consumption rates were recorded weekly.
Ophthalmologic examinations were conducted prior to treatment and at termination of treatment.
Hematology, serum chemistry, and urinalysis assessments were done during interim sacrifices at 52 and
78 weeks. Organ weights were evaluated and all animals received a complete necropsy.
Effects noted in study and corresponding doses: At the two highest dose groups, the following findings
were observed in rats: decreased body weights and body weight gains in both sexes, with concomitant
decrease in water consumption throughout the study and in food consumption during the first 13 weeks,
and changes in hematology and serum chemistry, but not urinalysis parameters. No treatment-related
ophthalmologic effects were observed throughout the study. Gross pathological effects were
unremarkable. There were no histopathologic changes except in the liver. A dose-related, statistically
significant, positive trend in the incidences of hepatic foci, areas of cellular alterations, and fatty deposits
were observed in all dose groups, but the lowest occurred at both week 78 and week 104. After 24 weeks
of nontreatment, the fatty deposits in the recovery group decreased, but there was no change in the
incidence of cellular foci or areas of cellular alterations.
In mice (Serota et al. 1986b), the liver was the only identifiable target organ. There was a marginal
increase in the fatty content of the liver in the highest dose group, although the significance of this finding
was not clear.
Dose and end point used for MRL derivation: 6 mg/kg/day. Histopathology was only observed in the
liver; therefore, the liver is the critical target organ. Marginal liver changes were only observed in mice at
the highest dose level tested. Statistically significant cellular changes (hepatic foci, areas of cellular
alterations) were observed in all dose groups in the rat except for the lowest. Therefore, the lowest dose
in the rat study was identified as the NOAEL. Based on measured mean drinking water consumption
rates, this dose was calculated to be 6 mg/kg/day.

A-12 METHYLENE CHLORIDE
APPENDIX A
[X] NOAEL [ ] LOAEL:
Uncertainty factors used in MRL derivation:
[ ] 1 [ ] 3 [ ] 10 (for use of a LOAEL)
[ ] 1 [ ] 3 [X] 10 (for extrapolation from animals to humans)
[ ] 1 [ ] 3 [X] 10 (for human variability)
Was a conversion factor used from ppm in food or water to a mg/body weight dose? If so, explain:
No.
If an inhalation study in animals, list conversion factors used in determining human equivalent dose:
Was a conversion used from intermittent to continuous exposure? If so, explain:
No.
Other additional studies or pertinent information that lend support to this MRL: These data are consistent
with the large body of data on methylene chloride toxicity and toxicokinetics.

B-1 METHYLENE CHLORIDE
APPENDIX B
USER'S GUIDE
Chapter 1
Public Health Statement
This chapter of the profile is a health effects summary written in non-technical language. Its intended
audience is the general public especially people living in the vicinity of a hazardous waste site or
chemical release. If the Public Health Statement were removed from the rest of the document, it would
still communicate to the lay public essential information about the chemical.
The major headings in the Public Health Statement are useful to find specific topics of concern. The
topics are written in a question and answer format. The answer to each question includes a sentence that
will direct the reader to chapters in the profile that will provide more information on the given topic.
Chapter 2
Tables and Figures for Levels of Significant Exposure (LSE)
Tables (2-1, 2-2, and 2-3) and figures (2-1 and 2-2) are used to summarize health effects and illustrate
graphically levels of exposure associated with those effects. These levels cover health effects observed at
increasing dose concentrations and durations, differences in response by species, minimal risk levels
(MRLs) to humans for noncancer end points, and EPA's estimated range associated with an upper- bound
individual lifetime cancer risk of 1 in 10,000 to 1 in 10,000,000. Use the LSE tables and figures for a
quick review of the health effects and to locate data for a specific exposure scenario. The LSE tables and
figures should always be used in conjunction with the text. All entries in these tables and figures
represent studies that provide reliable, quantitative estimates of No-Observed-Adverse- Effect Levels
(NOAELs), Lowest-Observed-Adverse-Effect Levels (LOAELs), or Cancer Effect Levels (CELs).
The legends presented below demonstrate the application of these tables and figures. Representative
examples of LSE Table 2-1 and Figure 2-1 are shown. The numbers in the left column of the legends
correspond to the numbers in the example table and figure.
LEGEND
See LSE Table 2-1
(1) Route of Exposure One of the first considerations when reviewing the toxicity of a substance using
these tables and figures should be the relevant and appropriate route of exposure. When sufficient
data exists, three LSE tables and two LSE figures are presented in the document. The three LSE
tables present data on the three principal routes of exposure, i.e., inhalation, oral, and dermal (LSE
Table 2-1, 2-2, and 2-3, respectively). LSE figures are limited to the inhalation (LSE Figure 2-1)
and oral (LSE Figure 2-2) routes. Not all substances will have data on each route of exposure and
will not therefore have all five of the tables and figures.

B-2 METHYLENE CHLORIDE
APPENDIX B
(2) Exposure Period Three exposure periods - acute (less than 15 days), intermediate (15–364 days),
and chronic (365 days or more) are presented within each relevant route of exposure. In this
example, an inhalation study of intermediate exposure duration is reported. For quick reference to
health effects occurring from a known length of exposure, locate the applicable exposure period
within the LSE table and figure.
(3) Health Effect The major categories of health effects included in LSE tables and figures are death,
systemic, immunological, neurological, developmental, reproductive, and cancer. NOAELs and
LOAELs can be reported in the tables and figures for all effects but cancer. Systemic effects are
further defined in the "System" column of the LSE table (see key number 18).
(4) Key to Figure Each key number in the LSE table links study information to one or more data points
using the same key number in the corresponding LSE figure. In this example, the study represented
by key number 18 has been used to derive a NOAEL and a Less Serious LOAEL (also see the 2
"18r" data points in Figure 2-1).
(5) Species The test species, whether animal or human, are identified in this column. Section 2.5,
"Relevance to Public Health," covers the relevance of animal data to human toxicity and Section
2.3, "Toxicokinetics," contains any available information on comparative toxicokinetics. Although
NOAELs and LOAELs are species specific, the levels are extrapolated to equivalent human doses
to derive an MRL.
(6) Exposure Frequency/Duration The duration of the study and the weekly and daily exposure
regimen are provided in this column. This permits comparison of NOAELs and LOAELs from
different studies. In this case (key number 18), rats were exposed to 1,1,2,2-tetrachloroethane via
inhalation for 6 hours per day, 5 days per week, for 3 weeks. For a more complete review of the
dosing regimen refer to the appropriate sections of the text or the original reference paper, i.e.,
Nitschke et al. 1981.
(7) System This column further defines the systemic effects. These systems include: respiratory,
cardiovascular, gastrointestinal, hematological, musculoskeletal, hepatic, renal, and dermal/ocular.
"Other" refers to any systemic effect (e.g., a decrease in body weight) not covered in these systems.
In the example of key number 18, 1 systemic effect (respiratory) was investigated.
(8) NOAEL A No-Observed-Adverse-Effect Level (NOAEL) is the highest exposure level at which no
harmful effects were seen in the organ system studied. Key number 18 reports a NOAEL of 3 ppm
for the respiratory system which was used to derive an intermediate exposure, inhalation MRL of
0.005 ppm (see footnote "b").
(9) LOAEL A Lowest-Observed-Adverse-Effect Level (LOAEL) is the lowest dose used in the study
that caused a harmful health effect. LOAELs have been classified into "Less Serious" and
"Serious" effects. These distinctions help readers identify the levels of exposure at which adverse
health effects first appear and the gradation of effects with increasing dose. A brief description of
the specific endpoint used to quantify the adverse effect accompanies the LOAEL. The respiratory
effect reported in key number 18 (hyperplasia) is a Less serious LOAEL of 10 ppm. MRLs are not
derived from Serious LOAELs.
(10) Reference The complete reference citation is given in chapter 8 of the profile.
(11) CEL A Cancer Effect Level (CEL) is the lowest exposure level associated with the onset of
carcinogenesis in experimental or epidemiologic studies. CELs are always considered serious

B-3 METHYLENE CHLORIDE
APPENDIX B
effects. The LSE tables and figures do not contain NOAELs for cancer, but the text may report
doses not causing measurable cancer increases.
(12) Footnotes Explanations of abbreviations or reference notes for data in the LSE tables are found in
the footnotes. Footnote "b" indicates the NOAEL of 3 ppm in key number 18 was used to derive an
MRL of 0.005 ppm.
LEGEND
See Figure 2-1
LSE figures graphically illustrate the data presented in the corresponding LSE tables. Figures help the
reader quickly compare health effects according to exposure concentrations for particular exposure
periods.
(13) Exposure Period The same exposure periods appear as in the LSE table. In this example, health
effects observed within the intermediate and chronic exposure periods are illustrated.
(14) Health Effect These are the categories of health effects for which reliable quantitative data exists.
The same health effects appear in the LSE table.
(15) Levels of Exposure concentrations or doses for each health effect in the LSE tables are graphically
displayed in the LSE figures. Exposure concentration or dose is measured on the log scale "y" axis.
Inhalation exposure is reported in mg/m
3
or ppm and oral exposure is reported in mg/kg/day.
(16) NOAEL In this example, 18r NOAEL is the critical endpoint for which an intermediate inhalation
exposure MRL is based. As you can see from the LSE figure key, the open-circle symbol indicates
to a NOAEL for the test species-rat. The key number 18 corresponds to the entry in the LSE table.
The dashed descending arrow indicates the extrapolation from the exposure level of 3 ppm (see
entry 18 in the Table) to the MRL of 0.005 ppm (see footnote "b" in the LSE table).
(17) CEL Key number 38r is 1 of 3 studies for which Cancer Effect Levels were derived. The diamond
symbol refers to a Cancer Effect Level for the test species-mouse. The number 38 corresponds to
the entry in the LSE table.
(18) Estimated Upper-Bound Human Cancer Risk Levels This is the range associated with the
upper-bound for lifetime cancer risk of 1 in 10,000 to 1 in 10,000,000. These risk levels are derived
from the EPA's Human Health Assessment Group's upper-bound estimates of the slope of the
cancer dose response curve at low dose levels (q
1
*).
(19) Key to LSE Figure The Key explains the abbreviations and symbols used in the figure.
B-4
SAMPLE
1
6
TABLE 2-1. Levels of Significant Exposure to [Chemical x] – Inhalation
Key to
figure
a
Species
Exposure
frequency/
duration System
NOAEL
(ppm)
Less serious (ppm)
LOAEL (effect)
Serious (ppm)
Reference
2 6 INTERMEDIATE EXPOSURE
5 6 7 8 9 10
6 Systemic 9 9 9 9 9 9
6 18 Rat 13 wk Resp 3
b
10 (hyperplasia) Nitschke et al.
5d/wk 1981
6hr/d
3
4
CHRONIC EXPOSURE
11
Cancer 9
38 Rat 18 mo 20 (CEL, multiple Wong et al. 1982
5d/wk organs)
7hr/d
39 Rat 89–104 wk 10 (CEL, lung tumors, NTP 1982
5d/wk nasal tumors)
6hr/d
40 Mouse 79–103 wk 10 (CEL, lung tumors, NTP 1982
5d/wk hemangiosarcomas)
6hr/d
a
The number corresponds to entries in Figure 2-1.
6
b
Used to derive an intermediate inhalation
Minimal Risk Level (MRL) of 5 x 10
-3
ppm; dose adjusted for intermittent exposure and divided by
12
an uncertainty factor of 100 (10 for extrapolation from animal to humans, 10 for human variability).
METHYLENE CHLORIDE
APPENDIX B
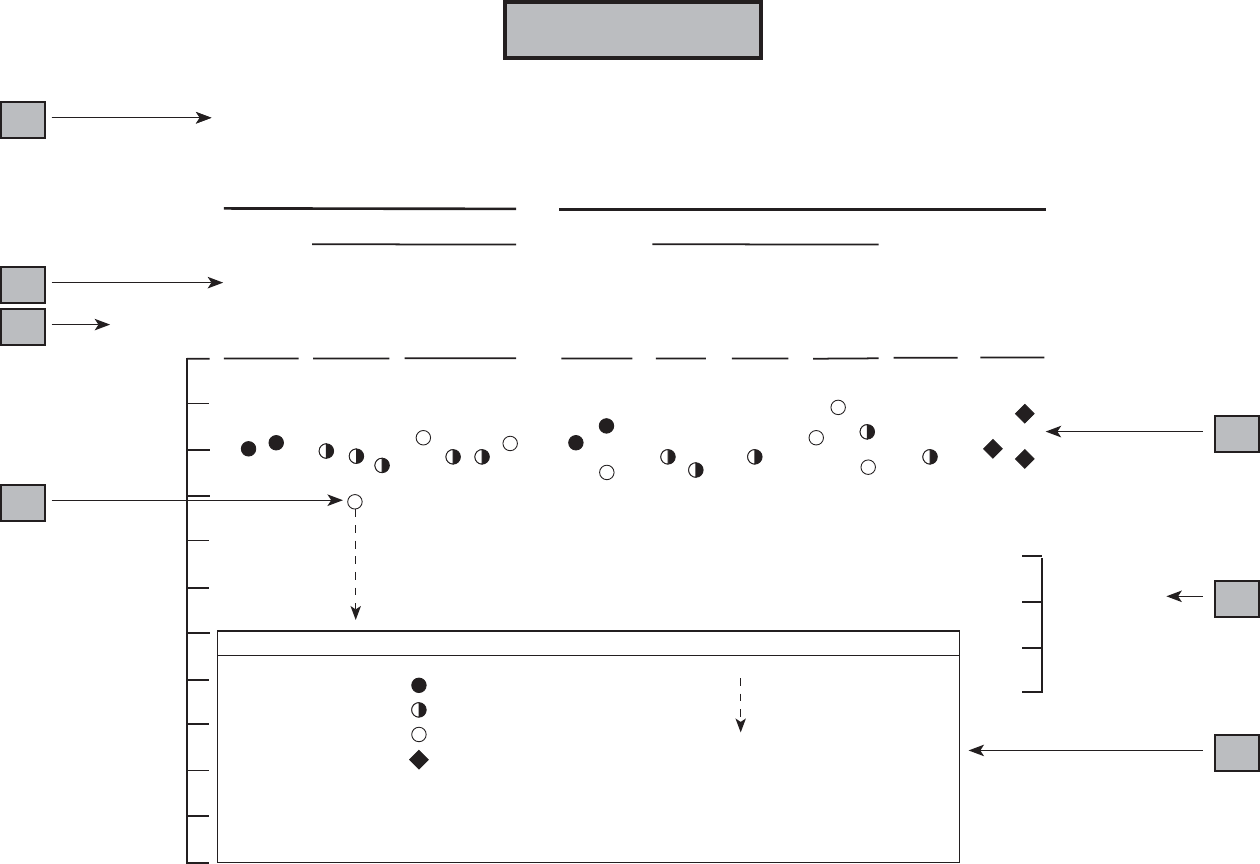
13
Death
Hematological
Respiratory
Death
Respiratory
Hematological
Hepatic
Reproductive
Cancer*
13
Figure 2-1. Levels of Significant Exposure to [Chemical X] – Inhalation
Acute
Intermediate
(≤14 days)
(15-364 days)
14
15
(ppm)
10000
1000
100
10
16
1
0.1
0.01
0.001
0.0001
0.00001
0.000001
0.0000001
Systemic Systemic
37m
30r
34r
20m 35m
17r
20m
31r
16r
24g
18r
22g 21r
28m 29r 27r
18r
22m
34r
33r
18r
* Doses represent the lowest dose tested per study that produced a tumorigenic response and do not imply
the existence of a threshold for the cancer end point.
Key
r Rat
m Mouse
h Rabbit
g Guinea Pig
k Monkey
The number next to each point
corresponds to entries in the
accompanying table.
Minimal risk level for effects
other than cancer
LOAEL for serious effects (animals)
LOAEL for less serious effects (animals)
NOAEL (animals)
CEL - Cancer Effect Level
38r
40m
39r
10
-4
10
-5
10
-6
10
-7
17
Estimated
18
Upper Bound
Human Cancer
Risk Levels
19
B-5 METHYLENE CHLORIDE
APPENDIX B
SAMPLE
B-6 METHYLENE CHLORIDE
APPENDIX B
Chapter 2 (Section 2.5)
Relevance to Public Health
The Relevance to Public Health section provides a health effects summary based on evaluations of
existing toxicologic, epidemiologic, and toxicokinetic information. This summary is designed to present
interpretive, weight-of-evidence discussions for human health end points by addressing the following
questions.
1. What effects are known to occur in humans?
2. What effects observed in animals are likely to be of concern to humans?
3. What exposure conditions are likely to be of concern to humans, especially around hazardous
waste sites?
The section covers end points in the same order they appear within the Discussion of Health Effects by
Route of Exposure section, by route (inhalation, oral, dermal) and within route by effect. Human data are
presented first, then animal data. Both are organized by duration (acute, intermediate, chronic). In vitro
data and data from parenteral routes (intramuscular, intravenous, subcutaneous, etc.) are also considered
in this section. If data are located in the scientific literature, a table of genotoxicity information is
included.
The carcinogenic potential of the profiled substance is qualitatively evaluated, when appropriate, using
existing toxicokinetic, genotoxic, and carcinogenic data. ATSDR does not currently assess cancer
potency or perform cancer risk assessments. Minimal risk levels (MRLs) for noncancer end points (if
derived) and the end points from which they were derived are indicated and discussed.
Limitations to existing scientific literature that prevent a satisfactory evaluation of the relevance to public
health are identified in the Data Needs section.
Interpretation of Minimal Risk Levels
Where sufficient toxicologic information is available, we have derived minimal risk levels (MRLs) for
inhalation and oral routes of entry at each duration of exposure (acute, intermediate, and chronic). These
MRLs are not meant to support regulatory action; but to acquaint health professionals with exposure
levels at which adverse health effects are not expected to occur in humans. They should help physicians
and public health officials determine the safety of a community living near a chemical emission, given the
concentration of a contaminant in air or the estimated daily dose in water. MRLs are based largely on
toxicological studies in animals and on reports of human occupational exposure.
MRL users should be familiar with the toxicologic information on which the number is based. Chapter
2.5, "Relevance to Public Health," contains basic information known about the substance. Other sections
such as 2.7, "Interactions with Other Substances,” and 2.8, "Populations that are Unusually Susceptible"
provide important supplemental information.
MRL users should also understand the MRL derivation methodology. MRLs are derived using a
modified version of the risk assessment methodology the Environmental Protection Agency (EPA)
provides (Barnes and Dourson 1988) to determine reference doses for lifetime exposure (RfDs).
B-7 METHYLENE CHLORIDE
APPENDIX B
To derive an MRL, ATSDR generally selects the most sensitive endpoint which, in its best judgement,
represents the most sensitive human health effect for a given exposure route and duration. ATSDR
cannot make this judgement or derive an MRL unless information (quantitative or qualitative) is available
for all potential systemic, neurological, and developmental effects. If this information and reliable
quantitative data on the chosen endpoint are available, ATSDR derives an MRL using the most sensitive
species (when information from multiple species is available) with the highest NOAEL that does not
exceed any adverse effect levels. When a NOAEL is not available, a lowest-observed-adverse-effect
level (LOAEL) can be used to derive an MRL, and an uncertainty factor (UF) of 10 must be employed.
Additional uncertainty factors of 10 must be used both for human variability to protect sensitive
subpopulations (people who are most susceptible to the health effects caused by the substance) and for
interspecies variability (extrapolation from animals to humans). In deriving an MRL, these individual
uncertainty factors are multiplied together. The product is then divided into the inhalation concentration
or oral dosage selected from the study. Uncertainty factors used in developing a substance-specific MRL
are provided in the footnotes of the LSE Tables.
C-1 METHYLENE CHLORIDE
APPENDIX C
ACRONYMS, ABBREVIATIONS, AND SYMBOLS
ACGIH American Conference of Governmental Industrial Hygienists
ADME Absorption, Distribution, Metabolism, and Excretion
atm atmosphere
ATSDR Agency for Toxic Substances and Disease Registry
BCF bioconcentration factor
BSC Board of Scientific Counselors
C Centigrade
CDC Centers for Disease Control
CEL Cancer Effect Level
CERCLA Comprehensive Environmental Response, Compensation, and Liability Act
CFR Code of Federal Regulations
CHO/HPRT Chinese Hamster ovary assay involving the hypoxanthine guanine phosphoribosyl
transferase gene
CI confidence interval
CLP Contract Laboratory Program
cm centimeter
CNS central nervous system
COHb carboxyhemoglobin
d day
DHEW Department of Health, Education, and Welfare
DHHS Department of Health and Human Services
DOL Department of Labor
ECG electrocardiogram
EEG electroencephalogram
EPA Environmental Protection Agency
EKG see ECG
F Fahrenheit
F
1
first filial generation
FAO Food and Agricultural Organization of the United Nations
FEMA Federal Emergency Management Agency
FIFRA Federal Insecticide, Fungicide, and Rodenticide Act
fpm feet per minute
ft foot
FR Federal Register
g gram
GC gas chromatography
gen generation
HPLC high-performance liquid chromatography
hr hour
IDLH Immediately Dangerous to Life and Health
IARC International Agency for Research on Cancer
ILO International Labor Organization
in inch
Kd adsorption ratio
kg kilogram
C-2 METHYLENE CHLORIDE
APPENDIX C
kkg metric ton
K
oc
organic carbon partition coefficient
K
ow
octanol-water partition coefficient
L liter
LC liquid chromatography
LC
Lo
lethal concentration, low
LC
50
lethal concentration, 50% kill
LD
Lo
lethal dose, low
LD
50
lethal dose, 50% kill
LOAEL lowest-observed-adverse-effect level
LSE Levels of Significant Exposure
m meter
mg milligram
min minute
mL milliliter
mm millimeter
mmHg millimeters of mercury
mmol millimole
mo month
mppcf millions of particles per cubic foot
MRL Minimal Risk Level
MS mass spectrometry
NIEHS National Institute of Environmental Health Sciences
NIOSH National Institute for Occupational Safety and Health
NIOSHTIC NIOSH's Computerized Information Retrieval System
ng nanogram
nm nanometer
NHANES National Health and Nutrition Examination Survey
nmol nanomole
NOAEL no-observed-adverse-effect level
NOES National Occupational Exposure Survey
NOHS National Occupational Hazard Survey
NPL National Priorities List
NRC National Research Council
NTIS National Technical Information Service
NTP National Toxicology Program
OR odds ratio
OSHA Occupational Safety and Health Administration
PBPK physiologically-based pharmacokinetic
PEL permissible exposure limit
pg picogram
pmol picomole
PHS Public Health Service
PMR proportionate mortality ratio
ppb parts per billion
ppm parts per million
ppt parts per trillion
REL recommended exposure limit
RfD Reference Dose
RTECS Registry of Toxic Effects of Chemical Substances
sec second

C-3 METHYLENE CHLORIDE
APPENDIX C
SCE sister chromatid exchange
SIC Standard Industrial Classification
SMR standard mortality ratio
STEL short term exposure limit
STORET STORAGE and RETRIEVAL
TLV threshold limit value
TSCA Toxic Substances Control Act
TRI Toxics Release Inventory
TWA time-weighted average
U.S. United States
UF uncertainty factor
yr year
WHO World Health Organization
wk week
> greater than
> greater than or equal to
= equal to
< less than
< less than or equal to
% percent
α alpha
β beta
δ delta
γ gamma
µm micrometer
µg microgram
